

Abstract
The broad study of histone deacetylases in chemistry, biology and medicine relies on tool compounds to derive mechanistic insights. A phylogenetic analysis of Class I and II HDACs as targets of a comprehensive, structurally diverse panel of inhibitors revealed unexpected isoform selectivity even among compounds widely perceived as non-selective. The synthesis and study of a focused library of cinnamic hydroxamates allowed the identification of a first non-selective HDAC inhibitor. These data will guide a more informed use of HDAC inhibitors as chemical probes and therapeutic agents.
Histone deacetylases (HDACs) regulate diverse cellular processes by modulating protein structure and function. Lysine acetylation is reversibly mediated by HDACs and acetyl transferases, establishing a dynamic post-translational modification of broad relevance to cell signaling and cell state1. As components of chromatin-modifying enzyme complexes, HDACs target the amino-terminal tails of histone proteins affecting chromatin conformation and gene-specific transcription1,2. Recent research has identified a significant number of non-histone protein substrates, extending the mechanistic relevance and research interest in HDACs well beyond the field of chromatin biology3.
The common classification of human deacetylases is based on molecular phylogenetic analysis of primary structure, subsequently grouped based on homology to yeast enzymes4. This approach yields four distinct classes that vary in size and function. Class I (HDAC1, 2, 3 and 8), Class IIa (HDAC4, 5, 7 and 9), Class IIb (HDAC6 and 10) and Class IV (HDAC11) HDACs contain predicted zinc-dependent deacetylase domains4. The Class III proteins form a structurally and mechanistically distinct class of NAD+-dependent hydrolases (Sirtuins; Sirt1-7)5.
Studies of human deacetylases have benefitted from the availability of small-molecule HDAC inhibitors (HDACi), most of which as a group obey a common “cap-linker-chelator” pharmacophore model6. The chelator refers to a suitable metal-binding biasing element (e.g. hydroxamate, o-aminoanilide) that engages zinc in the enzyme active site. A linker mimicking the lysine side chain spans the narrow channel of HDAC enzymes, connecting the zinc-binding feature to a simple aromatic or macrocyclic cap. The capping feature interacts with the rim-region of the active site cavity and establishes the appropriate three-dimensional conformation for presentation of the chelator7. Both chelator and cap features have been shown to convey target potency and isoform selectivity.
The remarkable demonstration of pro-differentiation and anti-proliferative effects in cancer model systems prompted the further development of natural product and tool HDACi into investigational agents for therapeutic use in humans. To date, two pharmaceutical HDAC inhibitors have been approved for use in humans [SAHA 1 (vorinostat), Merck Research Laboratories and FK-228 2 (romidepsin), Gloucester Pharmaceuticals], and more than ten additional compounds are in advanced clinical testing1,8. As such, there is considerable interest in HDACi as tool compounds for cellular biology and as therapeutic agents for the treatment of cancer, inflammatory conditions and infectious diseases.
Widely maintained is the perception that many of the currently used small-molecule inhibitors are non-selective8. Recent research has revealed unique aspects of Class IIa HDAC biochemistry, which calls into question the accuracy of prior homogeneous assays for reporting target potency9. This is becoming more problematic as the mechanistic understanding of Class IIa HDACs is expanding, enhanced by the availability of genetic probes of protein function such as silencing reagents and knock-out mice10-16. Key regulatory roles have been suggested in immune tolerance, cardiac remodeling and neuronal death. We therefore endeavored to derive a more complete knowledge of isoform-specific activity so as to guide a more thoughtful use of these compounds as chemical probes of deacetylase function in both the research and clinical setting.
RESULTS
Comparative study of HDAC inhibitors
Recently, we have reported the optimization of a miniaturized kinetic assay for the biochemical profiling of HDAC1, 2, 3, 6 and 8 17. However, implementation of this assay for Class IIa HDACs proved challenging due to the low catalytic turnover of the acetylated tripeptide substrate 3 as well as a recently reported Class IIa-specific substrate 4 (Fig 1a), both of which require a prohibitively large amount of enzyme9,18,19. During assay development, we observed diminished turnover by Class I HDACs of Boc-functionalized substrate 5 compared to tripeptide substrate 3 (Supplementary Fig 1)18. We therefore devised a new tripeptide substrate 6, which, like substrate 4, features the relatively labile and sterically more demanding trifluoroacetyl group that is readily hydrolyzed by the catalytically less avid Class IIa HDACs (Fig 1a,b). With substrate 6, Class IIa HDACs exhibit markedly faster kinetics, further reducing the requisite enzyme concentration (0.002-0.03 ng/μL; Supplementary Fig 2) and allowing a first, precise profiling of HDACi against HDAC1-9. Enzymatic activity of HDAC10 and HDAC11 could not be determined with these or other prepared substrates (data not shown).
