

Summary
Cell signaling networks display an extraordinary range of temporal and spatial plasticity. Our programmatic approach focuses on the construction of intracellular probes, including sensors, inhibitors, and functionally unique proteins that can be temporally and spatially controlled by the investigator even after they have entered the cell. We have designed and evaluated protein kinase sensors that furnish a fluorescent readout upon phosphorylation. In addition, since the sensors are inert (i.e. cannot be phosphorylated) until activated by light, they can be carried through the various stages of any given cell-based behavior without being consumed. Using this strategy, we have shown that PKCβ is essential for nuclear envelope breakdown and thus the transition from prophase to metaphase in actively dividing cells. Photoactivatable proteins furnish the means to initiate cellular signaling pathways with a high degree of spatial and temporal control. We have used this approach to demonstrate that cofilin serves as a component of the steering apparatus of the cell. Finally, inhibitors are commonly used to assess the participation of specific enzymes in signaling pathways that control cellular behavior. We have constructed a photo-deactivatable inhibitor, an inhibitory species that can be switched off with light. In the absence of light, the target enzyme is inactive due to the presence of the potent inhibitory molecule. Upon photolysis, the inhibitory molecule is destroyed and enzymatic activity is released.
Keywords: Signal Transduction, Photoactivation, Caged proteins, Kinase sensors, Inhibitors
Introduction
The remarkable complexity of life entails intricately tuned cascades of biochemical events in individual cells, tissues and whole organisms. Signal transduction pathways mediate the cellular response to environmental factors, such as pH, ions, electromagnetic radiation, and the wide assortment of cell-derived stimuli (neurotransmitters, growth factors, secondary messengers). With the sequencing of the human genome in 2001,[1, 2] there were great expectations that the information gained could be used to correlate gene expression with biochemical activity and thus with normal and aberrant (i.e. disease) cell-based behaviors. However, complete insight into the necessary events that initiate and maintain normal and diseased states requires not just DNA sequences, but also an understanding of the interplay of gene expression in the context of protein inhibition, activation, and recycling.
Advances in technology have lead to significant improvements in our understanding of intracellular chemistry, especially in the fields of epigenomics and proteomics. Although these disciplines have generated a vast array of information about gene expression and protein activity, they do not lend themselves to investigating the action of specific signaling molecules, genes, or proteins at the single cell level. Several methods have been developed to address how gene expression influences cellular events, including overexpression or knock down of the gene of interest. Molecular biology has also provided a unique set of fluorescent proteins, most notably an array green fluorescent protein (GFP) analogs, that can be used to produce fluorescent protein constructs that can monitor the location of a specific protein within an individual cell [3]. Furthermore, chemists have supplied numerous small molecule activators, inhibitors and sensors to this expanding biological toolbox, which have been used to alter or monitor protein activity. Despite the fact that these innovations have led to a better understanding of cellular events, these tools are generally unable to probe or manipulate the biochemistry of life with a high degree of spatial or temporal control.
Cellular states and events like homeostasis, mitosis and apoptosis all involve precise timing of gene transcription, protein activation, inactivation and degradation. Along with temporal control, cellular events are often spatially restricted to subcellular organelles, the cytoskeletal network, or cellular extensions, allowing activation of specific signaling networks in a spatially confined region of the cell. In order to address the spatiotemporal aspects involved in signaling cascades, photoactivatible or “caged” compounds have been developed for the precise time-dependent release of the bioactive molecule. Caged compounds are biologically inert until they absorb one or more photons of light, thereby liberating a bioactive molecule, be it a protein, a genetic coding sequence, an inhibitor, activator, or sensor. ATP and cAMP were the first caged compounds to be described [4, 5]. Caged compounds have been developed as effectors of (i) gene expression, (ii) protein expression, (iii) protein activation, (iv) fluorescence, (v) protein inhibition (vi) biochemical sensing [6–22]. Our lab has primarily focused on the design, synthesis, and characterization of caged compounds for elucidating the spatiotemporal dynamics of signaling pathways.
Chemistry of Photoactivatible Molecules
Temporal or spatial control of a bioactive species (gene, protein, etc) is readily afforded by introducing a photolabile group at a site on the molecule of interest required for biological activity. Caged compounds are commonly activated by absorbance of one or more photons, thereby resulting in the cleavage of the photolabile group and restoration of biological activity. More recently, the term caging has also been used in reference to photoswitches, which undergo isomerization resulting in a reversible conformational change that alters activity. Azobenzene moieties and spiropyran-based functionality have been utilized to generate photoswitches in peptides[23–28], peptidomimetics[29], proteins[6, 9, 30–36] and nucleic acids[16, 37, 38]. In addition, photoactivatible derivatives of GFPs have been described which display photoinduced fluorescence increases [39–48]. Recently a genetically-encoded caging strategy using a photosensory domain from phytochrome B has been described to cage WASP.[49]
Caged molecules require (i) the introduction of a photolabile moiety on a functional group that is essential for biological activity, (ii) biologically inertness, and (iii) light-driven formation of the active species in the absence of toxic byproducts. A large number of photolabile groups have been developed as caging moieties and the vast majority of these are photolyzed at wavelengths less than 365 nm [8, 12, 13, 20, 50]. The most commonly used photocleavable species are the ortho-nitrobenzyl, hydroxyphenacyl, coumarin, cinnamate moieties, and their derivatives. The mechanism of photolysis for the ortho-nitrobenzyl group involves absorption of a photon to generate an aci-nitro intermediate (Scheme 1). The latter is rapidly converted to a hemiacetal, which decomposes to furnish the free biologically active molecule and a nitrosobenzene byproduct (Scheme 1). Aldehyde byproducts from this reaction are electrophilic and therefore may have deleterious intracellular consequences. By contrast, ketone byproducts are generally considered to be biologically inert [5].
Scheme 1.

Mechanism of photolytic cleavage for the ortho-nitrobenzyl group.
Two key parameters that must be considered when dealing with photolabile groups are wavelength of activation and quantum yield (Φ) of uncaging. Caging moieties that suffer photolysis at wavelengths less than 300 nm are not regarded as biologically useful because of the toxicity associated with UV light. Although derivatives of the ortho-nitrobenzyl group have been developed that absorb at longer wavelengths (>360 nm) they display a striking decrease in quantum yield [51]. At these short wavelengths photons are absorbed by cellular chromophores and thus do not penetrate deeply enough into samples to be effective in whole tissues or live animals. Photolabile groups with longer uncaging wavelengths, such as coumarin derivatives that absorb at 400 nm[52, 53], or those that contain a large two photon cross section[54–59] have been identified. The field of uncaging awaits the development of multiple photolabile groups that are sensitive to longer wavelengths as well as the creation of additional two photon responsive caging groups that can be applied in live animals.
Photoactivatable Proteins
Perturbation of intracellular protein levels provides an understanding of how specific proteins influence cell behavior. However, changes in expression levels do not always correlate with changes in activity. Protein activity is regulated by an intricate network of signaling pathways that serve as both on and off signals for cellular processes. A host of techniques have been developed to address how an active or inactive protein influences cellular chemistry, including site-directed mutagenesis to generate constitutively active proteins or dominant negative constructs. However, these strategies do not address the time-dependent nature of signaling cascades. By contrast, a caged derivative of a protein allows for precise control over where and when the protein of interest is activated. Over the last two decades a number of methods have been utilized to create caged proteins: (i) covalent modification of an existing nucleophilic residue on the protein with a photolabile group, (ii) insertion of an appropriate reactive residue at the desired position via site-directed mutagenesis, (iii) enzyme-catalyzed site-specific introduction of reactive functionality[60], (iv) construction of semisynthetic proteins via expressed protein ligation[61–63], (v) insertion of caged amino acids via unnatural amino acid mutagenesis[64], and (vi) genetic methods that employ a photosensory domain[49].
The cAMP-dependent protein kinase (PKA) is arguably the best mechanistically and structurally understood of all protein kinases [65]. Binding of cAMP to the holoenzyme, a tetramer consisting of two regulatory and two catalytic subunits, releases the catalytic subunits that are now free to phosphorylate substrates. The catalytic subunit contains two cysteine residues. Cys199 is located at the edge of the active site and Cys343 is positioned on the surface far removed from the active site region.
We designed an active-site directed peptide (3) that selectively labels PKA at Cys199 [66]. The affinity of the active site directed peptide for PKA is relatively weak (KD ~ 1 mM). Consequently, following photolysis the released peptidic species does not serve as an effective inhibitor of PKA. The peptide backbone of 3 was prepared via standard Fmoc solid-phase synthesis (Scheme 2). The ethylene diamine moiety (see compound 1) serves as a linker to situate the caging group adjacent to Cys199 once the peptide is bound to the active site. The free primary amine on peptide 1 was coupled to the caging unit 2 to generate the desired active site directed affinity label 3. Cys199 of the catalytic subunit was modified in a time-dependent fashion by 3 (KI = 1.5 ± 0.3 mM) to generate caged (i.e. inactive) PKA 4. Electrospray ionization mass spectrometry revealed a single alkylation of PKA confirming the active site delivery of the caging group to Cys199. In vitro analysis of PKA revealed that 4 has less than 2% residual activity and a 25-fold enhancement in activity after photolysis. Although only 50% of the native activity is recovered after uncaging, the 25-fold difference in activity before and after photolysis proved sufficient for subsequent intracellular studies.
Scheme 2.
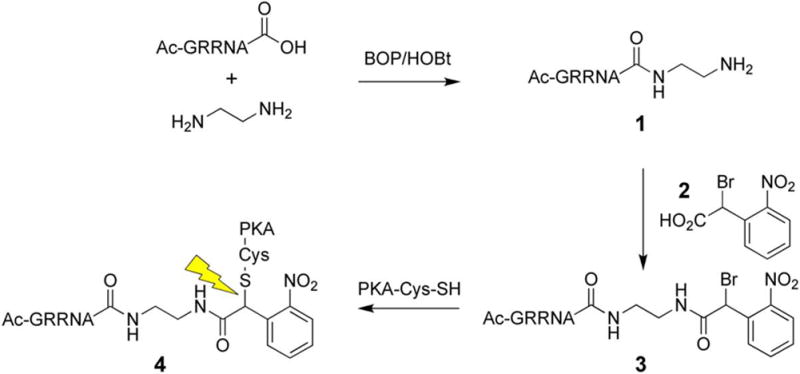
Synthesis of caged PKA.
