

Abstract
Congenital heart block (CHB) is an autoimmune disease associated with autoantibodies against intracellular ribo-nucleoproteins SSB/La and SSA/Ro. The hallmark of CHB is complete atrioventricular block. We have recently established that anti-SSA/Ro -SSB/La autoantibodies inhibit α1D L-type Ca current, ICa-L, and cross-react with the α1D Ca channel protein. This study aims at identifying the possible binding sites on α1D protein for autoantibodies from sera of mothers with CHB children. GST fusion proteins of the extracellular regions between the transmembrane segments (S5–S6) of each of the four α1D Ca channel protein domains I-IV were prepared and tested for reactivity with sera from mothers with CHB children and controls using ELISA. Sera containing anti-Ro/La autoantibodies from 118 mothers with CHB children and from 15 mothers with anti-Ro/La autoantibodies but have healthy children, and from 28 healthy mothers without anti-Ro/La autoantibodies and healthy children were evaluated. Seventeen of 118 (14.4%) sera from mothers with CHB children reacted with the extracellular loop of domain I S5–S6 region (E1). In contrast, only 2 of 28 (7%) of sera from healthy mothers (- anti-Ro/La) and healthy children reacted with E1 loop and none (0 of 15) of sera from healthy mothers (+ anti-Ro/La) and healthy children reacted with the E1 loop. Preincubation of E1 loop with the positive sera decreased the O.D reading establishing the specificity of the response. Electrophysiological characterization of the ELISA positive sera and purified IgG showed inhibition (44.1% and 49.8%, respectively) of the α1D ICa-L expressed in tsA201 cells. The inhibition was abolished when the sera were pre-incubated with E1 fusion protein. The results identified the extra-cellular loop of domain I S5–S6 of L-type Ca channel α1D subunit as a target for autoantibodies from a subset of mothers with CHB children. This novel finding provides insights into the potential development of therapeutic peptides that could bind to the pathogenic antibodies and prevent CHB.
Keywords: Autoantibodies, calcium, heart block, ion channels, sinoatrial node
Introduction
Autoimmune-associated congenital heart block (CHB) is a passively acquired autoimmune disease. Fetal injury is presumed to occur as a consequence of transplacental passage of maternal autoantibodies against the intracellular ribonucleoproteins 48-kDa SSB/La, 52-kDa SSA/Ro, and 60-kDa SSA/Ro [1]. The hallmark of CHB is characterized by irreversible atrioventricular (AV) block, not accompanied by any cardiac structural or anatomic abnormalities [1]. Sinus bradycardia has also been reported in animal models of CHB [2, 3] and in the clinical settings [4, 5]. Detection of CHB occurs at or after 16 weeks of gestation, when maternal antibodies effectively gain access to the fetal circulation. Because of the rarity and complex etiology of CHB, the incidence is not well established. A generally accepted mean incidence is 1:17,000 in the 1970s [6] and 1:11,000 in the latter decade [7, 8]. However, this incidence dramatically increases to about 5% in Lupus patients and to 18% in subsequent pregnancies. This indicates that the incidence of CHB in the latter decades [7, 9] was higher than previously reported likely due to more effective detection of CHB during pregnancy using fetal ultrasound and to the improved diagnostics. Current therapies include dexamethasone, plasmapheresis, sympathomimetics, and in-utero cardiac pacing. Unfortunately none have significantly altered mortality justifying the need for more basic and clinical investigations. CHB carries substantial morbidity and mortality approaching 30%, with 100% of affected children requiring lifelong pacemakers before entering adulthood [7, 10].
The association of anti-SSA/Ro -SSB/La autoantibodies with CHB is universally accepted. However, the mechanism by which these autoantibodies cause cardiac conduction abnormalities is not completely understood. Under physiologic conditions, the cognate antigens are normally intracellular and thus inaccessible to the circulating antibodies. Two plausible explanations can be considered: either the intracellular antigens SSA/Ro -SSB/La are trafficked to the cell surface or anti-SSA/Ro -SSB/La autoantibodies cross react with a surface antigen on the sarcolemma of cardiomyocytes. A considerable amount of evidence has been proposed to explain the accessibility of the intracellular antigens and circulating antibodies. These include viral infection [11], ultraviolet light exposure [12] and apoptosis [13] which all lead to the translocation of SSA/Ro -SSB/La to the cell surface. The second possibility is that maternal antibodies react with cell surface components. To date, sarcolemmal proteins have been identified including the muscarinic receptor [14], laminin [15], the serotonin receptor 5-HT4 [16], and the L-type Ca channels [2, 17–19].
Cardiac L-type Ca channels are divided into α1C and α1D Ca channels. The α1D Ca channel has been shown to localize exclusively to the supraventricular tissue including the conduction system [18, 20, 21] (SA and AV node) and has the biophysical properties that could account for its possible contribution to the diastolic depolarization and impulse generation in the SA node [18, 20, 21].
Accordingly, inhibition of the α1D L-type Ca current (ICa-L) by anti-SSA/Ro -SSB/La antibodies from mothers with CHB children would be predicted to result in supraventricular electrocardiographic abnormalities. Indeed, knockout of the α1D Ca channel in the mouse results in sinus bradycardia and AV block [18, 20–24].