Figure 1.
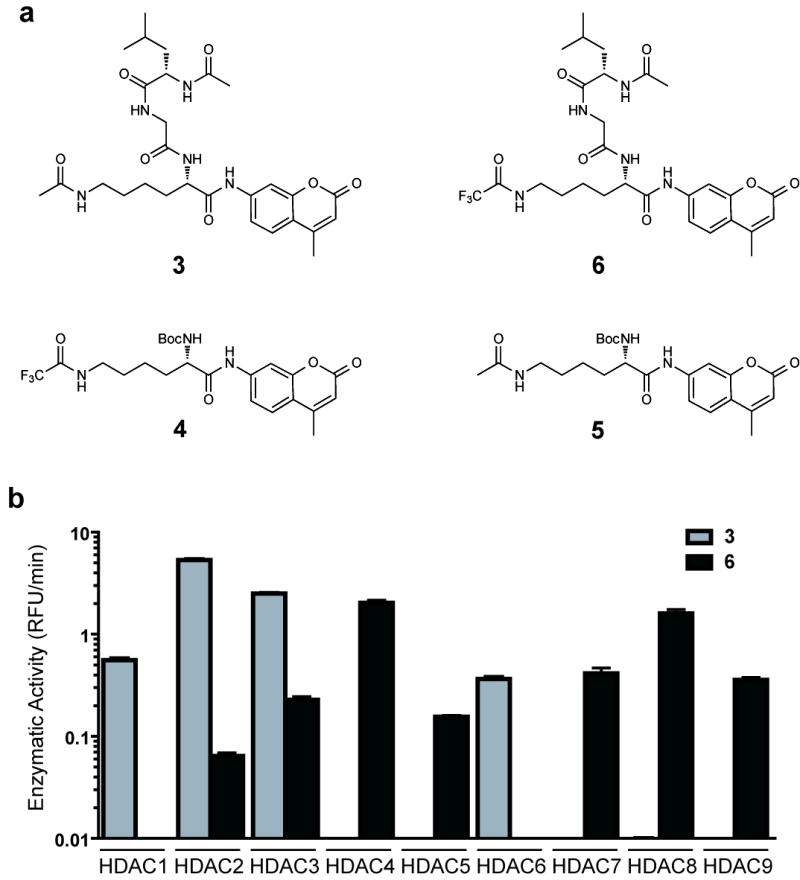
Development of a platform for biochemical profiling of human deacetylases. (a) Chemical structure of substrates 3 - 6. (b) Comparative enzymatic activity of HDAC1-9 with tripeptide substrate 3 and trifluoro acetyl-lysine tripeptide substrate 6, studied at equivalent substrate concentration (10 μM). Data represent mean values of three measurements ± s.d. Substrate 6 allows miniaturized, kinetic study of HDAC4, 5, 7, 8 and 9.
Collaboratively, we synthesized and assembled a panel of structurally-diverse small-molecule HDACi 1, 2, 7-20 that comprise most of the relevant literature-reported tool compounds and pharmaceutically developed clinical candidates (Supplementary Fig 3)1,8. We next conducted a high-throughput, precise profiling of HDACi potency against all Class I and II enzymes, in a miniaturized dose-ranging format (Supplementary Table 1).
A chemical genetic phylogeny of human histone deacetylases
Using statistical methods validated for assessing evolutionary relatedness, we constructed a chemical genetic phylogeny of deacetylases derived from these kinetic data (Fig 2a). This approach was selected to prompt inferences into biochemical, pharmacologic and structural relationships. The analysis revealed a number of unexpected findings. First and foremost, the Class IIa enzymes are not targeted by most HDACi at pharmacologically-relevant concentrations. None of the inhibitors tested demonstrated a preference for Class IIa enzymes. In fact, significant inhibitory activity was observed only several orders of magnitude above the Ki for Class I/IIb enzymes. Consequently, none of the inhibitors tested is suitable for use as a tool compound to study Class IIa enzymes in settings where Class I/IIb enzymes are functionally present (i.e. in cells). Indeed, this class of structurally diverse probes exhibits high functional redundancy for inhibition of HDAC1, HDAC2 and HDAC3. Data for a representative benzamide (MS-275 19) and hydroxamate (SAHA 1) are presented schematically in Fig 2b, c.
