

Abstract
Hepatopulmonary syndrome (HPS) following rat common bile duct ligation results from pulmonary molecular changes that may be influenced by circulating TNF-α and increased vascular shear stress, through activation of NF-κB or Akt. Increased pulmonary microvascular endothelin B (ETB) receptor and endothelial nitric oxide synthase (eNOS) levels contribute to nitric oxide production and the development of experimental HPS. Pentoxifylline (PTX), a phosphodiesterase and nonspecific TNF-α inhibitor, ameliorates experimental HPS when begun before hepatic injury. However, how PTX influences the molecular events associated with initiation of experimental HPS after liver injury is established is unknown. We assessed the effects of PTX on the molecular and physiological features of HPS in vivo and on shear stress or TNF-α-mediated events in rat pulmonary microvascular endothelial cells in vitro. PTX significantly improved HPS without altering portal or systemic hemodynamics and downregulated pulmonary ETB receptor levels and eNOS expression and activation. These changes were associated with a reduction in circulating TNF levels and NF-κB activation and complete inhibition of Akt activation. In rat pulmonary microvascular endothelial cells, PTX inhibited shear stress-induced ETB receptor and eNOS expression and eNOS activation. These effects were also associated with inhibition of Akt activation and were reproduced by wortmanin. In contrast, TNF-α had no effects on endothelial ETB and eNOS alterations in vitro. PTX has direct effects in the pulmonary microvasculature, likely mediated through Akt inhibition, that ameliorate experimental HPS.
Keywords: common bile duct ligation, endothelial cells, endothelin B receptor, endothelial nitric oxide synthase
THE HEPATOPULMONARY SYNDROME (HPS) results when intrapulmonary vascular dilatation causes hypoxemia in the setting of liver disease or portal hypertension (13). This syndrome is found in 10–20% of patients with cirrhosis, and its presence increases mortality (15). Despite a significant increase in our understanding of HPS over the last 20 years, its pathogenesis remains incompletely understood, and no medical therapies are available.
Chronic common bile duct ligation (CBDL) in the rat leading to biliary cirrhosis reproduces the pulmonary physiological abnormalities of human HPS and serves as an experimental model (2). A series of alterations in the pulmonary microvasculature, including endothelin-1 (ET-1)-mediated endothelial nitric oxide (NO) synthase (eNOS) activation and NO production via increased endothelial endothelin B (ETB) receptors (8–10, 19), and accumulation and activation of intravascular macrophages, leading to inducible NO synthase (iNOS)-derived NO production and heme oxygenase (HO)-1-derived carbon monoxide production (12, 17, 18), have been identified after CBDL and contribute to intrapulmonary vasodilatation. ETB-receptor overexpression after CBDL temporally correlates with the onset of a hyperdynamic circulation and increased vascular shear stress (10), a known modulator of ETB-receptor expression (11) and Akt activation (3). Circulating TNF-α levels also increase after CBDL, and inhibition of TNF-α begun before CBDL can also ameliorate experimental HPS.
Recently, pentoxifylline [1-(5-oxohexyl)-3,7-dimethylxanthine; PTX], a nonspecific phosphodiesterase inhibitor that may increase intracellular cAMP levels and is recognized to block effects mediated by TNF-α in inflammatory and endothelial cells through inhibition of NF-κB/IκB, has been reported to suppress TNF-α production and pulmonary intravascular macrophage iNOS/NO induction and prevent the onset of experimental HPS when begun before CBDL (16). In addition, PTX has recently been reported to inhibit Akt activation by blocking membrane translocation (7). PTX is attractive as a potential treatment for HPS due to its low cost and established safety profile. However, if PTX influences experimental HPS when administered in a more clinically relevant way, after hepatic injury is established following CBDL, and how it influences the pulmonary microvascular alterations that contribute to vasodilatation in this situation are not established.