Ca channels have been identified as autoantigens and also as targets for autoantibodies in a number of diseases [25, 26]. In this regard, we have recently established the direct interaction (inhibition) between the autoantibodies from mothers whose children have CHB and the α1D Ca channel protein [18]. We proposed that the resulting inhibition of the α1D ICa-L by maternal autoantibodies may contribute to the electrocardiographic abnormalities seen in CHB [18, 27]. The aim of the present study is to assess the possible antigenic sites in the α1D Ca channel to maternal autoantibodies. Since antibodies, under physiological conditions, do not cross intact cellular membranes, we hypothesized that the anti-SSA/Ro -SSB/La autoantibodies cross react with the extracellular region of the α1D Ca channel protein.
Methods
Patients’ sera
Sera were obtained from the U.S. based Research Registry for Neonatal Lupus established by the National Institute for Arthritis, Musculoskeletal and Skin Diseases (RRNL) previously described and characterized [7]. One hundred and sixty one sera were tested of which 118 were from mothers with anti-SSA/Ro –SSB/La antibodies and have children with CHB, 15 sera (with anti-SSA/Ro –SSB/La antibodies) from mothers whose children are healthy and did not have CHB and 28 from mothers (without anti-SSA/Ro –SSB/La antibodies) whose children are healthy and did not have CHB. The study was approved by the IRB and the subjects gave informed consent. IgG purification was performed as described previously [28].
GST Fusion Proteins
The Extra-cellular loops of I, II, III, IV Domains between S5–S6 (Figure 1) were amplified from Human Cav1.3 pGFP- plasmid (kindly provided by Dr J. Striessnig, Innsbruck, Austria) using the following primers:
Figure 1.

Panel A: Schematic structure of L-type Ca channel α1D subunit. α1 subunit is organized in four domains, I-IV, each consisting of transmembrane segments S1–S6 connected with a small stretch of amino acids. The extracellular loop between transmembrane segments S5–S6, E-Loop, and is the ion conductance pore and selectivity filter. The 4 extracellular loops in each domain that were prepared and tested in this study are shown. Panel B: Coomassie stain of total extract from BL21 E-coli following induction with 0.1 mM Isopropyl β-D-1-thiogalactopyranoside for 2 hours, and the successful expression of the extracellular loops E1-E4 of representative domains I-IV S5–S6 segments. Panel C: Immunoblot of the extracellular proteins E1-E4 on 15% SDS-PAGE after pulldown using 50% slurry of Glutathione Sepharose 4B and probed with monoclonal anti-GST antibody at 1:1000 dilution.
E1:
Forward: 5-CGCGGATCCATTGGAAAAATGCACAAAACA-3
Reverse: 5-CCGGAATTCCCATTCCCATCCTATCGCATC-3
E2:
Forward: 5-CGCGGATCCGGCAAGTTTAATTTTGATG-3
Reverse: 5-CCGGAATTCGATCATTCCTGAAGAGGATGG-3
E3:
Forward: 5-CGCGGATCCAAGGGGAAGTTCTATCGCTGT-3
Reverse: 5-CCGGAATTCCTCCACGCGGTGGTTGTAGAT-3
E4:
Forward: 5-CGCGGATCCTTTGGGAAAGTTGCCATGAG-3
Reverse: 5-CCGGAATTCAAAGTTGCTCCCACATGTAT-3
The E-Loops were then subcloned into an EcoR1/BamH1 sites in pGEX-6p1 vector (Amersham, Piscataway, NJ, USA). The sequence of all fusion proteins were verified by commercial sequencing (Genemed Synthesis, San Antonio, TX, USA). Recombinant vectors were screened for the presence of the E-Loops sizes of E1-264bp, E2-162bp, E3-270bp, and E4-204bp. Liquid cultures were induced by Isopropyl β-D-1-thiogalactopyranoside (IPTG) at a final concentration of 0.1–0.4 mM for 2 hours, centrifuged and lysed using lysis buffer (2.5mM ethylenediaminetetraacetic acid (EDTA) pH7.4, 2% Triton X-100, 2% [v/v] with protease inhibitor cocktail III) and pulled down using 50% slurry of Glutathione Sepharose 4B. Samples were run on 15% bis-acrylamide gels and stained with coomassie blue and pictures taken using Kodak Imager 2000R (Carestream Health Molecular Imaging, New Haven, CT, USA).
ELISA
Enzyme-linked immunosorbent assays (ELISA) were performed as described previously[29] using purified GST fusion E-loop proteins (E1-E4) and recombinant SSA/Ro52 protein as control. Ten μg/mL of each E-loops or SSA/Ro52 diluted in 1X PBS at a pH= 7.4 were incubated in 96-well ELISA plates overnight at 4 °C. The ELISA plates were blocked with 0.1% gelatin in PBS with 0.05% Tween, were incubated with sera diluted 1:10,000 in PBS with 0.05% Tween for 1 hour followed by incubation with goat anti-human IgG alkaline phosphatase conjugate (Sigma, St. Louis, MO, USA), and were developed with disodium p-nitrophenyl phosphate in diethanolamine buffer. All samples were run in triplicates. Results are expressed as the optical absorbance at 405 nm minus that of the reagent blank. A result was considered positive if it was greater than 2 SD above the averaged value obtained for normal healthy donors.