Figure 2.

Chemical phylogenetic analysis of HDACs identifies unexpected selectivity of HDAC inhibitors. (a) Hierarchical clustering of HDACs and a representative panel of structurally-diverse HDAC inhibitor tool and investigational compounds 1, 2, 7-20 weighted by inhibitory potency (Ki). Complete quantitative data are shown in Supplementary Table 1. Data based on mean values of triplicate measurements. (b,c) Chemical structure and enzymatic selectivity profile of (b) MS-275 19 and (c) SAHA 1, overlaying molecular phylogeny. HDAC dendrograms are adapted from Supplementary Figure 4. Circles are proportionate in size to Ki on a logarithmic scale, as shown.
Interrogating the bidirectional hierarchical clustering of small molecules and proteins, remarkable chemotype-deacetylase relationships emerge. Driving the striking clustering of HDACi are principally the linker-chelator motifs, as most clearly observed with the benzamide based inhibitors (i.e. o-aminoanilides MS-275 19, CI-994 20 and MGDC-0103 18, see Supplementary Information for structures). In the second dimension, a provocative correlation was observed when comparing this chemical phylogeny to the molecular phylogeny of HDAC1-11 (Supplementary Fig 4). In general, HDACs with known, high sequence identity exhibit correspondingly high degrees of relatedness in both analyses. Yet pharmacology appears to defy phylogeny for HDAC6 and HDAC8, between which class assignments are reversed. Here, the inhibitor sensitivity emulates the substrate preferences, as for all deacetylases studied (Fig 1b), rendering 6 also the preferred reagent for biochemical studies of HDAC8.
Inhibition of Class IIa deacetylases by acetylated lysine
Substrate 3 shows no appreciable turn-over with human Class IIa deacetylases, despite possessing the acetylated lysine feature present in post-translationally modified proteins. Having established a capacity to elicit sensitive measurements of Class IIa enzymatic activity using the non-natural trifluoroacetyl substrate 6, we asked whether substrate 3 and the related substrate 21 might inhibit HDAC4, 5, 7 and 9. Remarkably, dose-ranging studies of substrates 3 and 21 demonstrated potent inhibitory activity against Class IIa enzymes (IC50 0.6-2.3 μM; Fig 3). These results demonstrate that substrates 3 and 6 exhibit comparable affinity for Class IIa enzymes, suggesting that the trifluoroacetyl group is not a critical determinant of binding affinity. Additionally, Class IIa enzymes appear to bind ε–N-acetyl lysine peptides with comparable affinity to Class I and IIb HDACs, yet Class IIa enzymes fail to deacetylate acetyl-lysine in this context. These data importantly establish the plausibility that Class IIa deacetylase domains function as acetyl-lysine binding domains, rather than functional deacetylases.
Figure 3.

Inhibition of Class IIa HDACs by acetylated lysine based substrates. Inhibition of trifluoroacetyl-lysine substrate 6 processing by (a) acetyl-lysine substrate 3 (b) acetyl-lysine substrate 21 is presented in dose-response format for Class IIa HDACs. Data represent mean values of triplicate measurements ± s.d. IC50 curves were fit by logistic regression.
Discovery of a first non-selective HDAC inhibitor
Broad inhibitors of enzyme classes are of great use as chemical probes for cell biology, biochemistry and proteomics. The surprising observation that non-selective HDAC inhibitors do not exist prompted the desire to identify such an agent. As suggested by the chemical phylogenetics, the central clustering of cinnamic hydroxamates identifies this pharmacophore as most leveraged for non-selectivity. We and others have observed dramatic contributions to ligand potency and selectivity by the structure and conformation of HDACi capping features7,20. Thus, we endeavored to expand a library of capped cinnamic hydroxamic acids, based on a high-throughput, parallel synthesis scheme we have used previously with success in targeting individual HDACs 21,22. This approach involves the clean and efficient condensation of a hydrazide-based linker-chelator feature with a diverse collection of aldehydes to readily explore the chemical space of the capping group. Meta- and para-substituted hydrazide-functionalized cinnamic hydroxamic acids were prepared (see Synthetic Procedures in Supplementary Information) and condensed with a set of 160 aliphatic and aromatic aldehydes to yield a HDAC-biased library of 320 compounds (Fig 4a).
