

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) is the causative agent of Kaposi's sarcoma (KS), an AIDS-related endothelial cell malignancy that is the most common cancer in central and southern Africa. The KSHV viral G protein-coupled receptor (vGPCR) is a viral oncogene that conveys a survival advantage to endothelial cells and causes KS-like tumors in mouse models. In this study we investigate the role of Shp2, a protein tyrosine phosphatase in vGPCR signaling. Shp2 is vital to many cytokine-induced signaling pathways and is dysregulated in various infections and malignancies. It has also recently been implicated in angiogenesis. We find that vGPCR activity results in phosphorylation of regulatory tyrosines in Shp2 and that in turn, Shp2 is required for vGPCR-mediated activation of MEK, NFκB, and AP-1. Furthermore, both genetic and chemical inhibition of Shp2 abrogate vGPCR-induced enhancement of endothelial cell migration. This establishes Shp2 as an important point of convergence of KSHV vGPCR signaling and a potential molecular target in the design of an anti-KSHV therapeutic regimen.
Keywords: Kaposi's sarcoma, KSHV, vGPCR, G protein-coupled receptor, Shp2, Protein tyrosine phosphatase, angiogenesis
Introduction
Kaposi's sarcoma (KS) is a highly angiogenic endothelial cell tumor that is among the most common AIDS-related malignancies world-wide. The burden of disease varies geographically but is particularly striking in central and southern Africa. The etiologic agent, Kaposi's sarcoma-associated herpesvirus (KSHV), was discovered in 1994 and is required for all epidemiologic types of KS (Chang et al., 1994; Whitby et al., 1995). In addition to endothelial cells, KSHV can also be found in lymph nodes, peripheral blood B cells, and is a driving force in all forms of primary effusion lymphoma (PEL), a neoplasm that accounts for 3-5% of AIDS-related non-Hodgkins lymphomas (Cesarman et al., 1995; Moore et al., 1996; Nador et al., 1996; Said et al., 1996). As with other human herpesviruses, not all KSHV infected individuals display pathologic sequelae. It is clear that HIV infection or other non-HIV immunomodulatory co-factors must be present. It has been hypothesized that at least in its early stages, KS is a cytokine driven process (Della Bella et al., 2008; Monini et al., 1999). How the cytokine milieu in the tumor microenvironment is established and maintained and how it in turn supports viral propagation and tumor progression is still being defined. Lesions are a complex mix of proliferating KS “spindle” cells, inflammatory cells and aberrant angiogenesis. There are many viral proteins that affect these processes either directly or via paracrine mechanisms.
The KSHV-encoded chemokine receptor, vGPCR, has the potential to play roles in angiogenesis, cell cycle regulation, and possibly the viral life cycle (Bais et al., 1998; Cannon, Cesarman, and Boshoff, 2006). KSHV vGPCR is a constitutively active homologue of the human IL-8 receptor CXCR2 presumably pirated from the host genome at some point during co-evolution with subsequent acquisition of activating mutations (Arvanitakis et al., 1997; Cesarman et al., 1996; Guo et al., 1997). Unlike its mammalian counterpart, vGPCR signals promiscuously through multiple G protein subtypes to activate multiple arms of the MAPK/SAPK cascade, the Src family members and the PI3K/AKT pathway. Transcription factor activation by vGPCR includes AP-1, CREB, NFκB, and NFAT. Furthermore, in transgenic mice, vGPCR expression recapitulates the most striking phenotypic features of KS including multifocal spindle cell tumors including robust but aberrant angiogenesis, and an inflammatory cell component (Guo et al., 2003; Yang et al., 2000). Furthermore, vGPCR potentiates the tumor-forming capacity of other KSHV gene products and a conditional knock-out model shows that ongoing vGPCR expression is required to maintain vGPCR-derived tumors (Jensen et al., 2005; Montaner et al., 2003). These properties argue that vGPCR and its downstream signaling events are crucial to the biology of KS and are promising anti-KSHV targets. Targeting vGPCR for inhibition either directly or by manipulating downstream cellular pathways that conduct vGPCR signals potentially offers new therapeutic approaches for treating KS and other KSHV-mediated diseases. There have been no significant successes discovering or designing potent inhibitors of constitutively active GPCRs, so our work has focused on identifying key cellular vGPCR effectors within the very complex network it activates.
In this study we focus on the cytoplasmic protein tyrosine phosphatase (PTP) Shp2 and its role in vGPCR signaling. The PTPs are crucial to the dynamic process of protein tyrosine phosphorylation and dephosphorylation that regulates many basic cellular functions including growth, survival, and migration. The non-receptor PTPs include Shp1 and Shp2 which contain tandem amino-terminal SH2 domains, a catalytic domain and putative carboxy-terminal regulatory sites (Koch et al., 1991; Qu, 2000). Shp2 is ubiquitously expressed, while Shp1 is primarily found in hematopoietic cells and at low levels in endothelial cells (Adachi et al., 1996; Yi, Cleveland, and Ihle, 1991). Shp2 is a positive regulator of signaling pathways initiated by many growth factors, including PDGF, EGF, IL-3, EPO, and GMCSF (Huyer and Alexander, 1999; Neel and Tonks, 1997). Generally speaking Shp1 has opposing effects to Shp2; Shp1 attenuates signals from EPO, IL-3, and GM-CSF receptors and mediates inhibitory signals from γFc domains, TCR, BCR, and the NK inhibitory receptor (Burshtyn et al., 1996; Chen et al., 1996; D'Ambrosio et al., 1995; Yi et al., 1993). The importance of the PTPs in cell homeostasis is evidenced by their deregulation by human pathogens as diverse as protozoa, bacteria, and viruses (Migone et al., 1998; Nandan and Reiner, 2005; Nandan et al., 2002; Tsutsumi et al., 2006). Furthermore, overexpression of Shp2 and reduction of Shp1 are associated with human leukemogenesis (Oka et al., 2001; Xu et al., 2005). Very recent work shows a role for Shp2 in endothelial cell proliferation and vessel growth mediated by the PI3K-Akt and Erk1/2 pathways (Mannell et al., 2008). As discussed above, vGPCR is highly angiogenic and signals by these same pathways. To determine if Shp2 would be a feasible point at which to interfere with vGPCR signaling and its downstream biologic effects, we studied the phosphorylation status of Shp2 in the presence of vGPCR as well as the effects of pharmacologic and genetic inhibition of Shp2 on vGPCR signaling. In addition to work in HEK293 cells we also used lentiviral transduction to study primary human endothelial cells. Our cell type of choice is the blood outgrowth endothelial cell (BOEC). These cells (“also called late- EPC”) are derived from human blood and display the typical cobblestone morphology of endothelial cells, are uniformly positive for vWF and incorporate acetylated LDL. They are all positive for P1H12 (CD146), thromobomodulin, VEGFR2, VE-cadherin, PECAM-1, CD34, and integrin αv. Of note they are uniformly CD14, CD133 and CD45 negative. This phenotype is maintained through all subsequent passages (up to 1012-fold expansion). Stimulation with LPS or IL-1 results in increased ICAM-1 expression and the appearance of VCAM-1 and tissue factor expression. (Jiang et al., 2007; Lin et al., 2000). Furthermore, BOEC have recently been shown to be infected with KSHV in vivo and are able to support the production of new virions; they are therefore a very suitable cell type for KSHV molecular pathogenesis studies (Della Bella et al., 2008).
We have shown that the constitutive activity of vGPCR results in phosphorylation of Shp2 at its regulatory C-terminal tyrosines and that in turn, Shp2 is required for vGPCR activation of the MEK-ERK1/2 axis and the transcription factors AP-1 and NFκB as well as vGPCR-induced endothelial cell migration. These results establish Shp2 as a promising target to interfere with KSHV-induced changes in the endothelial cell phenotype.