In order to confirm the specificity of the positive sera in ELISA, an inhibition immunoassay was set up by titrating the positive sera preincubated with 10 μg/ml of the extracellular loops for 3 hrs at 37°C. The samples were centrifuged at 14,000 rpm for 5 minutes and processed as in the ELISA procedure above. Concurrently, the extracellular loop S5–S6 of domain I was used in parallel to detect the optical density before and after the preincubation. All samples were done in triplicates.
Detection of Extracellular fusion protein E1 using ELISA positive sera by Western blots
E1 GST fusion protein (5 μg) was resolved on a 15% SDS/PAGE. Blots were transferred, blocked with 5% milk and incubated with the ELISA positive sera (at 1:1000 dilution) identified in the ELISA at 4°C overnight. After 5 washings with PBS-Tween, peroxidase conjugated secondary antibodies diluted 1:2000 in 1% milk in PBS were added for 1 hr at room temperature. The blots were developed using ECL reagent (Amersham, Piscataway, NJ, USA).
Expression and electrophysiological recording of α1D Ca current in tsA201 cells
tsA201 cells were grown in culture media consisting of Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum, penicillin (100 IU/ml) and streptomycin (100 μg/ml) as previously reported [18]. The cells were grown at 37 ºC, 5% CO2 and transiently transfected with 4 μg of a mix of α1D, β2a, α2δ, and lymphocyte surface antigen (CD8-a) cDNAs (in pCMV6b vector, kindly provided by Drs J. Striessnig, Innsbruck, Austria; S. Seino, Kobe, Japan) by the Ca phosphate method as described previously [30]. Cells expressing surface CD8-a fixed the beads and were visually distinguishable from non-transfected cells under the microscope. Whole-cell patch-clamp recording was performed with the Axopatch 200B (Axon Instruments, Molecular Devices, Sunnyvale, CA, USA) at 48 hours after transfection. The internal solution contained (in mmol/L): 135 CsCl, 4 MgCl2, 4 ATP, 10 HEPES, 10 EGTA, and 1 EDTA, adjusted to pH 7.2 with tetraethylammonium hydroxide (TEAOH). The bath solution contained (in mmol/L): 135 choline chloride, 1 MgCl2, 2 CaCl2, and 10 HEPES, adjusted to pH 7.4 with TEAOH. Data were analyzed with pClamp version 9.0 (Axon Instruments). For α1D ICa-L current-voltage (I-V) relations, tsA201 cells were depolarized from a holding potential of −100 mV to test potentials between −80 and 60 mV with increments of 10 mV.
Statistical Analysis
Statistical comparisons between groups were evaluated using unpaired student t-test, Unpaired t-test with Welch's correction, Mann Whitney test and one-way ANOVA as appropriate. Patch clamp data are expressed as mean ± SEM. P values less than 0.05 were considered significant.
Results
Epitope mapping in the molecular structure of α1D L-type Ca Channel: GST fusion proteins
Figure 1A shows the schematic structure of the α1D Ca channel subunit with the E-Loops in bold. The α1 subunit of α1D L-type Ca channel consists of 4 domains I-IV each with 6 transmembrane spanning segments referred to as S1-S6. Each domain has 3 extracellular loops between S1–S2, S3–S4, and S5–S6, with S5–S6 loop being the largest. The four S5–S6 loops (referred to as E-loops throughout) join together to form the pore of the channel and the selectivity filter. Successful expression of the E-loops is shown in Figure 1B using total protein extract from transformed E-coli stained with coomassie blue and in Figure 1C by Western blot using anti-GST antibody. Lanes1, 2, 3, 4 and 5 represent purified GST alone, E1, E2, E3, and E4 proteins respectively. All loops were tested in the ELISA experiments, however, since reactivity was only detected with E1 loops, further purification of E3 and E4 loops was not pursued.
Reactivity of Extracellular loop E1 with patient sera using ELISA
Three sera groups were screened by ELISA for reactivity with the extracellular loops between S5–S6 (E-loops): Sera from 118 mothers whose children have CHB and are seropositive to SSA/Ro -SSB/La antibodies in one hand and sera from 28 mothers whose children are healthy (no CHB) and are seronegative to SSA/Ro -SSB/La antibodies, and sera from 15 mothers whose children are healthy (no CHB) and are seropositive to SSA/Ro -SSB/La antibodies on the other hand. Seventeen of 118 (14.4%) CHB sera, 2 of 28 (7%) seronegative sera from mothers whose children are healthy and 0 of 15 (0%) seropositive sera from mothers with healthy children reacted with the E1 loop. The difference between the CHB and the other 2 control groups was statistically significant (p<0.05; Unpaired t test P=0.0369, Unpaired t-test with Welch's correction P=0.003, and Mann Whitney test P=0.0088). None (0%) of CHB samples or the healthy sera group reacted with GST protein alone, E2, E3, and E4. Figure 2 shows the summary of the ELISA data with the horizontal line representing the cutoff of positive sera calculated by averaging the O.D. of the healthy sera plus two standard deviations. Although there were 2 positive sera from healthy controls, their O.D. value was low relative to the CHB positive sera.
Figure 2.