Figure 4.

Synthesis and testing of an HDAC-biased chemical library and identification of a non-selective HDAC inhibitor. (a) Library design of meta- and para-substituted hydroxamic acid HDAC inhibitors, utilizing parallel condensation of aldehydes, efficiently samples chemical diversity at the capping feature. (b) Biochemical profiling data for the para-substituted sub-library (n=160 compounds), presented in dose-response format for inhibition of HDAC5. Structural variation in the capping feature was observed to confer a broad range of potency, as illustrated with the most (IC50= 18 nM) and least (IC50 = 55 μM) potent small molecules tested. (c) Comparative biochemical profiling of meta- (red) and para-substituted (blue) sub-libraries for relative inhibition of HDAC2 and HDAC3. The complete library was studied and is displayed at a range of concentrations (0.03, 0.3, 3.0 and 30.0 μM). Compounds of this structural class do not discriminate between HDAC2 and HDAC3. (d) Comparative biochemical profiling of meta- (red) and para-substituted (blue) sub-libraries for relative inhibition of HDAC5 and HDAC7. The complete library was studied and is displayed at a range of concentrations (0.03, 0.3, 3.0 and 30.0 μM). Para-substituted cinnamic hydroxamic acids exhibit increased potency for HDAC5, relative to meta-substituted regioisomers. (e) Specificity profile of pandacostat 22 overlaying molecular phylogeny. HDAC dendrograms are adapted from Supplementary Figure 4. Circles are proportionate in size to Ki on a logarithmic scale, as shown. (f) Immunoblot of Jurkat cells treated with pandacostat for 24 hours and stained for acetylated histones (AcH3K18), acetylated alpha-tubulin (AcTub) or GAPDH. (g) Chemical structure of pandacostat 22.
The entire library was profiled against Class I and IIa HDACs in dose-ranging format to provide a richly annotated data set. The capping feature was confirmed to confer a dramatic effect on target potency, as shown in Figure 4b. Pair-wise comparison of potency for individual deacetylases revealed a substantial impact of linker substitution and geometry on target selectivity, particularly evident between HDAC5 and other Class IIa enzymes (Fig 4c,d; Supplementary Fig 5). Based on these profiling data, we selected four compounds with high potency against Class IIa HDACs relative to Class I inhibition. These compounds were resynthesized on 30 mg scale, purified by reversed phase HPLC and assayed in dense dose-response format for the accurate determination of potency and selectivity.
One compound was identified, which uniformly inhibited all profiled HDAC isoforms, in contrast to control compounds MS-275 19, SAHA 1 and trichostatin A 8 (Fig 4e; Supplementary Fig 6, 7). We term this compound pandacostat 22 (Fig 4g). Assessment of cellular permeability and non-selectivity were assessed by immunoblotting for changes in protein acetylation in treated cancer cell lines. Indeed, pandacostat confers hyperacetylation of Class I deacetylase targets (bulk histones) and the prominent HDAC6 target α-tubulin in a time- and dose-dependent manner (Fig 4f). Both biomarkers unambiguously demonstrate cytosolic and nuclear activity of Pandacostat. Intracellular on-target activity for Class IIa HDACs cannot as of yet be probed due the lack of bona fide markers for these isoforms.
DISCUSSION
Lysine acetylation has emerged as a regulatory mechanism for diverse cellular processes in developmental and disease biology. Recent global proteomic analyses have identified more than 1700 protein substrates of histone deacetylases3. Ongoing, detailed, mechanistic studies of substrate protein structure and function require well-annotated small molecule probes. Here, we report an effort to characterize the target selectivity profile of pharmaceutical and tool HDAC inhibitors. The initial motivation for this research was to ascertain subtle differences in enzyme potency which might avail research or therapeutic opportunities. Given the extensive characterization of HDAC inhibitors by academic scientists and pharmaceutical manufacturers, we were surprised to observe unexpected selectivity and significant target redundancy among this class of structurally-diverse compounds.