Results
KSHV vGPCR induces phosphorylation of Shp2 that requires Gαq and Src
Shp2 undergoes phosphorylation of tyrosine residues Y542 and Y580 in its C-terminus that may regulate phosphatase activity or enhance scaffolding functions (Feng, Hui, and Pawson, 1993; Lorenz et al., 1994; Minoo et al., 2004). We found in HEK293 that vGPCR expression constitutively enhances phosphorylation of Shp2 Y542 (phospho-Y580 levels were high at baseline and not affected, data not shown) (Fig. 1A). vGPCR is known to couple with both Gαi and Gαq subunits depending on cellular context. To define the proximal events by which Shp2 phosphorylation occurs, we used pertussis toxin (PTX) to inhibit Gαi and found that Gαi subunits are not required for either baseline or vGPCR-induced Shp2 phosphorylation. This makes is highly likely that Gαq coupling explains vGPCR's effects on Shp2. In the absence of a standard potent method to inhibit Gαq, we used gain-of-function experiments with constitutively-active mutant, Gq(Q209L)(Wu et al., 1992). Figure 1B shows that Gq(Q209L) causes phosphorylation of Shp2 at Y542. Together these data highly suggest that it is a vGPCR-Gαq-Shp2 axis that we are observing in HEK293. MEK is activated by vGPCR and is variably reported to signal upstream or downstream of Shp2 depending on experimental conditions. Pharmacologic inhibition of MEK (Fig. 1A) with PD98059 shows that ERK1/2 activity is not required for Shp2 phosphorylation caused by vGPCR. Pharmacologic inhibition of Src was also assessed for its effect since vGPCR can activate Src family members (Cannon, Philpott, and Cesarman, 2003). Src inhibition by the potent and specific inhibitor PP2 (4-Amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine) resulted in dramatic decrease of both baseline Shp2 Y542 phosphorylation and vGPCR-induced phosphorylation. A dominant-negative (DN) construct of Src also inhibited vGPCR-mediated phosphorylation, with no appreciable effect found on baseline phosphorylation (Fig 1C). The more modest effect of Src-DN compared to PP2 is likely due to the inherent difficulties in achieving complete inhibition using DN constructs. To further verify a role for Src we also used an shRNA construct from OpenBiosystems, TRCN0000038153 (shSrc) to knock down Src. Knockdown was not particularly efficient, but even with a very modest level of Src knockdown, levels of p542-Shp2 induced by vGPCR were blunted (Figure 1D). Together, these data show that in HEK 293 cells, vGPCR-induced phosphorylation of Shp2 is Gαi-independent, likely Gαq-dependent and is Src-dependent.
Figure 1.

vGPCR-induced Shp2 phosphorylation is Gαi-independent and Src-dependent, but Erk1/2 independent. HEK 293 cells were transfected with pcDNA3.1+ as control plasmid or with pcKSHV-vGPCR (A,C) or Gq(209L) plasmid (B) as shown. (A-C) After 48 hours protein lysates were Western blotted for phospho-542-Shp2; membranes were then stripped and re-probed for total Shp2. (A,B) Inhibitors were added 36 hours post-transfection where indicated. PTX (pertussis toxin, 100 ng/ml), PP2 (5 mM), PD (PD98059, 25 μM). (C) Cells were co-transfected with Src-DN plasmid where noted. (D) Cells were transfected with scrambled shRNA (sc) or shRNA targeted to Src (shSrc). After 48 hours, cells were transfected with vGPCR and protein harvested after 48 additional hours. Western blot was performed for p542-Shp2, stripped and re-probed for total Shp2. Total levels of Src are also shown to verify knockdown.
Shp2 is required for vGPCR-initiated signals that activate MEK, NFκB, and AP-1
Having found that vGPCR potentially affects Shp2 function via phosphorylation, we next assessed how inhibition of Shp2 would in turn affect vGPCR-mediated events further downstream. NSC-87877 is a novel pharmacologic inhibitor of Shp2 phosphatase activity (Chen et al., 2006). Figure 2 shows that pharmacologic inhibition of Shp2 inhibits vGPCR-mediated activation of MEK as measured by phospho-ERK1/2 levels. NFκB and AP-1 activation by vGPCR are also at least partially Shp2-dependent (Fig 2B). To support the above pharmacologic data, we tested several shRNA plasmid constructs against Shp2 from the pLKO library obtained from Open Biosystems. Several candidate knockdown sequences were tested and in transient assays we found that shRNA TRCN0000005003 (sh3) and shRNA TRCN0000005004 (sh4) knocked down Shp2, whereas shRNA TRCN0000005006 (sh6) and the scrambled shRNA (sc) did not (Fig 2C). In agreement with the pharmacologic data, knockdown of Shp2 blunted vGPCR-induced activation of MEK, NFκB, and AP-1 without affecting vGPCR expression levels (Fig 2D,E). Residual transcription factor activity could reflect either sub-total Shp2 inhibition or perhaps contribution of Shp2-independent pathways in these cells. Figure 2F also shows that knock-down and pharmacologic inhibition of Shp2 are additive and together reduce phospho-ERK1/2 levels to baseline. Together, these experiments argue strongly that Shp2 contributes broadly to vGPCR signaling. It is very possible that pathway cross-talk distal to Shp2 accounts for the broad effect; this is under study but nonetheless, the data support Shp2 as an important vGPCR effector.
Figure 2.
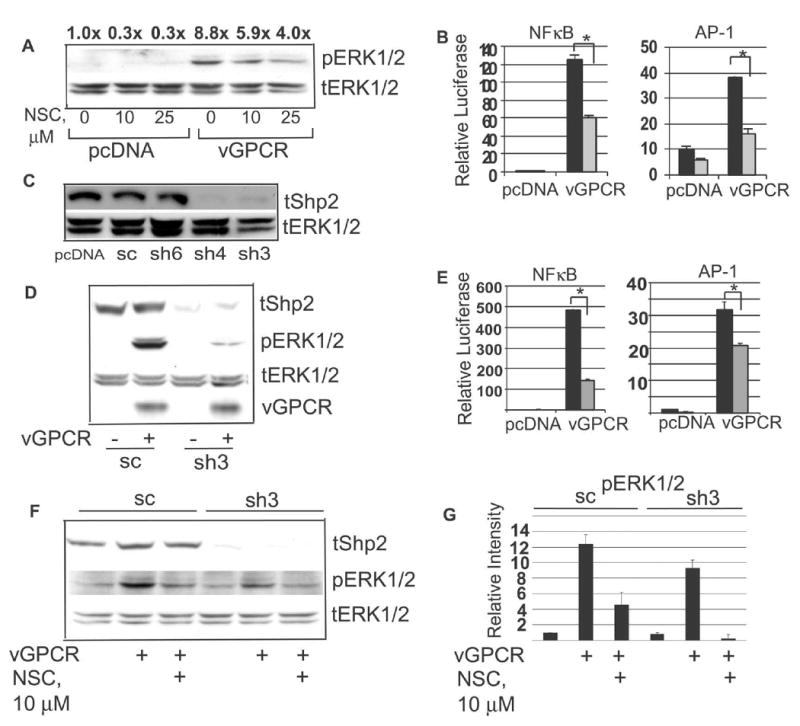
Shp2 is required for vGPCR activation of MEK, NFκB and AP-1. (A) HEK 293 cells were transfected with pcDNA3.1+ or pKSHV-vGPCR and incubated with NSC-87877 at doses shown overnight. Western blot for phospho-ERK1/2 was then done followed by blot for total ERK1/2. Numbers above bands represent density normalized to control, lane 1. (B) Cells were transfected with pcDNA3.1+ or vGPCR along with the indicated luciferase reporter construct and a constant amount of β-Gal plasmid. 25 μm NSC-87877 (grey bars) was added 5 hours later. Luciferase activity was assessed 24 hours post-transfection. Results were normalized to β–GAL levels. (C) HEK 293 were transfected with empty pcDNA3.1+, scrambled shRNA (sc) or one of three different candidate shRNA plasmid constructs (Open Biosystems) to knock down Shp2 which was then assessed by Western blot. (D) 48 hours after transfection with Shp2 shRNA (sh3), cells were transfected with vGPCR or empty plasmid. After 24 hours protein was harvested, transferred to PVDF which was then cut into three sections to immunoblot for total Shp2, total vGPCR, and phospho-ERK1/2; the latter was stripped and re-probed for total ERK1/2. (E) Shp2 was knocked down with shRNA as in (C), followed by transfection with vGPCR or control plasmid along with the indicated luciferase reporter plasmid and a β–GAL construct for normalization. (F) Cells stably expressing either scrambled shRNA (sc) or Shp2 shRNA (sh3) were subjected to a sub-maximal dose (10 μM) of NSC-87877 overnight and Western blot performed as above. (G) Density of phospho-ERK1/2 bands from 2F was normalized to corresponding total ERK1/2 band; each experimental point was then normalized to the control, lane 1. Bars = S.D. * denotes p≤0.05 by student's t –test.
