

Abstract
The cGMP-dependent protein kinase type I (PKG I) is an essential regulator of cellular function in blood vessels throughout the body. DT-2, a peptidic inhibitor of PKG, has played a central role in determining the molecular mechanisms of vascular control involving PKG and its signaling partners. Here, we report the development of (D)-amino acid DT-2 derivatives, namely the retro-inverso ri-(D)-DT-2 and the all (D)-amino acid analog, (D)-DT-2. Both peptide analogs were potent PKG Iα inhibitors with Ki values of 5.5 nM (ri-(D)-DT-2) and 0.8 nM ((D)-DT-2) as determined using a hyperbolic mixed-type inhibition model. Also, both analogs were proteolytically stable in vivo, showed elevated selectivity, and displayed enhanced membrane translocation properties. Studies on isolated arteries from the resistance vasculature demonstrated that intraluminally perfused (D)-DT-2 significantly inhibited vasodilation induced by 8-Br-cGMP. Furthermore, in vivo application of (D)-DT-2 established a uniform translocation pattern in the resistance vasculature, with exception of the brain. Thus, (D)-DT-2 caused significant increases in mean arterial blood pressure in unrestrained, awake mice. Further, mesenteric arteries isolated from (D)-DT-2 treated animals showed a markedly reduced dilator response to 8-Br-cGMP in vitro. Our results clearly demonstrate that (D)-DT-2 is a superior inhibitor of PKG Iα and its application in vivo leads to sustained inhibition of PKG in vascular smooth muscle cells. The discovery of (D)-DT-2 may help our understanding of how blood vessels constrict and dilate and may also aid the development of new strategies and therapeutic agents targeted to the prevention and treatment of vascular disorders such as hypertension, stroke and coronary artery disease.
Keywords: cGMP-dependent protein kinase, protein kinase inhibitors, DT-2, (D)-DT-2, smooth muscle
1. Introduction
Inhibitors of cyclic nucleotide-dependent protein kinases have served as valuable tools in identifying the fundamental roles of cGMP-dependent protein kinase (PKG) and cAMP-dependent protein kinase (PKA) in intracellular signaling. Both kinases have served as Rosetta stones in our understanding of a vast number of intracellular signaling mechanisms ranging from smooth muscle cell relaxation to neuronal synaptic plasticity [1–6]. Therefore, the search for potent inhibitors of these kinases has been extensively investigated. However, the structural similarities of PKG and PKA have posed a formidable obstacle in the design of selective inhibitors that specifically target cyclic nucleotide-dependent protein kinases and show little inhibitory potency to other basophilic Ser/Thr-kinases.
The multi-domain structure of PKG dictates the target sites for putative inhibitors. (Rp)-phosphorothioate analogs of cGMP are the only known inhibitors that bind to the cyclic nucleotide binding sites [7–10]. Although their mode of action is still not completely understood, studies have indicated that the binding of these derivatives fails to induce the conformational changes essential for releasing catalytic activity [11, 12]. A diverse pool of derivatives, moderate selectivity and cell membrane permeability are regarded as the major advantages of Rp-cGMPS analogs as tools in intact cell studies [10, 13, 14]. However, partial antagonism and limited potencies restrict their versatility [9, 15]. The catalytic domain of PKG contains two target sites for inhibitors: the ATP-binding site and the substrate binding site. In general, compounds mimicking ATP represent a diverse class of inhibitors, as has been known for all other major families of protein kinases [16–18]. Peptide inhibitors designed to block the substrate binding site of PKG have long remained elusive, partly because the sequence requirements for PKG inhibition do not follow a classic consensus sequence and the kinase appears to nonspecifically favor positively charged amino acids [19–21].
Recently, we developed a new class of potent and cell membrane permeable PKG peptide inhibitors [19]. We utilized SPOT-based combinatorial peptide libraries [22–26] to identify PKG selective inhibitor peptides of which DT-2 was the first example [27]. DT-2 shows PKG specificity and due to its membrane-permeable segment from HIV-1 tat(47–59) remarkable cellular translocation characteristics [28]. In addition, only DT-2 is able to inhibit basal kinase activity, suggesting that DT-2 competes with the N-terminal domain of PKG for the catalytic center of the enzyme [27]. Although DT-2 is a potent PKG inhibitor (Ki = 13 nM), its in vivo applications are likely limited due to proteolysis. To overcome this obstacle we here report the development of a proteolytically stable derivative of DT-2, namely (D)-DT-2. This (D)-amino acid derivative was more potent against PKG and showed fast and reliable translocation in smooth muscle cells in a variety of vascular beds. Consequently, (D)-DT-2 increased blood pressure in mice and arteries removed from animals treated with (D)-DT-2 were resistant against cGMP-mediated relaxation. We propose that our novel PKG inhibitors have the potential to shed new light on the central role of PKG in vascular biology.
2. Experimental Procedures
2.1. Peptide synthesis
Solid-phase synthesis of the peptides (D)-DT-2 and retro-inverso-DT-2 (ri-DT-2) in the form of C-terminal carboxamides was carried out on TentaGel S RAM resin (Rapp Polymere, Tübingen, Germany) with a Pioneer automatic peptide synthesizer (Applied Biosystems) employing Fmoc chemistry with TBTU-activation and a fourfold excess of amino acids. Side chain protections were as follows: Tyr: t-Bu; Gln and His: Trt; Arg: Pbf; Lys: Boc. Coupling time was 1 h. ri-DT-2 was acetylated N-terminally with acetic acid anhydride (5% in DMF) prior to cleavage. Peptides were cleaved from the resin and deprotected by a 3 hour treatment with TFA containing 3% triisopropylsilane and 2% water (10 ml/g resin). After precipitation with t-butylmethyl ether, the resulting crude peptides were purified by preparative HPLC on a 250x40 mm Nucleosil 100-7 C18 column (Macherey-Nagel, Düren, Germany) with water/acetonitrile gradients containing 0.1% TFA and characterized by analytical HPLC in the same solvent system on a 50x2 mm Gemini 5μ C18 column (Phenomenex) and MALDI-MS.