Most striking was the general lack of inhibitory activity against Class IIa enzymes. The lack of potency of ortho-aminoanilides for Class IIa HDACs was not surprising based on prior studies of HDAC6 and HDAC8, which suggested extraordinary selectivity for HDAC1, 2 and 3. However, the overall lack of potency of hydroxamic acid-based inhibitors was highly unexpected. We interpret this observation based on the available crystal structures of HDAC4 (2VQM) and HDAC7 (3C0Z, 3C10) bound to hydroxamate inhibitors. None of the ligand-protein complexes shows the expected bidentate chelation geometry of the central zinc cation, as observed in the structures of ligand-bound human HDAC8 (1T64, 1T69) and bacterial homologs (e.g. 1ZZ1). According to DFT calculations, the tight bidentate complexation is a result of the deprotonation of the hydroxamic acid upon ligand binding23. The observed geometry in the published structures, however, is more in line with weaker monodentate binding mode of the neutral form of the hydroxamic acid (Supplementary Fig 8)23.
Common to all Class IIa HDACs is the substitution of a tyrosine residue in the active site, which is conserved in Class I enzymes, as a histidine. Furthermore, it has been shown for HDAC4 and HDAC7 that the mutation of the respective histidine to tyrosine markedly increases the biochemical activity of both enzymes19,24,25. Interestingly, in the Class I structures, this tyrosine forms a hydrogen bond to the hydroxamic acid carbonyl, which will increase binding affinity through hydrogen bond formation and as we speculate, sufficiently lower the pKa of the bound chelator facilitating deprotonation and consequently tighter binding. Consistent with this model is the 100-fold increased affinity observed with the hydroxamate LAQ-824 for the H976Y HDAC4 gain of function mutant 9,24,25. These observations may explain, in part, the differential potency of hydroxamate-based HDAC inhibitors and provide useful guidance for Class IIa-selective inhibitor design.
The data presented herein underscore a particularly pressing need for Class IIa-selective inhibitors. Reports in the literature have identified putative Class IIa-selective chemotypes, though many of these data must be revisited with the new knowledge of Class IIa enzyme biochemistry. In this manner, substrate 6 may prove a useful research tool for high-throughput screening and follow-up chemistry. In addition, highly potent, selective inhibitors of HDAC6 are equally underrepresented in this set of chemical probes. Clinically, the apparent redundancy of pharmaceutical compounds for HDAC1, HDAC2 and HDAC3 is troubling, but resonates with the class-associated toxicities shared commonly by structurally dissimilar compounds. A historical explanation likely accounts for this redundancy. Current pharmaceutical HDAC inhibitors matured in anti-cancer medicinal chemistry programs, where driving biomarkers of histone hyperacetylation and single-agent cytotoxicity evolved lead candidates for clinical development. Consequently, the Class I targets linked to bulk chromatin acetylation and transcriptional networks are over-represented in the target profile of these agents.
From an evolutionary perspective, Class IIa HDACs predate Class I HDACs and histone proteins4, thus establishing the rationale that Class IIa enzymes process cytosolic protein or non-protein substrates9,19. Our data suggest another plausible function of Class IIa enzymes. Given the observed enzymatic inhibition of the Class IIa enzymes by the simple acetyl-lysine tripeptide 3, we propose that a major function of Class IIa HDACs may be the recognition of acetyl-lysine in a sequence-dependent context. That is, Class IIa HDACs may function as receptors rather than enzymes, in some settings. The dissociation constant of substrate 3 for Class IIa HDACs is 3.7-12 μM, which is within the range of binding affinities reported for bromodomains (10-100uM)26. Like Class IIa deacetylases, bromodomains are found in complex with chromatin-modifying enzymes and regulate transcription. This preliminary observation is the focus of ongoing research in our laboratories.
