

Abstract
The investigation of an intramolecular Diels-Alder reaction, en route to a projected total synthesis of maoecrystal V, is described.
Our laboratory is devoted to the total synthesis and biological evaluation of structurally challenging natural products possessing promising biological activity. In this context, we took note of a recent report by Sun and coworkers of a novel natural product, termed maoecrystal V (1), which, due to its potent inhibitory activity against HeLa cells (IC50 = 20 ng/mL), is believed to hold promise as a lead anticancer agent. 1 As shown in Figure 1, maoecrystal V is a densely functionalized diterpene, possessing an unprecedented ent-kaurane skeleton, two contiguous quaternary carbon stereocenters, and a tertiary alcohol, embedded within a complex ring system. The structural complexity of 1, coupled with its biological activity, prompted us to undertake a program directed toward its total synthesis. We report herein the development of some interesting chemistry en route to a projected total synthesis of maoecrystal V (1).
Figure 1.
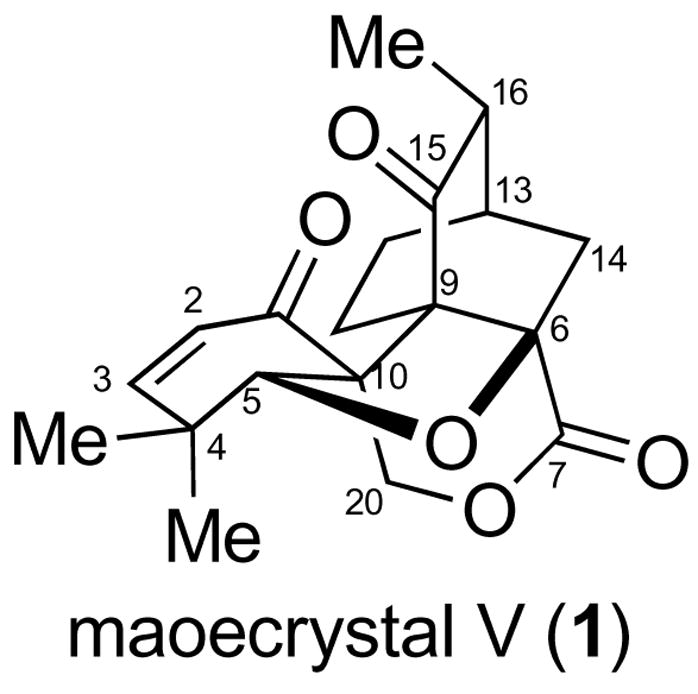
Maoecrystal V (1).
Using the logic of “pattern recognition” 2 to guide our synthetic design, we discerned a bicyclo [2.2.2]-octane core in maoecrystal V (red, 1) and hypothesized that this motif could be synthesized from an intramolecular Diels-Alder (IMDA) cyclization (cf. 3→4).3 It was further supposed that following construction of 4, the tetrahydrofuran moiety would be installed (4→5). A series of functional group transformations providing access to maoecrystal V (Scheme 1) from 5 seemed to be feasible.
Scheme 1.

A proposed route to maoecrystal V.
As outlined in Scheme 2, the synthesis commenced with a palladium-catalyzed ketone α-arylation of 6,4 followed by etherification with TMSCHN2, to provide 8 (90% overall yield). This intermediate was subjected to the Stork-Danheiser protocol,5 with Bu3SnCH2OMOM,6 to afford compound 9 in 75% yield. Following replacement of the MOM ether with a pivaloate ester, intermediate 10 was in hand. Luche reduction of 10 provided an allylic alcohol, which was protected as its MOM ether (11). Hydrolysis of 11 served to liberate the hydroxymethyl intermediate, which was then alkylated with α-iodomethyl tributylstannane to furnish the Wittig-Still precursor (12).7 Through careful control of the reaction temperature, we were able to achieve the desired [2,3] rearrangement, to provide alcohol 2 in 88% yield. It is appreciated that this bond reorganization establishes the requisite quaternary carbon center in a highly stereoregulated fashion.
Scheme 2.