ELISA results of 161 serum samples against the GST fusion proteins of the E1 loop of the α1D L-type Ca channel. Seventeen of 118 (14.4%) samples from the CHB group were ELISA positive and above the O.D. cutoff point of 0.1 which represents the average for the healthy sera plus 2 standard deviation. In contrast, sera (with anti-SSA/Ro and SSB/La antibodies) from mothers with healthy children did not react (0/15) with the E1 loop, while in the sera (without anti-SSA/Ro and SSB/La antibodies) from mothers with healthy children, only 2 of 18 (7%) were ELISA positive.
Specificity of ELISA positive sera using competition immunoassay
A significant decrease of O.D was observed when ELISA positive sera (in Figure 2) were preincubated with E1 protein, indicating the specificity of ELISA results. Figure 3 shows the representative O.D.’s from 4 control sera and 8 positive sera shown in Figure 2, which after the preincubation resulted in an O.D. below the cutoff of 0.1 O.D. The inhibition immunoassay showed O.D. values that are 48% to 72% less than the original values in the ELISA experiments. In fact, all the CHB sera except CHB serum # 4 had an O.D. value below the cutoff of 0.1 after the inhibition immunoassay. Under the same conditions, the healthy sera (without anti-SSA/Ro and -SSB/La) showed very limited reactivity (2–8%). These results confirmed that the E1 loop is able to inhibit the reactivity of the anti-SSA/Ro and -SSB/La autoantibodies by 48–72% in the CHB sera group only.
Figure 3.

Competitive ELISA assay using 4 healthy controls sera and 8 CHB samples with the highest O.D. out of the 17 ELISA positive sera in Figure 2. Sera were incubated with 10 μg of E1 loop at 37°C for 3 hours, centrifuged and used as the probe in the ELISA experiment. The O.D. of each of the 8 CHB sera decreased by more than half to a value below the cutoff of 0.1 nm.
Detection of reactivity of E1-loop with ELISA positive sera by Western blot
Further validation of the ELISA results was sought by Western Blot. Three representative samples from the ELISA positive sera recognized the E1 GST fusion protein, whereas the ELISA negative sera did not react with the E1 loop (Figure 4). The reactivity in the Western blot against the extracellular loop S5–S6 of Domain I, further validates the ELISA results.
Figure 4.

Cross reactivity of the ELISA positive sera with the E1 protein by Western blot. Five micrograms of the E1 protein was resolved on a 15% SDS-PAGE and probed using a healthy control serum and 3 ELISA positive sera with the highest O.D. from the congenial heart block (CHB).
ELISA positive sera inhibited a1D ICa-L in tsA201 cells
We next tested whether the positive sera identified by ELISA can inhibit the α1D Ca channel expressed in tsA201 cells. Mammalian tsA201 cells were used because α1D ICa-L cannot be separated biophysically or pharmacologically from α1C ICa-L in the native cardiac myocytes. ELISA positive sera inhibited the α1D ICa-L by 44.1% at −10 mV (control: −4.9 ± 1.1 pA/pF, n = 7 vs. −2.8 ± 0.6 pA/pF, P < 0.05, n = 7). Figure 5A shows current-voltage relationships for α1D ICa-L density during control and with serum #1. Similarly, the application of purified IgG from serum #1 inhibited the α1D ICa-L by 49.8% at −10 mV (control: −5.49 ± 0.5 pA/pF, n = 8 vs. −2.75 ± 0.6 pA/pF, P < 0.05, n = 8) (Figure 5B). Preincubation of the ELISA positive sera with 10 μg/ml E1 loop, prevented the inhibition of α1D ICa-L indicating the specificity of effect of ELISA positive sera on α1D ICa-L, as shown in Figure 5C. ELISA negative sera did not significantly affect α1D ICa-L (control: −5.6 ± 1.3 pA/pF, n = 6 vs. healthy sera: −4.8 ± 1.4 pA/pF, P = not significant (NS), n = 6); as shown in Figure 5D. Table 1 shows the averaged data from 2 high O.D. ELISA positive sera and IgG vs. ELISA negative sera used in the patch clamp experiments.
Figure 5.
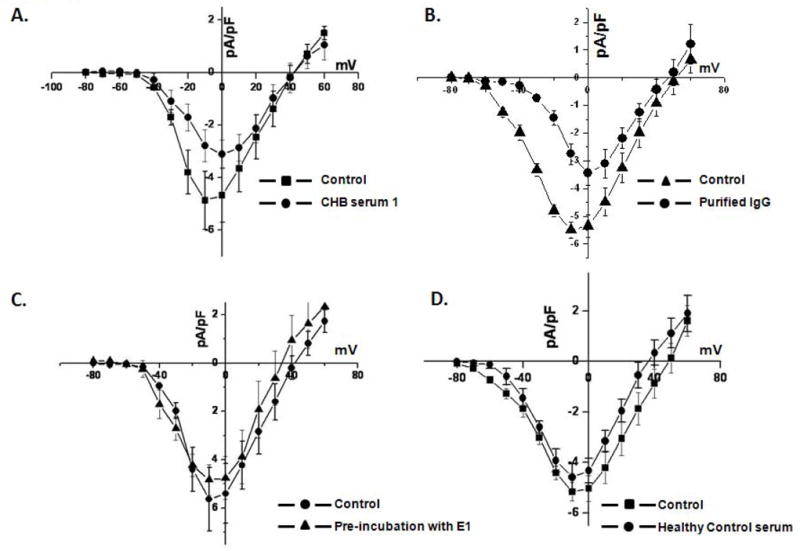
A. Effect of ELISA positive sera on the α1D ICa-L expressed in tsA201 cells. α1D ICa-L was recorded using whole cell mode of the patch clamp technique with 2 mmol/L Ca as a charge carrier. Panel A shows the current-voltage relationships for the α1D ICa-L densities during control and in the presence of ELISA positive serum 1. Panel B shows the current-voltage relationships for the α1D ICa-L densities during control and purified IgG from serum 1. Panel C shows the current-voltage relationships of the α1D ICa-L densities before and after the application of preincubated serum 1 with E1 fusion protein. Panel D shows the current voltage relationships for the α1D ICa-L densities before and after application of the healthy control serum.
