

Abstract
Vascular endothelial growth factor (VEGF) signaling is critical for tumor angiogenesis. However, therapies based on the inhibition of VEGF receptors have shown modest results in patients with cancer. Surprisingly little is known about mechanisms underlying the regulation of VEGFR1 and VEGFR2 expression, the main targets of these drugs. Here, analysis of tissue microarrays revealed an inversely reciprocal pattern of VEGF receptor regulation in the endothelium of human squamous cell carcinomas (high VEGFR1, low VEGFR2), as compared to the endothelium of control tissues (low VEGFR1, high VEGFR2). Mechanistic studies demonstrated that VEGF signals through the Akt/ERK pathway to inhibit constitutive ubiquitination and induce rapid VEGFR1 accumulation in endothelial cells. Surprisingly, VEGFR1 is primarily localized in the nucleus of endothelial cells. In contrast, VEGF signals through the JNK/c-Jun pathway to induce endocytosis, nuclear translocation, and downregulation of VEGFR2 via ubiquitination. VEGFR1 signaling is required for endothelial cell survival, while VEGFR2 regulates capillary tube formation. Notably, the antiangiogenic effect of Bevacizumab (anti-VEGF antibody) requires the normalization of VEGFR1 and VEGFR2 levels in human squamous cell carcinomas vascularized with human blood vessels in immunodeficient mice. Collectively, this work demonstrate that VEGF-induced angiogenesis requires the inverse regulation of VEGFR1 and VEGFR2 in tumor-associated endothelial cells.
Keywords: Angiogenesis, Apoptosis, Receptor Tyrosine Kinase, Differentiation, Tumor Microenvironment
Introduction
Vascular endothelial growth factor (VEGF) is a key regulator of physiologic angiogenesis, and plays a major role in the pathobiology of cancer (1-3). Lack of one vegf allele is sufficient to cause death early in embryonic development due to abnormal vessel development (4,5) and autocrine VEGF signaling is required to maintain vascular homeostasis in adult healthy tissues (6). The recognition of the prominent role for VEGF in cancer led to the development of antiangiogenic therapies targeting this pathway (3,7). Indeed, these therapies have become standard of care for several malignancies, including metastatic colorectal cancer (3,8). However, resistance to antiangiogenic therapies has been described in xenograft tumor models and in clinical trials (9-11). The understanding of mechanisms underlying the regulation and function of VEGF receptors is imperative to determine how vascular homeostasis is maintained in health and how these receptors contribute to the pathobiology of angiogenesis dependent diseases, such as cancer. Furthermore, the understanding of the cellular localization and regulation of these receptors in healthy tissues and in tumors is critical for the improvement of the outcomes of therapies targeted to the inhibition of VEGF signaling.
The VEGF receptors, VEGFR1 and VEGFR2, are structurally related members of the receptor tyrosine kinase (RTK) family that mediate critical signaling pathways in endothelial cells (12-14). VEGFR1 is a receptor for VEGF-A, VEGF-B and PlGF, and is believed to have low kinase activity (15,16). In contrast, VEGFR2 is considered a highly active receptor that mediates broad signaling cascades and regulates many endothelial cell functions including proliferation, migration, and differentiation (15,17). Ligand-mediated degradation of RTKs is an important regulatory step of signaling transduction. It is known that epidermal growth factor (EGF) causes downregulation of EGFR by ubiquitination (18). VEGF dependent VEGFR2 downregulation appears to involve endocytosis in either bovine aortic endothelial cells or human endothelial cells (19,20). Notably, autophosphorylation of the kinase domain of VEGFR2 is necessary for receptor internalization (21). While ligand-mediated downregulation of VEGFR2 has been described, mechanisms underlying the reciprocal upregulation of VEGFR1 in endothelial cells exposed to VEGF and the impact of this reversal of expression levels to endothelial cell proliferation, sprouting, survival, and tumor angiogenesis have not been characterized. In this study, we showed that VEGF-induced upregulation of VEGFR1 is dependent upon Akt and ERK signaling, while the downregulation of VEGFR2 is mediated by the JNK-c-Jun pathway. Gene silencing and functional studies demonstrated that VEGFR1 signaling is critical for endothelial cell survival, while VEGFR2 signaling is required for capillary tube formation. Notably, the anti-angiogenic effect of the monoclonal anti-VEGF antibody bevacizumab in vivo correlates with the reversal of VEGFR1 and VEGFR2 levels in the vascular endothelial cells of squamous cell carcinomas.
Results
VEGF reverses VEGFR1 and VEGFR2 protein levels in endothelial cells
A tissue microarray containing a panel of human squamous cell carcinomas (22) was used to evaluate VEGFR1 and VEGFR2 protein levels in tumor-associated endothelial cells. We found more VEGFR1-positive blood vessels in human squamous cell carcinomas than in control tissues, i.e. oral mucosa (Figure 1a and b). In contrast, VEGFR2-positive vessels were more prevalent in oral mucosa than within the tumor environment (Figure 1a and b). Notably, while in human tumors the majority of blood vessels are VEGFR1-positive, in the oral mucosa most vessels are VEGFR2-positive (Figure 1b). As expected, human squamous cell carcinomas strongly express VEGF (Figure 1a). To evaluate the spatial relationship between VEGF receptor expression and VEGF, we performed immunostaining of serial and sequential tissue sections from xenografted human squamous cell carcinomas grown in SCID mice. Confirming the data from the tissue microarrays, we observed strong immunostaining for VEGF and VEGFR1, and weaker staining for VEGFR2 in the xenograft tumors (Supplementary Figure 1). To begin to understand mechanisms involved in the regulation of VEGFR1 and VEGFR2 expression, we exposed primary human dermal microvascular endothelial cells (HDMEC) to increasing concentrations of fetal bovine serum (FBS). While FBS downregulated VEGFR2, it potently induced VEGFR1 in HDMEC (Figure 1c). Knowing that ligands exert regulatory functions on their own receptors, that FBS contains VEGF, and that human squamous cell carcinomas express high levels of VEGF, we evaluated the effect of recombinant VEGF on the regulation of VEGFR1 and VEGFR2. VEGF induced expression of VEGFR1 while downregulating VEGFR2 in two primary endothelial cell types, i.e. HDMEC and human pulmonary endothelial cells (HPEC) (Figure 1d). Treatment with anti-VEGF antibody inhibited VEGF-regulatory effects on VEGFR1 and VEGFR2 in a dose-dependent manner, demonstrating specificity of responses (Figure 1e). To further verify the specificity of VEGF receptor regulation, we evaluated the effect of EGF, which is known to downregulate its tyrosine kinase receptor (18). EGF downregulated EGFR in squamous cell carcinoma cells (Figure 1g), but had no effect on the expression levels of VEGFR1 and VEGFR2 in endothelial cells (Figure 1f and h). Likewise, the VEGF receptor ligand placental growth factor (PlGF), and the angiogenic cytokine IL-6 (Interleukin-6), had no effect on VEGFR1 or VEGFR2 expression (Figure 1f and i).
Figure 1.