Activation of PKA by cAMP or injection of free catalytic subunit in fibroblasts is known to induce conversion from an extended to rounded cellular morphology as well as loss of actin-containing stress fibers [67]. Rat embryo fibroblasts (REF) cells were microinjected with 3 – 7 μM of caged PKA catalytic subunit or its free active counterpart. After microinjection, cells were illuminated with a 200 W Hg arc lamp and placed in an incubator for 1 h. The cells were subsequently fixed for immunofluorescence with rhodamine-labeled phalloidin, which binds to F-actin found in stress fibers. Microinjected, but non-illuminated, REF cells display a similar morphology and stress fiber network as their non-microinjected counterparts (Fig a). However, upon photolysis, cells containing caged PKA exhibit membrane ruffling, a rounded morphology (Fig 1b), and a loss of stress fibers. These are all characteristics of an activated PKA signaling cascade. Similar results were obtained when free catalytic subunit was microinjected (Fig 1c). In addition, photolysis without microinjection of the caged enzyme, failed to produce any changes in cellular morphology.
Figure 1.
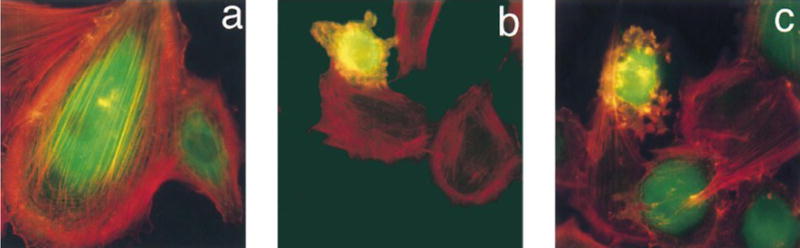
Changes in morphology and stress fiber formation in REFs stained with phalloidin for F-actin (Red) and FITC-IgG for microinjected cells (green). (A) REFs microinjected with caged PKA 4, (B) REFs microinjected with caged PKA 4 followed by photouncaging, (C) REFs microinjected with free PKA catalytic subunit. Reprinted with permission from [66]. Copyright 1998 American Chemical Society.
In addition to regulation by small molecule activators such as cAMP, many members of signaling pathways are turned on or off by protein kinases or phosphatases. Consequently, photoactivation of a caged signaling protein may be transient at best due to the presence these regulatory enzymes. In order to circumvent this potential difficulty, we constructed a caged signaling protein that, upon photolysis, generates a constitutively active species that is not regulated (bio-orthogonal) by the endogenous biochemistry (i.e. kinases and phosphatases) of the cell.
Epidermal growth factor (EGF) stimulates cell motility, a behavior that is dependent, at least in part, on the intracellular protein cofilin. However, the specific role played by this protein kinase-regulated species in growth factor-directed motility is unclear. The initial step in chemotaxis is the formation of a lammelipod, a membrane protrusion driven by actin polymerization [i.e. the conversion of monomeric G (globular) actin into polymeric F (filamentous) actin]. Cofilin is known to control actin dynamics in living cells, by altering the equilibrium between the G and F actin states. Cofilin both cleaves and promotes the depolymerization of F-actin. The latter suggests that, in its active state, cofilin should block lammelipod formation by breaking down F-actin and thus inhibit motility. On the other hand, the newly cleaved F-actin contains “barbed ends” that, in the presence of a sufficient supply of G-actin, serve as initiation sites for filament elongation. Under these conditions, active cofilin should enhance lammelipod formation by inducing actin polmerization and thus promote cell motility. In short, cell free in vitro studies have implicated cofilin in both F-actin polymerization and depolymerization depending upon the supply of G-actin [68, 69]. Based on these in vitro results, it is simply unclear whether intracellular cofilin activity blocks or promotes lammelipod formation. Cofilin activity is regulated by phosphorylation at Ser3. LIM kinase catalyzed phosphorylation inactivates cofilin, whereas dephosphorylation restores activity (Fig 2).
Figure 2.

Design and synthesis of caged cofilin.
In order to address the role of cofilin in cell motility, we generated a photo-regulated cofilin that could be switched on at anytime and anywhere in a live cell [70, 71]. Site-directed mutagenesis of Ser3 to Cys in cofilin generates a protein that maintains a high rate of F-actin severing but cannot be phosphorylated by LIM kinase, thereby rendering cofilin constitutively active. Caged cofilin was synthesized by covalently modifying Cys3 with the same ortho-nitrobenzyl moiety (2) used to cage PKA, which introduces a negatively charge carboxyl group that mimics the electrostatic state of the inactive phosphorylated cofilin (Fig 2). Mass spectroscopy confirmed covalent modification of the Ser-to-Cys cofilin at a single site, a modification that is removed upon irradiation at 365 nm. SDS-PAGE of sedimentation assays revealed that the caged cofilin is unable to bind to F-actin. However, upon photolysis F-actin binding is restored. The ability of the caged versus uncaged cofilin to sever F-actin was tested using two assays: (i) a spectrofluorimetric assay for F-actin depolymerization and (ii) microscopic imaging of F-actin cleavage using a fluorescently labeled actin. Caged cofilin has no effect on F-actin depolymerization (versus buffer alone), but photolysis restores up to 80% of the F-actin severing activity (Fig 3).
Figure 3.
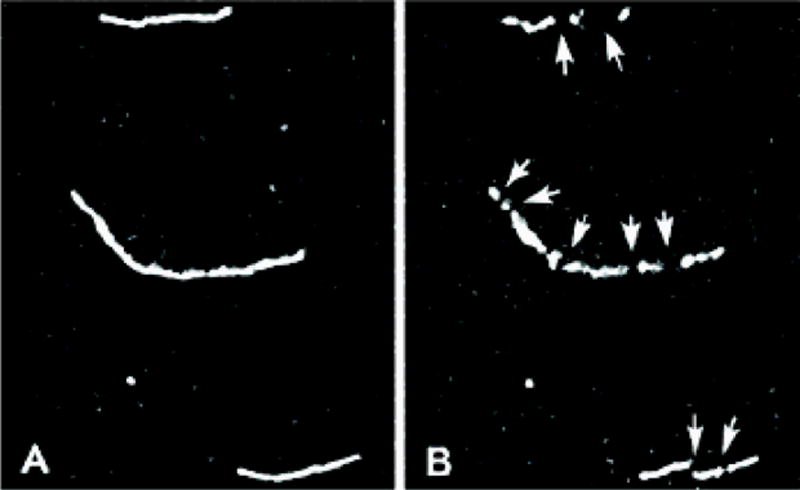
Cleavage of F-actin filaments. (A) Rhodamine-labeled F-actin filaments (B) in the presence of photoactivated cofilin. Reprinted with permission from [70]. Copyright 2002 American Chemical Society.
The caged cofilin construct was microinjected into MTLn3 cells to assess the effect of spatially- and temporally-confined cofilin activity on actin polymerization and depolymerization, leading edge protrusion, and motility. A 36% increase in the cellular level of F-actin was observed after whole cell photoactivation of caged cofilin for 0.5 s using a 100 W Hg arc lamp directed through the 40X oil objective. As expected, the increase in cellular F-actin after photolysis of caged cofilin also increases the production of barbed ends. Photoactivation of cofilin in MTLn3 cells produced an increase in the size of the lamellipods as well as the velocity of their formation. In order to assess if cofilin activity affects localized protrusions of lamellipodia or can influence the directionality of motility, uncaging was carried out in a 3 μm diameter spot and cell movement monitored by time lapse photography. Cell protrusions were observed near the spot of irradiation in approximately 80% of all cells microinjected with caged cofilin compared to just 4% of non-injected cells (Fig 4). Cells that were not loaded with caged cofilin show a directionless random walking in serum, whereas 70% of cells that have photoreleased cofilin move in the direction of spot illumination. These intracellular experiments confirm that cofilin produces free barbed ends, leads to cell protrusions, and sets the direction of motility.
Figure 4.

Local protrusions caused by activation of cofilin. (A) MTLn3 cells in serum after microinjection of caged cofilin (B) Spatial uncaging of cofilin (C) 1 min after uncaging (D) DAIS perimeter time-lapse images taken before and after photoactivation; showing site of photoactivation (blue), protrusion (green) and retraction (red). From M. Ghosh, X. Song, G. Mouneimne, M. Sidani, D.S. Lawrence, J.S. Condeelis, Cofilin promotes actin polymerization and defines the direction of cell motility, 304, 2004, 743–746. Reprinted with permission from AAAS.
A wide variety of caged proteins have been described. However, only a small fraction of these proteins have actually been evaluated in living cells (or cell lysates). Furthermore, the overwhelming majority of these cell-based experiments were primarily performed to validate the light-dependent activity of the caged protein rather than to address a biological question. Given the potential advantages associated with these light activated species, it is somewhat surprising that caged proteins have not found more widespread application. The limited number of cellular studies is partly due to the difficulties associated with the preparation of these caged species. As noted above, a number of methods have been developed to introduce a photocleavable moiety at a specific site on the protein to be caged. However, mere modification at a specific site on the protein of interest does not necessarily furnish an ideal caged protein for intracellular studies. For example, issues that often need to be addressed are (i) acquisition of caged protein that, upon photolysis, exhibits a dramatic change in activity (i.e. from little or no activity to nearly fully active), (ii) introduction of appropriate post-translational modifications (including phosphorylation) that may be required for intracellular function, (iii) cellular delivery of the caged protein, (iv) and elimination (or reduction) of endogenous wild type activity that the caged protein is designed to replace. However, in our opinion, the primary reason for the limited application of caged proteins as biological tools is the absence of input from biologists in the design of these species. The preparation of caged proteins is currently an art form practiced primarily by members of the chemical community. By contrast, the design of specific photosensitive protein constructs to address the temporal and spatial consequences of protein action in living cells is best achieved in consultation with biologists.