We present, for the first time, the kinetic study of the biochemically active HDACs and a comprehensive library of tool and pharmaceutical deacetylase inhibitors. These data are derived from robust assays and a novel substrate, which allow for the rapid and efficient study of Class IIa HDACs. Our studies have revealed the unexpected selectivity of previously perceived “non-selective” HDAC inhibitors. From literature-reported crystallographic data and ab initio calculations, we provide a rationale for the diminished potency that will guide future ligand development for Class IIa HDACs. Recognizing the broad, potential utility of a non-selective HDACi, we synthesized a library of Class IIa-biased inhibitors and identified the first pan-HDACi reported, to date with activity in biochemical and cellular assay systems. This tool compound is expected to have great utility to the research community. In studying the chemical phylogenetics of HDACs, we illustrate how a structurally-diverse set of small-molecule probes may be used for the functional classification of a protein family.
METHODS
Biochemical HDAC assay
The inhibitory effect of compounds on HDAC1-9 function was determined in vitro using an optimized homogenous assay performed in 384-well plate format. In this assay, recombinant, full-length HDAC protein (BPS Biosciences) was incubated with fluorophore conjugated substrates 3 and 6 at a concentration equivalent to the substrate Km (3; 6 μM for HDAC1, 3 μM for HDAC2, 6 μM for HDAC3 and 16 μM for HDAC6; concentrations of 6 for HDAC4, 5, 7, 8, 9 are provided in Supplementary Fig 2f). Reactions were performed in assay buffer (50 mM HEPES, 100 mM KCl, 0.001% Tween-20, 0.05% BSA, 200 μM TCEP, pH 7.4) and followed for fluorogenic release of 7-amino-4-methylcoumarin from substrate upon deacetylase and trypsin enzymatic activity. Fluorescence measurements were obtained approximately every five minutes using a multilabel plate reader and plate-stacker (Envision; Perkin-Elmer). Data were analyzed on a plate-by-plate basis for the linear range of fluorescence over time. The first derivative of data obtained from the plate capture corresponding to the mid-linear range was imported into analytical software (Spotfire DecisionSite and GraphPad Prism). Replicate experimental data from incubations with inhibitor were normalized to controls.
Immunoblotting
Human T-cells (Jurkat) were incubated with compound (as indicated, DMSO < 0.2%) for 24 hours. Clarified cellular lysates were prepared from washed cell pellets and protein content was quantified using the Bradford dye assay. Standardized amounts (5.5 μg sample) were loaded into wells and resolved by denaturing gel electrophoresis. Following membrane transfer, immunoblots were prepared using commercially-available primary antibodies recognizing acetyl-tubulin (T7451; Sigma), acetyl-H3K18 (#9675, Cell Signaling Technology) and GAPDH (#14C10; Cell Signaling Technology). Chemiluminescent detection was performed with appropriate secondary antibodies: anti-rabbit IgG conjugated to horseradish peroxidase (#NA9340; ECL) and anti-mouse IgG conjugated to horseradish peroxidase (#170-6516; BIO RAD).
Statistical methods
Biochemical inhibition of HDAC enzymes by small-molecule inhibitors was measured as described above. Data were analyzed by logistic regression with determination of IC50 and standard deviation (Spotfire DecisionSite and GraphPad Prism). Calculation of Ki was determined using a derivation of the standard formula Ki = [Inhibitor]/((V0/Vi)*(1+S/Km))-[Substrate]/Km)-1. Bidirectional hierarchical clustering was performed on biochemical profiling data (Ki) for each HDAC1-9 by generating a pairwise distance matrix using the unweighted pair group method with arithmetic mean and a Euclidean distance similarity measure (Spotfire DecisionSite).
Phylogenetic analysis
Amino acid sequences for full length human histone deacetylases were obtained from the NHLBI (HDAC1 Accession No. Q13547; HDAC2 Accession No. Q92769; HDAC3 Accession No. O15379; HDAC4 Accession No. P56524; HDAC5 Accession No. Q9UQL6; HDAC6 Accession No. Q9UBN7; HDAC7 Accession No. Q8WUI4; HDAC8 Accession No. Q9BY41; HDAC9 Accession No. Q9BY41, HDAC10 Accession No. Q969S8, HDAC11 Accession No. Q96DB2). Multiple sequence alignment of full-length human HDAC1-11 was performed using MAFFT (v6.240). 27-30 The E-INS-i algorithm was selected as suitable for sequences containing large unalignable regions and the BLOSUM62 scoring matrix was used as suitable for highly evolutionarily conserved sequences. Gap opening penalty and offset value were set to default parameters. A maximum parsimony tree was first generated using RAxML (RAxML-VI-HPC31) on the CIPRES Portal 2.0 (http://www.phylo.org/portal2/home.action). Maximum likelihood-based inference of an evolutionary tree was next performed with RAxML (RAxML-VI-HPC) again on the CIPRES Portal 2.0. Settings were selected for protein sequence and a BLOSUM62 substitution matrix. Rapid bootstrapping (1000 replicates) was conducted while searching for the best scoring maximum likelihood tree in one run. For this analysis, we specified 25 distinct rate categories, a random seed value for parsimony inferences using default parameters and 1000 bootstrap iterations. Initial rearrangements and constraints were not specified. All phylogenetic trees were visualized as unrooted radial phylograms, with Dendroscope. Bootstrap support values are provided on the ML tree.