VEGF inversely regulates VEGFR1 and VEGFR2 in endothelial cells. (a) Immunohistochemical analysis of VEGFR1, VEGFR2, and VEGF expression (brown staining) in representative specimens from human squamous cell carcinomas or normal human oral mucosa. Arrows point to blood vessels. Scale bars: left panels, 50 μm (100×); right panels, 20 μm (400×). (b) Quantification of VEGFR1 and VEGFR2 positive blood vessels (100×) in normal human oral mucosa and human squamous cell carcinomas (HSCC) from the human tissue microarrays used in panel (a). * Number of VEGFR1-positive versus VEGFR2-positive vessels in the oral mucosa (p<0.001); ** Number of VEGFR1-positive versus VEGFR2-positive vessels in HSCC (p<0.001); *** VEGFR1-positive in oral mucosa versus VEGFR1-positive vessels in HSCC (p<0.001); **** VEGFR2-positive in oral mucosa versus VEGFR2-positive vessels in HSCC (p=0.005). (c) Primary human dermal microvascular endothelial cells (HDMEC) were serum-starved overnight and then were incubated with increasing concentrations of FBS for 24 hours. (d) HDMEC or primary human pulmonary endothelial cells (HPEC) were serum-starved overnight, and treated with 50 ng/ml VEGF165 for indicated time points. (e) HDMEC were starved overnight, pre-incubated with 0-5 μg/ml anti-VEGF neutralizing antibody for 1 hour, then treated with 50 ng/ml VEGF165 for additional 2 hours in presence of the anti-VEGF antibody. Isotype-matched IgG was used as negative control. (f and g) HDMEC (f) or UM-SCC-1 cells (g) were serum-starved and treated with 50 ng/ml EGF, 50 ng/ml VEGF165, or 20 ng/ml IL-6 for 24 hours. Alternatively, cells were cultured in 5% FBS. (h and i) HDMEC were treated with 50 ng/ml EGF, 50 ng/ml VEGF165 (h), 50 ng/ml PlGF (i), or left untreated for the indicated time points. VEGFR1, VEGFR2, and EGFR expression was detected by Western blots. (c-i) The molecular weight for the VEGFR1 bands is approximately 200 kDa, and for the VEGFR2 bands is approximately 220 kDa. Numbers depicted in Western blots represent quantification of band density normalized initially by GAPDH, and then normalized by untreated controls.
The reversal of VEGFR1 and VEGFR2 protein levels in endothelial cells happened within minutes of exposure to VEGF (Figure 1h), which suggested that this process was not regulated at the transcriptional level. Reverse transcriptase polymerase chain reaction (RT-PCR) and real-time PCR (qPCR) analyses confirmed this hypothesis showing that VEGF had no effect on VEGFR1 or VEGFR2 mRNA within the same timeframe (Figure 2a and b). We then reasoned that intracellular receptor trafficking and differential regulation of ubiquitination might be involved in the process of VEGF-induced regulation of VEGFR1 and VEGFR2. Confocal microscopy showed that VEGFR2 is translocated to the nucleus within 30 minutes of VEGF treatment (Figure 2c). Surprisingly, most of the VEGFR1 is found in the nucleus of untreated or VEGF-treated HDMEC (Figure 2c). Cell fractionation studies confirmed that VEGFR1 is mainly expressed in the nucleus, while VEGFR2 is in the cytoplasmic/cell membrane fraction in untreated cells (Figure 2d). VEGFR1 remained in the nucleus upon treatment with VEGF, while VEGFR2 translocated from the cytoplasmic/cell membrane fraction to the nucleus (Figure 2d). EGF and IL-6 did not affect VEGFR2 localization (Figure 2e). Treatment with Concanavalin-A revealed that VEGF-induced VEGFR2 nuclear translocation depends on endocytosis (Figure 2f). However, blockade of nuclear translocation did not prevent VEGF-induced VEGFR2 downregulation (Figure 2f). Interestingly, only the high molecular weight VEGFR2 isoform translocated into the nucleus upon treatment with VEGF (Figure 2d-g). To determine the glycosylation state of the VEGFR2 isoform translocated to the nucleus, we treated cell lysates with an enzymatic cocktail that eliminates all N-linked and most O-linked forms. Interestingly, we observed that both VEGFR2 isoforms are glycosylated (Figure 2g), suggesting that the nuclear translocation is not differentially regulated by VEGFR2's glycosylation.
Figure 2.

VEGFR1 is localized in the nucleus of endothelial cells, while VEGFR2 undergoes endocytosis and nuclear translocation upon exposure to VEGF. (a and b) Serum-starved HDMEC were treated with 50 ng/ml EGF, 50 ng/ml VEGF165, 20 ng/ml IL-6, or left untreated for 1 hour. RT-PCR (a) and real-time PCR (b) were performed to evaluate VEGFR1 and VEGFR2 mRNA expression levels. Data for the real-time PCR was normalized against untreated controls. (c) Representative confocal microscopy images of VEGFR1 and VEGFR2 in untreated HDMEC or HDMEC exposed to 50 ng/ml rhVEGF165 for 30 min. VEGFR1 and VEGFR2 were detected with FITC-conjugated antibodies (green color), and DAPI was used for identification of cell nuclei (blue color). The exposure time for the panels depicting VEGFR2 expression in VEGF-treated HDMEC was increased to allow for visualization of the staining and evaluation of the cellular localization of this receptor. (d-f) VEGFR1 and VEGFR2 sub-cellular distribution (CE, cytoplasmic/cell membrane extract; NE, nuclear extract) were detected by cell fractionation followed by Western blots. GAPDH was used as control for cytoplasmic/cell membrane fraction, and hnRNP as control for nuclear fraction. (d) HDMEC were treated with 0 or 50 ng/ml VEGF165 for 30 min. (e) HDMEC were treated with 50 ng/ml EGF, 50 ng/ml rhVEGF165, 20 ng/ml IL-6, or left untreated for 30 min. Expression of phospho-STAT3 in HDMEC treated with IL-6 was used as positive control for nuclear translocation. (f) Starved HDMEC were preincubated with the inhibitor of endocytosis Concanavalin A (ConA) for 30 min, then treated with 50 ng/ml VEGF165 in presence of 5 μg/ml ConA for additional 30 min. (g) HDMEC were starved overnight, and treated with 50 ng/ml VEGF165 for 20 min to induce VEGFR2 translocation. VEGFR2 was deglycosylated with an enzyme cocktail constituted of PNGase, sialidase, O-Glycosidase, β-Galactosidase and Glucosaminidase. Western blot with rabbit anti-VEGFR2 antibody was performed from both nuclear (NE) and cytoplasmic (CE) extracts.
VEGF-induced VEGFR2 downregulation requires signaling through the JNK-c-Jun pathway and activity of the ubiquitin/proteasome system
To evaluate the role of the ubiquitin-proteasome system on VEGF-induced VEGFR2 downregulation, HDMEC were exposed to VEGF in presence of specific inhibitors. We observed that inhibition of ubiquitin or proteasome activity partially prevented VEGF-induced VEGFR2 downregulation, but not nuclear translocation (Figure 3a and b). It is known that JNK targets ubiquitination of regulatory proteins (23). HDMEC exposed to VEGF showed strong phosphorylation of JNK-2, and to some extent JNK-1, resulting in c-Jun activation (Figure 3c and d). Notably, phosphorylation of JNK correlated with a marked decrease in VEGFR2 protein levels (Figure 3c and d). Indeed, blockade of JNK activity inhibited VEGF-mediated VEGFR2 downregulation and partially blocked VEGFR1 upregulation in HDMEC and HPEC (Figure 3e and f). However, inhibition of JNK activity did not affect VEGF-induced VEGFR2 nuclear translocation (Figure 3g). We also evaluated two additional means of directly inducing JNK activity, UV radiation and Anisomycin (24). Both strategies were sufficient to downregulate VEGFR2 protein levels in endothelial cells, bypassing the requirement for VEGF (Supplementary Figure 2a and b). Of note, blockade of JNK and c-Jun phosphorylation with SP600125 prevented Anisomycin-induced downregulation of VEGFR2 (Supplementary Figure 2b). Taken together, these data demonstrated that signaling through the JNK/c-Jun pathway and activity of the ubiquitin-proteasome system are necessary for VEGF-induced downregulation of VEGFR2 protein levels in primary endothelial cells.
Figure 3.