Light-Driven Small Molecule Activation of Proteins
In addition to caged proteins, caged cell-permeable small molecule activators of protein function have been developed. However, although the activity of caged low molecular weight compounds can be unleashed with a high degree of temporal control, spatial control at or below the cellular level is not feasible due to the high diffusion rate of small molecules. As noted in the Introduction, one of the first caged compounds ever reported was that of cAMP[4], the activator of the cAMP-dependent protein kinase. In addition, since this initial report, a number of caged neurotransmitters, hormones, small molecule agonists, and peptides have been used to activate signaling cascades [8, 12, 17, 20].
The majority of caged peptides have been synthesized by modifying key side chain functionality, which usually requires an off-resin, multistep process that is not amendable to all amino acid groups. We designed an on-resin caging strategy that places the photolabile unit on the backbone amide nitrogen adjacent to a key binding site residue [72]. The backbone-bound photolabile group disrupts proper alignment of active site-targeted functionality, rendering the peptide ineffective. Two other reports have described peptides caged on the backbone, one at a backbone-substituted glycine and one using an N-benzylated protecting group[73, 74].
Src family tyrosine kinases are comprised of an SH1 domain for catalysis and SH2 and SH3 domains that are involved in enzyme regulation. SH2 domains bind peptides that contain a phosphoTyr (pTyr) residue embedded within an appropriate amino acid sequence. SH2 domain-targeted peptides activate members of the Src kinase family. A caged Lck SH2 peptide ligand was synthesized using standard Fmoc solid phase synthesis on Rink resin as shown in Scheme 3. The free amine of 5 was covalently modified with 4,5-dimethoxy-2-nitrobenzaldehyde (Ar-CHO) to generate the corresponding imine, which was subsequently reduced with NaBH3CN to furnish 6. The latter was ultimately converted into the caged SH2 ligand 8. The affinity of 8 (Kd = 127 μM) for Lck SH2 domain is 50-fold poorer than the corresponding uncaged species (Kd = 2.6 μM), as determined by competition assay with a previously described dapoxyl labeled peptide [75]. This backbone caging strategy should be applicable to a variety of peptides and has been used to cage peptide substrates for PKA as well as chymotrypsin. Two other SH2 domain-targeted peptides have been developed which are caged at the phosphotyrosine group [76, 77].
Scheme 3.
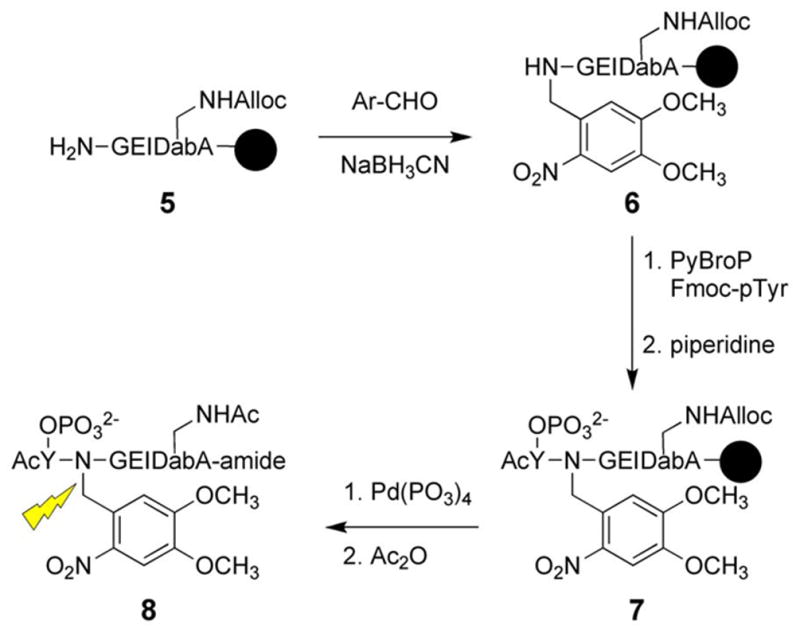

Synthesis of caged SH2 ligand.
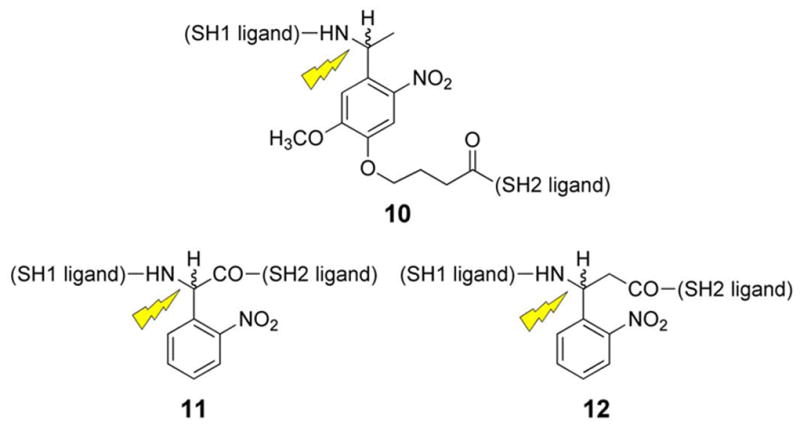
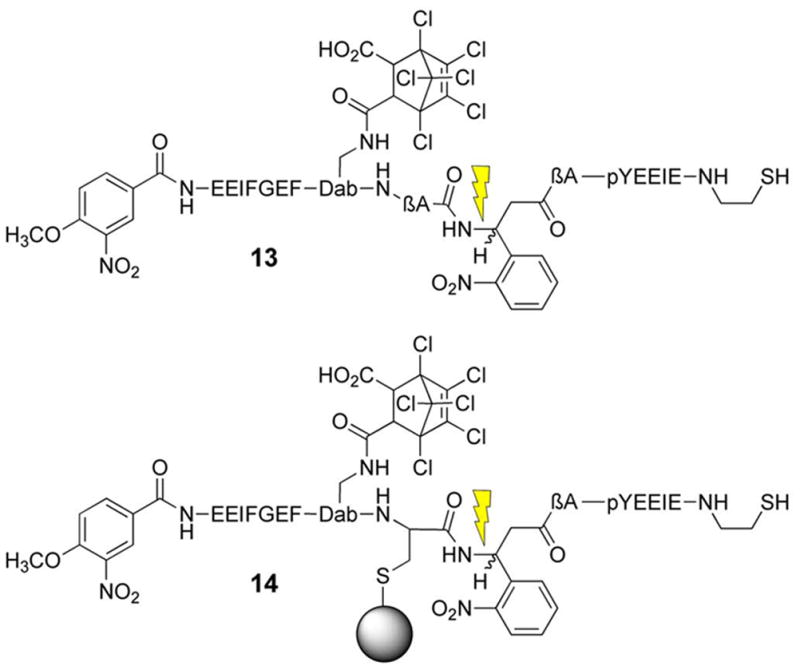
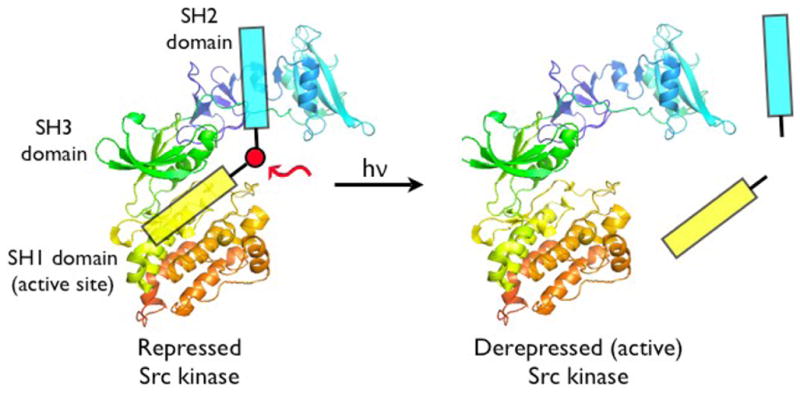
The above SH2 domain-directed strategy employs a peptide that converts an inactive form of an enzyme into the active state. We’ve also developed an alternative approach in which an activated form of the enzyme is trapped by a bivalent inhibitor, which holds the enzyme in inhibitory stasis. Photolysis cleaves the bivalent inhibitor in half, eliminating inhibitory potency and restoring enzymatic activity (Fig 5) [78]. Our lab has previously constructed a high affinity bivalent inhibitor for the Src kinase (9) (Ki = 26 nM), which simultaneously binds to both the SH1 and SH2 domains. Compound 9 displays a significantly enhanced affinity for the Src kinase relative to its monovalent active site-directed counterpart (SH1 ligand IC50 = 1.9 μM) [79]. A small library of caged bivalent inhibitors was prepared with three different photolabile moieties (10 – 12) placed at different locations in the peptide. The caged bivalent peptide with the highest affinity for Src (IC50 = 18 nM) contains a photolabile group (13) positioned in the middle of the sequence that links the SH1- and SH2-targeted ligands. Further analysis of this peptide using a real time fluorescent assay[80, 81] revealed that compound 13 inhibits Src kinase (pure enzyme or in cell lysates) almost completely at the appropriate concentration (nM/μM boundary). Photolysis destroys this inhibitory activity. As noted above, low molecular weight species, such as small caged peptides, display a high rate of diffusion. Therefore, spot illumination of a caged low molecular compound in a living cell will not furnish a long-lived spatially focused burst of an activated compound. The latter will simply diffuse rapidly from the illuminated region. Although caging technology is commonly regarded as furnishing the investigator with temporal and spatial control over biological activity, in reality spatial control is only afforded with those molecules that diffuse slowly on the experimental time scale. With the latter in mind, we inserted a cysteine residue into 13 and attached the peptide to PierceUltra link beads (14). Incubation of the Src kinase with the beads before and after photolysis provided a direct assessment of Src’s ability to bind to the caged and uncaged peptide beads, respectively. As expected, photolysis releases Src from the beads, which suggests that spatial control is feasible by tethering caged low molecular species to slowly diffusing supramolecular entities (e.g. a bead, organelle, membrane, etc).