Fluorescein peptide labeling was carried out by incubating 10 mg peptide, containing an extra Cys followed by β-Ala at the N-terminus, in 1 mL of 1 M phosphate buffer, pH 7.4, with 60 uL of a 0.1 M stock solution of fluorescein-5-maleimide (Molecular Probes, Eugene, OR, USA) in dimethylsulfoxide at 4°C overnight in the dark. Determination of the exact peptide concentrations of stock solutions (approximately 6–10 mM in water) and further characterization of the products was carried out by amino acid analysis.
(D)-DT-2 has been made commercially available through BioLog (www.biolog.de).
2.2. Kinetics
PKG Iα was expressed using SF9 insect cells (Invitrogen) in suspension as reported previously [27]. Kinetic constants were determined using a [γ-32P]ATP transfer assay as previously reported [25, 29] with the following modifications. PKG Iα at a concentration of 2 nM was incubated with the PKG specific substrate TQAKRKKSLAMA [22] at 16 μM.
Inhibition constants were calculated using a variety of graphical and mathematical methods. First, IC50 values were determined by keeping the concentration of substrate (TQAKRKKSLAMA) constant and varying the amount of inhibitor. The obtained data were plotted as v vs. log([I]) and the Ki was calculated from the IC50 using the Cheng-Prusoff equation [30]. Secondly, Dixon plot analysis was employed as an alternative to obtain Ki values and to determine the mode of kinase inhibition. The change in velocity for three increasing substrate concentrations was measured under varying inhibitor amounts. The resulting data were plotted as 1/v vs. [I] and the initial slopes for each substrate were replotted as slope vs. 1/[S] [30]. Ki values were then calculated from the slope of the linear regression using equation 1.
| Equation 1 |
To ascertain αKi and βVmax values, we measured the change in v from several constant inhibitor concentrations and varied the amount of substrate [30]. The resulting data were plotted as v vs. log([S]) (Schild-plot). The Km, app and Vmax patterns are indicative of the specific inhibition model. Furthermore, from these measurements we replotted %Vmax vs. [I] according to equation 2 to establish βVmax, and replotted Km, app vs. [I] (equation 3) to determine αKi.
| Equation 2 |
| Equation 3 |
The replots of %Vmax vs. [I] and Km,app vs. [I] were hyperbolic (see Figure 4B, C) from which the asymptotic βVmax and αKi values were determined.
Figure 4. Proposed inhibitory mechanism for (D)-DT-2.

(A) Hyperbolic mixed type inhibition model showing the catalytically productive inhibitor complex ESI as part of the non-competitive component (grey) of the model. Determination of (B) βVmax and (C) αKi was accomplished by plotting Vmax or Km against *I+. The equations used to derive α and β factors are shown in Methods.
To investigate the stability of the peptides against degradation, the inhibitors were incubated in 2 μg/mL trypsin for 5 minutes. The trypsin/peptide mixture was than heat inactivated for 10 minutes at 60 °C before adding it to the enzyme reaction mixture.
2.4. Animal and Tissue preparation
Male C57BL6 mice were euthanized with an injection of sodium pentobarbital (120 mg/kg, intraperiotoneal (i.p.)) and subsequent decapitation, in accordance with the University of Vermont Institutional Animal Care and Use Committee and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The majority of the small intestine was removed and placed in cold (4°C) physiological saline solution (PSS) containing 119 mM NaCl, 4.7 mM KCl, 24 mM NaHCO3, 1.2 mM KH2PO4, 0.03 mM EDTA, 1.2 mM MgSO4, 1.6 mM CaCl2, and 10.6 mM glucose, pH 7.4. Mesenteric arteries with a diameter of 60 to 100 μm were dissected, connective tissue was removed, and the arteries were then treated as indicated below.
2.5. Internalization of fluorescent labeled peptides in vitro and in vivo
Posterior cerebral arteries isolated from rat brain (Figure 6) and mesenteric arteries from male C57BL6 mice (Figure 5B) were incubated with 2 μM Fluorescein labeled peptide, diluted in PSS for 45 min. at room temperature. After washing 4 times with PSS the arteries are imaged on a BioRad MRC 1024ES Confocal Fluorescence Microscope.
Figure 6. Kinetic profiles of Fluo-(D)-DT-2 uptake in intact cerebral arteries.

In vitro staining of Fluo-(D)-DT-2 in cerebral arteries after incubation with Fluo-(D)-DT-2 at (A) 10 min, (B) 30 minutes and (C) 60 minutes. Nuclear stains (DRAQ5) emphasizing nuclear exclusion or nuclear staining for Fluo-(D)-DT-2 at (D) 10 minutes incubation and (E) 60 minutes incubation.
Figure 5. Resistance to proteolysis and cellular uptake of (D)-DT-2 in intact vessels.