In primary endothelial cells, vGPCR induces phosphorylation of Shp2 via both Gαi and Gαq
Although vGPCR can signal via both Gαq and Gαi subunits, these interactions and their effects are cell-type specific; for example we have found Gαi is required for vGPCR signaling in B cells but has minimal contribution in HEK293 (reference 39 and unpublished data). Although KSHV can infect many cell types in vitro, endothelial and hematopoietic cells appear to be the main targets in vivo. We therefore extended our studies into BOEC cells. BOEC are derived from circulating endothelial cell progenitors, display a micro-vascular phenotype, and as noted above have recently been found to harbor KSHV infection in vivo (Della Bella et al., 2008; Lin et al., 2002; Lin et al., 2000). To introduce exogenous DNA into these primary cells, we used lentiviral transduction. Figure 3A shows that with increasing multiplicity of infection (MOI) of lenti-vGPCR, increasing amounts of vGPCR are detectable by Western blot at 48 hours and Figure 3B confirms the expected spindled morphology induced by vGPCR. We found that vGPCR expression caused very high levels of Shp2 phosphorylation at Y542 (Fig. 3C); we also found that unlike in HEK293, vGPCR caused moderate increased phosphorylation of Y580 (data not shown). MEK is activated by vGPCR in most cell types studied so far, so pERK levels were also assayed as a positive control for vGPCR activity. Interestingly, in distinction to HEK293, phosphorylation of Shp2 in BOEC was partly inhibited by exposure to pertussis toxin overnight, supporting a role for Gαi coupling (Fig. 3D, top). Furthermore, we found that under serum-starved conditions, the effects of pertussis were consistently more potent, causing reduction of phospho-Shp2 to near baseline levels (bottom). The residual Shp2 phosphorylation in full medium suggested that non-Gαi G protein coupling may be contributing as well. Indeed, using a vGPCR mutant that signals only via Gαq (gift John Nicholas), we saw Shp2 phosphorylation (Fig. 3E) (Liu et al., 2004). Since lentiviral preps vary in concentration and even rigorous quantification is not always perfectly accurate, for this experiment, we chose to use two MOI for each construct. This allowed us to select points that expressed as near as possible the same amount of each vGPCR construct. We then normalized phospho-Shp2 levels to loading controls and vGPCR. Results of 3D and 3E argue for contribution of both Gαq and Gαi to vGPCR-induced Shp2 phosphorylation. Importantly, we found that very short exposure to pertussis inhibits Shp2 phosphorylation; this effect is too fast to attribute to a change cytokine levels so argues that via Gαi, vGPCR acts on Shp2 autologously (i.e. within the same cell) (Fig. 3F). Furthermore, since Gαi inhibition in serum-starved conditions so potently reverses vGPCR induced Shp2 phosphorylation, the contribution of Gαq is likely via autocrine/paracrine effectors known to be upregulated by vGPCR. Given these results we hypothesize that in full serum conditions the role of the vGPCR-Gαq pathway far outweighs and likely makes irrelevant the vGPCR-Gαi contribution to Shp2 activation. However, the ability of vGPCR to signal to Shp2 via Gαi may help propagate vGPCR's pro-growth and survival effects in otherwise poor growth conditions that may happen in KS tumors in vivo. Of note, as in HEK293, Src is absolutely required for both baseline and vGPCR-induced Shp2 phosphorylation (Fig. 3D).
Figure 3.
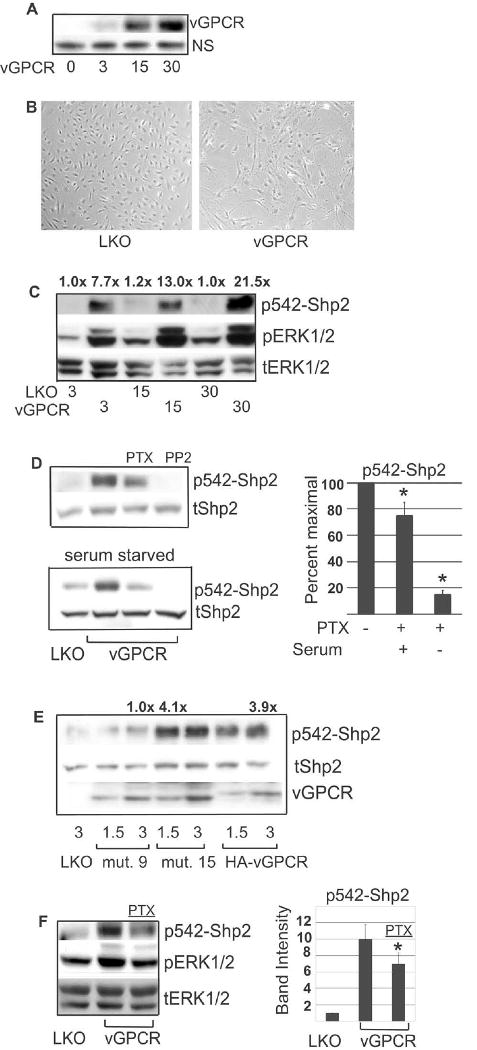
Lentiviral transduction of vGPCR into BOEC results in a Gαq- and Gαi-dependent increase in phosphorylated form of Shp2. (A) BOEC cells were transduced with lenti-vGPCR at MOI shown. After 48 hours, RIPA lysates were immunoblotted for vGPCR. ‘NS’ designates a non-specific band. (B) Micrograph showing lenti-vGPCR induced change in morphology from cobblestone to spindled. (C) BOEC were transduced with control lentivirus (LKO) or lenti-vGPCR at MOI shown. At 48 hours RIPA lysates were transferred to PVDF which was cut and simultaneously immunoblotted for p542-Shp2, and pERK1/2; blot was stripped and re-probed for total ERK1/2 as a loading control. Numbers above bands represent p542-Shp2 band density normalized to corresponding loading control and then normalized to control, lane 1. (D) BOEC were lenti-transduced with control virus (LKO) or vGPCR at an MOI of 3. Forty eight hours later, cells were serum-starved (bottom panel) or not (top panel). At 60 hours post-transduction PTX (100 nM), or the Src inhibitor PP2 (5 μM) were added and protein lysates harvested and immunoblotted for p542-Shp2 at 72 hours post-transduction. Blots were stripped and re-probed for total Shp2. Graph to right represents p542-Shp2 band densities normalized to loading control and then normalized to peak levels in the corresponding lane 2. Bands were undetectable in PP2 lanes and not represented graphically. (E) BOEC were lenti-transduced with LKO, HA-vGPCR, vGPCR mutant #9 (inactive) or vGPCR mutant #15 (Gq-only), each at two different MOI as shown. After 72 hours cells were harvested for protein and immunoblotting done for p542-Shp2, total Shp2 and vGPCR. Lanes 3, 4 and 7 had most similar amounts of vGPCR construct and were used for densitometry in which p542-Shp2 and vGPCR bands were normalized to tShp2 loading control and then p542-Shp2 normalized to corresponding vGPCR band. Densities were normalized to inactive mutant, lane 3, and fold change shown numerically above bands. (F) Lenti-vGPCR infected BOEC were serum- starved overnight 60 hours post-transduction. 30 minutes before harvesting protein, 100 nM PTX was added where indicated. p542-Shp2 bands were quantified and normalized to tERK1/2 loading control and then to control, lane1, and represented graphically. PTX, pertussis toxin. Bars= S.D. *= p≤0.05
KSHV vGPCR-induced MEK activation in BOEC requires Shp2
Having established that vGPCR signaling induces Shp2 phosphorylation, we next investigated whether Shp2 was necessary to downstream vGPCR signaling in BOEC. Figure 4A shows that the Shp2-specific pharmacologic inhibitor, PHPS1, blunts vGPCR-induced MEK activity as represented by ERK1/2 phosphorylation. PHPS1 was used here because it does not cross react with Shp1 as can NSC-87877. This was not an issue in HEK293 which do not express Shp1; however, we and others have found low levels of Shp1 expression in endothelial cells. IP-10 was also used as a control in that it is an inverse-agonist known to inhibit vGPCR by approximately 30% as measured by IP3 production (Geras-Raaka et al., 1998). To support the pharmacologic data, we also inhibited Shp2 by various genetic means. Figure 4B shows that Shp2 dominant-negatives Shp2-c/s and DSH2, the former containing an inactivating substitution in the catalytic domain and the latter a carboxy-truncated mutant lacking the catalytic domain altogether inhibit ERK-1/2 phosphorylation caused by vGPCR. Figure 4C shows a similar effect from shRNA knockdown of Shp2. In some cases slight increases in pERK1/2 are detectable with vGPCR but these levels remain well below those induced in control cells and in some cases pERK1/2 is undetectable. We found Shp2-c/s to be particularly potent at suppressing both baseline and induced MEK activity. Due to rather large doses of lentivirus required to inhibit or knock down Shp2, relatively less lenti-vGPCR could be used and protein levels fall below our ability to detect them by Western blot. However, Figure 2D reassures that lack of Shp2 does not affect vGPCR levels. Since vGPCR is known to have potent paracrine effects via increased secretion of various cytokines, we tested whether Shp2 was necessary in BOEC to convey these vGPCR-mediated effects. Figure 4D shows that Shp2 inhibition by both shRNA and dominant-negative constructs inhibits MEK activation by vGPCR-conditioned medium in serum-starved BOEC. This is somewhat expected since Shp2 is known to be involved in signaling by many cytokines as discussed above. Levels of pERK1/2 induced by vGPCR-conditioned medium are not generally as high as those in vGPCR expressing cells presumably because the latter are subject to both paracrine/autocrine and direct vGPCR signaling. The increase in ERK-1/2 phosphorylation by vGPCR-conditioned medium is very modest so it is unclear if this would result in a biologically significant response, but it is feasible that even these small paracrine effects of vGPCR may have an important role in modifying the KS tumor microenvironment in vivo. It should also be pointed out that although there is still some vGPCR-induced ERK-1/2 phosphorylation when Shp2 is knocked down, the resulting levels are at or below baseline thus supporting the notion that by inhibiting Shp2, vGPCR's effects can also be inhibited. So together, these results show that Shp2 is a key step in vGPCR-initiated MEK activation, including that via paracrine stimuli.