Figure 5.
Photocleavable caged Src kinase activator. Reprinted with permission from [78]. Copyright 2008 American Chemical Society.
Light-Dependent Peptide Antagonists of Enzymatic Activity
Inhibition of specific enzymes or disruption of protein-protein interactions provides a means to correlate intracellular biochemistry with cellular behavior. However, many pathways involve multiple timing requirements involving enzymatic activity, feed back loops, or subcellular spatial localization. The spatiotemporal dynamics of these signaling pathways cannot be fully understood without the ability inhibit a protein of interest with spatial and temporal control. Photoactivatible inhibitors provide an off switch for intracellular protein activity without altering endogenous protein expression levels [82–86]. The first example of caged peptides designed to interfere with intracellular protein-protein interactions were described by Walker et al in 1998 [87]. Antagonists that disrupt calmodulin binding to myosin light chain kinase (MLCK) were derived from the calmodulin binding and autoinhibitory domains of MLCK. The corresponding caged derivatives of these antagonist display 50-fold weaker affinities than their uncaged counterparts for MLCK. Furthermore, these species block cell motility in a light-dependent fashion. Since this initial report, several other groups have utilized caged protein antagonists to investigate the role of specific proteins in cell-based actions [77, 88, 89].
In conjunction with our caged PKA on switch construct (4), we synthesized a caged PKA inhibitor that can serve as the corresponding off switch for PKA-dependent signaling pathways. The design of the caged PKA antagonist was based on an active site-targeted sequence (Gly-Arg-Thr-Gly-Arg-Arg-Asn-Ala-Ile-amide) derived from an endogenous protein inhibitor of PKA known as PKI [90, 91]. The Ala residue in this sequence occupies the site reserved for the phosphorylatable Ser or Thr in PKA substrates. The Arg plays a key role in promoting active site binding [92, 93]. Therefore modification of the Arg side chain with a photolabile moiety should sharply reduce the inhibitory potency of the nonapeptide. The caged PKI derivative 18 was prepared using the novel guanylating moiety 16, which was derived from S-methylthiourea and a cloroformate derivative (15) of the common ortho-nitrobenzyl caging group (Scheme 4). Guanylation of the ornithine residue in peptide 17 furnished the Arg caged PKI inhibitor 18. To the best of our knowledge, this represents the first (and thus far, only) report of a peptide caged at an Arg residue. Photolysis of 18 furnishes the active inhibitor 19 with a quantum yield of 0.023.
Scheme 4.
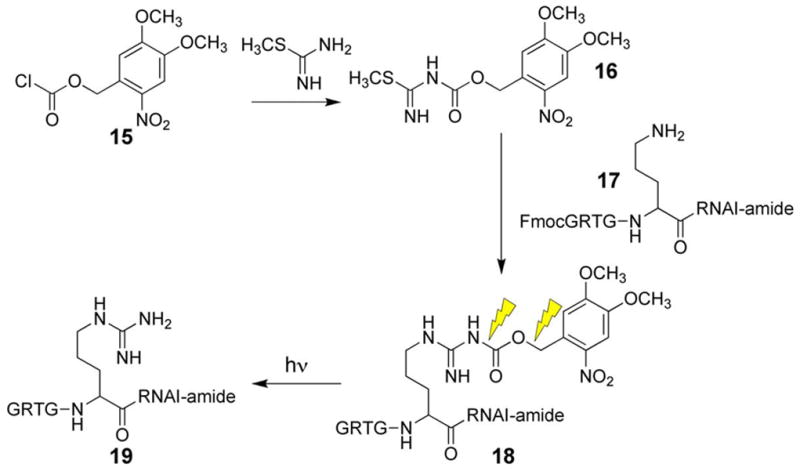
Synthesis of caged PKA inhibitor.
Enzymatic analysis of 18 confirmed that this caged species is a weaker PKA inhibitor (Ki = 20 μM) than its uncaged counterpart 19 (Ki = 420 nM) by almost two orders of magnitude. Peptide 18 was microinjected into REF cells at an approximate intracellular concentration of 5 μM, below the Ki of the caged inhibitor. The caged inhibitor did not prevent the morphological changes associated with activation of PKA by a cell permeable analog of cAMP, 8-(4-chlorophenylthio)-cAMP (CPT-cAMP). However, upon UV irradiation at 300 – 400 nm, the uncaged inhibitor blocked the CPT-cAMP-induced morphological changes. Caged PKA 4 and the caged PKI inhibitor 18 can potentially serve as an on/off switch for addressing precise temporal questions that pertain to PKA-mediated signaling cascades. However, in order for both derivatives to be separately controlled in the same experiment the caged constructs need to undergo photolysis at different wavelengths. Unfortunately, the latter is not feasible with currently available caging moieties.
Caged Sensors of Enzymatic Activity
The timing (both start point and duration) of intracellular enzymatic activity in response to a stimulus can be useful in establishing a relationship between biochemical action and cellular behavior. A large number of fluorescent probes have been developed that furnish real-time visualization of intracellular enzymatic activity. Although powerful, these tools are at the mercy of the biochemical activity of the cell, which may or may not be significant prior to stimulation or may vary as a function of the cell cycle. In addition, whether a probe of protein activity is microinjected or simply cell permeable, it takes time to load the agent into the cell. Consequently, if basal activity is present, it becomes difficult to establish a precise t = 0. Furthermore, it can be challenging to assess enzymatic activity during the course of a biological event if the sensor becomes significantly depleted. Caged sensors provide investigators with the means to load the reagent at their leisure and subsequently activate it once the cell has reached the appropriate stage for analysis.
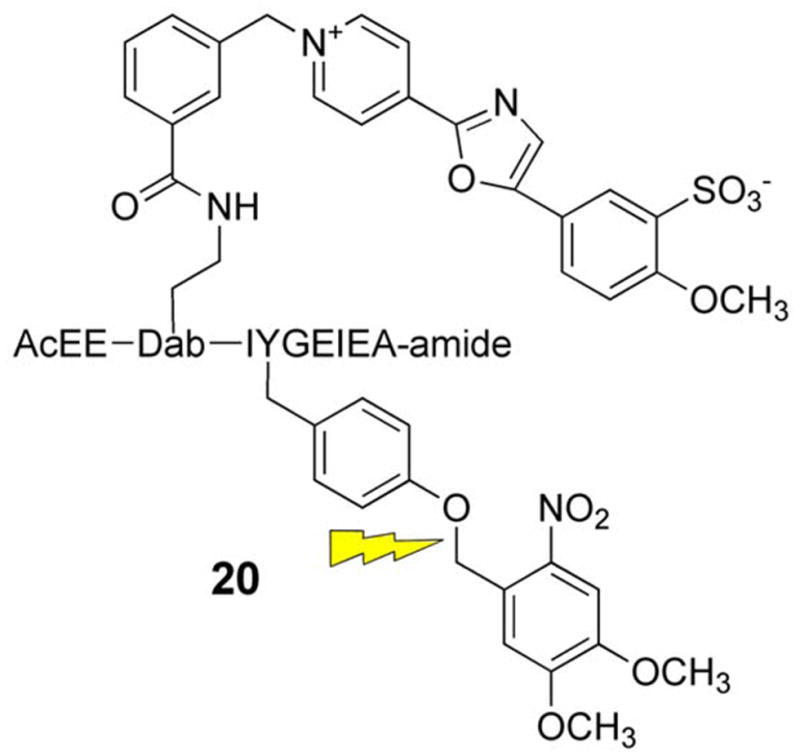
We have described a pyrene-based Src kinase sensor that displays a significant enhancement in fluorescence upon phosphorylation [80]. However, the short excitation and emission wavelengths associated with pyrene limits its usefulness in living cells. Therefore, a small library of Src kinase peptide substrates containing Oregon green, cascade blue, or cascade yellow was prepared. Src kinase-catalyzed phosphorylation of these substrates induces up to a several fold enhancement in fluorescence. We subsequently prepared a caged version of one of these species by covalently modifying the phosphorylatable tyrosine side chain with an ortho-nitrobenzyl group (20). The caged sensor fails to serve as a Src substrate but can be unleashed upon photolysis, where the duration of photolysis is directly correlated with the amount of active sensor available for phosphorylation. The caged Src sensor 20 was microinjected into the human carcinoma cell line A549, which is known to have high expression levels of Src. As expected, time-dependent visualization of Src kinase activity is photolysis-driven (Fig 6).
Figure 6.

Light-induced time-dependent change in fluorescence intensity in A549 cells microinjected with caged Src sensor 20 (A) 1 min after uncaging and (B) 25 min after uncaging. Reprinted with permission from [81]. Copyright 2006 American Chemical Society.