Table 1.
Peak current densities for α1D transfected tsA201 cells before and after the addition of respective sera.
| No Antibody Density pA/pF | With Antibody Density pA/pF (P-value) | |
|---|---|---|
| CHB Serum 1 (n=7) | −4.9 ± 1.1 | −2.8 ± 0.6 (p=0.021) |
| CHB Serum 2 (n=6) | −5.2 ± 0.4 | −3.6 ± 0.4 (p=0.044) |
| Purified IgG (n=8) | −5.49 ± 0.5 | −2.75± 0.6 (p=0.036) |
| Healthy ELISA negative Serum (n=6) | −5.6 ± 1.3 | −4.8 ± 1.4 (p=0.49) |
Discussion
In this study, we expressed and purified GST fusion proteins corresponding to the extracellular loop S5–S6 of each of the four domains that form the pore of the Ca channel α1D subunit and tested their reactivity with sera from mothers who have children with CHB. The results demonstrate that a fraction (14.4%) of sera containing anti-SSA/Ro and SSA/La autoantibodies from mothers whose children have CHB reacted specifically with the extracellular loop of S5–S6 of the first, but not the second, third or forth domain of the α1D subunit as demonstrated by both ELISA and Western blots. Furthermore, the ELISA positive sera inhibited the expressed α1D Ca current in tsA201 cells. The presence of anti-α1D Ca channel antibodies in the sera of mothers with CHB children suggest that additional risk factors may contribute to the pathogenesis of CHB.
Relevance of α1D L-type Ca channel to CHB
In previous publications, we and others established an active [2, 28] and passive [3] animal model of CHB, reproduced the clinical complete AV block in isolated Langendorff perfused fetal hearts [2, 17], and correlated these findings with maternal anti-SSA/Ro -SSB/La autoantibodies’ specific inhibition and cross-reactivity with the L- and T-type Ca channel [18, 27] without affecting other ion channels such as Na and K channels [17, 27] indicating specificity for Ca channels. We showed that maternal antibodies’ inhibition was higher for α1C and α1D Ca channels (from 40–60%) compared to α1H T-type Ca channels (19%) [2, 27]. This is consistent with the higher (70%) homology between the E1 loop of the α1D Ca channel and that of the α1C Ca channel compared with only 48% homology between E1 loop of the α1D Ca channel and that of the α1H T-type Ca channel. Based on these experimental findings, we proposed that L-type Ca channels play a major role in the pathogenesis of CHB.
In the cardiovascular system, voltage-gated L-type Ca channels are essential for the generation of normal cardiac rhythm, for induction of rhythm propagation through the SA node, AV node and for the excitation-contraction coupling of the atrial and ventricular muscles [31]. Cardiac L-type Ca channels are heterologomeric complexes of up to four subunits: α1, β, α2/δ subunits. The α1 subunit contains the voltage sensor domain, the selectivity filter, the ion-conducting pore, and the binding sites for the Ca channel blockers [31]. Interestingly, in the heart, the expression of α1D L-type Ca channel is restricted to the SA node, AV node and atria, and is not expressed in the adult ventricles [18, 20, 21] consistent with a unique and discrete role in the supraventricular tissue. Moreover, genetic deletion of the α1D Ca channel in the mouse, leads to various degrees of AV block and sinus bradycardia [21], phenotypes similar to those found in children with CHB. In this regard, we recently demonstrated that autoantibodies from mothers with CHB children inhibited the α1D Ca current expressed in tsA201 cells by binding directly to the α1D Ca channel protein [18]. However, the exact binding site (s) of these autoantibodies on the α1D protein remained unknown.
Pathogenic Antibodies and Sarcolemmal Antigens
In this study, we show that a fraction of sera containing anti-SSA/Ro and SSA/La autoantibodies from mothers whose children have CHB reacted with the extracellular loop of S5–S6 of the first domain of α1D Ca channel protein and inhibited α1D Ca current.