Figure 4.
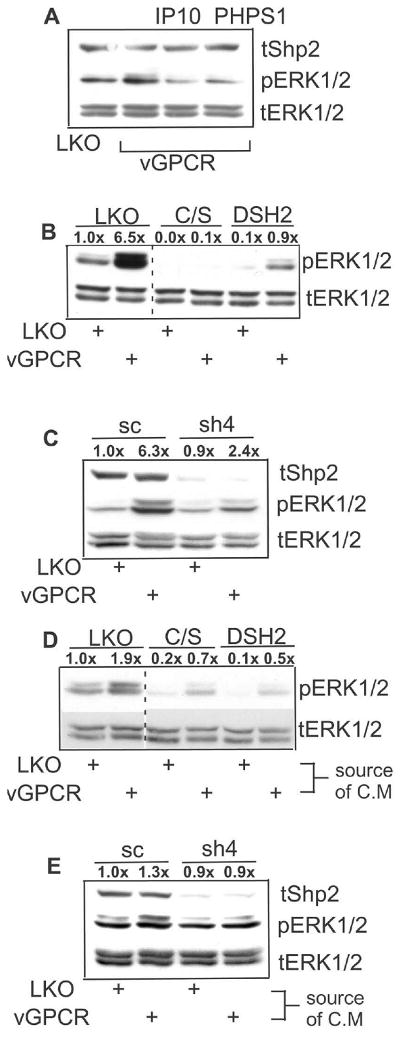
KSHV vGPCR-induced MEK activation in BOEC requires intact Shp2. BOEC cells were lenti-transduced with control virus (LKO) or lenti-vGPCR (MOI 3). At 48 hours RIPA lysates were immunoblotted for phospho-ERK1/2, stripped and re-probed for total ERK1/2. (A) At 36 hours post-transduction, IP-10 (100 nM) or the Shp2 inhibitor PHPS1 (10 μM) was added. (B) 48 hours prior to lenti-vGPCR transduction, BOEC were transduced where shown with control lenti-LKO, lenti-Shp2-c/s, or -DSH2, dominant negatives of Shp2. (C) Experiment performed as in (B) but using scrambled (sc) control versus lenti-sh4. (D) BOEC were transduced with control lentivirus, lenti-Shp2-c/s, or lenti-DSH2 as shown. After 48 hours cells were serum-starved for 17 hours in M199 (0.5% FBS), then exposed for 20 minutes to conditioned medium from BOEC that had been previously transfected with either control LKO lentivirus or lenti-vGPCR. Protein lysates were immunoblotted for pERK1/2 and total ERK1/2. (E) Experiment performed as in (D) but using lenti-sc as control versus lenti-sh4. Densitometry was performed and numbers over bands indicate fold intensity changes as normalized to loading controls and then to control bands.
Shp2 is required for vGPCR-mediated AP-1 and NFκB activation
In addition to MEK activation, vGPCR activates transcription factors (TF) such as AP-1 and NFκB. Both play roles in cell survival, growth and migration. NFκB in particular is upregulated by various KSHV gene products, and is a survival factor in both KSHV-infected B cells and endothelial cells. We therefore wanted to establish if Shp2 is required for activation of these TF by vGPCR specifically in endothelial cells. Given technical difficulties with simultaneous co-transduction multiple lentiviruses in our BOEC, we developed a tetracycline(tet)-inducible lentivirus expressing vGPCR using vectors from Clontech. This allowed us to split cells after lenti-transduction thereby controlling for transduction efficiency rather than relying on an additional reporter construct such as β-GAL. Figure 5A shows that with increasing MOI of tetracycline-inducible lenti-vGPCR, expression of vGPCR is enhanced. Figure 5B shows that both genetic and pharmacologic inhibition of Shp2 inhibit vGPCR-induced NFκB and AP-1 activity.
Figure 5.
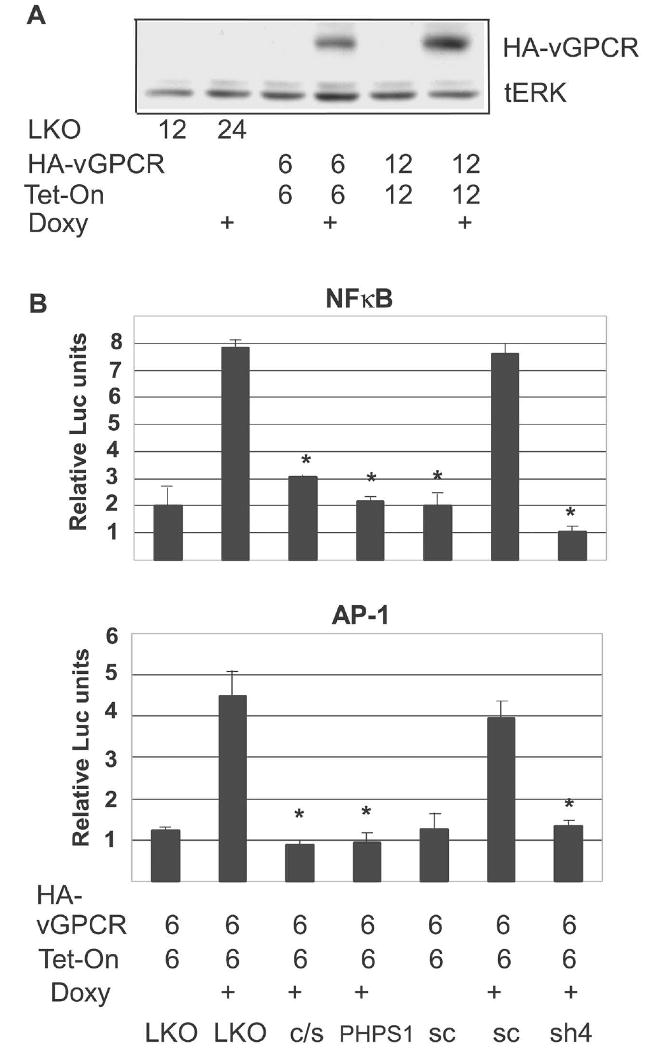
Shp2 is required for vGPCR-mediated activation of NFκB and AP-1 in BOEC. (A) BOEC were transfected with control virus or with pLVX-Tight-HA-vGPCR (HA-vGPCR) and pLVX-Tet-On (Tet-On) at MOI shown. After 48 hours cells were split equally and doxycycline (2 μg/ml) was added where indicated for an additional 48 hours. Protein lysates were harvested and immunoblotted for HA-vGPCR total ERK1/2 as a loading control. (B) BOEC were co-transduced with lenti-NFκB-luc reporter (top panel) along with pLVX-Tight-HA-vGPCR and pLVX-Tet-On. After 24 hours, cells were split, transduced again with control LKO versus Shp2 dominant-negative lentiviruses, or lenti-scrambled versus lenti-sh4. Doxycycline (doxy) was added to cause vGPCR expression where indicated for 48 hours, after which luciferase assay was performed. PHPS1 (10 μM) was added 17 hours before harvesting where shown. Bottom Panel: Identical to top except lenti-AP1-luc reporter was used. Values were normalized to protein concentration. Bars= S.D. * denotes p≤0.05 compared to maximal activation.