The aim of the present study was to evaluate if PTX influences experimental HPS when administered after the onset of liver injury following CBDL and to characterize the mechanisms through which PTX modifies intrapulmonary vasodilatation. We assessed the effects of PTX on the physiological and molecular changes of HPS in vivo after CBDL and evaluated the effects of PTX on cultured rat pulmonary microvascular endothelial cells (RPMVECs) in vitro. Our findings demonstrate that PTX markedly diminishes the severity of experimental HPS and support that direct effects in pulmonary endothelial cells, mediated in part by inhibiting Akt activation, are important in influencing the development of experimental HPS.
MATERIALS AND METHODS
Animal models
Male Sprague-Dawley rats (200–250 g, Charles River Laboratories, Wilmington, MA) underwent sham surgery or CBDL, as previously described (4). PTX (50 mg · kg−1 · day−1; Sigma, St. Louis, MO) was given orally in drinking water for 2 wk beginning 1 wk after CBDL, based on dosing used in prior studies (16). Body weight, mean systemic arterial pressure, portal venous pressure, and spleen weight were measured to evaluate systemic effects and liver abnormalities. The study was approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham, and conforms to National Institutes of Health guidelines on the care and use of laboratory animals. Five to eight animals were used in each group.
Measurement of plasma TNF-α and ET-1 levels
Plasma samples were serially diluted with sterile endotoxin-free water and heat treated to destroy inhibitors that can interfere with activation. TNF-α levels were measured with a commercially available solid-phase sandwich enzyme-linked immunosorbent assay, according to the protocol supplied by the manufacturer (R&D Systems, Minneapolis, MN). ET-1 concentrations were measured with a commercially available RIA kit (Phoenix Pharmaceuticals, Mountain View, CA), as previously described, following the manufacturer’s instructions.
Arterial blood gas analysis
Arterial blood drawn at rest was analyzed on an ABL 520 radiometer (Radiometer America, Westlake, OH) in the Clinical Laboratory, UAB Hospital, as previously described (4). The alveolar-arterial oxygen gradient was calculated as 150 − (PCO2/0.8) − PO2.
Microsphere protocol
An established technique was used to evaluate intrapulmonary vasodilatation. Cross-linked colored polystyrene-divinylbenzene microspheres (2.5 × 106; size range 5.5–10 μm; Interactive Medical Technologies, Irvine, CA) were injected into animals through a femoral vein catheter, after an aliquot of microspheres was removed to verify the numbers and sizes injected. Microspheres passing through the pulmonary microcirculation were measured in a blood sample withdrawn from a femoral arterial catheter beginning at the time of femoral vein injection. Numbers and sizes of microspheres were assessed using a Leitz microscope (Wetzlar, Germany) with a color video imaging and digital analysis system (Image Pro 3.0, Media Cybernetics, Silver Spring, MD) and counted directly. Total numbers of microspheres passing through the microcirculation were calculated as previously described (4).
Western blot analysis
Antibodies for eNOS (BD Biosciences, San Jose, CA) and its phosphorylation at Ser1177 (peNOSser1177; Cell Signaling Technology, Danvers, MA), ETB receptor (EMD Biosciences, San Diego, CA), ED1 (specific marker for monocytes/macrophages, Santa Cruz Biotechnology, Santa Cruz, CA), HO-1 (Stressgen Bioreagents, Ann Arbor, MI), and iNOS (Santa Cruz Biotechnology), total and phosphorylation of IκB-α (pIκB-αser32/36; Cell Signaling Technology) and Akt (pAktser473; Cell Signaling Technology) were used to measure their levels in lung from sham and CBDL animals. Tissue was homogenized in RIPA buffer in the presence of protease inhibitors. Protein (35 μg) from homogenates was fractionated on SDS-PAGE gel and transferred to nitrocellulose membranes (Amersham, Piscataway, NJ). Incubation with primary antibodies was followed by addition of horseradish peroxidase-conjugated secondary antibodies and detection with enhanced chemiluminescence.