Although only 17 of 118 (14.4%) sera from mothers with CHB children reacted with the E1 loop, the difference between CHB and control healthy group was statistically significant. This level of reactivity is similar to that reported by Kamel et. al. [29] against a peptide corresponding to a portion of the extracellular loop of the sarcolemmal 5-HT4 receptor. Specifically, they showed that of the 75 sera from mothers of children with CHB, 12 were reactive (16%) with the 5-HT4 peptide. Takamori et al. [32] showed that 6 sera samples (20%) from 30 Lambert-Eaton Myasthenic syndrome patients reacted to synthetic peptides against Ca channels using immunoprecipitation, 5 of which were positive for antibodies to domain II S5–S6 linker peptide. It is not completely clear why the reactivity is infrequent in this study and the other studies cited above [29, 32]. One plausible explanation for these observations is that the pathogenesis of CHB is multifactorial. In this regard, the low incidence of CHB and the universal association of anti-SSA/Ro -SSB/La antibodies with CHB suggest that maternal autoantibodies are necessary but not sufficient to cause the disease. The fact that no reactivity (0/15) with seropositive sera from mothers with healthy children was detected to the E1 loop in the ELISA experiment points to the potential involvement of other factors such us fetal, maternal and environment factors individually or in combination which may be necessary for the full expression of the disease.
Proposed Pathogenesis of CHB
Considerable progress has been made in understanding the pathogenesis by which anti-SSA/Ro and SSB/La autoantibodies mediate CHB [33–35]. One critical challenge is that SSA/Ro -SSB/La autoantigens are intracellular proteins and are inaccessible to the circulating autoantibodies under physiological conditions. Apoptosis is one proposed mechanism for the SSA/Ro -SSB/La antigen translocation to the surface of cardiomyocytes where they are accessible to maternal anti-SSA/Ro -SSB/La autoantibodies to initiate an inflammatory response and subsequent activation of cardiac fibroblasts [36]. Although apoptosis in the normal conduction tissue has been shown to occur [36], the percentage of myocytes undergoing this physiological deletion during embryogenesis remains minimal albeit apoptotic cells are normally cleared rapidly thus limiting detection. Others have also proposed that abnormal intracellular Ca homeostasis followed by cell death is another plausible mechanism in the pathogenesis of CHB [37]. Here, we propose that circulating maternal autoantibodies bind to and react with an extracellular portion of L-type Ca channels which are known to play an essential role in the electrogenesis and conduction at the SA and AV node of the human fetal heart [20]. These autoantibodies bind to the extracellular region of Ca channels and inhibit the vital entry of Ca needed for SA and AV nodes action potential genesis, conduction and excitation-coupling of the fetal myocytes during development. We speculate that this chronic inhibition may lead to internalization of the channel thereby decreasing the channel density at the cell surface which in turn may lead to the abnormal intracellular Ca levels and to the subsequent cell death. Cell death per se can lead directly to fibrosis and calcification of the conduction system and/or to the surface expression of the autoantigens SSA/Ro and SSB/La which can then be accessible to their cognate antibodies triggering an inflammatory reaction, fibrosis and calcification of the conduction system.
Therapeutic Significance
The identification of the necessary or essential factors is only part of the challenge in defining the pathology of CHB. The potential novel therapeutic development in identifying the epitope(s) that could bind to the pathogenic autoantibodies is obvious. In this study, preincubating E1 fusion protein with ELISA positive patients’ sera abolished the sera’s inhibition of α1D Ca current in tsA201 cells. These results may lead to the development of short peptides that could serve as a decoy to the maternal autoantibodies in the fetal circulation thereby preventing the interaction with the Ca channel in the fetal cardiac cells and subsequently prevent the development of CHB.
Study Limitations
The inability to detect reactivity to S5–S6 loops of domain II, III, or IV as well as the low cross reactivity to S5–S6 loop of Domain I could be due to the possibility that the small fusion proteins are unable to assume the antigenic conformation required for recognition by the autoantibodies as occurs in native cardiac cells. Plating of fusion proteins to the ELISA plates might thus be a limiting factor. The peptides could attach at different angles and expose different epitopes. We recognize that the complexity of the etiology and pathogenesis of CHB makes it challenging and difficult to account for all the clinical aspects of CHB by the “Ca channel hypothesis” or “the apoptosis hypothesis” and that further studies are warranted.