Signaling pathways required for VEGF-induced inverse regulation of VEGFR1 and VEGFR2 in primary endothelial cells. (a) Starved HDMEC were pretreated with 5 μM ubiquitin aldehyde inhibitor (Ub) or 10 μM proteasome inhibitor MG132 for 1 hour, and then treated with 50 ng/ml VEGF for 30 min in presence of Ub or MG132. VEGFR2 expression was detected by Western blot. (b) Cell fractionation was used to determine sub-cellular distribution of VEGFR2 in HDMEC treated with 50 ng/ml VEGF165 in presence of Ub or MG132. (c-g) Western blots to evaluate the effect of JNK-c-Jun signaling on expression of VEGFR1 and VEGFR2. (c,d) HDMEC were exposed to 50 ng/ml VEGF165 for times indicated (c), or for 1 hour (d). HDMEC (e and g) or HPEC (f) were preincubated for 1 hour with 10 μM JNK inhibitor SP600125, followed by treatment with 50 ng/ml VEGF165 in presence of SP600125. (h) Starved HDMEC were treated with 10 μM MG132 for 2 hours. Sub-cellular distribution of VEGFR1 was detected by Western blot. (i) HDMEC were pretreated with 5 μM Ub for 1 hour, and treated with 50 ng/ml VEGF165 in presence of Ub for 30 min. VEGFR1 expression was detected by Western blots. (j) HDMEC were treated with 50 ng/ml VEGF165 for indicated time points. Western blots were performed for VEGFR1, VEGFR2, P-Akt, Akt, P-ERK1/2 and ERK1/2. (k and l) HDMEC were preincubated with 20 μM LY294002 (k) or 20 μM PD98059 (l) for 1 hour, followed by treatment with 50 ng/ml VEGF165 in presence of inhibitors for indicated time points. Western blots were performed to evaluate VEGFR1, VEGFR2, P-Akt, Akt, P-ERK1/2 and ERK1/2 expression.
VEGF signals through PI3k-Akt to inhibit ubiquitination and rapidly increase VEGFR1 levels in endothelial cells
Since VEGFR1 expression was upregulated within minutes of exposure to VEGF, we hypothesized that VEGFR1 was continuously being degraded by the ubiquitin-proteasome system to maintain its homeostatic levels. This hypothesis was confirmed by the observation that inhibition of either ubiquitin or proteasome activity was sufficient to rapidly induce VEGFR1 expression (Figure 3h and i). Then, we evaluated the function of the PI3K/Akt and ERK pathways in VEGF-induced VEGFR1 expression in HDMEC (Figure 3j-l). Inhibition of either Akt or ERK1/2 phosphorylation suppressed VEGF-mediated VEGFR1 upregulation, but had no effect on VEGFR2 protein levels (Figure 3k and l).
VEGFR1 signaling is required for the anti-apoptotic effect of VEGF, while VEGFR2 signaling is required for VEGF-induced capillary tube formation
It is known that VEGF protects endothelial cells against apoptosis induced by growth factor starvation (Figure 4a) (25). To evaluate the relative role of VEGFR1 and VEGFR2 in endothelial cell survival, we used shRNA-mediated gene silencing with lentiviral vectors (Figure 4b). Unexpectedly, shRNA-mediated downregulation of VEGFR2 resulted in upregulation of VEGFR1 protein levels, while downregulation of VEGFR1 did not affect VEGFR2 protein (Figure 4b). VEGFR1 knockdown had a stronger impact on endothelial cell density than VEGFR2 silencing (Figure 4c). These findings correlated with a 2-3-fold increase in apoptosis of serum-starved VEGFR1-knockdown endothelial cells, while VEGFR2 knockdown had no effect on cell survival (Figure 4d). To confirm these data using a second apoptotic assay, we analyzed activity of the effector of apoptosis caspase-3 (Figure 4f and g). We observed that VEGFR1, but not VEGFR2, knockdown is sufficient to enhance caspase-3 activity in starved endothelial cells (Figure 4f and g). Knockdown of either VEGFR1 or VEGFR2 did not have any effect on endothelial cell cycle (Figure 4e). Notably, with complete endothelial cell medium (5% FBS), we observed a decrease in capillary tube formation only when VEGFR2 expression was knockdown in endothelial cells (Figure 4h and i). However, under a low serum concentration (2.5%), downregulation of either receptor led to a significant inhibition of capillary tube formation (Figure 4h and i). Together, these data demonstrate that while VEGFR1 signaling primarily regulates endothelial cell survival, VEGFR2 signaling is involved in vascular differentiation and capillary-like tube formation.
Figure 4.

VEGFR1 signaling is critical for endothelial cell survival, while VEGFR2 regulates capillary tube formation. (a) HDMEC cells were cultured in serum-free medium supplemented with 0 or 50 ng/ml VEGF165 for 4 days. Cell number was determined every day under light microscopy at 100×. (b) HDMEC were stably transduced with lentiviruses encoding two different shRNA sequences (“a” & “b”) for VEGFR1 and VEGFR2, or a scrambled sequence control (shRNA-C), and selected with puromycin. VEGFR1 and VEGFR2 expression were evaluated by Western blots. (c) HDMEC transduced with lentiviruses encoding shRNA-VEGFR1, shRNA-VEGFR2, or shRNA-C were cultured for 3 days. Cell number was determined every day under light microscopy at 100×. (d and e) HDMEC stably transduced with shRNA-VEGFR1, shRNA-VEGFR2, or shRNA-C were cultured for 48 hours, and retrieved for flow cytometric analysis after staining with propidium iodide. (d) Apoptotic cells were identified as sub-G0/G1 populations. Data is expressed as percentage of apoptotic cells. (e) Percentage of viable cells in G1, S or G2/M phase of the cell cycle. (f and g) shRNA-transduced HDMEC were cultured in EGM2-MV for 48 hours and caspase-3 activity was determined at 37°C by the conversion of the substrate, Ac-DEVD-AMC. Caspase-3 inhibitor, Ac-DEVD-CHO, was used to verify specificity of reaction, and recombinant human caspase-3 used as a positive control. (h and i) Capillary-like tube formation assay with HDMEC cultured in growth factor reduced Matrigel for 24 hours. (h) Graph depicting the number of capillary branches and (i) representative photomicrographs (40×) of HDMEC transduced with shRNA-VEGFR1, shRNA-VEGFR2, or shRNA-C and cultured with complete EGM2-MV medium (containing 5% FBS), or serum-decreased medium (containing 2.5% FBS). One asterisk indicates p<0.05, as compared to shRNA-Control transduced cells. Two asterisks depict p<0.05 when endothelial cells transduced with shRNA-VEGFR1 are compared to cells transduced with shRNA-VEGFR2.
VEGF was shown to enhance endothelial cell survival by inducing expression of Bcl-2 (Figure 5a) (25). We observed that VEGFR1 signaling is required for VEGF-induced upregulation of Bcl-2 expression in endothelial cells (Figure 5b). Surprisingly, VEGFR2 knockdown correlated with baseline increase in Bcl-2 expression and c-Jun phosphorylation in unstimulated endothelial cells (Figure 5c and d). However, VEGF does not induce Bcl-2 or phospho-c-Jun levels above baseline in VEGFR2-knockdown HDMEC (Figure 5c and d). JNK-c-Jun signaling is known to control expression of proteins involved in the regulation of cell survival (26). Here, we observed that blockade of c-Jun phosphorylation inhibited VEGF-induced Bcl-2 expression, survival, and the angiogenic potential of HDMEC (Figure 5e-g).
Figure 5.