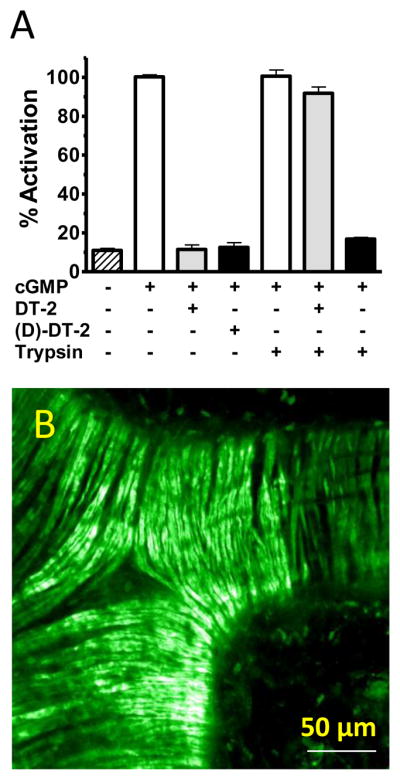
(A) Trypsin pre-incubated (D)-DT-2 inhibits PKG. PKG activity (white) was assayed for DT-2 (gray) and (D)-DT-2 (black) the presence and absence of trypsin. (B) In vitro internalization of Fluo-(D)-DT-2. Mouse mesenteric arteries were incubated with 2 μM (1 nmole) Fluo-(D)-DT-2 for 30 minutes. Live confocal fluorescence imaging was performed without fixation. Scale bar = 50 μm
Male C57BL6 mice were injected with a total of 300 μg peptide (150 μg intraperiotoneal (i.p.) and 150 μg intravenous (i.v.) via the tail vein). The mice were euthanized after 45 minutes as described above. During this period no adverse effects of the peptide were observed. Arteries from various tissues (mesentery, skeletal muscle and brain) were isolated and imaged as described above. For in vitro studies arteries from mouse mesentery and rat brain were isolated and incubated with 2 μM peptide in PSS.
2.6. Long-term telemetric blood pressure analysis
Mean arterial blood pressures (MAP) was measured on awake C57BL6xSV129 hybrid background mice implanted with the DataScience transponder system. Briefly, a radiotelemetric device (Data Sciences International, St. Paul, MN) was used for long-term analysis of mean arterial blood pressure (MAP), heart rate (HR) and physical activity in conscious male mice. Mice were three to four months old and did not significantly differ in body size and weight (26.2±1.3 g and 23.5±0.7 g, respectively). The mice were anaesthetized using a conventional isoflurane inhalation regime and placed on a heating pad. A ventral midline incision was performed prior to careful isolation of the left common carotid artery. For ligation and retraction, 2 silk ligatures were passed under the vessel, one about 0.8 cm caudal to the bifurcation of the interior and exterior carotid artery and another about 0.5 cm rostral from the caudal ligature. A small incision in the carotid artery was made for insertion of the catheter. The inserted catheter tip was advanced to the thoracic aorta and fixed with suture. A subcutaneous pocket was performed along the right flank for placing the transmitter body. After subcutaneous fixation of the transmitter the incision was closed using 6-0 silk. Inhalation anesthesia was stopped and mice were kept under infrared light for 2 hours. Their food and water intake, wound healing and morbidity was kept under surveillance. Five days after surgery, mice have regained normal locomotor activity. MAP, HR and physical activity were recorded at days 5–7 after surgery. Radiotelemetric data were continuously sampled for 1 minute at 5-min intervals and stored using the Dataquest ART data acquisition system (DSI).
2.7. Arterial diameter measurements
Diameter measurements were conducted using a DMT organ culture myograph (Danish Myo Technology A/S). Mesenteric arteries from male C57BL6 mice (initial internal diameter 50 – 100 μm) were isolated in cold PSS, and cannulated on glass pipettes within the pressure myograph chamber. The chamber bath was continuously superfused with warmed (37°C), gassed (20% O2/5% CO2/75%N2) PSS. Pipettes were connected to a PSS-containing reservoir, such that intravascular pressure could be adjusted using a pressure-manometer. The setup also allowed continuous flow of PSS through the vessel. Arterial diameters were measured using a video dimension analyzer (VediView, DMT). Arteries were tested for viability with 60 mM KCl, (those attaining less than 30 % constriction were discarded). Maximal (passive) arterial diameters were obtained via treatment with Ca2+-free PSS containing EGTA (2 mM) and the Ca2+ channel blocker diltiazem (10 μM). For experiments testing the acute of PKG inhibitors, vessels were intraluminally perfused with various PKG inhibitors at concentrations 10-fold above Ki, pressurized to 80 mm Hg and allowed to develop myogenic tone. Arteries were subsequently dose-dependently dilated with 8-Br-cGMP. For experiments testing the sustained effects of PKG inhibitors, male C57BL6 mice were injected with a total of 300 μg peptide (150 μg IP and 150 μg tail vein). The mice were euthanized after 45 minutes as described above and mesenteric arteries were isolated in cold PSS and studied using the myograph system as described above.
3. Results
3.1. Kinetic properties of stereo-isomers of DT-2
Although DT-2 is a potent and, by virtue of its membrane permeability, a very effective cellular inhibitor of PKG, the peptidic structure of DT-2 makes it prone to degradation in vivo. To obtain more stable inhibitors based on DT-2, we first synthesized the retro-inverso analog ri-DT-2 (Figure 1A). It has been shown that such derivatives have the potential to remain biologically active because they maintain the relative three-dimensional orientation of the side chains (only the orientation of the backbone is reversed) (Figure 1B) [31–33]. As Figure 1C demonstrates, IC50 values obtained for ri-(D)-DT-2 clearly showed that the retro-inverso analog was in fact much more potent then DT-2. However the plain (D)-amino-acid analog (D)-DT-2, synthesized as a control compound, showed a remarkable and additional increase in inhibitory potency. This finding was completely unexpected. To further support this extraordinary pattern of PKG inhibition by (D)-amino acid derivatives of DT-2, and to obtain the first clues as to the mechanism of PKG inhibition by these new compounds, we conducted Dixon-plot analyses on all three DT-2 type compounds (Figure 2). In addition, we included W45, a less potent precursor of DT-2 known for its purely competitive mode of PKG inhibition (Figure 2A) [19]. Conjointly, all three DT-2 type compounds also showed competitive inhibition, albeit restricted to initial inhibitor concentrations (Figure 2B–D). From these initial slopes (Figure 2E) inhibition constants (Ki) shown in Table 1 were calculated and found to be similar to Ki values obtained from the IC50 studies using the Cheng-Prusoff equation [30]. Remarkably, the Ki improved approximately 20-fold from 12.5 nM for DT-2 to 5.5 nM for ri-(D)-DT-2 and 0.5 nM for (D)-DT-2. More careful analysis of the Dixons plots revealed however a significant deviation from a competitive model as the inhibitory potencies of the compounds increased. Whereas W45 is purely and DT-2 predominantly competitive in nature (Figure 2A, B), a non-competitive component that is a deviation from a linear 1/v versus [I] correlation, becomes apparent for ri-(D)-DT-2 and (D)-DT-2 at higher inhibitor concentrations (Figure 2C, D).