Acknowledgments
This study is supported by NIH R01 HL-077494 and VA MERIT grants to Dr. Boutjdir, VA MREP grant to Dr. Qu, NIAMS Contract No. AR-4-2271 to J.P. Buyon. We thank Dr Buyon of the Research Registry for Neonatal Lupus for kindly providing the sera.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Buyon J. Neonatal Lupus Syndrome. In: Lahita R, editor. Systemic Lupus Erythromatosus. Elsevier Academic Press; San Diego, CA: 2004. pp. 449–484. [Google Scholar]
- 2.Boutjdir M, Chen L, Zhang ZH, Tseng CE, DiDonato F, Rashbaum W, Morris A, el-Sherif N, Buyon JP. Arrhythmogenicity of IgG and anti-52-kD SSA/Ro affinity-purified antibodies from mothers of children with congenital heart block. Circ Res. 1997;80:354–62. doi: 10.1161/01.res.80.3.354. [DOI] [PubMed] [Google Scholar]
- 3.Mazel JA, El-Sherif N, Buyon J, Boutjdir M. Electrocardiographic abnormalities in a murine model injected with IgG from mothers of children with congenital heart block. Circulation. 1999;99:1914–8. doi: 10.1161/01.cir.99.14.1914. [DOI] [PubMed] [Google Scholar]
- 4.Brucato A, Cimaz R, Catelli L, Meroni P. Anti-Ro-associated sinus bradycardia in newborns. Circulation. 2000;102:E88–9. doi: 10.1161/01.cir.102.11.e88. [DOI] [PubMed] [Google Scholar]
- 5.Menon A, Silverman ED, Gow RM, Hamilton RM. Chronotropic competence of the sinus node in congenital complete heart block. Am J Cardiol. 1998;82:1119–21. A9. doi: 10.1016/s0002-9149(98)00569-4. [DOI] [PubMed] [Google Scholar]
- 6.Michaelsson M, Engle MA. Congenital complete heart block: an international study of the natural history. Cardiovasc Clin. 1972;4:85–101. [PubMed] [Google Scholar]
- 7.Buyon JP, Hiebert R, Copel J, Craft J, Friedman D, Katholi M, Lee LA, Provost TT, Reichlin M, Rider L, et al. Autoimmune-associated congenital heart block: demographics, mortality, morbidity and recurrence rates obtained from a national neonatal lupus registry. J Am Coll Cardiol. 1998;31:1658–66. doi: 10.1016/s0735-1097(98)00161-2. [DOI] [PubMed] [Google Scholar]
- 8.Hamilton RM. Disorders of heart rate and rhythm. Springer-Verlag Ltd; London: G.R. Neonatal Heart Disease. [Google Scholar]
- 9.Siren MK, Julkunen H, Kaaja R. The increasing incidence of isolated congenital heart block in Finland. J Rheumatol. 1998;25:1862–4. [PubMed] [Google Scholar]
- 10.Michaelsson M, Riesenfeld T, Jonzon A. Natural history of congenital complete atrioventricular block. Pacing Clin Electrophysiol. 1997;20:2098–101. doi: 10.1111/j.1540-8159.1997.tb03636.x. [DOI] [PubMed] [Google Scholar]
- 11.Baboonian C, Venables PJ, Booth J, Williams DG, Roffe LM, Maini RN. Virus infection induces redistribution and membrane localization of the nuclear antigen La (SS-B): a possible mechanism for autoimmunity. Clin Exp Immunol. 1989;78:454–9. [PMC free article] [PubMed] [Google Scholar]
- 12.LeFeber WP, Norris DA, Ryan SR, Huff JC, Lee LA, Kubo M, Boyce ST, Kotzin BL, Weston WL. Ultraviolet light induces binding of antibodies to selected nuclear antigens on cultured human keratinocytes. J Clin Invest. 1984;74:1545–51. doi: 10.1172/JCI111569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clancy RM, Neufing PJ, Zheng P, O'Mahony M, Nimmerjahn F, Gordon TP, Buyon JP. Impaired clearance of apoptotic cardiocytes is linked to anti-SSA/Ro and -SSB/La antibodies in the pathogenesis of congenital heart block. J Clin Invest. 2006;116:2413–22. doi: 10.1172/JCI27803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bacman S, Sterin-Borda L, Camusso JJ, Hubscher O, Arana R, Borda ES. Circulating antibodies against neurotransmitter receptor activities in children with congenital heart block and their mothers. Faseb J. 1994;8:1170–6. doi: 10.1096/fasebj.8.14.7958624. [DOI] [PubMed] [Google Scholar]
- 15.Horsfall AC, Li JM, Maini RN. Placental and fetal cardiac laminin are targets for cross-reacting autoantibodies from mothers of children with congenital heart block. J Autoimmun. 1996;9:561–8. doi: 10.1006/jaut.1996.0075. [DOI] [PubMed] [Google Scholar]
- 16.Eftekhari P, Salle L, Lezoualc'h F, Mialet J, Gastineau M, Briand JP, Isenberg DA, Fournie GJ, Argibay J, Fischmeister R, et al. Anti-SSA/Ro52 autoantibodies blocking the cardiac 5-HT4 serotoninergic receptor could explain neonatal lupus congenital heart block. Eur J Immunol. 2000;30:2782–90. doi: 10.1002/1521-4141(200010)30:10<2782::AID-IMMU2782>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 17.Boutjdir M, Chen L, Zhang ZH, Tseng CE, El-Sherif N, Buyon JP. Serum and immunoglobulin G from the mother of a child with congenital heart block induce conduction abnormalities and inhibit L-type calcium channels in a rat heart model. Pediatr Res. 1998;44:11–9. doi: 10.1203/00006450-199807000-00002. [DOI] [PubMed] [Google Scholar]
- 18.Qu Y, Baroudi G, Yue Y, Boutjdir M. Novel molecular mechanism involving alpha1D (Cav1.3) L-type calcium channel in autoimmune-associated sinus bradycardia. Circulation. 2005;111:3034–41. doi: 10.1161/CIRCULATIONAHA.104.517326. [DOI] [PubMed] [Google Scholar]