Supplementary Material
Acknowledgments
We thank Stuart Schreiber and the Broad Chemical Biology Program for research space and support, Jon Clardy, Tim Mitchison, Ralph Weissleder, Olaf Wiest, Timothy Lewis and Rebecca Maglathlin for support, thoughtful discussions and access to key instrumentation and reagents; Alexandros Stamatakis for helpful discussions on phylogenetics; Chris Johnson, Galina Beletsky and Stephen Jonston for analytical support;. This work was supported by grants from the National Cancer Institute (1K08CA128972; JEB), the American Society of Hematology (JEB), the Multiple Myeloma Research Foundation (JEB), the Burroughs-Wellcome Foundation (JEB), the National Institutes of Health T32CA079443 (MLG) and P01CA078048 (RM), the National Science Foundation DEB 0733029 (TW) and ITR 0331453 (TW). The project has been funded in part, with funds from the National Cancer Institute’s Initiative for Chemical Genetics (Contract No. N01-CO-12400). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Service, nor does the mention of trade names, commercial products or organizations imply endorsement by the US government.
Footnotes
Competing Financial Interests Statement JEB and RM are scientific founders of and shareholders in SHAPE Pharmaceuticals (Cambridge, MA) and Acetylon Pharmaceuticals (Boston, MA).
Author Contributions JEB developed biochemical methods, analyzed data, designed and synthesized the cinnamic hydroxmate library, provided research funding, supervised, prepared the manuscript and mentors NHW, MLG and EFG. NHW developed the Class IIa biochemical methods, analyzed data and synthesized pandacostat. MLG synthesized and purified pandacostat and analyzed data. EFG developed the Class I and IIb biochemical methods and analyzed data. SJH designed experiments and provided reagents. TW advised on phylogenetic analysis. RM designed and synthesized substrates and tool HDAC inhibitors, synthesized the cinnamic hydroxmate library, developed methods, analyzed data, provided research funding, prepared the manuscript, and mentors MLG. The corresponding authors (JEB and RM) certify that all authors have agreed to all the content in the manuscript, including the data as presented.
References
- 1.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 2.Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn’t fit all. Nat Rev Mol Cell Biol. 2007;8:284–295. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- 3.Choudhary C, et al. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 4.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 5.Smith BC, Hallows WC, Denu JM. Mechanisms and molecular probes of sirtuins. Chem Biol. 2008;15:1002–1013. doi: 10.1016/j.chembiol.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sternson SM, Wong JC, Grozinger CM, Schreiber SL. Synthesis of 7200 small molecules based on a substructural analysis of the histone deacetylase inhibitors trichostatin and trapoxin. Org Lett. 2001;3:4239–4242. doi: 10.1021/ol016915f. [DOI] [PubMed] [Google Scholar]
- 7.Bowers AA, et al. Synthesis and Conformation-Activity Relationships of the Peptide Isosteres of FK228 and Largazole. J Am Chem Soc. 2009;131:2900–2905. doi: 10.1021/ja807772w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 9.Jones P, et al. Probing the elusive catalytic activity of vertebrate class IIa histone deacetylases. Bioorg Med Chem Lett. 2008;18:1814–1819. doi: 10.1016/j.bmcl.2008.02.025. [DOI] [PubMed] [Google Scholar]
- 10.Zhou X, Richon VM, Rifkind RA, Marks PA. Identification of a transcriptional repressor related to the noncatalytic domain of histone deacetylases 4 and 5. Proc Natl Acad Sci U S A. 2000;97:1056–1061. doi: 10.1073/pnas.97.3.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parra M, Kasler H, McKinsey TA, Olson EN, Verdin E. Protein kinase D1 phosphorylates HDAC7 and induces its nuclear export after T-cell receptor activation. J Biol Chem. 2005;280:13762–13770. doi: 10.1074/jbc.M413396200. [DOI] [PubMed] [Google Scholar]