Synthesis of intermediate 2.
Reagents and conditions: (a) Pd(OAc)2, 2-di-t-butylphosphino-2′-methylbiphenyl, K3PO4,, THF, 80 °C, 12 h, 91%; (b) TMSCHN2, Hunig’s base, CH3CN/MeOH = 9:1, 6 h, 100%; (c) Bu3SnCH2OMOM, BuLi, THF, −78 °C to −40 °C, 30 min, 0.5% HCl work-up, 75%; (d) HCl/MeOH, 50 °C, 75%; (e) PivCl, Py, DCM, 12 h, 96%; (f) NaBH4, CeCl3, MeOH, 0 °C, 2 h, 93%; (g) MOMCl, Hunig’s base, DCM, 12 h, 95%; (h) DIBAL-H, −78 °C, DCM, 30 min, 95%; (i) KH, 18-crown-6, ICH2SnBu3, 0 °C, THF, 6 h, 90%; (j) n-BuLi, −78 °C to −20 °C, THF, 6 h, 88%.
With advanced intermediate 2 in hand, we were now approaching the possibility of examining the feasibility of the key IMDA cyclization To our surprise, when 13 subjected to Birch reduction conditions, the exomethylene group was also reduced, giving rise to 13 as a 1:3 α/β mixture of methyl epimers. It seems not unlikely that electron transfer from Birch reduction intermediates serves to facilitate reduction of the exomethylene group.
Although the reduction of the undesired exomethylene group rendered this particular synthetic route toward maoecrystal V problematic, we nevertheless felt that intermediate 13 could serve as a valuable model substrate, with which to probe the viability of the key proposed IMDA reaction. Accordingly, acidic hydrolysis of 13 furnished 14, which was then subjected to esterification with a range of acid chlorides, to provide a series of IMDA precursors, 15a–c. When compounds 15a and 15b were subjected to thermal reaction conditions, we observed no cycloadduct, but only isomerized diene (17). However, it was found that 15c readily underwent cyclization, under thermal conditions, to furnish the Diels-Alder cycloadduct 16. Subsequent fluoro-assisted deprotection afforded 18 in 48% overall yield from 15c. The assignment of the stereochemistry of the Diels-Alder adduct 18 rests on NMR analysis. Thus, a key NOESY experiment revealed that the IMDA cycloaddition had, in fact, proceeded with the opposite sense of facial selectivity than that required for the plan. To account for the observed facial selectivity, we postulate the predominance of transition structure A (Figure 2), wherein the dienophile approaches the diene from the back face of the rotamer shown in 15c. The preference for this rotamer in a Curtin-Hammet sense8 is not clear. This IMDA outcome surely complicates its application to a total synthesis of 1. Nonetheless, programs to reach 1 from the general logic adumbrated above can still be imagined and are being evaluated9.
Figure 2.

Transition structures for the Diels-Alder reaction.
Supplementary Material
Scheme 3.

Intramolecular Diels-Alder reaction of 15c.
Reagents and conditions: (a) Li, NH3(l), t-BuOH/THF, −78 °C, 20 min, −33 °C, 40 min; (b) 1 N HCl, 0 °C, THF/MeOH (10/1), 8 h, 2 steps, 78%; (c) acid chloride, Py, DCM, 0 °C. 15a : 70%, 15b : 75%, 15c : 72%; (d) TBSOTf, TEA, DCM, 0 °C, 15 h, 81%; (e) 180 °C, sealed tube, toluene, 12 h; (f) TBAF, 0 °C, THF, 2 steps, 48%.
Acknowledgments
Support was provided by the NIH (HL25848 and CA103823 to SJD). F.P. thanks Eli Lilly for a graduate fellowship. We thank Prof. Shengping Zheng (Hunter College) and Dr. Fay Ng and for helpful discussions. Special thanks to Ms. Rebecca Wilson for scientific advice, as well as editorial consultation. We acknowledge Ms. Dana Ryan for assistance with the preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Li S, Wang J, Niu X, Shen Y, Zhang H, Sun H, Li M, Tian Q, Lu Y, Cao P, Zheng Q. Org Lett. 2004;6:4327–4330. doi: 10.1021/ol0481535. [DOI] [PubMed] [Google Scholar]
- 2.Wilson RM, Danishefsky SJ. J Org Chem. 2007;72:4293–4305. doi: 10.1021/jo070871s. [DOI] [PubMed] [Google Scholar]
- 3.a) Nicolaou KC, Snyder SA, Montagnon T, Vassilikogiannakis G. Angew Chem Int Ed. 2002;41:1668–1698. doi: 10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]; b) Liao C, Peddinti RK. Acc Chem Res. 2002;35:856–866. doi: 10.1021/ar000194n. [DOI] [PubMed] [Google Scholar]; c) Ihara M. Chem Pharm Bull. 2006;54:765–774. doi: 10.1248/cpb.54.765. [DOI] [PubMed] [Google Scholar]
- 4.Fox JM, Huang X, Chieffi A, Buchwald S. J Am Chem Soc. 2000;122:1360–1370. [Google Scholar]
- 5.Stork G, Danheiser R. J Org Chem. 1973;38:1775. [Google Scholar]
- 6.a) Still WC. J Am Chem Soc. 1978;100:1481–1487. [Google Scholar]; b) Danheiser R, Romines K, Koyama H, Gee S, Johnson CR, Medich JR. Organic Synthesis. 1993;71:133–139. [Google Scholar]
- 7.Still C, Mitra A. J Am Chem Soc. 1978;100:1927–1928. [Google Scholar]
- 8.Seeman JI. Chem Rev. 1983;83:84–134. [Google Scholar]
- 9.For another IMDA approach to the maoecrystal V core structure, please see: Gong J, Lin G, Li C, Yang Z. Org Lett. ASAP; 2009.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.