- 19.Garcia S, Nascimento JH, Bonfa E, Levy R, Oliveira SF, Tavares AV, de Carvalho AC. Cellular mechanism of the conduction abnormalities induced by serum from anti-Ro/SSA-positive patients in rabbit hearts. J Clin Invest. 1994;93:718–24. doi: 10.1172/JCI117025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mangoni ME, Couette B, Bourinet E, Platzer J, Reimer D, Striessnig J, Nargeot J. Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc Natl Acad Sci U S A. 2003;100:5543–8. doi: 10.1073/pnas.0935295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Z, He Y, Tuteja D, Xu D, Timofeyev V, Zhang Q, Glatter KA, Xu Y, Shin HS, Low R, et al. Functional roles of Cav1.3 (alpha1D) calcium channels in atria: insights gained from gene-targeted null mutant mice. Circulation. 2005;112:1936–44. doi: 10.1161/CIRCULATIONAHA.105.540070. [DOI] [PubMed] [Google Scholar]
- 22.Matthes J, Yildirim L, Wietzorrek G, Reimer D, Striessnig J, Herzig S. Disturbed atrio-ventricular conduction and normal contractile function in isolated hearts from Cav1.3-knockout mice. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:554–62. doi: 10.1007/s00210-004-0940-7. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Z, Xu Y, Song H, Rodriguez J, Tuteja D, Namkung Y, Shin HS, Chiamvimonvat N. Functional Roles of Ca(v)1.3 (alpha(1D)) calcium channel in sinoatrial nodes: insight gained using gene-targeted null mutant mice. Circ Res. 2002;90:981–7. doi: 10.1161/01.res.0000018003.14304.e2. [DOI] [PubMed] [Google Scholar]
- 24.Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, Zheng H, Striessnig J. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell. 2000;102:89–97. doi: 10.1016/s0092-8674(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 25.Lennon VA, Kryzer TJ, Griesmann GE, O'Suilleabhain PE, Windebank AJ, Woppmann A, Miljanich GP, Lambert EH. Calcium-channel antibodies in the Lambert-Eaton syndrome and other paraneoplastic syndromes. N Engl J Med. 1995;332:1467–74. doi: 10.1056/NEJM199506013322203. [DOI] [PubMed] [Google Scholar]
- 26.Smith RG, Hamilton S, Hofmann F, Schneider T, Nastainczyk W, Birnbaumer L, Stefani E, Appel SH. Serum antibodies to L-type calcium channels in patients with amyotrophic lateral sclerosis. N Engl J Med. 1992;327:1721–8. doi: 10.1056/NEJM199212103272405. [DOI] [PubMed] [Google Scholar]
- 27.Xiao GQ, Hu K, Boutjdir M. Direct inhibition of expressed cardiac L- and T-type calcium channels by IgG from mothers whose children have congenital heart block. Circulation. 2001;103:1599–604. doi: 10.1161/01.cir.103.11.1599. [DOI] [PubMed] [Google Scholar]
- 28.Miranda-Carus ME, Boutjdir M, Tseng CE, DiDonato F, Chan EK, Buyon JP. Induction of antibodies reactive with SSA/Ro-SSB/La and development of congenital heart block in a murine model. J Immunol. 1998;161:5886–92. [PubMed] [Google Scholar]
- 29.Kamel R, Eftekhari P, Clancy R, Buyon JP, Hoebeke J. Autoantibodies against the serotoninergic 5-HT4 receptor and congenital heart block: a reassessment. J Autoimmun. 2005;25:72–6. doi: 10.1016/j.jaut.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 30.Qu Y, Baroudi G, Yue Y, El-Sherif N, Boutjdir M. Localization and modulation of {alpha}1D (Cav1.3) L-type Ca channel by protein kinase A. Am J Physiol Heart Circ Physiol. 2005;288:H2123–30. doi: 10.1152/ajpheart.01023.2004. [DOI] [PubMed] [Google Scholar]
- 31.Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev. 2005;57:411–25. doi: 10.1124/pr.57.4.5. [DOI] [PubMed] [Google Scholar]
- 32.Takamori M. Lambert-Eaton myasthenic syndrome as an autoimmune calcium channelopathy. Biochem Biophys Res Commun. 2004;322:1347–51. doi: 10.1016/j.bbrc.2004.08.040. [DOI] [PubMed] [Google Scholar]
- 33.Boutjdir M. Molecular and ionic basis of congenital complete heart block. Trends Cardiovasc Med. 2000;10:114–22. doi: 10.1016/s1050-1738(00)00059-1. [DOI] [PubMed] [Google Scholar]
- 34.Friedman DM, Rupel A, Buyon JP. Epidemiology, etiology, detection, and treatment of autoantibody-associated congenital heart block in neonatal lupus. Curr Rheumatol Rep. 2007;9:101–8. doi: 10.1007/s11926-007-0003-4. [DOI] [PubMed] [Google Scholar]
- 35.Wahren-Herlenius M, Sonesson SE. Specificity and effector mechanisms of autoantibodies in congenital heart block. Curr Opin Immunol. 2006;18:690–6. doi: 10.1016/j.coi.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 36.Clancy RM, Buyon JP. Autoimmune-associated congenital heart block: dissecting the cascade from immunologic insult to relentless fibrosis. Anat Rec A Discov Mol Cell Evol Biol. 2004;280:1027–35. doi: 10.1002/ar.a.20072. [DOI] [PubMed] [Google Scholar]
- 37.Salomonsson S, Sonesson SE, Ottosson L, Muhallab S, Olsson T, Sunnerhagen M, Kuchroo VK, Thoren P, Herlenius E, Wahren-Herlenius M. Ro/SSA autoantibodies directly bind cardiomyocytes, disturb calcium homeostasis, and mediate congenital heart block. J Exp Med. 2005;201:11–7. doi: 10.1084/jem.20041859. [DOI] [PMC free article] [PubMed] [Google Scholar]