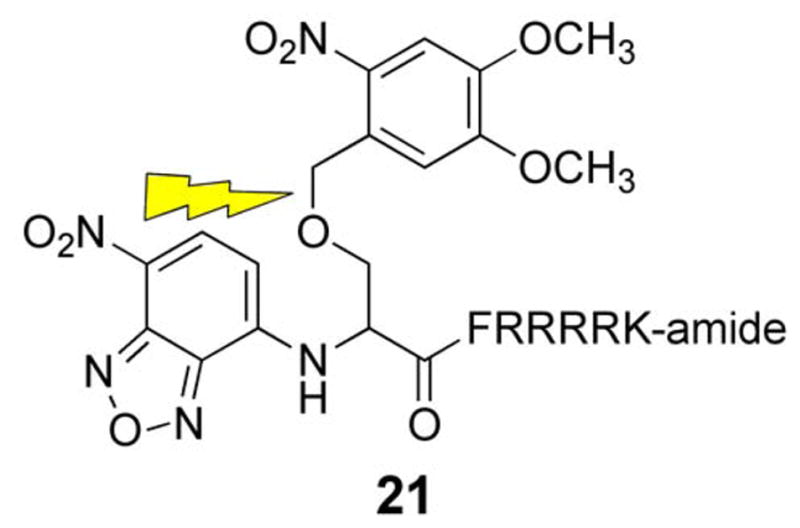
Protein kinase C (PKC) is implicated in many cellular processes including the G2/M transition of mitosis, particularly around the time of nuclear envelope breakdown (NEB) [94–98]. We’ve previously described a fluorophore-labeled PKC substrate that displays a 2.5 fold increase in fluorescence upon phosphorylation [99]. However, since PKC is active in interphase cells, the sensor is consumed before the activity of PKC can be tested during mitosis. Therefore, a caged derivative (21) was prepared so that PKC activity can be precisely monitored at the time of NEB [100, 101]. In vitro assays with PKC demonstrated that peptide 21 doesn’t serve as a substrate. However, after photolysis, a robust fluorescence increase is observed and the amount fluorescence corresponds to the amount of sensor uncaged. Further analysis of 21 revealed a maximal conversion to the uncaged sensor of 60% with a quantum yield of 0.06.
To address the question of when PKC is active during the G2/M transition during mitosis, 21 was microinjected into PtK2 cells along with a 70 kDa Texas red dextran. The latter is excluded from the nucleus and thus provides a readily observable measure of NEB. PtK2 cells remain relatively flat during mitosis (unlike most cell lines, which tend to round up), which allows for ready identification of the stages of mitosis. Photoactivation of 21 during prophase results in a time-dependent increase in fluorescence just prior to NEB. This is consistent with phosphorylation of the peptide substrate, a direct barometer of PKC activity. Interestingly, the observed real time fluorescence enhancement subsequently levels at the initiation of NEB. The latter may represent a corresponding decrease in PKC activity during the nuclear envelope breakdown event. However, the apparent absence of peptide phosphorylation during this stage could also be due to complete phosphorylation of all photouncaged molecules. In order to address the latter possibility, compound 21 was photo-converted to the active substrate after NEB (Fig 7B). No fluorescence increase was observed under these conditions. These results are consistent with PKC activity prior to but not during or after NEB. Furthermore, the introduction of PKC inhibitors during prophase not only blocks the expected fluorescence increase but also prevents NEB. Consequently, PKC activity during prophase is required for NEB. The active sensor is an excellent substrate for PKC α, β and γ. However, only α and β are expressed in Ptk2 cells. In order to assess which PKC isoform is active just prior to NEB, highly selective inhibitors for PKC α and β were co-injected with 21. These studies revealed that PKC β is responsible for both the observed phosphorylation of the PKC sensor as well as NEB itself.
Figure 7.
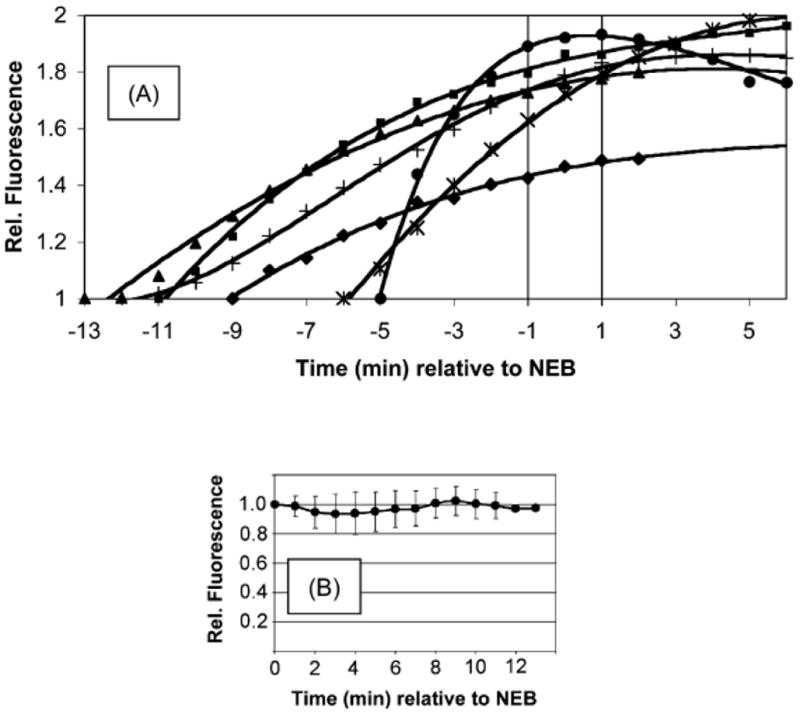
Protein kinase activity prior to and after NEB. (A) Time course of phosphorylation of 21 prior to NEB and (B) after NEB. Reprinted from Chemistry & Biology, 14, Z. Dai, N.G. Dulyaninova, S. Kumar, A.N. Bresnick, D.S. Lawrence, Visual Snapshots of intracellular kinase activity at the onset of mitosis, 1254–1260, copyright (2007) with permission from Elsevier.
Alteration of Gene Expression
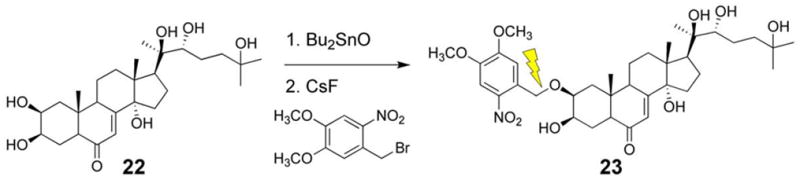
Perturbation of signaling pathways in living organisms provides a means to correlate biochemical activity with cell behavior within the context of a community of cells. However, spatiotemporal control generally conveys a significantly different meaning at the organismal level than at the cellular level. Spatial effects in the former commonly refer to differences in the environment encompassing a cell or group of cells, be they different organs or microenvironments within a specific organ. Temporal effects at the organismal level are generally concerned with longer-lived phenomena than those within an individual cell. In addition, there exist unique challenges associated with reagent delivery in multicellular organisms. For example, although microinjection of a caged protein into single cells in culture is routine, the corresponding delivery of intact proteins into cells in a multicellular organism is far from routine. Indeed, given the looser time constraints associated with organismal biology (versus cell biology), “protein delivery” is generally effected via introduction of genes (e.g. via viral delivery), which are subsequently transcribed and translated. Alternatively, transgenic animals have been constructed that possess genetic constructs that respond to small molecule effectors. In 1996, No et al reported an especially effective small molecule gene expression system, which has low levels of basal expression in the absence of the inducing agent, but robust uniform expression and no toxicity in its presence [102]. The transgenic construct consists of a constitutively expressed ecdysone receptor that, in combination with an endogenously expressed retinoid X receptor, forms a heterodimer with the insect hormone ecdysone (22) to induce expression of the transgene. Ecdysone, and the more potent plant homolog ponasterone A, induce organism-wide expression of the specified gene in transgenic organisms. We reasoned that it may be feasible to limit expression of the transgene to a single cell (or a small cluster of cells) by using a combination of a caged ecdysteroid and a highly focused light source.
We synthesized a photoactivatible ecdysone (23) designed to work in conjunction with the ecdysteroid-inducible gene expression system [103]. The biological activity of ecdysone requires a free hydroxyl group at C2. Caged ecdysone was prepared by generating the dibutylstannylene acetal at the C2–C3 diol, followed by selective alkylation with an ortho-nitrobenzyl derivative in the presence of CsF (Scheme 5). Covalent modification proceeds exclusively at the equatorial C2 position to generate 23.
Scheme 5.
Synthesis of caged ecdysone.
Our initial studies focused on the light-driven expression of luciferase. A transfected 293T cell line was constructed, which constitutively express the ecdysone receptor, and contains a luciferase gene driven by an ecdysone response element in its promotor. Ecdysone 22 induces a 90-fold increase in luciferase expression, whereas, the caged form 23 exhibits a 6-fold increase. By contrast, photolysis furnishes a 55-fold increase over non-stimulated luciferase expression with maximal activity occurring after 16 h. Several hundred-fold changes in gene expression are observed with the more potent ponasterone A analog of ecdysone. Spatially discrete luciferase expression was monitored by incubating 293T cells with 23 followed by spatially focused photolysis. As can be seen in Figure 8, luciferase expression is spatially restricted to illuminated cells. These results validate the notion a small cell-permeable photosensitive compound (23) can be used to spatially control gene expression in a multicellular environment.
Figure 8.

Light-driven, spatially discrete luciferase espression. (A) Transfected 293T cells exposed to 22 (B) Transfected 293T cells exposed to 23 and spot illuminated (C) Transfected 293T cells exposed to 23 outside illumination area. Reprinted from Chemistry & Biology, 9, W. Lin, C. Albanese, R.G. Pestell, D.S. Lawrence, Spatially discrete, light-driven protein expression, 1347–1353. Copyright (2002) with permission from Elsevier.
Future Directions
Both the timing and location of signaling pathways can have significant consequences in terms of biological behavior. We have constructed activators, inhibitors, and sensors of signaling pathways that provide the means to control when and where a pathway is perturbed or probed. Further advances in the field of caged biological molecules are required to sequentially switch on and off signaling pathways or to activate multiple signaling pathways at multiple time points. In order to achieve these possibilities, a family of caging moieties must be developed that respond to unique wavelengths of light. Progress in the transfection of cells with peptides and proteins is required to eliminate the need for microinjection. The latter will enable the use of common techniques, such as western blots and flow cytometry, to analyze the biological response. Finally, the acquisition of two-photon caging moieties, particularly those that can be photoreleased in the 800 nm range, would be of profound utility for analysis in tissues and live animals.