Shp2 is required for vGPCR-induced BOEC migration
As discussed above, vGPCR expression in endothelial cells pheno-mimics KSHV infection in vitro and in mouse models. Since effects seen in signaling assays do not necessarily translate into phenotypic effects, we wanted to know if Shp2 inhibition interferes with vGPCR-induced endothelial cell migration. Using a standard scratch wound assay, we found that both genetic and pharmacologic inhibition of Shp2 inhibited vGPCR-induced migration of BOEC. Cells were grown to confluence and within each experiment we confirmed that cell density was within 10% across all points. A scratch was then made and full medium replaced with low serum medium to minimize the effects of cell division. At 10 hour post-wounding, the microphotographs show the vGPCR-expressing cells oriented themselves perpendicularly to the wound and far more had entered into the wound as compared to control cells. Shp2 knockdown with lenti-sh4 had a significant inhibitory effect as did the PHPS1 which was more potent and inhibited migration even below that of the control cells which did display some baseline migration.
Discussion
KS tumors are highly angiogenic lesions of proliferating endothelial cells accompanied by a prominent inflammatory cell component. Although KSHV vGPCR is not the only viral product with the potential to influence these processes, it is a potent and constitutively active GPCR that conveys a survival advantage to endothelial cells in vitro and causes KS-like lesions in transgenic mice. Therefore, understanding how it signals will shed light not only on the role of vGPCR in KS tumor formation, but will also inform as to which cellular pathways are deregulated by KSHV more generally; perhaps by vGPCR at certain points in viral reproduction, but via different viral proteins with overlapping functions at other times. Indeed, given its lytic transcription pattern, is likely that the role of vGPCR changes with the viral life cycle and perhaps with KS lesion stage. Therefore, identifying key points at which cellular signaling control is disrupted by vGPCR will establish promising targets to interfere with KSHV replication and pathogenesis. To that end, we sought to determine whether the cytoplasmic phosphatase, Shp2, is an effector of vGPCR.
Given the increasing interest in the cytoplasmic PTPs as molecular targets for cancer therapy and very recent data supporting a role of Shp2 in normal angiogenesis, we investigated whether Shp2 is required for at least some aspects of vGPCR signaling; either via direct activation of Shp2 by vGPCR or indirectly by vGPCR-induced secreted factors, or elements of both. It is estimated that 81 human genes encode PTPs, which is very similar to the number for PTKs; and as mentioned above, PTPs are dysregulated in a diverse set of malignant and infectious human diseases (Alonso et al., 2004; Cheng et al., 2007; Migone et al., 1998; Nandan and Reiner, 2005; Nandan et al., 2002; Tsutsumi et al., 2006). For all these reasons, PTPs are increasingly the target of medicinal chemistry research (Hellmuth et al., 2008; Noren-Muller et al., 2006; Qu, 2000).
Our work in HEK293 and in BOEC shows that vGPCR causes phosphorylation of Shp2 at putative regulatory sites. Importantly, the proximal events in this pathway differed between cell types in that a vGPCR-Gαi axis was easily demonstrated in BOEC but not in HEK293. Presumably this is due to differences in expression levels of proximal effectors. Nonetheless, a vGPCR-Gαq axis was also at work in BOEC, although since PTX potently inhibits the vGPCR effect in serum-starved conditions, it may be that the role of Gαq is indirect in that it is required for vGPCR-mediated transcriptional upregulation of secreted cytokines which in turn signal via Shp2. We have also shown that Shp2 is required for successful conduction of various vGPCR-mediated signaling events. Although inhibition of Shp2 did not always completely abrogate vGPCR function, it is important to note that without Shp2, vGPCR's signaling effects were very near to baseline levels. Consistent with recent findings that Shp2 is involved in angiogenesis, we found that vGPCR-induced endothelial cell migration is Shp2-dependent.
Although this study argues for Shp2 as a potentially important convergence point for vGPCR signaling and as a promising target to disrupt KSHV-induced pathology, several questions remain and are under study. For instance, there is some debate over whether Shp2 can function as a scaffolding molecule (via its SH2 domains) regardless of an intact catalytic domain, or whether all Shp2 functions require its intact phosphatase activity. There is also some controversy regarding the relationship of potential regulatory tyrosine and serine sites in Shp2 and its catalytic activity; with some data showing that these phosphorylations are required and other not. We are currently investigating how vGPCR affects Shp2 phosphatase activity. We are also further exploring the mechanism of vGPCR signaling to Shp2 which is only partially elucidated here in that we looked only at canonical heterotrimeric G protein coupling. Non-canonical vGPCR signaling is an emerging field and there is a very small literature describing GPCRs that contain putative immunoreceptor tyrosine inhibitory motifs (ITIM). ITIM motifs bind to and activate Shp1 and Shp2 among other phosphatases, by inducing a conformational change (Poole and Jones, 2005).
We have identified a putative ITIM motif on the intracellular tail of vGPCR; our preliminary mutational studies and protein-protein binding assays support this as a bone fide ITIM motif.
The overall role of vGPCR to the biology of KS and the KSHV life cycle is not entirely clear. Although many investigators had early argued for a primarily paracrine role for vGPCR in tumor development, Lira's group has recently developed a mouse model in which vGPCR and LacZ are expressed simultaneously and conditionally, allowing mapping and phenotyping of vGPCR-expressing cells (Grisotto et al., 2006). Using this model they argue that vGPCR also has a direct proliferative effect on endothelial precursor cells. Of course paracrine effects are likely also at work; indeed, vGPCR-expressing hematopoietic cells in vitro express cytokines known to be growth factors for endothelial cells, most prominent of which is VEGF (Cannon, Philpott, and Cesarman, 2003). It is possible then that cells undergoing lytic KSHV infection not only produce new virions but via vGPCR help to condition the microenvironment such that it is conducive to ongoing KSHV infection and replication. A direct role for vGPCR in angiogenesis or survival is somewhat harder to explain in that vGPCR is a lytic KSHV gene product expressed in cells destined to produce new virions and die. There is however, some evidence that the vGPCR may be regulated outside of the canonical latent-to-lytic switch by cellular factors (Liang and Ganem, 2004). Another direct role could occur early in de novo KSHV infection when in vitro models show that there is a lytic burst of viral gene transcription prior to the establishment of latency. It is possible in this setting that vGPCR conveys survival and growth effects to offset a cellular response to viral infection. As a very proximal effector of vGPCR, Shp2 could play a role in any or all of these potential vGPCR functions. Of course studying the broader role of PTPs in the context of KSHV infection is also important in that there may be additional viral mechanisms to perturb these pathways that ultimately contribute to KSHV-induced pathology.
Materials and Methods
Plasmids and Chemicals
PD98059, NSC-87877, and pertussis toxin (PTX) were from Calbiochem. PHPS1 is from Asinex (Moscow, Russia). IP-10 is from Peprotech (Rocky Hill, NJ). PP2 (4-Amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine) was from Calbiochem. pcKSHV-vGPCR has been previously described (Arvanitakis et al., 1997). The pSG5-vGPCR.15 mutant was kind gift of John Nicholas and the HA-vGPCR expression construct was a gift of Pinghui Feng. pCMV-Shp2 WT and pCMV-Shp2-c/s were from Benjamin Neel via Addgene (Cambridge, MA). The C-terminal deletion Shp2 mutant (DSH2) was created by PCR using pCMV-Shp2 WT as template and the following primers: 5′-ATGGATCCACCATGACA TCGCGGAGATGGTT-3′ and 5′-ATGCGGCCGCACGAGTCGTGTTAAG-3′. Plasmids to generate lentivirus encoding shRNA sequences to Shp2 were obtained from Open Biosystems; sh3 (clone Id TRCN0000005003), sh4 (clone Id TRCN0000005004), sh6 (clone Id TRCN0000005006). Constructs from the Open Biosystems library are designed by algorithm to target a given transcript but need to be tested and validated by the end user. As demonstrated in the Results, we found sh3 and sh4 to be effective, but sh6 ineffective. For Src knockdown we used clone ID TRCN0000038153. Luciferase reporter constructs include pAP1(PMA)-TA-Luc, pNFκB-TA-Luc (Clontech).