Figure 1. Stereochemistry and IC50 profiles of the DT-2 family of PKG inhibitors.

(A) (L)- and (D)- amino acid sequences of DT-2, ri-(D)-DT-2 and (D)-DT-2 are shown in capital and small letter formats. (B) DT-2 and (D)-DT-2 differ in the side chain orientations relative to the plane of the peptide backbones. (C) The IC50 curves using the substrate TQAKRKKSLAMA [19].
Figure 2.

Dixon-plot analyses of (A) W45, a competitive precursor of DT-2, (B) DT-2, (C) ri-(D)-DT-2 and (D) (D)-DT-2. The velocities of three substrate concentrations, 4 μM (■), 8 μM (▲) and 16 μM (▼) TQAKRKKSLAMA were analyzed under varying inhibitor concentrations. (E) Re-plots of the initial 1/v vs. [I] slopes taken from A, B, C, and D.
Table 1. Inhibition constants of novel PKG blockers using unnatural amino acids.
Various inhibition parameters of (D)-amino acid analogs of DT-2 are compared with the precursor DT-2 [7].
| PKG | PKA | PKA/PKG | |||
|---|---|---|---|---|---|
| Peptide | IC50, nM | Ki(CP), nM | Ki(Dixon), nM | Ki(Dixon), μM | |
| DT-2 | 332 ± 53 | 13.9 ± 2.2 | 12.5 ± 3 | 16.5 ± 3.8 | 1,320 |
| riD-DT-2 | 54 ± 17 | 2.8 ± 1.2 | 5.5 ± 0.3 | 9.9 ± 2.2 | 1,800 |
| D-DT-2 | 12 ± 2.3 | 0.5 ± 0.1 | 0.8 ± 0.24 | 12 ± 3.3 | 15,000 |
Ki(D)=Dixon-plot analysis, Ki(CP)=Cheng-Prusoff inhibition constants derived from IC50 values (see Experimental Procedures).
To ascertain the degree of the non-competitive component and develop a comprehensive inhibitory model for ri-(D)-DT-2 and (D)-DT-2 we performed Schild-plot analyses for these compounds (Figure 3). By comparing the exclusively competitive inhibitor W45 (Figure 3A) to (D)-DT-2 (Figure 3B), Schild-plot analyses confirmed the non-competitive properties of the (D)-amino acid analogs of DT-2 (data for ri-(D)-DT-2 not shown). Moreover, the coupled decrease in Vmax with the right-shift of the Michaelis-Menten constants (Km) revealed the signature model of a mixed competitive/non-competitive scenario in which the ESI complex is catalytically productive. This hyperbolic, partial mixed-type inhibitor model is depicted in Figure 4A. It requires the introduction of α- and β-factors, because the rates of substrate and/or inhibitor binding as well as the rates of product formation from the ES and ESI complexes must be different as previously reported [30]. We determined the α- and β-factors for all three DT-2 compounds (see Methods) and found noticeable contribution only for (D)-DT-2. Here, the α- and β-factors were found to be 5.3 and 0.28, respectively (Figure 4B, C). These findings illustrate that the contribution of product formation (substrate phosphorylation) from the ESI complex is approximately 1/3 of the total. Likewise, the ratio of the inhibitor complexes ESI:EI does not exceed 1:5. In conclusion, although (D)-DT-2 showed a deviation from a competitive inhibition model, the overall Ki constant based on a purely competitive model was not significantly affected by the input of the hyperbolic, partial mixed-type inhibitor model. The value of the mixed model lays in the potential consequences as to the molecular mechanism of (D)-DT-2 mediated PKG inhibition.
Figure 3.

Schild-plot analyses of (A) W45 and (B) (D)-DT-2. Michaelis-Menten plots were determined for the PKG substrate TQAKRKKSLAMA in the presence of various inhibitor concentrations. The so obtained Km,app and Vmax values were used for further analysis (see Figure 5).
It should also be noted that ri-(D)-DT-2 and (D)-DT-2, similar to DT-2, displayed exceptional selectivity for PKG compared to PKA. (D)-DT-2 in particular was exquisitely selective with a PKG:PKA ratio of approximately 15,000 fold (Table 1). Thus, compounds such as (D)-DT-2 are especially valuable to study the designated roles of PKG in controlling vasomotor reactivity in vivo. Since (D)-DT-2 provided a more promising overall inhibitory profile than ri-(D)-DT-2, all subsequent experiments were performed with the former compound.