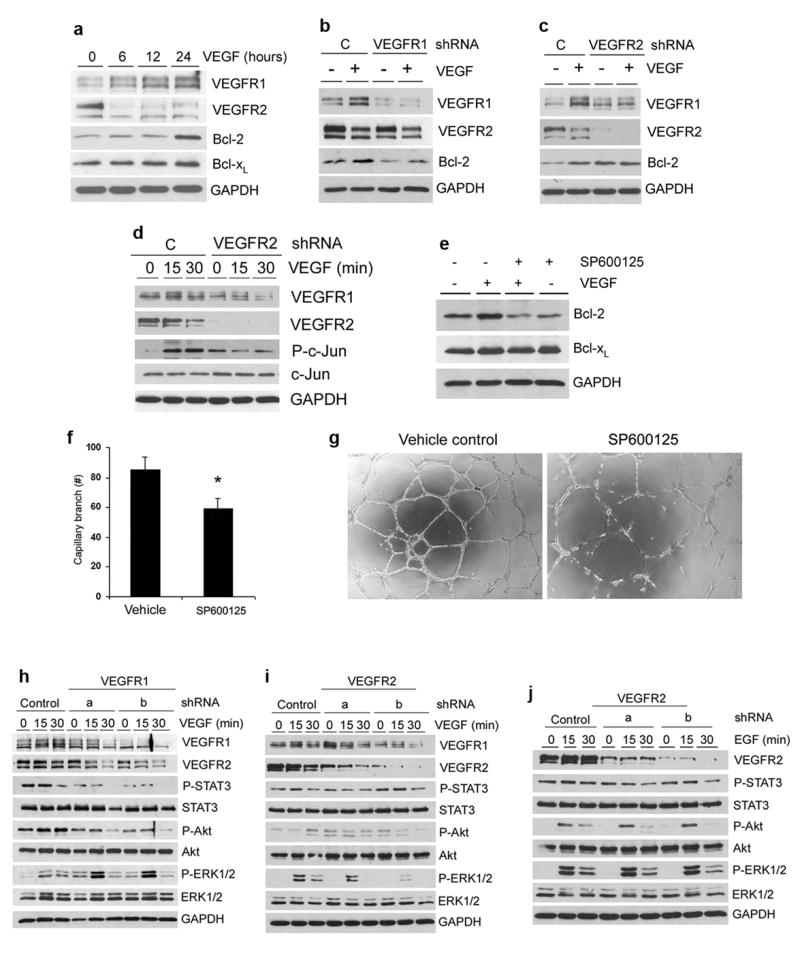
VEGF signals through the JNK-c-Jun axis to induce Bcl-2 and enhance the angiogenic potential of endothelial cells. (a) Starved HDMEC were incubated with 50 ng/ml VEGF for indicated time points. VEGFR1, VEGFR2 and Bcl-2 expression was determined by Western blot. (b and c) HDMEC stably transduced with shRNA-VEGFR1 (b) or shRNA-VEGFR2 (c) were cultured with 50 ng/ml VEGF165 for 24 hours. VEGFR1, VEGFR2 and Bcl-2 expression was detected by Western blot. (d) HDMEC stably transduced with shRNA-VEGFR2 were treated with 50 ng/ml VEGF165 for indicated time points. P-c-Jun and c-Jun expression levels were detected by Western blot. (e) HDMEC cells were preincubated with 5 μM SP600125 for 1 hour, then treated with 50 ng/ml VEGF in presence of SP600125 overnight. Bcl-2 and Bcl-xL expression levels were detected by Western blot. (f) Graph depicting the number of capillary branches and (g) representative photomicrographs (40×) of HDMEC cultured in presence of 0 or 5 μM SP600125 for 24 hours. Data are represented as mean +/- SD from three independent experiments. Asterisk indicates p<0.05. VEGFR1 signaling is required for maintaining basal activity of STAT3 and Akt, while VEGFR2 is required for VEGF-induced ERK phosphorylation in endothelial cells. (h-j) HDMEC stably transduced with shRNA-VEGFR1 (h), shRNA-VEGFR2 (i and j) or control shRNA-C were cultured in medium supplemented with 50 ng/ml VEGF165 (h and i) or 50 ng/ml EGF (j). P-STAT3, STAT3, P-Akt, Akt, P-ERK1/2, and ERK1/2 expression levels were detected by Western blots.
It is known that STAT3, Akt, and ERK mediate survival and differentiation signals in endothelial cells (27-29). We observed that the basal levels of phosphorylation of STAT3 and Akt are reduced in VEGFR1 knockdown endothelial cells, but not in VEGFR2 knockdown cells (Figure 5h and i). Notably, VEGF's ability to induce Akt phosphorylation involves signaling through VEGFR1 (Figure 5h). VEGFR1 knockdown (but not VEGFR2) increased the basal levels of phosphorylated ERK (Figure 5h). VEGFR1 knockdown further enhanced the induction of ERK phosphorylation in response to a 15-minute exposure to VEGF, as compared with vector control endothelial cells treated with VEGF (Figure 5h). However, the level of ERK phosphorylation in response to VEGF in VEGFR2 knockdown cells was similar (shRNA-VEGFR2a), or lower (shRNA-VEGFR2b), than in control cells (Figure 5i). Knockdown of either VEGFR1 (data not shown) or VEGFR2 had no effect on EGF-mediated signaling, confirming the specificity of the responses to VEGF (Figure 5j). Functional assays revealed that PI3K/Akt signaling is required for capillary tube formation (Supplementary Figure 3). STAT3 inhibition mediated similar effects, but the decrease in the number of capillary tubes was not as pronounced. In contrast, blockade of ERK1/2 signaling inhibited capillary tube formation without a comparable decrease in cell number (Supplementary Figure 3). Together, these data demonstrated that VEGFR1 signaling is involved in the regulation of STAT3 and Akt homeostatic activity, which have been shown to be required for endothelial cell survival (30,31). On the other hand, VEGF signals through VEGFR2 to activate ERK1/2, which is required for capillary tube formation (29).
Bevacizumab normalizes VEGFR1 and VEGFR2 protein levels in tumor-associated endothelial cells
To evaluate mechanisms underlying the regulation of VEGFR1 and VEGFR2 protein levels mediated by tumor cells in vitro, HDMEC were exposed to conditioned medium (CM) of VEGF-expressing human squamous cell carcinoma cells (UM-SCC-1) (Figure 6a). The expected increase in endothelial cell number and capillary tube formation mediated by UM-SCC-1 CM correlated with upregulation of VEGFR1 and downregulation of VEGFR2 (Figure 6b-d). UM-SCC-1 CM induced phosphorylation of c-Jun, Akt, ERK1/2, and STAT3, and upregulation of Bcl-2 expression in endothelial cells (Figure 6d-f and 7c). VEGFR1 signaling participates in UM-SCC-1-induced phosphorylation of STAT3 and Akt, but not in ERK1/2 phosphorylation (Figure 6f). In contrast, VEGFR2 signaling is involved in UM-SCC-1-induced ERK1/2 phosphorylation, but has no effect on STAT3 and Akt activity (Figure 6g).
Figure 6.

Effect of the growth factor milieu secreted by squamous cell carcinomas on VEGFR1 and VEGFR2 expression and signaling in endothelial cells. (a) ELISA for secreted VEGF165 in HDMEC and UM-SCC-1 cells. (b) Graph depicting the number of cells when HDMEC were cultured with UM-SCC-1 conditioned medium (CM; no FBS) or with control medium (DMEM; no FBS). (c) Capillary tube assay with HDMEC cultured in growth factor reduced Matrigel for 24 hours. HDMEC were cultured with UM-SCC-1 conditioned medium (CM) or control DMEM medium. Representative images of capillary tubes at 40×. (d) HDMEC were treated with UM-SCC-1 conditioned medium (CM) for the indicated time points, and VEGFR1, VEGFR2, P-c-Jun, and c-Jun expression levels were evaluated by Western blots. (e-g) HDMEC (e), HDMEC transduced with shRNA-VEGFR1 (f), HDMEC-transduced with shRNA-VEGFR2 (g), or shRNA-C controls were starved overnight, and treated with UM-SCC-1 conditioned medium for indicated time points. VEGFR1, VEGFR2, P-STAT3, STAT3, P-Akt, Akt, P-ERK1/2, and ERK1/2 were detected by Western blots. Asterisk indicates p<0.05.