Cell Culture and Conditioned Medium
HEK293 cells were maintained in DMEM plus 40 mg/L gentamicin with 10% FBS (Invitrogen) at 37°C, 5% CO2. BOEC cells were obtained from Dr. Robert Hebbel, University of Minnesota and maintained in EGM-2 plus Bullet Kit (Lonza, Walkersville, MD) on collagen-coated plates and dishes (Lin et al., 2000). To produce conditioned medium, BOEC were transduced with control lentivirus or lenti-vGPCR; at 48 hours cells were washed and M199 (Invitrogen) plus 0.5% FBS was added. 48 hours later medium was filtered through a 45 μm syringe filter and either used immediately or aliquoted and frozen at -80° C for future use.
Transfections
Transfections of HEK293 or HEK293T were performed on exponentially growing cells using a 3:1 ratio of Fugene6 (Roche) to plasmid DNA as per manufacturer's recommendations.
Luciferase assays
Cells were transfected or transduced with luciferase reporter plasmids or lentiviruses respectively, as well as other constructs appropriate to each experiment. After 24-48 hours, lysates were prepared using the provided lysis reagent as per the manufacturer's directions (Promega, Madison, WI). Assays were performed using 10 μl of lysate and 50 μl of beetle luciferin in a Luminoskan Ascent FL (Thermo Fisher Scientific, Waltham, MA) using a 10-s read time. For HEK293, cells were co-transfected with a β-galactosidase plasmid and luciferase results normalized to β-gal levels as per manufacturer's protocol (Promega, Madison, WI). In the case of BOEC transductions, cells were divided after transfection, so efficiency was inherently controlled for and luciferase levels were normalized to protein levels; protein was quantified using DC Protein Assay Kit II (Bio-Rad, Hercules, CA).
Western Blotting and Antibodies
Both HEK293 and BOEC cells were lysed with standard RIPA buffer with phosphatase and protease inhibitor cocktails (Sigma). Protein was quantitated by the Bradford method and loaded onto 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) gels. Semidry transfer to PVDF (Millipore, Billerica, MA) was performed using transfer buffer, 48 mM Tris, 39 mM glycine, 0.037% SDS, and 20% methanol. Blots were probed with 1:1000 dilution of primary antibody (Ab) overnight at 4°C in the case of phospho-specific antibodies, or at room temperature for 1 hour for all others. HRP-conjugated secondary Ab was added after washing and detected by an enhanced chemiluminescence system (ECL) (Amersham, Little Chalfont, UK). Antibodies used: anti-Shp2(C-18) (Santa Cruz, Santa Cruz, CA); antibodies to phosphorylated forms of AKT (9271), ERK1/2 (9101), Try542-Shp2 (3751) and Tyr580-Shp2 (3754) were all from Cell signaling Technologies (Danvers, MA) as was anti-AKT (9272). Anti-ERK1/2 (06182) was from Millipore/Upstate (Billerica, MA). Anti-HA antibody (16B12) was from Covance (Princeton, NJ) and anti-n-terminal vGPCR was a kind gift of Gary Hayward of Johns Hopkins University. Western blot autoradiographs were scanned with an Epson V700 Photo scanner. Images were cropped and brightness/contrast adjusted with Adobe Photoshop 6.0. Notation for figures was done with Deneba Canvas 8.0. Band density was quantitated from film using a Kodak Image Station 2000R and software Kodak MI v4.0.4.
Lentivirus Production and Infection
Transgenes were PCR-cloned to contain an upstream BamHI site and a downstream NotI site and ligated into BamH1/NotI digested pHR-CSGW (gift of Adrian Thrasher). For luciferase reporter lentiviruses, pHR-CSGW was digested with EcoR1, blunt ended, and then digested with NotI to remove both the SFFV promoter and the GFP transgene. Into this site was ligated the sequence from the transcription blocker through the end of the luciferase coding region from the AP1 and NFκB reporter plasmids (see above), using primers, 5′-CGGATATCCGGCCGCAAT AAAATATCTT-3′ and 5′- ATAAGAATGCGGCCGCCCGACTCTAGAATTACAC-3′. To obtain pseudotyped lentivirus (recombinant HIV-1 with vesicular stomatitis virus G (VSV-G) envelope protein), we used the gene delivery and production system developed by Naldini et al (Naldini et al., 1996). In short, HEK293T cells were transfected with pHR-CSGW-transgene, p8.74 and pMD.G in a 1.5:1:1 ratio. At 72 hours, supernatants were filtered through a 0.45 μm syringe filter then ultracentrifuged for 2 hours at 22,000 rpm. Pellets were resuspended in Opti-Mem, aliquoted and frozen. Titering was done by Western blot of 3 μl and 15 μl of lentivirus for p24 using antibody from Abcam (ab9071) (Cambridge, MA). A sample of lentivirus that expresses GFP and had been previously titered by flow cytometry was loaded as a control. BOEC were transduced at approximately 50% confluence by resuspending lentivirus in 20 μl of Opti-Mem which was then added to cells and left on for 48-72 hours, depending on experimental set-up. For tetracycline-inducible lentivirus experiments, HA-vGPCR was cloned into pLVX-Tight-Puro and cells were infected with equal MOI of this virus and pLVX-Tet-On (Clontech) along with the transcription factor reporter lentivirus. After 48 hours, cells were split and subjected to further transduction with control, dominant-negative or knock-down lentis; doxycycline was added where stated in figure legends. In the case of shRNA knock-down, plasmid to generate lentivirus encoding various candidate knock-down sequences were obtained from Open Biosystems (see Materials above).
Scratch Assay
BOEC cells were seeded into 6 well plates and allowed to reach confluency. A 200 μl pipette tip was used to wound cells in a straight line across the well. Full medium was than replaced with M199 with 0.5% FBS to minimize the effects of cell division. Cells were periodically examined and micrographs obtained with an Axiovert 200M, A-plan 10×/0.25 objective, and Axiocam (Zeiss). The width of the wound and number of cells that had migrated into wound were quantitated using NIH ImageJ (http://rsb.info.nih.gov/ij/). BOEC were previously transduced or chemically treated as appropriate for each experiment.
Figure 6.
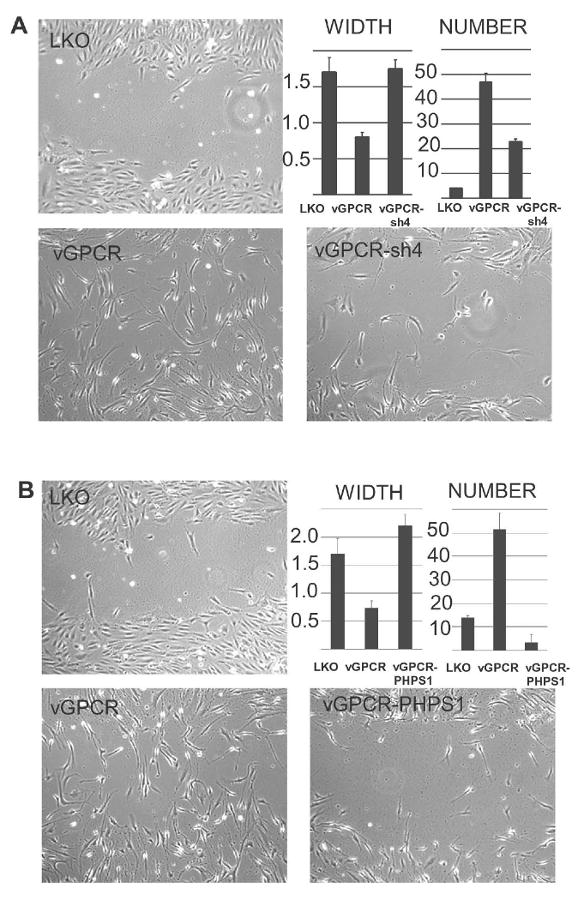
Shp2 is required for vGPCR-induced migration in BOEC. (A) BOEC were lenti- transduced with control virus LKO or vGPCR with and without lenti-sh4 to knockdown Shp2. After 48 hours cells reached confluence and a scratch “wound” was made with a pipette tip and medium changed to low-serum (0.1% FBS) without additional supplements. The width of the scratch and the number of cells migrating back into the wound were quantified using NIH ImageJ, 10 hours post-wounding (width measured in arbitrary units). (B) Shp2 was inhibited by PHPS1 (10 μM) where indicated for 12 hours prior to wounding. Error bars represent S.D. One representative of 3 experiments is shown.