3.2. In vitro effects of (D)-DT-2: Resistance to proteolysis, cellular uptake and constrictor effects on isolated blood vessels
In order to conduct long term in vitro and in vivo studies, for any peptidic inhibitor, the conceptual stumbling block of proteolysis must be addressed. However, (D)-DT-2 is completely resistant to trypsin degradation, as is expected for a peptide entirely made of (D)-amino-acids. As Figure 5A shows, both DT-2 and (D)-DT-2 abolished the cGMP-stimulated PKG activity. In the presence of trypsin however, only (D)-DT-2 retained its inhibitory power (trypsin treatment alone had no effect on PKG activity). Next, we examined the membrane translocation properties of (D)-DT-2 and found them to be of superior quality. Incubation of small arterial segments (20–80 μm diameter) from rat and mouse mesentery with fluorescein-labeled (D)-DT-2 (Fluo-(D)-DT-2, see Methods) for 30 minutes revealed substantial peptide translocation into smooth muscle cells with clearly defined cytosolic distribution (Figure 5B). We previously demonstrated that DT-2 exhibits favorable internalization properties in terms of overall kinetics and intracellular distribution [28]. Compared with Fluo-DT-2, however the internalization properties of Fluo-(D)-DT-2 were even more pronounced. We further compared the in vitro internalization of Fluo-(D)-DT-2 using different vascular beds from rat and mouse and found a generally efficient uptake and cytosolic distribution in all types of vascular smooth muscle cells. As demonstrated in Figures 6A, D, utilizing anterior and posterior arteries from rat brain, as little as 10 minute incubation times were sufficient to obtain live confocal fluorescence images showing the outlines of individual smooth muscle cells with no detectable nuclear translocation of Fluo-(D)-DT-2. After 30 to 45 minutes, Fluo-(D)-DT-2 showed optimal cytosolic distribution with minimal nuclear translocation (Figure 6B). Measurable nuclear staining was only detected at 60 minutes and longer incubations times (Figure 6C, E). These results reinforce the value of DT-2 type compounds as intra cellular PKG inhibitors.
We previously demonstrated that the cellular translocation properties of DT-2 were of great value in ascertaining the role of PKG in regulating vasomotor reactivity in functional experiments [27]. Figure 7 illustrates that this effect is even more pronounced for (D)-DT-2. Under control conditions, cannulated, third order arteries from mouse mesentery showed up to 50% myogenic reactivity when subjected to 80 mm Hg intraluminal pressure (Figure 7A). These constricted arteries (60–110 μm luminal diameters) were then relaxed with the membrane permeable cGMP analog, 8-Br-cGMP, in a dose-dependent manner. Importantly, the 8-Br-cGMP induced vasodilation was substantially inhibited when arteries were intraluminally perfused with (D)-DT-2, and to a lesser extent by DT-2, for the duration of the experiment (Figure 7B, C). Total relaxation was quantified as the ratio of maximal myogenic tone at 80 mm Hg to maximal (passive) dilation obtained by treating the arteries with Ca2+-free PSS containing the Ca2+ channel blocker diltiazem. On average, DT-2 and (D)-DT-2 inhibited the maximal 8-Br-cGMP induced vasodilation by approximately 30% and 55%, respectively (Figure 7D).
Figure 7. Effects of PKG inhibitors on 8-Br-cGMP induced dilation of mesenteric resistance arteries.

Vessels were superfused with (A) saline), (B) DT-2, or (C) (D)-DT-2 and pressurized to 80 mm Hg. Following development of tone, increasing concentrations of 8-Br-cGMP were titrated. At the end of each experiment vessel viability was tested using high K+ (maximal constriction) and Ca2+ fresh PSS (maxima dilation. (D) The bar graph summarizes DT-2 and (D)-DT-2 mediated constrictions relative to maximal relaxation observed in Ca2+ free PSS.
3.3. In vivo effects of (D)-DT-2: vascular tissue distribution, blood pressure and functional correlations
The stability, potency and membrane translocation properties of (D)-DT-2 make this PKG inhibitor an ideal tool to probe the functions of PKG in intact tissues. To study the cellular translocation properties of (D)-DT-2 in the intact animal, we administered the fluorescein-tagged peptide (Fluo-(D)-DT-2) into mice through the tail vein injection. 30–40 minutes after application, small resistance arteries from various tissues were removed and the internalization into endothelial and smooth muscle cells examined. These experiments showed two major results. First, peptide translocation in the medial layer of vessels from different vascular beds such as mesentery and hind limbs is very rapid and efficient (Figure 8A, B). Second, the blood brain barrier appears to prevent peptide translocation into smooth muscle cells in cerebral arteries (only endothelial cells were stained) (Figure 8C). Similar staining patterns were obtained with DT-2 (data not shown), although DT-2 internalization efficiency and staining intensity was generally lower as compared to (D)-DT-2. The fluorescein-labeled peptide W45, lacking the tat-internalization sequence showed no uptake (Figure 8D).
Figure 8. In vivo uptake of Fluo-(D)-DT-2.
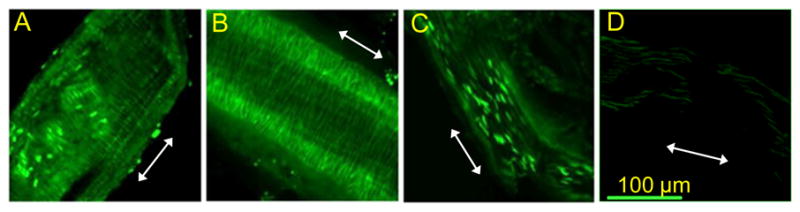
C57BL/6 mice were injected with 20 μl, 1mM peptide (20 nmoles) by tail vein injection and left in the circulation for 30 minutes. Live confocal imaging was performed on small resistance arteries immediately after harvest. A: mesenteric artery, B: femoral artery, C: posterior cerebral artery. D: injection of Fluo-W45, a PKG specific inhibitor sequence without tat-translocation sequence showed no uptake in arteries from mesentery. Arrows depict vessel orientation. Scale bar= 100 μm
Given the superiority of DT-2 analogs as effective, potent and cell-permeable inhibitors of PKG, we next examined the role of PKG in the regulation of vasodilation in vivo by determining the changes in mean arterial blood pressure (MAP) in response to DT-2 and (D)-DT-2 in vivo. Following intraperiotoneal application of either peptide, an immediate and highly significant increase in MAP of up to 18 mmHg was observed (Figure 9). However, this increase in MAP was transient with DT-2 (Figure 9A), but stable with (D)-DT-2 (Figure 9B), while injection of saline had no effect (Figure 9C). Interestingly, the DT-2 MAP increase returned to baseline within 20–30 minutes, while MAP remained elevated, using (D)-DT-2 over the entire 70 minute interval of the experiment. The time intervals labeled A, B, C in Figure 9D illustrate this finding. For DT-2, MAP is only significantly elevated in the first 10–20 minute interval, however (D)-DT-2 increases MAP significantly at all time intervals. These results suggest rapid degradation of DT-2 in the blood circulation. In contrast, (D)-DT-2 does not present a target for rapid proteolysis (see Figure 5A), hence the inhibitor remains active over time. These results demonstrate that (D)-DT-2 constitutes a significant advance in the use of our PKG inhibitors for in vivo applications.