Figure 7.

The antiangiogenic activity of bevacizumab correlates with the normalization of VEGFR1 and VEGFR2 expression levels in xenografted squamous cell carcinomas. (a) HDMEC were cultured without fetal bovine serum (FBS), and treated with 0-100 μg/ml Bevacizumab for 24 hours. VEGFR1 and VEGFR2 expression were detected by Western blot. (b-c) HDMEC were preincubated with Bevacizumab for 2 hours, and then treated with UM-SCC-1 (b) or OSCC3 (c) conditioned medium (CM) for additional 24 hours in presence of increasing concentrations of Bevacizumab. VEGFR1, VEGFR2 and Bcl-2 were detected by Western blot. (d) The endogenous expression levels of VEGFR1 and VEGFR2 were evaluated in untreated HDMEC, UM-SCC-1, and UM-SCC-11B by Western blot. VE-cadherin and E-cadherin were used as markers of endothelial cells and cells from epithelial origin, respectively. (e-g) Mice bearing engineered human tumors vascularized with human blood vessels received two i.p. injections of 15 or 30 mg/kg Bevacizumab or vehicle control with a one-day interval between injections. The day after completion of treatment, tumors were retrieved, homogenized, and VEGFR1 and VEGFR2 expression was evaluated by Western blot (e). (f) Graph depicting the number of microvessels per microscopic power field (200×) from 8 tumors per experimental condition. Asterisk represents p<0.05. (g) Representative photomicrographs (400×) of tumors treated with Bevacizumab or vehicle treated controls. Tissue sections were immunostained for Factor VIII to identify blood vessels (brown, DAB-positive cells).
To understand the effect of VEGF on the regulation of VEGFR1 and VEGFR2 in tumor-associated endothelial cells, we used the humanized anti-VEGF monoclonal antibody bevacizumab (Avastin) that is approved for treatment of several cancers (3). In absence of FBS, bevacizumab had no effect on VEGFR1 or VEGFR2 protein levels (Figure 7a). Bevacizumab inhibited the effect of FBS or tumor cell CM (UM-SCC-1 and OSCC3), and restored baseline levels of VEGFR1, VEGFR2, and Bcl-2 in human endothelial cells in vitro (Figure 7b and c; Supplementary Figure 4). To evaluate the effect of bevacizumab on VEGFR1 and VEGFR2 protein levels in vivo, we engineered human tumors vascularized with human blood vessels in immunodeficient mice, as described (32,33). UM-SCC-1 cells were selected because they express high levels of VEGF (Figure 6a) but have undetectable levels of VEGFR1 or VEGFR2 (Figure 7d). The anti-angiogenic effect of 15 mg/kg bevacizumab (Figure 7f and g) correlated with the normalization of VEGF receptor expression levels within the tumors, i.e. downregulation of VEGFR1 and upregulation of VEGFR2 (Figure 7e).
Discussion
The understanding of mechanisms underlying VEGF signaling has become critically important in recent years due to the increasing use of therapeutic inhibitors of this pathway for the treatment of cancer. It has become evident that while some patients benefit from such therapy, a significant subset of patients is refractory to treatment. This might be due to the development of evasive resistance to VEGF inhibition by “turning on” alternative angiogenic pathways, or by enhancing recruitment of circulating endothelial progenitor cells (11,34,35). In both cases, the activity and regulation of VEGF receptor signaling pathways play a critical role.
We observed that recombinant human VEGF, FBS, or conditioned medium from VEGF-expressing tumor cell lines induce a “rheostat-like” pattern of regulation of VEGF receptors, enhancing VEGFR1 while decreasing VEGFR2 protein levels in primary endothelial cells. Notably, the sub-cellular distribution of VEGFR1 and VEGFR2 is fundamentally different. Our studies demonstrate that VEGFR1 is found primarily in the nucleus and does not move from there upon stimulation with VEGF. Of note, a recent study demonstrated by fluorescence microscopy the presence of VEGFR1 in the Golgi of human umbilical vein endothelial cells (HUVEC) (36). The data presented here does not exclude the possibility that some VEGFR1 can be found in the cytoplasmic fraction of HDMEC. However, our cell fractionation and confocal microscopy studies demonstrated unequivocally that the majority of VEGFR1 is present in the nucleus. In contrast, VEGFR2 clearly changes location from the cell membrane/cytoplasmic fraction to the nucleus when endothelial cells are treated with VEGF. Western blots for VEGFR2 typically show two bands (representing two isoforms) in unstimulated endothelial cells. However, upon stimulation with VEGF we observed that only the larger isoform is translocated to the nucleus. The most significant decrease in expression mediated by VEGF is observed with the larger isoform, through mechanisms that are currently under investigation in our laboratory. Notably, blockade of endocytosis prevented VEGF-induced nuclear translocation of VEGFR2, but did not prevent its downregulation. In contrast, inhibition of the ubiquitin/proteasome system prevented VEGF-induced VEGFR2 downregulation, but did not prevent nuclear translocation. These data suggest that the processes of nuclear translocation and VEGFR2 downregulation are mechanistically independent.
It is known that JNK targets the ubiquitination of regulatory proteins, including ATF2 (23). We observed here that VEGF induced potent activation of the JNK-c-Jun pathway, and that JNK activity is associated with ubiquitination of VEGFR2. On the other hand, inhibition of the ubiquitin or proteasome activity is sufficient to enhance expression of VEGFR1 in primary endothelial cells. These findings suggest that endothelial cells are continuously synthesizing VEGFR1 and that ubiquitin/proteasome activity is necessary to maintain its homeostatic levels. Notably, the regulation of VEGFR1 protein levels is dependent on Akt and ERK1/2 phosphorylation. Since these two kinases typically inhibit the degradation of proteins by the ubiquitin/proteasome system, we postulate that VEGF induces phosphorylation of Akt and ERK1/2, which in turn prevents the degradation of VEGFR1 by the ubiquitin/proteasome system. Collectively, these data suggest that VEGF orchestrates an intricate process mediated by the Akt/ERK and JNK/c-Jun that protects VEGFR1 while “tagging” VEGFR2 for degradation, leading to rapid reversal of the protein levels of these two receptors. We propose that this mechanism is involved in the well-characterized ability of the vascular endothelium to quickly adapt to sudden changes in the microenvironment.
We and other colleagues have reported that VEGF enhances the survival of endothelial cells by regulating the activity of the anti-apoptotic Bcl-2 and inhibiting caspase-mediated cell death (25,37). At that time, it was believed that this process was mediated by VEGFR2 signaling. The availability of specific shRNA constructs to selectively downregulate expression of each VEGF receptor allowed us to revisit these data. We demonstrate here that endothelial cell survival is regulated primarily by VEGFR1 signaling, while VEGFR2 signaling regulates the differentiation of endothelial cells into capillary tubes.
Our ability to discriminate the function of VEGFR1 and VEGFR2 in vivo is hindered by the fact that both receptors play critical and complementary roles in angiogenesis (38,39). However, in attempt to validate the concepts tested here within relevant tumor models, we exposed the endothelial cells to the conditioned medium of VEGF-producing squamous cell carcinomas. As expected, the tumor cells enhanced the survival and angiogenic potential of endothelial cells. This was correlated with an increase in the expression of VEGFR1, Bcl-2 and phospho-c-Jun, which is consistent with the increase in the survival typically observed in tumor-associated endothelial cells (25).