Acknowledgments
We would like to thank Robert Hebbel and Julia Nguyen for supplying us with low-passage BOEC and technical advice on their maintenance in culture. The work was supported by NIH grant K08-AI53971. The authors have no conflicts to declare.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adachi M, Fischer EH, Ihle J, Imai K, Jirik F, Neel B, Pawson T, Shen S, Thomas M, Ullrich A, Zhao Z. Mammalian SH2-containing protein tyrosine phosphatases. Cell. 1996;85(1):15. doi: 10.1016/s0092-8674(00)81077-6. [DOI] [PubMed] [Google Scholar]
- Alonso A, Sasin J, Bottini N, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Protein tyrosine phosphatases in the human genome. Cell. 2004;117(6):699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- Arvanitakis L, Geras-Raaka E, Varma A, Gershengorn MC, Cesarman E. Human herpesvirus KSHV encodes a constitutively active G-protein-coupled receptor linked to cell proliferation. Nature. 1997;385(6614):347–50. doi: 10.1038/385347a0. [DOI] [PubMed] [Google Scholar]
- Bais C, Santomasso B, Coso O, Arvanitakis L, Raaka EG, Gutkind JS, Asch AS, Cesarman E, Gershengorn MC, Mesri EA. G-protein-coupled receptor of Kaposi's sarcoma-associated herpesvirus is a viral oncogene and angiogenesis activator. Nature. 1998;391(6662):86–9. doi: 10.1038/34193. [DOI] [PubMed] [Google Scholar]
- Burshtyn DN, Scharenberg AM, Wagtmann N, Rajagopalan S, Berrada K, Yi T, Kinet JP, Long EO. Recruitment of tyrosine phosphatase HCP by the killer cell inhibitor receptor. Immunity. 1996;4(1):77–85. doi: 10.1016/s1074-7613(00)80300-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon M, Cesarman E, Boshoff C. KSHV G protein-coupled receptor inhibits lytic gene transcription in primary-effusion lymphoma cells via p21-mediated inhibition of Cdk2. Blood. 2006;107(1):277–84. doi: 10.1182/blood-2005-06-2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon M, Philpott NJ, Cesarman E. The Kaposi's Sarcoma-Associated Herpesvirus G Protein-Coupled Receptor Has Broad Signaling Effects in Primary Effusion Lymphoma Cells. J Virol. 2003;77(1):57–67. doi: 10.1128/JVI.77.1.57-67.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med. 1995;332(18):1186–91. doi: 10.1056/NEJM199505043321802. see comments. [DOI] [PubMed] [Google Scholar]
- Cesarman E, Nador RG, Bai F, Bohenzky RA, Russo JJ, Moore PS, Chang Y, Knowles DM. Kaposi's sarcoma-associated herpesvirus contains G protein-coupled receptor and cyclin D homologs which are expressed in Kaposi's sarcoma and malignant lymphoma. J Virol. 1996;70(11):8218–23. doi: 10.1128/jvi.70.11.8218-8223.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science. 1994;266(5192):1865–9. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- Chen HE, Chang S, Trub T, Neel BG. Regulation of colony-stimulating factor 1 receptor signaling by the SH2 domain-containing tyrosine phosphatase SHPTP1. Mol Cell Biol. 1996;16(7):3685–97. doi: 10.1128/mcb.16.7.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Sung SS, Yip ML, Lawrence HR, Ren Y, Guida WC, Sebti SM, Lawrence NJ, Wu J. Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Mol Pharmacol. 2006;70(2):562–70. doi: 10.1124/mol.106.025536. [DOI] [PubMed] [Google Scholar]
- Cheng J, Kydd AR, Nakase K, Noonan KM, Murakami A, Tao H, Dwyer M, Xu C, Zhu Q, Marasco WA. Negative regulation of the SH2-homology containing protein-tyrosine phosphatase-1 (SHP-1) P2 promoter by the HTLV-1 Tax oncoprotein. Blood. 2007;110(6):2110–20. doi: 10.1182/blood-2006-11-058388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ambrosio D, Hippen KL, Minskoff SA, Mellman I, Pani G, Siminovitch KA, Cambier JC. Recruitment and activation of PTP1C in negative regulation of antigen receptor signaling by Fc gamma RIIB1. Science. 1995;268(5208):293–7. doi: 10.1126/science.7716523. [DOI] [PubMed] [Google Scholar]
- Della Bella S, Taddeo A, Calabro ML, Brambilla L, Bellinvia M, Bergamo E, Clerici M, Villa ML. Peripheral Blood Endothelial Progenitors as Potential Reservoirs of Kaposi's Sarcoma-Associated Herpesvirus. PLoS ONE. 2008;3(1):e1520. doi: 10.1371/journal.pone.0001520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng GS, Hui CC, Pawson T. SH2-containing phosphotyrosine phosphatase as a target of protein-tyrosine kinases. Science. 1993;259(5101):1607–11. doi: 10.1126/science.8096088. [DOI] [PubMed] [Google Scholar]
- Geras-Raaka E, Varma A, Ho H, Clark-Lewis I, Gershengorn MC. Human interferon-gamma-inducible protein 10 (IP-10) inhibits constitutive signaling of Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor. J Exp Med. 1998;188(2):405–8. doi: 10.1084/jem.188.2.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisotto MG, Garin A, Martin AP, Jensen KK, Chan P, Sealfon SC, Lira SA. The human herpesvirus 8 chemokine receptor vGPCR triggers autonomous proliferation of endothelial cells. J Clin Invest. 2006;116(5):1264–73. doi: 10.1172/JCI26666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo HG, Browning P, Nicholas J, Hayward GS, Tschachler E, Jiang YW, Sadowska M, Raffeld M, Colombini S, Gallo RC, Reitz MS., Jr Characterization of a chemokine receptor-related gene in human herpesvirus 8 and its expression in Kaposi's sarcoma. Virology. 1997;228(2):371–8. doi: 10.1006/viro.1996.8386. [DOI] [PubMed] [Google Scholar]
- Guo HG, Sadowska M, Reid W, Tschachler E, Hayward G, Reitz M. Kaposi's sarcoma-like tumors in a human herpesvirus 8 ORF74 transgenic mouse. J Virol. 2003;77(4):2631–9. doi: 10.1128/JVI.77.4.2631-2639.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmuth K, Grosskopf S, Lum CT, Wurtele M, Roder N, von Kries JP, Rosario M, Rademann J, Birchmeier W. Specific inhibitors of the protein tyrosine phosphatase Shp2 identified by high-throughput docking. Proc Natl Acad Sci U S A. 2008;105(20):7275–80. doi: 10.1073/pnas.0710468105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyer G, Alexander DR. Immune signalling: SHP-2 docks at multiple ports. Curr Biol. 1999;9(4):R129–32. doi: 10.1016/s0960-9822(99)80080-3. [DOI] [PubMed] [Google Scholar]
- Jensen KK, Manfra DJ, Grisotto MG, Martin AP, Vassileva G, Kelley K, Schwartz TW, Lira SA. The Human Herpes Virus 8-Encoded Chemokine Receptor Is Required for Angioproliferation in a Murine Model of Kaposi's Sarcoma. J Immunol. 2005;174(6):3686–94. doi: 10.4049/jimmunol.174.6.3686. [DOI] [PubMed] [Google Scholar]
- Jiang A, Pan W, Milbauer LC, Shyr Y, Hebbel RP. A practical question based on cross-platform microarray data normalization: are BOEC more like large vessel or microvascular endothelial cells or neither of them? J Bioinform Comput Biol. 2007;5(4):875–93. doi: 10.1142/s0219720007002989. [DOI] [PubMed] [Google Scholar]
- Koch CA, Anderson D, Moran MF, Ellis C, Pawson T. SH2 and SH3 domains: elements that control interactions of cytoplasmic signaling proteins. Science. 1991;252(5006):668–74. doi: 10.1126/science.1708916. [DOI] [PubMed] [Google Scholar]
- Liang Y, Ganem D. RBP-J (CSL) is essential for activation of the K14/vGPCR promoter of Kaposi's sarcoma-associated herpesvirus by the lytic switch protein RTA. J Virol. 2004;78(13):6818–26. doi: 10.1128/JVI.78.13.6818-6826.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Chang L, Solovey A, Healey JF, Lollar P, Hebbel RP. Use of blood outgrowth endothelial cells for gene therapy for hemophilia A. Blood. 2002;99(2):457–62. doi: 10.1182/blood.v99.2.457. [DOI] [PubMed] [Google Scholar]