Figure 9. DT-2 and (D)-DT-2 increase blood pressure.

Mean arterial blood pressures (MAP) was recorded from alert C57BL6xSV129 hybrid background mice implanted with a telemetric DataScience transponder system after i.p. injection of 256 nmoles DT-2 (A, n=8), 134 nmoles (D)-DT-2 (B, n=9) or saline (C, n=8) for a total of 70 minutes. D; Statistical analysis was performed on three time intervals (a) 10–20 minutes, (b) 30–40 minutes and (c) 10–70 min after injection. DT-2 increased MAP significantly only in the first time interval. Blood pressure returned to normal after 20–40 minutes. In contrast, (D)-DT-2 increased blood pressure significantly by 15–25 mmHg for all three intervals.
To establish an additional functional correlate of the vascular effects of in vivo exposure to (D)-DT-2, we evaluated the 8-Br-cGMP induced dilator responses of arteries isolated from (D)-DT-2 treated mice in vitro (Figure 10). In these experiments we observed a near maximal dilator response to 50 μM 8-Br-cGMP in myogenically active arteries from saline injected control animal (Figure 10A, C). In contrast, arteries from mice treated with (D)-DT-2 (10 nmoles) for 30 minutes showed an almost complete resistance to 8-Br-cGMP mediated dilation (Figure 10B, C). Interestingly, cerebral arteries isolated from treated animals did not show any change in 8-Br-cGMP induced dilation compared to those from untreated animals (data not shown). This finding complements our results shown in Figure 8, suggesting that the blood-brain barrier prevents the peptide inhibitor from entering smooth muscle of the cerebral vasculature.
Figure 10. In vivo treatment with (D)-DT2 suppresses vasodilation induced by 8-Br-cGMP in vitro.

(A) A mesenteric arteriole isolated from an untreated mouse relaxes to near maximal diameter in response to 8-Br-cGMP. (B) The 8-Br-cGMP response is greatly reduced in an arteriole isolated from a treated (10 nmoles (D)-DT-2, i.p.) mouse. (C) 8-Br-cGMP induces an 80% dilation in arteries from untreated mice, but only a 12% dilation in arteries from mice treated with (D)-DT2 in vivo. It should be noted that only very small mesenteric arteries (30–50 μm starting diameter) developed sufficient pressure induced tone to reveal the in vitro vasodilator response to 8-Br-cGMP.
4. Conclusions and Perspectives
Potent and selective PKG inhibitors may aid our understanding of how blood vessels constrict and dilate, and therefore be critical in the development of new strategies and therapeutic agents aimed at prevention and treatment of vascular disorders such as hypertension, stroke, and coronary artery disease. In this study we developed the (D)-amino acid derivatives ri-(D)-DT-2 and (D)-DT-2. Although ri-(D)-DT-2 was more potent then DT-2, the all (D)-amino-acid analog, (D)-DT-2, which had been initially synthesized as a negative control showed a remarkable increase in inhibitory potency. The Ki shifted approximately 20-fold from 13 nM to 0.8 nM. This finding was unanticipated. These results clearly demonstrate that stereo-specificity is not a prerequisite for PKG inhibition. Thus, altering the oligomeric backbone of DT-2 has the potential to result in PKG inhibitors with significantly improved inhibitory potency, resistance against proteolysis, and membrane permeability. Therefore, our studies may open new avenues for future inhibitor design. Obviously, the primary goal is no longer to achieve inhibitory potency but to generate new structures with improved stability, tailored cell-type specific internalization properties and, generally favorable pharmacological properties normally not seen with peptides or peptidomimetics. Future work will show if DT-2 derivatives with alternate backbone designs or organic scaffolds carrying guanidinium-type charges can also potently inhibit PKG and thus, may provide improved potential pharmaceutical lead structures.
One of the hallmarks of the DT-2 family of PKG inhibitors is their cellular translocation properties. Compared with Fluo-DT-2, the internalization properties of Fluo-(D)-DT-2 were even more pronounced. In order to conduct in vivo studies, the membrane permeability is a welcome benefit and we demonstrated the superiority of (D)-DT-2 in this regard when the peptide presented itself in the cytosol of smooth muscle cells in all major vascular beds of the resistance vasculature. We did not observe any translocation in cerebral arteries, a sign of an intact blood-brain barrier. The in vivo implications of (D)-DT-2 application were then found to be of profound significance. We showed that (D)-DT-2 causes vasoconstriction in isolated arteries. Next, we demonstrated that (D)-DT-2 mediated PKG inhibition and subsequent vasoconstriction in vivo caused a sustained increase in arterial blood pressure and that mesenteric arteries isolated from (D)-DT-2 treated animals had a reduced dilator response to 8-Br-cGMP. These findings clearly indicated that (D)-DT-2 is ideally suited for in vivo studies and that its application results in sustained inhibition of PKG in vascular smooth muscle cells. Furthermore, these results provide a solid foundation for further studies to test the possibility that PKG activity is a major regulator of peripheral resistance.