Acknowledgments
The National Institutes of Health (NS048406, CA79954, and GM086217) is gratefully acknowledged for generous financial support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blocker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowski J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 2.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, Clark AG, Nadeau J, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng Z, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge W, Gong F, Gu Z, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke Z, Ketchum KA, Lai Z, Lei Y, Li Z, Li J, Liang Y, Lin X, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue B, Sun J, Wang Z, Wang A, Wang X, Wang J, Wei M, Wides R, Xiao C, Yan C, Yao A, Ye J, Zhan M, Zhang W, Zhang H, Zhao Q, Zheng L, Zhong F, Zhong W, Zhu S, Zhao S, Gilbert D, Baumhueter S, Spier G, Carter C, Cravchik A, Woodage T, Ali F, An H, Awe A, Baldwin D, Baden H, Barnstead M, Barrow I, Beeson K, Busam D, Carver A, Center A, Cheng ML, Curry L, Danaher S, Davenport L, Desilets R, Dietz S, Dodson K, Doup L, Ferriera S, Garg N, Gluecksmann A, Hart B, Haynes J, Haynes C, Heiner C, Hladun S, Hostin D, Houck J, Howland T, Ibegwam C, Johnson J, Kalush F, Kline L, Koduru S, Love A, Mann F, May D, McCawley S, McIntosh T, McMullen I, Moy M, Moy L, Murphy B, Nelson K, Pfannkoch C, Pratts E, Puri V, Qureshi H, Reardon M, Rodriguez R, Rogers YH, Romblad D, Ruhfel B, Scott R, Sitter C, Smallwood M, Stewart E, Strong R, Suh E, Thomas R, Tint NN, Tse S, Vech C, Wang G, Wetter J, Williams S, Williams M, Windsor S, Winn-Deen E, Wolfe K, Zaveri J, Zaveri K, Abril JF, Guigo R, Campbell MJ, Sjolander KV, Karlak B, Kejariwal A, Mi H, Lazareva B, Hatton T, Narechania A, Diemer K, Muruganujan A, Guo N, Sato S, Bafna V, Istrail S, Lippert R, Schwartz R, Walenz B, Yooseph S, Allen D, Basu A, Baxendale J, Blick L, Caminha M, Carnes-Stine J, Caulk P, Chiang YH, Coyne M, Dahlke C, Mays A, Dombroski M, Donnelly M, Ely D, Esparham S, Fosler C, Gire H, Glanowski S, Glasser K, Glodek A, Gorokhov M, Graham K, Gropman B, Harris M, Heil J, Henderson S, Hoover J, Jennings D, Jordan C, Jordan J, Kasha J, Kagan L, Kraft C, Levitsky A, Lewis M, Liu X, Lopez J, Ma D, Majoros W, McDaniel J, Murphy S, Newman M, Nguyen T, Nguyen N, Nodell M, Pan S, Peck J, Peterson M, Rowe W, Sanders R, Scott J, Simpson M, Smith T, Sprague A, Stockwell T, Turner R, Venter E, Wang M, Wen M, Wu D, Wu M, Xia A, Zandieh A, Zhu X. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 3.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 4.Engels J, Schlaeger EJ. Synthesis, structure, and reactivity of adenosine cyclic 3′,5′-phosphate-benzyltriesters. J Med Chem. 1977;20:907–911. doi: 10.1021/jm00217a008. [DOI] [PubMed] [Google Scholar]
- 5.Kaplan JH, Forbush B, 3rd, Hoffman JF. Rapid photolytic release of adenosine 5′-triphosphate from a protected analogue: utilization by the Na:K pump of human red blood cell ghosts. Biochemistry. 1978;17:1929–1935. doi: 10.1021/bi00603a020. [DOI] [PubMed] [Google Scholar]
- 6.Banghart MR, Volgraf M, Trauner D. Engineering light-gated ion channels. Biochemistry. 2006;45:15129–15141. doi: 10.1021/bi0618058. [DOI] [PubMed] [Google Scholar]
- 7.Curley K, Lawrence DS. Caged regulators of signaling pathways. Pharmacol Ther. 1999;82:347–354. doi: 10.1016/s0163-7258(98)00055-2. [DOI] [PubMed] [Google Scholar]
- 8.Goeldner M, Givens R. Dynamic studies in biology: phototriggers, photoswitches and caged biomolecules. Wiley-VCH; Weinheim: 2005. [Google Scholar]
- 9.Gorostiza P, Isacoff EY. Optical switches for remote and noninvasive control of cell signaling. Science. 2008;322:395–399. doi: 10.1126/science.1166022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lawrence DS. The preparation and in vivo applications of caged peptides and proteins. Curr Opin Chem Biol. 2005;9:570–575. doi: 10.1016/j.cbpa.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 11.Lee HM, Larson DR, Lawrence DS. Illuminating the Chemistry of Life: Design, Synthesis, and Applications of “Caged” and Related Photoresponsive Compounds. ACS Chem Biol. 2009 doi: 10.1021/cb900036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marriott G, Ottl J. Synthesis and applications of heterobifunctional photocleavable cross-linking reagents. Methods Enzymol. 1998;291:155–175. doi: 10.1016/s0076-6879(98)91012-6. [DOI] [PubMed] [Google Scholar]
- 13.Pelliccioli AP, Wirz J. Photoremovable protecting groups: reaction mechanisms and applications. Photochem Photobiol Sci. 2002;1:441–458. doi: 10.1039/b200777k. [DOI] [PubMed] [Google Scholar]
- 14.Politz JC. Use of caged fluorochromes to track macromolecular movement in living cells. Trends Cell Biol. 1999;9:284–287. doi: 10.1016/s0962-8924(99)01585-8. [DOI] [PubMed] [Google Scholar]
- 15.Shigeri Y, Tatsu Y, Yumoto N. Synthesis and application of caged peptides and proteins. Pharmacol Ther. 2001;91:85–92. doi: 10.1016/s0163-7258(01)00148-6. [DOI] [PubMed] [Google Scholar]
- 16.Tang X, Dmochowski IJ. Regulating gene expression with light-activated oligonucleotides. Mol Biosyst. 2007;3:100–110. doi: 10.1039/b614349k. [DOI] [PubMed] [Google Scholar]
- 17.Dorman G, Prestwich GD. Using photolabile ligands in drug discovery and development. Trends Biotechnol. 2000;18:64–77. doi: 10.1016/s0167-7799(99)01402-x. [DOI] [PubMed] [Google Scholar]
- 18.Ellis-Davies GC. Caged compounds: photorelease technology for control of cellular chemistry and physiology. Nat Methods. 2007;4:619–628. doi: 10.1038/nmeth1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kramer RH, Chambers JJ, Trauner D. Photochemical tools for remote control of ion channels in excitable cells. Nat Chem Biol. 2005;1:360–365. doi: 10.1038/nchembio750. [DOI] [PubMed] [Google Scholar]
- 20.Mayer G, Heckel A. Biologically active molecules with a “light switch”. Angew Chem Int Ed Engl. 2006;45:4900–4921. doi: 10.1002/anie.200600387. [DOI] [PubMed] [Google Scholar]
- 21.Young DD, Deiters A. Photochemical control of biological processes. Org Biomol Chem. 2007;5:999–1005. doi: 10.1039/b616410m. [DOI] [PubMed] [Google Scholar]
- 22.Casey JP, Blidner RA, Monroe WT. Caged siRNAs for Spatiotemporal Control of Gene Silencing. Mol Pharm. 2009 doi: 10.1021/mp900082q. [DOI] [PubMed] [Google Scholar]
- 23.Aemissegger A, Hilvert D. Synthesis and application of an azobenzene amino acid as a light-switchable turn element in polypeptides. Nat Protoc. 2007;2:161–167. doi: 10.1038/nprot.2006.488. [DOI] [PubMed] [Google Scholar]
- 24.Kneissl S, Loveridge EJ, Williams C, Crump MP, Allemann RK. Photocontrollable peptide-based switches target the anti-apoptotic protein Bcl-xL. Chembiochem. 2008;9:3046–3054. doi: 10.1002/cbic.200800502. [DOI] [PubMed] [Google Scholar]
- 25.Renner C, Moroder L. Azobenzene as conformational switch in model peptides. Chembiochem. 2006;7:868–878. doi: 10.1002/cbic.200500531. [DOI] [PubMed] [Google Scholar]
- 26.Woolley GA. Photocontrolling peptide alpha helices. Acc Chem Res. 2005;38:486–493. doi: 10.1021/ar040091v. [DOI] [PubMed] [Google Scholar]
- 27.Zhang F, Sadovski O, Woolley GA. Synthesis and characterization of a long, rigid photoswitchable cross-linker for promoting peptide and protein conformational change. Chembiochem. 2008;9:2147–2154. doi: 10.1002/cbic.200800196. [DOI] [PubMed] [Google Scholar]
- 28.Fujimoto K, Amano M, Horibe Y, Inouye M. Reversible photoregulation of helical structures in short peptides under indoor lighting/dark conditions. Org Lett. 2006;8:285–287. doi: 10.1021/ol0526524. [DOI] [PubMed] [Google Scholar]
- 29.Pearson D, Alexander N, Abell AD. Improved photocontrol of alpha-chymotrypsin activity: peptidomimetic trifluoromethylketone photoswitch enzyme inhibitors. Chemistry. 2008;14:7358–7365. doi: 10.1002/chem.200800082. [DOI] [PubMed] [Google Scholar]
- 30.Banghart M, Borges K, Isacoff E, Trauner D, Kramer RH. Light-activated ion channels for remote control of neuronal firing. Nat Neurosci. 2004;7:1381–1386. doi: 10.1038/nn1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fang J, Sakata T, Marriott G, Iwasa KH. Probing conformational changes of prestin with thiol-reactive optical switches. Biophys J. 2008;95:3036–3042. doi: 10.1529/biophysj.108.132878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fortin DL, Banghart MR, Dunn TW, Borges K, Wagenaar DA, Gaudry Q, Karakossian MH, Otis TS, Kristan WB, Trauner D, Kramer RH. Photochemical control of endogenous ion channels and cellular excitability. Nat Methods. 2008;5:331–338. doi: 10.1038/nmeth.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gorostiza P, Isacoff E. Optical switches and triggers for the manipulation of ion channels and pores. Mol Biosyst. 2007;3:686–704. doi: 10.1039/b710287a. [DOI] [PubMed] [Google Scholar]