- Lin Y, Weisdorf DJ, Solovey A, Hebbel RP. Origins of circulating endothelial cells and endothelial outgrowth from blood. J Clin Invest. 2000;105(1):71–7. doi: 10.1172/JCI8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Sandford G, Fei G, Nicholas J. Galpha protein selectivity determinant specified by a viral chemokine receptor-conserved region in the C tail of the human herpesvirus 8 g protein-coupled receptor. J Virol. 2004;78(5):2460–71. doi: 10.1128/JVI.78.5.2460-2471.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz U, Ravichandran KS, Pei D, Walsh CT, Burakoff SJ, Neel BG. Lck-dependent tyrosyl phosphorylation of the phosphotyrosine phosphatase SH-PTP1 in murine T cells. Mol Cell Biol. 1994;14(3):1824–34. doi: 10.1128/mcb.14.3.1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannell H, Hellwiga N, Gloea T, Plankc C, Sohnb H, Groessera L, Walzoga B, Pohla U, Krötza F. Inhibition of the Tyrosine Phosphatase SHP-2 Suppresses Angiogenesis in vitro and in vivo. Journal of Vascular Research. 2008;45(2):153. doi: 10.1159/000110081. [DOI] [PubMed] [Google Scholar]
- Migone TS, Cacalano NA, Taylor N, Yi T, Waldmann TA, Johnston JA. Recruitment of SH2-containing protein tyrosine phosphatase SHP-1 to the interleukin 2 receptor; loss of SHP-1 expression in human T-lymphotropic virus type I-transformed T cells. Proc Natl Acad Sci U S A. 1998;95(7):3845–50. doi: 10.1073/pnas.95.7.3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minoo P, Zadeh MM, Rottapel R, Lebrun JJ, Ali S. A novel SHP-1/Grb2-dependent mechanism of negative regulation of cytokine-receptor signaling: contribution of SHP-1 C-terminal tyrosines in cytokine signaling. Blood. 2004;103(4):1398–407. doi: 10.1182/blood-2003-07-2617. [DOI] [PubMed] [Google Scholar]
- Monini P, Colombini S, Sturzl M, Goletti D, Cafaro A, Sgadari C, Butto S, Franco M, Leone P, Fais S, Leone P, Melucci-Vigo G, Chiozzini C, Carlini F, Ascherl G, Cornali E, Zietz C, Ramazzotti E, Ensoli F, Andreoni M, Pezzotti P, Rezza G, Yarchoan R, Gallo RC, Ensoli B. Reactivation and persistence of human herpesvirus-8 infection in B cells and monocytes by Th-1 cytokines increased in Kaposi's sarcoma. Blood. 1999;93(12):4044–58. see comments. [PubMed] [Google Scholar]
- Montaner S, Sodhi A, Molinolo A, Bugge TH, Sawai ET, He Y, Li Y, Ray PE, Gutkind JS. Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates Kaposi's sarcomagenesis and can promote the tumorigenic potential of viral latent genes. Cancer Cell. 2003;3(1):23–36. doi: 10.1016/s1535-6108(02)00237-4. [DOI] [PubMed] [Google Scholar]
- Moore PS, Gao SJ, Dominguez G, Cesarman E, Lungu O, Knowles DM, Garber R, Pellett PE, McGeoch DJ, Chang Y. Primary characterization of a herpesvirus agent associated with Kaposi's sarcomae. J Virol. J Virol. 1996 Dec;7070(12)(1):9083. 549–58. doi: 10.1128/jvi.70.1.549-558.1996. published erratum appears. 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nador RG, Cesarman E, Chadburn A, Dawson DB, Ansari MQ, Sald J, Knowles DM. Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi's sarcoma-associated herpes virus. Blood. 1996;88(2):645–56. [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272(5259):263–7. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- Nandan D, Reiner NE. Leishmania donovani engages in regulatory interference by targeting macrophage protein tyrosine phosphatase SHP-1. Clin Immunol. 2005;114(3):266–77. doi: 10.1016/j.clim.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Nandan D, Yi T, Lopez M, Lai C, Reiner NE. Leishmania EF-1alpha activates the Src homology 2 domain containing tyrosine phosphatase SHP-1 leading to macrophage deactivation. J Biol Chem. 2002;277(51):50190–7. doi: 10.1074/jbc.M209210200. [DOI] [PubMed] [Google Scholar]
- Neel BG, Tonks NK. Protein tyrosine phosphatases in signal transduction. Curr Opin Cell Biol. 1997;9(2):193–204. doi: 10.1016/s0955-0674(97)80063-4. [DOI] [PubMed] [Google Scholar]
- Noren-Muller A, Reis-Correa I, Jr, Prinz H, Rosenbaum C, Saxena K, Schwalbe HJ, Vestweber D, Cagna G, Schunk S, Schwarz O, Schiewe H, Waldmann H. Discovery of protein phosphatase inhibitor classes by biology-oriented synthesis. Proc Natl Acad Sci U S A. 2006;103(28):10606–11. doi: 10.1073/pnas.0601490103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T, Yoshino T, Hayashi K, Ohara N, Nakanishi T, Yamaai Y, Hiraki A, Sogawa CA, Kondo E, Teramoto N, Takahashi K, Tsuchiyama J, Akagi T. Reduction of hematopoietic cell-specific tyrosine phosphatase SHP-1 gene expression in natural killer cell lymphoma and various types of lymphomas/leukemias : combination analysis with cDNA expression array and tissue microarray. Am J Pathol. 2001;159(4):1495–505. doi: 10.1016/S0002-9440(10)62535-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole AW, Jones ML. A SHPing tale: perspectives on the regulation of SHP-1 and SHP-2 tyrosine phosphatases by the C-terminal tail. Cell Signal. 2005;17(11):1323–32. doi: 10.1016/j.cellsig.2005.05.016. [DOI] [PubMed] [Google Scholar]
- Qu CK. The SHP-2 tyrosine phosphatase: signaling mechanisms and biological functions. Cell Res. 2000;10(4):279–88. doi: 10.1038/sj.cr.7290055. [DOI] [PubMed] [Google Scholar]
- Said W, Chien K, Takeuchi S, Tasaka T, Asou H, Cho SK, de Vos S, Cesarman E, Knowles DM, Koeffler HP. Kaposi's sarcoma-associated herpesvirus (KSHV or HHV8) in primary effusion lymphoma: ultrastructural demonstration of herpesvirus in lymphoma cells. Blood. 1996;87(12):4937–43. [PubMed] [Google Scholar]
- Tsutsumi R, Takahashi A, Azuma T, Higashi H, Hatakeyama M. Focal adhesion kinase is a substrate and downstream effector of SHP-2 complexed with Helicobacter pylori CagA. Mol Cell Biol. 2006;26(1):261–76. doi: 10.1128/MCB.26.1.261-276.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitby D, Howard MR, Tenant-Flowers M, Brink NS, Copas A, Boshoff C, Hatzioannou T, Suggett FE, Aldam DM, Denton AS. Detection of Kaposi sarcoma associated herpesvirus in peripheral blood of HIV-infected individuals and progression to Kaposi's sarcoma. Lancet. 1995;346(8978):799–802. doi: 10.1016/s0140-6736(95)91619-9. [DOI] [PubMed] [Google Scholar]
- Wu DQ, Lee CH, Rhee SG, Simon MI. Activation of phospholipase C by the alpha subunits of the Gq and G11 proteins in transfected Cos-7 cells. J Biol Chem. 1992;267(3):1811–7. [PubMed] [Google Scholar]
- Xu R, Yu Y, Zheng S, Zhao X, Dong Q, He Z, Liang Y, Lu Q, Fang Y, Gan X, Xu X, Zhang S, Zhang X, Feng GS. Overexpression of Shp2 tyrosine phosphatase is implicated in leukemogenesis in adult human leukemia. Blood. 2005;106(9):3142–9. doi: 10.1182/blood-2004-10-4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Chen S, Leach M, Manfra D, Homey B, Wiekowski M, Sullivan L, Jenh C, Narula S, Chensue S, Lira S. Transgenic expression of the chemokine receptor encoded by human herpesvirus 8 induces an angioproliferative disease resembling Kaposi's sarcoma. J Exp Med. 2000;191(3):445–54. doi: 10.1084/jem.191.3.445. see comments. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi T, Cleveland JL, Ihle JN. Identification of novel protein tyrosine phosphatases of hematopoietic cells by polymerase chain reaction amplification. Blood. 1991;78(9):2222–8. [PubMed] [Google Scholar]
- Yi T, Mui AL, Krystal G, Ihle JN. Hematopoietic cell phosphatase associates with the interleukin-3 (IL-3) receptor beta chain and down-regulates IL-3-induced tyrosine phosphorylation and mitogenesis. Mol Cell Biol. 1993;13(12):7577–86. doi: 10.1128/mcb.13.12.7577. [DOI] [PMC free article] [PubMed] [Google Scholar]