The importance of cGMP and PKG as mediators of vascular tone regulation cannot be underestimated given the fact that cGMP acts directly downstream of the nitric oxide (NO) signaling pathway. While NO/cGMP initiates the signaling cascade of smooth muscle dilation, PKG as the subsequent receptor provides a major intracellular check- and branch point with a multitude of downstream substrates and regulatory targets. Deciphering the roles of various PKG activities with the aid of our DT-2 family of PKG inhibitors is likely to lead to advances in the treatment of a spectrum of cardiovascular/smooth muscle diseases (septic shock, hypertension, erectile dysfunction, vasospasm).
Acknowledgments
We would like to thank Kristina Laskovski for excellent technical assistance. We would like to thank the UVM College of Medicine Animal Facility as well as the laboratories of Drs. Nelson, Wellman, Osol and Gokina for providing mouse tissues used in some of the studies. This work was supported by NIH grant RO1-HL68891 and by the Totman Trust for Medical Research.
Abbreviations
- cGMP
cyclic guanosine 3′, 5′-mono-phosphate
- i.p.
intraperiotoneal
- i.v.
intravenous
- Ki
inhibition constant
- MAP
mean arterial blood pressure
- Km
Michaelis-Menten constant
- MPP
Membrane permeable peptides
- PKA
cAMP-dependent protein kinase
- PKG
cGMP-dependent protein kinase
- Vmax
maximal (enzyme) velocity
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Beene DL, Scott JD. A-kinase anchoring proteins take shape. Curr Opin Cell Biol. 2007;19:192–198. doi: 10.1016/j.ceb.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feil R, Feil S, Hofmann F. A heretical view on the role of NO and cGMP in vascular proliferative diseases. Trends Mol Med. 2005;11:71–75. doi: 10.1016/j.molmed.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 3.Hofmann F, Feil R, Kleppisch T, Schlossmann J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol Rev. 2006;86:1–23. doi: 10.1152/physrev.00015.2005. [DOI] [PubMed] [Google Scholar]
- 4.Kim C, Vigil D, Anand G, Taylor SS. Structure and dynamics of PKA signaling proteins. Eur J Cell Biol. 2006;85:651–654. doi: 10.1016/j.ejcb.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 5.Lincoln TM, Wu X, Sellak H, Dey N, Choi CS. Regulation of vascular smooth muscle cell phenotype by cyclic GMP and cyclic GMP-dependent protein kinase. Front Biosci. 2006;11:356–367. doi: 10.2741/1803. [DOI] [PubMed] [Google Scholar]
- 6.Taylor SS, Kim C, Cheng CY, Brown SH, Wu J, Kannan N. Signaling through cAMP and cAMP-dependent protein kinase: diverse strategies for drug design. Biochim Biophys Acta. 2008;1784:16–26. doi: 10.1016/j.bbapap.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Butt E, Eigenthaler M, Genieser HG. (Rp)-8-pCPT-cGMPS, a novel cGMP-dependent protein kinase inhibitor. Eur J Pharmacol. 1994;269:265–268. doi: 10.1016/0922-4106(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 8.Butt E, Pohler D, Genieser HG, Huggins JP, Bucher B. Inhibition of cyclic GMP-dependent protein kinase-mediated effects by (Rp)-8-bromo-PET-cyclic GMPS. Br J Pharmacol. 1995;116:3110–3116. doi: 10.1111/j.1476-5381.1995.tb15112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gjertsen BT, Mellgren G, Otten A, Maronde E, Genieser HG, Jastorff B, Vintermyr OK, McKnight GS, Doskeland SO. Novel (Rp)-cAMPS analogs as tools for inhibition of cAMP-kinase in cell culture. Basal cAMP-kinase activity modulates interleukin-1 beta action. J Biol Chem. 1995;270:20599–20607. doi: 10.1074/jbc.270.35.20599. [DOI] [PubMed] [Google Scholar]
- 10.Schwede F, Maronde E, Genieser H, Jastorff B. Cyclic nucleotide analogs as biochemical tools and prospective drugs. Pharmacol Ther. 2000;87:199–226. doi: 10.1016/s0163-7258(00)00051-6. [DOI] [PubMed] [Google Scholar]
- 11.Dostmann WR. (RP)-cAMPS inhibits the cAMP-dependent protein kinase by blocking the cAMP-induced conformational transition. FEBS Lett. 1995;375:231–234. doi: 10.1016/0014-5793(95)01201-o. [DOI] [PubMed] [Google Scholar]
- 12.Wu J, Jones JM, Nguyen-Huu X, Ten Eyck LF, Taylor SS. Crystal structures of RIalpha subunit of cyclic adenosine 5′-monophosphate (cAMP)-dependent protein kinase complexed with (Rp)-adenosine 3′,5′-cyclic monophosphothioate and (Sp)-adenosine 3′,5′-cyclic monophosphothioate, the phosphothioate analogues of cAMP. Biochemistry. 2004;43:6620–6629. doi: 10.1021/bi0302503. [DOI] [PubMed] [Google Scholar]
- 13.Dostmann WR, Taylor SS, Genieser HG, Jastorff B, Doskeland SO, Ogreid D. Probing the cyclic nucleotide binding sites of cAMP-dependent protein kinases I and II with analogs of adenosine 3′,5′-cyclic phosphorothioates. J Biol Chem. 1990;265:10484–10491. [PubMed] [Google Scholar]