- 34.Lester HA, Krouse ME, Nass MM, Wassermann NH, Erlanger BF. A covalently bound photoisomerizable agonist: comparison with reversibly bound agonists at Electrophorus electroplaques. J Gen Physiol. 1980;75:207–232. doi: 10.1085/jgp.75.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raymo FM, Tomasulo M. Electron and energy transfer modulation with photochromic switches. Chem Soc Rev. 2005;34:327–336. doi: 10.1039/b400387j. [DOI] [PubMed] [Google Scholar]
- 36.Volgraf M, Gorostiza P, Numano R, Kramer RH, Isacoff EY, Trauner D. Allosteric control of an ionotropic glutamate receptor with an optical switch. Nat Chem Biol. 2006;2:47–52. doi: 10.1038/nchembio756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liang X, Takenaka N, Nishioka H, Asanuma H. Molecular design for reversing the photoswitching mode of turning ON and OFF DNA hybridization. Chem Asian J. 2008;3:553–560. doi: 10.1002/asia.200700384. [DOI] [PubMed] [Google Scholar]
- 38.Young DD, Deiters A. Light-regulated RNA-small molecule interactions. Chembiochem. 2008;9:1225–1228. doi: 10.1002/cbic.200800051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miyawaki A. Fluorescent proteins in a new light. Nat Biotechnol. 2004;22:1374–1376. doi: 10.1038/nbt1104-1374. [DOI] [PubMed] [Google Scholar]
- 40.Chudakov DM, Verkhusha VV, Staroverov DB, Souslova EA, Lukyanov S, Lukyanov KA. Photoswitchable cyan fluorescent protein for protein tracking. Nat Biotechnol. 2004;22:1435–1439. doi: 10.1038/nbt1025. [DOI] [PubMed] [Google Scholar]
- 41.Gurskaya NG, Verkhusha VV, Shcheglov AS, Staroverov DB, Chepurnykh TV, Fradkov AF, Lukyanov S, Lukyanov KA. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat Biotechnol. 2006;24:461–465. doi: 10.1038/nbt1191. [DOI] [PubMed] [Google Scholar]
- 42.Henderson JN, Gepshtein R, Heenan JR, Kallio K, Huppert D, Remington SJ. Structure and mechanism of the photoactivatable green fluorescent protein. J Am Chem Soc. 2009;131:4176–4177. doi: 10.1021/ja808851n. [DOI] [PubMed] [Google Scholar]
- 43.Matsuda T, Miyawaki A, Nagai T. Direct measurement of protein dynamics inside cells using a rationally designed photoconvertible protein. Nat Methods. 2008;5:339–345. doi: 10.1038/nmeth.1193. [DOI] [PubMed] [Google Scholar]
- 44.Patterson GH. Photoactivation and imaging of photoactivatable fluorescent proteins. Curr Protoc Cell Biol. 2008;Chapter 21(Unit 21–26) doi: 10.1002/0471143030.cb2106s38. [DOI] [PubMed] [Google Scholar]
- 45.Patterson GH, Lippincott-Schwartz J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 2002;297:1873–1877. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- 46.Patterson GH, Lippincott-Schwartz J. Selective photolabeling of proteins using photoactivatable GFP. Methods. 2004;32:445–450. doi: 10.1016/j.ymeth.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 47.Schneider M, Barozzi S, Testa I, Faretta M, Diaspro A. Two-photon activation and excitation properties of PA-GFP in the 720–920-nm region. Biophys J. 2005;89:1346–1352. doi: 10.1529/biophysj.104.054502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Testa I, Parazzoli D, Barozzi S, Garre M, Faretta M, Diaspro A. Spatial control of pa-GFP photoactivation in living cells. J Microsc. 2008;230:48–60. doi: 10.1111/j.1365-2818.2008.01951.x. [DOI] [PubMed] [Google Scholar]
- 49.Leung DW, Otomo C, Chory J, Rosen MK. Genetically encoded photoswitching of actin assembly through the Cdc42-WASP-Arp2/3 complex pathway. Proc Natl Acad Sci U S A. 2008;105:12797–12802. doi: 10.1073/pnas.0801232105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adams SR, Tsien RY. Controlling cell chemistry with caged compounds. Annu Rev Physiol. 1993;55:755–784. doi: 10.1146/annurev.ph.55.030193.003543. [DOI] [PubMed] [Google Scholar]
- 51.Aujard I, Benbrahim C, Gouget M, Ruel O, Baudin JB, Neveu PL. Jullien, o-nitrobenzyl photolabile protecting groups with red-shifted absorption: syntheses and uncaging cross-sections for one- and two-photon excitation. Chemistry. 2006;12:6865–6879. doi: 10.1002/chem.200501393. [DOI] [PubMed] [Google Scholar]
- 52.Hagen V, Frings S, Wiesner B, Helm S, Kaupp UB, Bendig J. [7-(Dialkylamino)coumarin-4-yl]methyl-Caged Compounds as Ultrafast and Effective Long-Wavelength Phototriggers of 8-Bromo-Substituted Cyclic Nucleotides. Chembiochem. 2003;4:434–442. doi: 10.1002/cbic.200300561. [DOI] [PubMed] [Google Scholar]
- 53.Lin W, Lawrence DS. A strategy for the construction of caged diols using a photolabile protecting group. J Org Chem. 2002;67:2723–2726. doi: 10.1021/jo0163851. [DOI] [PubMed] [Google Scholar]
- 54.Furuta T, Wang SS, Dantzker JL, Dore TM, Bybee WJ, Callaway EM, Denk W, Tsien RY. Brominated 7-hydroxycoumarin-4-ylmethyls: photolabile protecting groups with biologically useful cross-sections for two photon photolysis. Proc Natl Acad Sci U S A. 1999;96:1193–1200. doi: 10.1073/pnas.96.4.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davis MJ, Kragor CH, Reddie KG, Wilson HC, Zhu Y, Dore TM. Substituent effects on the sensitivity of a quinoline photoremovable protecting group to one- and two-photon excitation. J Org Chem. 2009;74:1721–1729. doi: 10.1021/jo802658a. [DOI] [PubMed] [Google Scholar]
- 56.Gug S, Bolze F, Specht A, Bourgogne C, Goeldner M, Nicoud JF. Molecular engineering of photoremovable protecting groups for two-photon uncaging. Angew Chem Int Ed Engl. 2008;47:9525–9529. doi: 10.1002/anie.200803964. [DOI] [PubMed] [Google Scholar]
- 57.Nikolenko V, Poskanzer KE, Yuste R. Two-photon photostimulation and imaging of neural circuits. Nat Methods. 2007;4:943–950. doi: 10.1038/nmeth1105. [DOI] [PubMed] [Google Scholar]
- 58.Gagey N, Neveu P, Benbrahim C, Goetz B, Aujard I, Baudin JB, Jullien L. Two-photon uncaging with fluorescence reporting: evaluation of the o-hydroxycinnamic platform. J Am Chem Soc. 2007;129:9986–9998. doi: 10.1021/ja0722022. [DOI] [PubMed] [Google Scholar]
- 59.Momotake A, Lindegger N, Niggli E, Barsotti RJ, Ellis-Davies GC. The nitrodibenzofuran chromophore: a new caging group for ultra-efficient photolysis in living cells. Nat Methods. 2006;3:35–40. doi: 10.1038/nmeth821. [DOI] [PubMed] [Google Scholar]
- 60.Zou K, Cheley S, Givens RS, Bayley H. Catalytic subunit of protein kinase A caged at the activating phosphothreonine. J Am Chem Soc. 2002;124:8220–8229. doi: 10.1021/ja020405e. [DOI] [PubMed] [Google Scholar]
- 61.Hahn ME, Muir TW. Photocontrol of Smad2, a multiphosphorylated cell-signaling protein, through caging of activating phosphoserines. Angew Chem, Int Ed Engl. 2004;43:5800–5803. doi: 10.1002/anie.200461141. [DOI] [PubMed] [Google Scholar]
- 62.Pellois JP, Hahn ME, Muir TW. Simultaneous triggering of protein activity and fluorescence. J Am Chem Soc. 2004;126:7170–7171. doi: 10.1021/ja0499142. [DOI] [PubMed] [Google Scholar]
- 63.Pellois JP, Muir TW. A ligation and photorelease strategy for the temporal and spatial control of protein function in living cells. Angew Chem, Int Ed Engl. 2005;44:5713–5717. doi: 10.1002/anie.200501244. [DOI] [PubMed] [Google Scholar]
- 64.Ellman J, Mendel D, Anthony-Cahill S, Noren CJ, Schultz PG. Biosynthetic method for introducing unnatural amino acids site-specifically into proteins. Methods Enzymol. 1991;202:301–336. doi: 10.1016/0076-6879(91)02017-4. [DOI] [PubMed] [Google Scholar]
- 65.Taylor SS, Buechler JA, Yonemoto W. cAMP-dependent protein kinase: framework for a diverse family of regulatory enzymes. Annu Rev Biochem. 1990;59:971–1005. doi: 10.1146/annurev.bi.59.070190.004543. [DOI] [PubMed] [Google Scholar]
- 66.Curley K, Lawrence DS. Photoactivation of a Signal Transduction Pathway in Living Cells. J Am Chem Soc. 1998;120:8573–8574. [Google Scholar]
- 67.Roger PP, Rickaert F, Huez G, Authelet M, Hofmann F, Dumont JE. Microinjection of catalytic subunit of cyclic AMP-dependent protein kinase triggers acute morphological changes in thyroid epithelial cells. FEBS Lett. 1988;232:409–413. doi: 10.1016/0014-5793(88)80781-6. [DOI] [PubMed] [Google Scholar]