- 12.Mottet D, et al. Histone deacetylase 7 silencing alters endothelial cell migration, a key step in angiogenesis. Circ Res. 2007;101:1237–1246. doi: 10.1161/CIRCRESAHA.107.149377. [DOI] [PubMed] [Google Scholar]
- 13.Renthal W, et al. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–529. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- 14.Tsankova NM, et al. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- 15.Bolger TA, Yao TP. Intracellular trafficking of histone deacetylase 4 regulates neuronal cell death. J Neurosci. 2005;25:9544–9553. doi: 10.1523/JNEUROSCI.1826-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohen TJ, et al. The histone deacetylase HDAC4 connects neural activity to muscle transcriptional reprogramming. J Biol Chem. 2007;282:33752–33759. doi: 10.1074/jbc.M706268200. [DOI] [PubMed] [Google Scholar]
- 17.Bowers A, et al. Total synthesis and biological mode of action of largazole: a potent class I histone deacetylase inhibitor. J Am Chem Soc. 2008;130:11219–11222. doi: 10.1021/ja8033763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riester D, Wegener D, Hildmann C, Schwienhorst A. Members of the histone deacetylase superfamily differ in substrate specificity towards small synthetic substrates. Biochem Biophys Res Commun. 2004;324:1116–1123. doi: 10.1016/j.bbrc.2004.09.155. [DOI] [PubMed] [Google Scholar]
- 19.Lahm A, et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acad Sci U S A. 2007;104:17335–17340. doi: 10.1073/pnas.0706487104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong JC, Sternson SM, Louca JB, Hong R, Schreiber SL. Modular synthesis and preliminary biological evaluation of stereochemically diverse 1,3-dioxanes. Chem Biol. 2004;11:1279–1291. doi: 10.1016/j.chembiol.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 21.Vegas AJ, et al. Fluorous-based small-molecule microarrays for the discovery of histone deacetylase inhibitors. Angew Chem Int Ed Engl. 2007;46:7960–7964. doi: 10.1002/anie.200703198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patel V, et al. Identification and Characterization of Small Molecule Inhibitors of a Class I Histone Deacetylase from Plasmodium falciparum. J Med Chem. 2009;52:2185–2187. doi: 10.1021/jm801654y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang D, Helquist P, Wiest O. Zinc binding in HDAC inhibitors: a DFT study. J Org Chem. 2007;72:5446–5449. doi: 10.1021/jo070739s. [DOI] [PubMed] [Google Scholar]
- 24.Schuetz A, et al. Human HDAC7 harbors a class IIa histone deacetylase-specific zinc binding motif and cryptic deacetylase activity. J Biol Chem. 2008;283:11355–11363. doi: 10.1074/jbc.M707362200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bottomley MJ, et al. Structural and functional analysis of the human HDAC4 catalytic domain reveals a regulatory structural zinc-binding domain. J Biol Chem. 2008;283:26694–26704. doi: 10.1074/jbc.M803514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mujtaba S, Zeng L, Zhou MM. Structure and acetyl-lysine recognition of the bromodomain. Oncogene. 2007;26:5521–5527. doi: 10.1038/sj.onc.1210618. [DOI] [PubMed] [Google Scholar]
- 27.Katoh K, Kuma K, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005;33:511–518. doi: 10.1093/nar/gki198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Briefings in Bioinformatics. 2008;9:286–298. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- 30.Wang LS, et al. The Impact of Multiple Protein Sequence Alignment on Phylogenetic Estimation. IEEE/ACM Transactions on Computational Biology and Bioinformatics. Sept. 2009. IEEE computer Society Digital Library. IEEE Computer Society, http://doi.ieeecomputersociety.org/10.1109/TCBB.2009.68. [DOI] [PubMed]
- 31.Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML Web servers. Systems Biology. 2008;57:758–771. doi: 10.1080/10635150802429642. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.