The inhibition of tumor angiogenesis mediated by VEGF signaling blockade with bevacizumab correlated with a reversal in VEGFR1 and VEGFR2 protein levels. These data raise the intriguing hypothesis that the anti-tumor effect of anti-VEGF therapies requires the normalization of VEGFR1 and VEGFR2 protein levels. This work suggests that the process of normalization of VEGFR1/VEGFR2 can potentially constitute a novel conceptual target for cancer treatment. Moreover, the reversal of VEGFR1 and VEGFR2 can potentially be useful as a molecular biomarker for the effectiveness of anti-VEGF therapies.
Collectively, our studies demonstrated that VEGFR1 is the dominant receptor in the tumor microenvironment, and that VEGFR1 is required for endothelial cell survival. These findings support the concept that VEGFR1 is the preferable therapeutic target in cancer. However, the unexpected finding that this receptor is localized in the nucleus of angiogenic endothelial cells suggests that highly permeable drugs are required to achieve clinically relevant inhibition of VEGFR1 signaling in vivo. Moreover, this work revealed a novel mechanism by which a ligand inversely regulates two receptors to promote angiogenesis. Inversely reciprocal regulation of receptors by a single ligand might be a common mechanism shared by cells to regulate the function of other ligand/receptor systems in physiology and in disease.
Materials and Methods
Cell culture and reagents
Primary human dermal microvascular endothelial cells (HDMEC; Cambrex, Walkersville, MD) and primary human pulmonary endothelial cells (Cambrex) were cultured in endothelial growth medium-2 for microvascular cells (EGM2-MV; Cambrex). Head and neck squamous cell carcinoma cells (UM-SCC-1 and UM-SCC-11B, gift from T. Carey, University of Michigan; OSCC3, gift from M. Lingen, University of Chicago) were cultured in Dulbecco's modified Eagle's medium (DMEM, Invitrogen, Carlsbad, CA) supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. HDMEC and UM-SCC-1 cells were starved overnight, and treated with 0-50 ng/ml rhEGF, 0-50 ng/ml rhVEGF165, or 0-50 ng/ml rhPlGF (R & D Systems, Minneapolis, MN), 0-50 ng/ml rhVEGF-E (Fitzgerald, Concord, MA), or 0-20 ng/ml rhIL-6 (Biosource, Camarillo, CA) for indicated time points. Alternatively, HDMEC were treated with 0-5 μg/ml goat anti-human VEGF antibody (R & D Systems); 0-20 μM LY294002 (PI3K inhibitor; A.G. Scientific Inc., San Diego, CA); 0-30 μM PD98059 (MEK-1 inhibitor, upstream of ERK1/2, Cell Signaling Technology, Inc, Danvers, MA); 0-10 μM SP600125 (JNK inhibitor; A.G. Scientific Inc.); 0-5 μg/ml Anisomycin (Sigma; St. Louis, MO); 0-5 μM Ubiquitin aldehyde inhibitor (Upstate, Temecula, CA); 0-10 μM proteasome inhibitor MG132 (Sigma); 0-10 μg/ml Concanavalin A (Con A, Sigma). At indicated time points, cells were collected, and Western blot or RT-PCR was performed.
Sub-cellular fractionation
Cells were resuspended in cytoplasmic extraction buffer (Panomics, Redwood, CA) containing protease inhibitors, and incubated for 3 min on ice. The suspension was centrifuged at 13,000 rpm for 5 min at 4°C. The supernatant was collected as cytoplasmic/cell membrane extract (CE). The nuclear extract (NE) was obtained by suspending the nuclear pellet in nuclear extraction buffer (Panomics) for 40 min on ice with occasional vortexing and clarified by centrifuging at 13,000 rpm for 10 min at 4°C. The supernatant was collected as NE.
Confocal microscopy
HDMEC were seeded in Lab-Tek chamber slides (Nalge Nunc International; Noperville, IL), starved overnight, and treated with 0-50 ng/ml rhVEGF165 for 15-30 min. Cells were fixed with methanol or 4% paraformaldehyde/PBS, treated with 3% H2O2 for 10 min, and then incubated with 1-2 μg/ml rabbit anti-human VEGFR2 (Santa Cruz), VEGFR1 (Abcam Inc, Cambridge, MA) for 1 hour at room temperature or overnight at 4°C. ProLong Gold anti-fade reagent with DAPI (Invitrogen) was used to identify nuclei. Images were captured in a Zeiss Confocal microscope (Microscopy and Image Analysis laboratory, University of Michigan). Here, and throughout this manuscript, at least three independent experiments were performed to verify reproducibility of results.
In vitro capillary tube formation
5 × 104 HDMEC cells were seeded in 24 well plates (Corning Incorporated, Corning, NY) coated with growth factor reduced Matrigel (BD Biosciences, Bedford, MA). When indicated, UM-SCC-1 conditioned medium, SP600125, AG490, Ly294002, or PD98059 were used at the concentrations described above. After 24 hours, capillary tubes were counted in random fields from 3 wells/condition.
VEGFR1 and VEGFR2 gene silencing
HEK 293T cells were transiently co-transfected with Lentivirus packaging vector psPAX2, pMD2.G, and shRNA-C (control), shRNA-VEGFR1 or shRNA-VEGFR2 (Vector Core, University of Michigan) by the calcium phosphate method. Supernatants containing lentiviruses were used to infect HDMEC overnight. HDMEC were selected with 1 μg/ml puromycin (InVivogen, San Diego, CA) for at least one week, and downregulation of target genes was examined by Western blot.
SCID Mouse Model of Human Tumor Angiogenesis
Xenograft human tumors vascularized with human blood vessels were generated under an UCUCA approved protocol, as described (32). Briefly, highly porous poly-L(lactic) acid (Boehringer Ingelheim, Ingelheim, Germany) scaffolds were seeded with 9 × 105 HDMEC and 1 × 105 UM-SCC-1 cells. SCID mice (CB.17.SCID; Taconic, Germantown, New York) were anesthetized with ketamine and xylazine, and two scaffolds were implanted in the subcutaneous space of the dorsal region of each mouse. When tumors reached 1 cm3, mice were injected intraperitonealy twice with 15 or 30 mg/kg bevacizumab (humanized anti-VEGF monoclonal antibody; Genentech) or vehicle control, with a 48-hour interval between each injection. Mice were euthanized the day after last injection, tumors were homogenized with NP-40 protein lysis buffer at 4°C, and Western blots were performed for VEGFR1 and VEGFR2. Tumor microvessel density was determined following identification of blood vessels by immunohistochemistry with a polyclonal anti-human factor VIII antibody (Lab Vision, Fremont, CA), as previously described (32).
Western Blots
Cells were lysed in 1% Nonidet P-40 (NP-40) lysis buffer (50 mM Tris-HCL, PH 7.4, 200 mM NaCl and 2 mM MgCl2 containing protease inhibitors. Membranes were incubated with the following primary antibodies for 1 hour at room temperature or overnight at 4°C: rabbit anti-human VEGFR1, rabbit anti-human VEGFR2, mouse anti-human phosphor-c-Jun, mouse anti-human phosphor-JNK, mouse anti-human JNK, rabbit hnRNP, mouse anti-human VE-Cadherin, and rabbit anti-human E-cadherin (Santa Cruz Biotechnology, Santa Cruz, CA); mouse anti-human phosphor STAT3, rabbit anti-human STAT3, mouse anti-human ERK1/2, rabbit anti-human phosphor-ERK1/2, rabbit anti-human phosphor-AKT, rabbit anti-human AKT, and rabbit anti-human c-Jun (Cell Signaling); mouse anti-GAPDH (Chemicon, Billerca, MA), hamster anti-human Bcl-2, mouse anti-human Bcl-xL (BD Biosciences, San Jose, CA). The molecular weight for the VEGFR1 bands is approximately 200 kDa, and for the VEGFR2 bands is approximately 220 kDa. The glycosylation status of VEGFR2 was evaluated with the Enzymatic CarboRelease Kit (QA-Bio, Palm Desert, CA), according to manufacturer's instructions. HDMEC were starved overnight, and treated with 50 ng/ml VEGF for 20 min. Cell fractionation was performed, as described below. Cell lysates (60-100 μg) were deglycosylated, and VEGFR2 was detected by Western blot with rabbit anti-VEGFR2 antibody (Santa Cruz). Affinity-purified second antibodies conjugated with horseradish peroxidase (Jackson ImmunoResearch Laboratories, West Grove, PA) were used, and immunoreactive proteins were visualized by SuperSignal West Pico chemiluminescent substrate (Thermo Scientific, Rockford, IL) and exposed to x-ray film.