- 14.Ogreid D, Dostmann W, Genieser HG, Niemann P, Doskeland SO, Jastorff B. (Rp)- and (Sp)-8-piperidino-adenosine 3′,5′-(cyclic)thiophosphates discriminate completely between site A and B of the regulatory subunits of cAMP-dependent protein kinase type I and II. Eur J Biochem. 1994;221:1089–1094. doi: 10.1111/j.1432-1033.1994.tb18828.x. [DOI] [PubMed] [Google Scholar]
- 15.Dostmann WR. Inhibitors of cyclic nucleotide dependent protein kinases. Academic Press; San Diego: 2003. [Google Scholar]
- 16.Hidaka H, Kobayashi R. Protein kinase inhibitors. Essays Biochem. 1994;28:73–97. [PubMed] [Google Scholar]
- 17.Ono-Saito N, Niki I, Hidaka H. H-series protein kinase inhibitors and potential clinical applications. Pharmacol Ther. 1999;82:123–131. doi: 10.1016/s0163-7258(98)00070-9. [DOI] [PubMed] [Google Scholar]
- 18.Hidaka H, Inagaki M, Kawamoto S, Sasaki Y. Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide dependent protein kinase and protein kinase C. Biochemistry. 1984;23:5036–5041. doi: 10.1021/bi00316a032. [DOI] [PubMed] [Google Scholar]
- 19.Dostmann WR, Taylor MS, Nickl CK, Brayden JE, Frank R, Tegge WJ. Highly specific, membrane-permeant peptide blockers of cGMP-dependent protein kinase Ialpha inhibit NO-induced cerebral dilation. Proc Natl Acad Sci U S A. 2000;97:14772–14777. doi: 10.1073/pnas.97.26.14772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glass DB, Krebs EG. Phosphorylation by guanosine 3′:5′-monophosphate-dependent protein kinase of synthetic peptide analogs of a site phosphorylated in histone H2B. J Biol Chem. 1982;257:1196–1200. [PubMed] [Google Scholar]
- 21.Kennelly PJ, Krebs EG. Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J Biol Chem. 1991;266:15555–15558. [PubMed] [Google Scholar]
- 22.Dostmann WR, Nickl C, Thiel S, Tsigelny I, Frank R, Tegge WJ. Delineation of selective cyclic GMP-dependent protein kinase Ialpha substrate and inhibitor peptides based on combinatorial peptide libraries on paper. Pharmacol Ther. 1999;82:373–387. doi: 10.1016/s0163-7258(98)00063-1. [DOI] [PubMed] [Google Scholar]
- 23.Frank R. Spot-synthesis: an easy thechnique for the positionally addressable, parallel chemical synthesis on a membrane support. Tetrahedron. 1992;48:9217–9232. [Google Scholar]
- 24.Martens W, Greiser-Wilke I, Harder TC, Dittmar K, Frank R, Orvell C, Moennig V, Liess B. Spot synthesis of overlapping peptides on paper membrane supports enables the identification of linear monoclonal antibody binding determinants on morbillivirus phosphoproteins. Vet Microbiol. 1995;44:289–298. doi: 10.1016/0378-1135(95)00023-4. [DOI] [PubMed] [Google Scholar]
- 25.Tegge W, Frank R, Hofmann F, Dostmann WR. Determination of cyclic nucleotide-dependent protein kinase substrate specificity by the use of peptide libraries on cellulose paper. Biochemistry. 1995;34:10569–10577. doi: 10.1021/bi00033a032. [DOI] [PubMed] [Google Scholar]
- 26.Tegge WJ, Frank R. Analysis of protein kinase substrate specificity by the use of peptide libraries on cellulose paper (SPOT-method) Methods Mol Biol. 1998;87:99–106. doi: 10.1385/0-89603-392-9:99. [DOI] [PubMed] [Google Scholar]
- 27.Taylor MS, Okwuchukwuasanya C, Nickl CK, Tegge W, Brayden JE, Dostmann WR. Inhibition of cGMP-dependent protein kinase by the cell-permeable peptide DT-2 reveals a novel mechanism of vasoregulation. Mol Pharmacol. 2004;65:1111–1119. doi: 10.1124/mol.65.5.1111. [DOI] [PubMed] [Google Scholar]
- 28.Foley KF, De Frutos S, Laskovski KE, Tegge W, Dostmann WR. Culture conditions influence uptake and intracellular localization of the membrane permeable cGMP-dependent protein kinase peptide inhibitor DT-2. Front Biosci. 2005;10:1302–1312. doi: 10.2741/1620. [DOI] [PubMed] [Google Scholar]
- 29.Ruth P, Landgraf W, Keilbach A, May B, Egleme C, Hofmann F. The activation of expressed cGMP-dependent protein kinase isozymes I alpha and I beta is determined by the different amino-termini. Eur J Biochem. 1991;202:1339–1344. doi: 10.1111/j.1432-1033.1991.tb16509.x. [DOI] [PubMed] [Google Scholar]
- 30.Segel IH. Enzyme Kinetics. Wiley Inter-science Publication; New York: 1993. [Google Scholar]
- 31.D’Ursi AM, Giannecchini S, Di Fenza A, Esposito C, Armenante MR, Carotenuto A, Bendinelli M, Rovero P. Retroinverso analogue of the antiviral octapeptide C8 inhibits feline immunodeficiency virus in serum. J Med Chem. 2003;46:1807–1810. doi: 10.1021/jm034012h. [DOI] [PubMed] [Google Scholar]
- 32.Sakurai K, Chung HS, Kahne D. Use of a retroinverso p53 peptide as an inhibitor of MDM2. J Am Chem Soc. 2004;126:16288–16289. doi: 10.1021/ja044883w. [DOI] [PubMed] [Google Scholar]
- 33.Staubitz P, Peschel A, Nieuwenhuizen WF, Otto M, Gotz F, Jung G, Jack RW. Structure-function relationships in the tryptophan-rich, antimicrobial peptide indolicidin. J Pept Sci. 2001;7:552–564. doi: 10.1002/psc.351. [DOI] [PubMed] [Google Scholar]