- 68.Condeelis J. Life at the leading edge: the formation of cell protrusions. Annu Rev Cell Biol. 1993;9:411–444. doi: 10.1146/annurev.cb.09.110193.002211. [DOI] [PubMed] [Google Scholar]
- 69.Carlier MF, Laurent V, Santolini J, Melki R, Didry D, Xia GX, Hong Y, Chua NH, Pantaloni D. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J Cell Biol. 1997;136:1307–1322. doi: 10.1083/jcb.136.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ghosh M, Ichetovkin I, Song X, Condeelis JS, Lawrence DS. A new strategy for caging proteins regulated by kinases. J Am Chem Soc. 2002;124:2440–2441. doi: 10.1021/ja017592l. [DOI] [PubMed] [Google Scholar]
- 71.Ghosh M, Song X, Mouneimne G, Sidani M, Lawrence DS, Condeelis JS. Cofilin promotes actin polymerization and defines the direction of cell motility. Science. 2004;304:743–746. doi: 10.1126/science.1094561. [DOI] [PubMed] [Google Scholar]
- 72.Nandy SK, Agnes RS, Lawrence DS. Photochemically-activated probes of protein-protein interactions. Org Lett. 2007;9:2249–2252. doi: 10.1021/ol070238t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Johnson ECB, Kent SBH. Synthesis, stability and optimized photolytic cleavage of 4-methoxy-2-nitrobenzyl backbone-protected peptides. Chem Commun. 2006:1557–1559. doi: 10.1039/b600304d. [DOI] [PubMed] [Google Scholar]
- 74.Tatsu Y, Nishigaki T, Darszon A, Yumoto N. A caged sperm-activating peptide that has a photocleavable protecting group on the backbone amide. FEBS Lett. 2002;525:20–24. doi: 10.1016/s0014-5793(02)03000-4. [DOI] [PubMed] [Google Scholar]
- 75.Wang Q, Lawrence DS. Phosphorylation-driven protein-protein interactions: a protein kinase sensing system. J Am Chem Soc. 2005;127:7684–7685. doi: 10.1021/ja050789j. [DOI] [PubMed] [Google Scholar]
- 76.Zou K, Miller WT, Givens RS, Bayley H. Caged Thiophosphotyrosine Peptides. Angew Chem Int Ed Engl. 2001;40:3049–3051. doi: 10.1002/1521-3773(20010817)40:16<3049::AID-ANIE3049>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 77.Humphrey D, Rajfur Z, Vazquez ME, Scheswohl D, Schaller MD, Jacobson K, Imperiali B. In situ photoactivation of a caged phosphotyrosine peptide derived from focal adhesion kinase temporarily halts lamellar extension of single migrating tumor cells. J Biol Chem. 2005;280:22091–22101. doi: 10.1074/jbc.M502726200. [DOI] [PubMed] [Google Scholar]
- 78.Li H, Hah JM, Lawrence DS. Light-mediated liberation of enzymatic activity: “small molecule” caged protein equivalents. Journal of the American Chemical Society. 2008;130:10474–10475. doi: 10.1021/ja803395d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hah JM, Sharma V, Li H, Lawrence DS. Acquisition of a “Group A”-selective Src kinase inhibitor via a global targeting strategy. J Am Chem Soc. 2006;128:5996–5997. doi: 10.1021/ja060136i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang Q, Cahill SM, Blumenstein M, Lawrence DS. Self-reporting fluorescent substrates of protein tyrosine kinases. J Am Chem Soc. 2006;128:1808–1809. doi: 10.1021/ja0577692. [DOI] [PubMed] [Google Scholar]
- 81.Wang Q, Dai Z, Cahill SM, Blumenstein M, Lawrence DS. Light-regulated sampling of protein tyrosine kinase activity. J Am Chem Soc. 2006;128:14016–14017. doi: 10.1021/ja065852z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Perdicakis B, Montgomery HJ, Abbott GL, Fishlock D, Lajoie GA, Guillemette JG, Jervis E. Photocontrol of nitric oxide production in cell culture using a caged isoform selective inhibitor. Bioorg Med Chem. 2005;13:47–57. doi: 10.1016/j.bmc.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 83.Montgomery HJ, Perdicakis B, Fishlock D, Lajoie GA, Jervis E, Guy Guillemette J. Photo-control of nitric oxide synthase activity using a caged isoform specific inhibitor. Bioorg Med Chem. 2002;10:1919–1927. doi: 10.1016/s0968-0896(02)00050-0. [DOI] [PubMed] [Google Scholar]
- 84.Kehayova PD, Bokinsky GE, Huber JD, Jain A. A caged hydrophobic inhibitor of carbonic anhydrase II. Org Lett. 1999;1:187–188. doi: 10.1021/ol990526s. [DOI] [PubMed] [Google Scholar]
- 85.Rossi FM, Margulis M, Hoesch RE, Tang CM, Kao JP. Caged probes for studying cellular physiology: application of o-nitromandelyloxycarbonyl (Nmoc) caging method to glutamate and a Ca(2+)-ATPase inhibitor. Methods Enzymol. 1998;291:431–443. doi: 10.1016/s0076-6879(98)91027-8. [DOI] [PubMed] [Google Scholar]
- 86.Sidky AO, Baimbridge KG. Calcium homeostatic mechanisms operating in cultured postnatal rat hippocampal neurones following flash photolysis of nitrophenyl-EGTA. J Physiol. 1997;504(Pt 3):579–590. doi: 10.1111/j.1469-7793.1997.579bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Walker JW, Gilbert SH, Drummond RM, Yamada M, Sreekumar R, Carraway RE, Ikebe M, Fay FS. Signaling pathways underlying eosinophil cell motility revealed by using caged peptides. Proc Natl Acad Sci U S A. 1998;95:1568–1573. doi: 10.1073/pnas.95.4.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nguyen A, Rothman DM, Stehn J, Imperiali B, Yaffe MB. Caged phosphopeptides reveal a temporal role for 14–3–3 in G1 arrest and S-phase checkpoint function. Nat Biotechnol. 2004;22:993–1000. doi: 10.1038/nbt997. [DOI] [PubMed] [Google Scholar]
- 89.Tatsu Y, Shigeri Y, Ishida A, Kameshita I, Fujisawa H, Yumoto N. Synthesis of caged peptides using caged lysine: application to the synthesis of caged AIP, a highly specific inhibitor of calmodulin-dependent protein kinase II. Bioorg Med Chem Lett. 1999;9:1093–1096. doi: 10.1016/s0960-894x(99)00140-7. [DOI] [PubMed] [Google Scholar]
- 90.Glass DB, Cheng HC, Mende-Mueller L, Reed J, Walsh DA. Primary structural determinants essential for potent inhibition of cAMP-dependent protein kinase by inhibitory peptides corresponding to the active portion of the heat-stable inhibitor protein. J Biol Chem. 1989;264:8802–8810. [PubMed] [Google Scholar]
- 91.Wood JS, Koszelak M, Liu J, Lawrence DS. A Caged Protein Kinase Inhibitor. J Am Chem Soc. 1998;120:7145–7146. [Google Scholar]
- 92.Kemp BE, Graves DJ, Benjamini E, Krebs EG. Role of multiple basic residues in determining the substrate specificity of cyclic AMP-dependent protein kinase. J Biol Chem. 1977;252:4888–4894. [PubMed] [Google Scholar]
- 93.Knighton DR, Zheng JH, Ten Eyck LF, Xuong NH, Taylor SS, Sowadski JM. Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253:414–420. doi: 10.1126/science.1862343. [DOI] [PubMed] [Google Scholar]
- 94.Black JD. Protein kinase C-mediated regulation of the cell cycle. Front Biosci. 2000;5:D406–423. doi: 10.2741/black. [DOI] [PubMed] [Google Scholar]
- 95.Murray NR, Burns DJ, Fields AP. Presence of a beta II protein kinase C-selective nuclear membrane activation factor in human leukemia cells. J Biol Chem. 1994;269:21385–21390. [PubMed] [Google Scholar]
- 96.Hocevar BA, Burns DJ, Fields AP. Identification of protein kinase C (PKC) phosphorylation sites on human lamin B. Potential role of PKC in nuclear lamina structural dynamics. J Biol Chem. 1993;268:7545–7552. [PubMed] [Google Scholar]
- 97.Goss VL, Hocevar BA, Thompson LJ, Stratton CA, Burns DJ, Fields AP. Identification of nuclear beta II protein kinase C as a mitotic lamin kinase. J Biol Chem. 1994;269:19074–19080. [PubMed] [Google Scholar]
- 98.Thompson LJ, Fields AP. betaII protein kinase C is required for the G2/M phase transition of cell cycle. J Biol Chem. 1996;271:15045–15053. doi: 10.1074/jbc.271.25.15045. [DOI] [PubMed] [Google Scholar]
- 99.Yeh RH, Yan X, Cammer M, Bresnick AR, Lawrence DS. Real time visualization of protein kinase activity in living cells. J Biol Chem. 2002;277:11527–11532. doi: 10.1074/jbc.M111300200. [DOI] [PubMed] [Google Scholar]
- 100.Dai Z, Dulyaninova NG, Kumar S, Bresnick AR, Lawrence DS. Visual snapshots of intracellular kinase activity at the onset of mitosis. Chem Biol. 2007;14:1254–1260. doi: 10.1016/j.chembiol.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Veldhuyzen WF, Nguyen Q, McMaster G, Lawrence DS. A light-activated probe of intracellular protein kinase activity. J Am Chem Soc. 2003;125:13358–13359. doi: 10.1021/ja037801x. [DOI] [PubMed] [Google Scholar]
- 102.No D, Yao TP, Evans RM. Ecdysone-inducible gene expression in mammalian cells and transgenic mice. Proc Natl Acad Sci U S A. 1996;93:3346–3351. doi: 10.1073/pnas.93.8.3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lin W, Albanese C, Pestell RG, Lawrence DS. Spatially discrete, light-driven protein expression. Chem Biol. 2002;9:1347–1353. doi: 10.1016/s1074-5521(02)00288-0. [DOI] [PubMed] [Google Scholar]