ELISA
Supernatants of 24-hour cell cultures were collected and centrifuged to eliminate cellular debris. VEGF expression was determined with a human VEGF ELISA kit (Quantikine, R & D System) according to the manufacturer's instructions.
Reverse Transcriptase PCR and Real Time PCR
Total RNA was prepared in Trizol (Invitrogen), according to manufacturer's instructions. RT-PCR was performed by SuperScript III Platinum one step RT-PCR kit (Invitrogen). The primers used in this study were: VEGFR1, sense 5′-CCT CAC TGC CAC TCT AAT TGT C-3′, antisense 5′-ACA GTT TCA GGT CCT CTC CTT-3′; VEGFR2, sense 5′-CTC ATG TCT GAA CTC AAG ATC C-3′, antisense 5,-CCA GAA TCC TCT TCC ATG CTC A-3′; GAPDH, sense 5′- GAC CCC TTC TTC ATT GAC CTC AAC T-3′, antisense 5′-CAC CAC CTT CTT CTT GAT GTC ATC-3′. PCR products were submitted to electrophoresis in agarose gels. Primers for Real Time PCR were obtained from TaqMan Gene Expression Assays (AB Applied Biosystems, Foster, CA, USA): VEGFR1 Hs01052937_ml; VEGFR2 Hs00176676_ml; and GAPDH Hs99999905_ml. cDNA products were diluted 10 times, 1 μl was used for Real-time PCR with Tap Man Universal PCR Master Mix (AB Applied Biosystems). The reactions were done in 96 well plates in triplicate using AB17700 Sequence Detection System (AB Applied Biosystems). The data were normalized by the data of GAPDH.
UV and Anisomycin treatment
For ultraviolet (UV) treatment, HDMEC were pre-incubated with 0-10μM SP600125 (A.G. Scientific) for 1 hour, and treated with UV (254 nM, 150-200 μW/cm2) for 1 min. Alternatively, cells were starved overnight, and treated with 0-2.5 μg/ml Anisomycin (Sigma) for indicated time points (after 1 hour pre-incubation with 0-10 μM SP600125). At the end of the experimental period, cells were collected, Western blot or RT-PCR was performed.
Flow Cytometry
HDMEC stably transduced with shRNA-C, shRNA-VEGFR1 or shRNA-VEGFR2 were cultured for 48 hours. Cells were counted, fixed in 70% ethanol for 1 hour at 4°C, and resuspended in propidium iodide solution containing 0.1% sodium citrate, 25 μg/ml propidium iodide (Sigma), 100 μg/ml RNase A, and 0.1% Triton X-100. Cells were analyzed for DNA content by flow cytometry (FACSCalibur; BD Biosciences; San Jose, CA). The proportion of cells in sub-G0/G1, G1, S, G2/M phase was determined using Modfit cell cycle analysis software (Verity Software, Topsham, ME).
Caspase-3 activity assay
shRNA-transduced HDMEC were cultured with EGM2-MV (Lonza) for 48 hours. Cell extracts were prepared and caspase-3 activity assay was carried out at 37°C using 10 μM Ac-DEVD-AMC (Alexis, San Diego, CA, USA). 20 μM caspase-3 inhibitor, Ac-DEVD-CHO (Alexis), was used to determine specificity of reactions, and purified recombinant human caspase-3 (Alexis) was used as a positive control. Caspase-3 activity was detected by luminescence microplate reader (TECAN, GENios, Austria) using 360 nm excitation and 465 nm emission wavelength.
Tumor cell conditioned medium
UM-SCC-1 or OSCC3 were cultured for 24 hours in serum-free medium (EBM2, Cambrex), and the supernatants were collected as conditioned medium (CM). HDMEC were starved overnight and treated with UM-SCC-1 or OSCC3 CM in presence of 0-50 μg/ml bevacizumab (Avastin; Genentech, San Francisco, CA) or vehicle for 24 hours. VEGFR1, VEGFR2, and Bcl-2 expression were detected by Western blot, as described above.
Statistical Analysis
Statistical analysis was performed using one-way ANOVA followed by post-hoc tests for multiple group comparison with SigmaStat 2.0 statistical software (SPSS, Chicago, IL, USA). The level of significance was determined at P<0.05.
Supplementary Material
Acknowledgments
We thank Dr. Zhihong Dong for his assistance with the SCID Mouse Model of Human Tumor Angiogenesis, and Dr. Seungwon Kim for critically reading this manuscript and providing invaluable suggestions. This work was supported by grant R01-CA112390 from the NIH/NCI (LME); grant R01-DE12322 from the NIH/NIDCR (MWL); grant P50-CA97248 (University of Michigan Head and Neck SPORE) from the NIH/NCI (JEN), and grants R01-DE14601, R01-DE15948, R01-DE16586, R21-DE19279 from the NIH/NIDCR (JEN).
Abbreviations
- VEGF
vascular endothelial growth factor
- VEGFR1
vascular endothelial growth factor receptor 1
- VEGFR2
vascular endothelial growth factor receptor 2
- RTK
receptor tyrosine kinase
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- PIGF
placental growth factor
- IL-6
interleukin-6
- PI3K/Akt
phosphoinositol 3-kinase/Akt
- ERK
extracellular signal-regulated kinase
- JNK
c-Jun N-terminal kinase
- STAT3
signal transducer and activator of transcription 3
- HDMEC
human dermal microvascular endothelial cells
- HPEC
human pulmonary endothelial cells
- FBS
fetal bovine serum
- CM
conditioned medium
References
- 1.Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–1309. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- 2.Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005;69(Suppl. 3):4–10. doi: 10.1159/000088478. [DOI] [PubMed] [Google Scholar]
- 3.Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008;8:579–591. doi: 10.1038/nrc2403. [DOI] [PubMed] [Google Scholar]
- 4.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 5.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea KS, et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 6.Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, et al. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130:691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358:2039–2049. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hurwitz H, Fehrenbacher L, Hainsworth JD, Heim W, Berlin J, Holmgren E, et al. Bevacizumab in combination with fluorouracil and leucovorin: an active regimen for first-line metastatic colorectal cancer. J Clin Oncol. 2005;23:3502–3508. doi: 10.1200/JCO.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 9.Kerbel RS, Yu J, Tran J, Man S, Viloria-Petit A, Klement G, et al. Possible mechanisms of acquired resistance to anti-angiogenic drugs: implications for the use of combination therapy approaches. Cancer Metastasis Rev. 2001;20:79–86. doi: 10.1023/a:1013172910858. [DOI] [PubMed] [Google Scholar]
- 10.Miller KD, Sweeney CJ, Sledge GW. Can tumor angiogenesis be inhibited without resistance? EXS. 2005;94:95–112. doi: 10.1007/3-7643-7311-3_7. [DOI] [PubMed] [Google Scholar]
- 11.Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;4:299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 12.Shibuya M, Yamaguchi S, Yamane A, Ikeda T, Tojo A, Matsushime H, et al. Nucleotide sequence and expression of a novel human receptor-type tyrosine kinase gene (flt) closely related to the fms family. Oncogene. 1990;5:519–524. [PubMed] [Google Scholar]
- 13.Matthews W, Jordan CT, Gavin M, Jenkins NA, Copeland NG, Lemischka IR. A receptor tyrosine kinase cDNA isolated from a population of enriched primitive hematopoietic cells and exhibiting close genetic linkage to c-kit. Proc Natl Acad Sci. 1991;88:9026–9030. doi: 10.1073/pnas.88.20.9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signaling - in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 15.Waltenberger J, Claesson WL, Siegbahn A, Shibuya M, Heldin CH. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J Biol Chem. 1994;269:26988–26995. [PubMed] [Google Scholar]
- 16.Takahashi H, Shibuya M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin Science. 2005;109:227–241. doi: 10.1042/CS20040370. [DOI] [PubMed] [Google Scholar]
- 17.Gille H, Kowalski J, Li B, LeCouter J, Moffat B, Zioncheck TF, et al. Analysis of biological effects and signaling properties of Flt-1 (VEGFR-1) and KDR (VEGFR-2). A reassessment using novel receptor-specific vascular endothelial growth factor mutants. J Biol Chem. 2001;276:3222–3230. doi: 10.1074/jbc.M002016200. [DOI] [PubMed] [Google Scholar]
- 18.Huang F, Kirkpatrick D, Jiang X, Gygi S, Sorkin A. Differential regulation of EGF receptor internalization and degradation by multi-ubiquitination within the kinase domain. Mol Cell. 2006;21:737–748. doi: 10.1016/j.molcel.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 19.Duval M, Bédard GS, Delisle C, Gratton JP. Vascular Endothelial Growth Factor-dependent down-regulation of Flk-1/KDR Involves Cbl-mediated Ubiquitination: consequences on nitric oxide production from endothelial cells. J Biol Chem. 2003;278:20091–20097. doi: 10.1074/jbc.M301410200. [DOI] [PubMed] [Google Scholar]
- 20.Ewan LC, Jopling HM, Jia H, Mittar S, Bagherzadeh A, Howell GJ, et al. Intrinsic tyrosine kinase activity is required for Vascular Endothelial Growth Factor Receptor 2 ubiquitination, sorting, and degradation in endothelial cells. Traffic. 2006;7:1270–1280. doi: 10.1111/j.1600-0854.2006.00462.x. [DOI] [PubMed] [Google Scholar]
- 21.Dougher M, Terman BI. Autophosphorylation of KDR in the kinase internalization. Oncogene. 1999;18:1619–1627. doi: 10.1038/sj.onc.1202478. [DOI] [PubMed] [Google Scholar]
- 22.Radhakrishnan R, Solomon M, Satyamoorthy K, Martin LE, Lingen MW. Tissue microarray - a high-throughput molecular analysis in head and neck cancer. J Oral Pathol Med. 2008;37:166–176. doi: 10.1111/j.1600-0714.2007.00606.x. [DOI] [PubMed] [Google Scholar]
- 23.Fuchs SY, Xie B, Adler V, Fried VA, Davis RJ, Ronai Z. c-Jun NH2-terminal kinases target the ubiquitination of their associated transcription factors. J Biol Chem. 1997;272:32163–32168. doi: 10.1074/jbc.272.51.32163. [DOI] [PubMed] [Google Scholar]
- 24.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein-and UV-responsive protein kinase that binds and potentiates the c-jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 25.Nör JE, Christensen J, Mooney DJ, Polverini PJ. Vascular Endothelial Growth Factor-mediated angiogenesis is associated with Bcl-2 upregulation and enhanced endothelial cell survival. Am J Pathol. 1999;154:375–384. doi: 10.1016/S0002-9440(10)65284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu C, Minemoto Y, Zhang J, Liu J, Tang F, Bui TN, et al. JNK suppresses apoptosis via phosphorylation of the pro-apoptotic Bcl-2 family protein BAD. Mol Cell. 2004;13:329–340. doi: 10.1016/s1097-2765(04)00028-0. [DOI] [PubMed] [Google Scholar]
- 27.Kim KW, Mutter RW, Cao C, Albert JM, Shinohara ET, Sekhar KR, et al. Inhibition of signal transducer and activator of transcription 3 activity results in down-regulation of Survivin following irradiation. Mol Cancer Ther. 2006;5:2659–2665. doi: 10.1158/1535-7163.MCT-06-0261. [DOI] [PubMed] [Google Scholar]
- 28.Kitamura T, Asai N, Enomoto A, Maeda K, Kato T, Ishida M, et al. Regulation of VEGF-mediated angiogenesis by the Akt/PKB substrate Girdin. Nat Cell Biol. 2008;10:329–337. doi: 10.1038/ncb1695. [DOI] [PubMed] [Google Scholar]
- 29.Mavria G, Vercoulen Y, Yeo M, Paterson H, Karasarides M, Marais R, et al. ERK-MAPK signaling opposes Rho-kinase to promote endothelial cell survival and sprouting during angiogenesis. Cancer Cell. 2006;9:33–44. doi: 10.1016/j.ccr.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 30.Hilfiker-Kleiner D, Hilfiker A, Fuchs M, Kaminski K, Schaefer A, Schieffer B, et al. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ Res. 2004;95:187–195. doi: 10.1161/01.RES.0000134921.50377.61. [DOI] [PubMed] [Google Scholar]
- 31.Nishimura K, Li W, Hoshino Y, Kadohama T, Asada H, Ohgi S, et al. Role of AKT in cyclic strain-induced endothelial cell proliferation and survival. Am J Physiol Cell Physiol. 2006;290:C812–821. doi: 10.1152/ajpcell.00347.2005. [DOI] [PubMed] [Google Scholar]
- 32.Nör JE, Peters MC, Christensen JB, Sutorik MM, Linn S, Khan MK, et al. Engineering and characterization of functional human microvessels in immunodeficient mice. Lab Invest. 2001;81:453–463. doi: 10.1038/labinvest.3780253. [DOI] [PubMed] [Google Scholar]
- 33.Kaneko T, Zhang Z, Mantellini MG, Karl E, Zeitlin B, Verhaegen M, et al. Bcl-2 orchestrates a crosstalk between endothelial cells and tumor cells that promotes tumor growth. Cancer Res. 2007;67:9685–9693. doi: 10.1158/0008-5472.CAN-07-1497. [DOI] [PubMed] [Google Scholar]
- 34.Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, et al. HIF1-alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karl E, Zhang Z, Dong Z, Neiva KG, Soengas MS, Koch AE, et al. Unidirectional Crosstalk Between Bcl-xL and Bcl-2 Enhances the Angiogenic Phenotype of Endothelial Cells. Cell Death Diff. 2007;14:1657–1666. doi: 10.1038/sj.cdd.4402174. [DOI] [PubMed] [Google Scholar]
- 36.Mittar S, Ulyatt C, Howell GJ, Bruns AF, Zachary I, Walker JH, Ponnambalam S. VEGFR1 receptor tyrosine kinase localization to the Golgi apparatus is calcium-dependent. Exp Cell Res. 2009;315:877–89. doi: 10.1016/j.yexcr.2008.12.020. [DOI] [PubMed] [Google Scholar]
- 37.Gerber HP, Dixit V, Ferrara N. Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J Biol Chem. 1998;273:13313–13316. doi: 10.1074/jbc.273.21.13313. [DOI] [PubMed] [Google Scholar]
- 38.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 39.Shibuya M, Claesson WL. Signal transduction by VEGF receptors in regulation angiogenesis and lymphangiogenesis. Exp Cell Res. 2006;312:549–560. doi: 10.1016/j.yexcr.2005.11.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.