Measurement of lung cAMP levels
cAMP assay was performed using a commercially available EIA kit (Cayman Chemical, Ann Arbor, MI), following the manufacturer’s instructions.
NOS activity assay
Total NOS activity was measured in lungs by determining the biochemical conversion of L-[3H]arginine to L-[3H]citrulline using a commercial NOS assay kit (Calbiochem, San Diego, CA), according to instructions (18).
HO activity assay
HO enzyme activity in rat lung was quantified by assessing bilirubin generation using a modification of established techniques, as previously described (18).
Cell culture
RPMVECs (VEC Technolgies, Rensselaer, NY) were maintained and subcultured in MCDB-131 complete medium at 37°C with 5% CO2. Cells (passages 5–10) were seeded in 100-mm tissue culture dishes and grown to confluency in the complete medium before exposure to shear stress. The number of viable cells was determined by Trypan blue exclusion.
Shear stress studies
A confluent RPMVEC monolayer grown in a 100-mm dish was exposed to nonpulsatile, laminar shear stress by rotating a Teflon cone (0.5° cone angle). Cells were exposed to an arterial level of shear stress (15 dyn/cm2) (1) for indicated time periods. In some sets of experiments, endothelial cells were pretreated with PTX (0.001 M) or wortmanin (10 nM), both before exposure to shear stress. Cell lysates were used for detecting ETB-receptor levels, total eNOS and peNOSser1177, total IκB-α and pIκB-αser32/36, and total Akt and pAktser473 by Western blot analysis, as described above. Low-serum (0.5% FBS) and endotoxin-free media were used in all flow experiments.
Statistical analysis
All measurements are expressed as means ± SE. Data were analyzed using Student’s t-test or ANOVA with Bonferroni correction for multiple comparisons between groups. P < 0.05 was considered statistical significance.
RESULTS
Effects of PTX administration on the development of HPS after CBDL
To evaluate how PTX influences HPS after CBDL in vivo, PTX was administered through drinking water for 2 wk to 1-wk CBDL animals. At 3 wk, systemic and portal hemodynamics and the physiological features of HPS were assessed (Table 1). PTX administration was associated with a significant decrease in alveolar-arterial oxygen gradient and intrapulmonary shunting relative to untreated animals, reflecting marked improvement in HPS. This improvement occurred without altering either systemic or portal hemodynamics, supporting that the vascular effects occurred predominantly within the lung. PTX treatment did not significantly influence water or food intake in the animal groups.
Table 1.
Effects of pentoxifylline administration on systemic alterations and the development of hepatopulmonary syndrome
| Control | 3-wk CBDL | 3-wk CBDL+PTX | |
|---|---|---|---|
| MSAP, mmHg | 116.9±4.0 | 97.7±2.4* | 97.7±4.0* |
| PVP, mmHg | 7.6±0.9 | 14.1±0.7* | 13.9±1.0* |
| A-aPO2, Torr | 6.9±0.5 | 20.2±2.2* | 10.1±1.4† |
| Intrapulmonary shunt fraction, % | 6.4±1.7 | 18.8±3.5* | 9.0±1.5† |
| Plasma TNF-α, ng/ml | ND | 766.0±68.3* | 460.3±66.5† |
| Lung cAMP, pmol/g protein | 1,658.2±273.9 | 3,501.8±569.2* | 3,146.7±538.1* |
Values are expressed as means ± SE (n = 5–8 for each group). CBDL, common bile duct ligation; PTX, pentoxifylline; MSAP, mean systemic arterial pressure; PVP, portal venous pressure; A-aPO2, alveolar-arterial oxygen gradient; ND, nondetectable,
P < 0.05 compared with control animals,
P<0.05 compared with CBDL animals.
Effects of PTX on pulmonary cAMP levels and TNF-α after CBDL
To explore the mechanisms of PTX effects in the lung after CBDL, we evaluated for evidence of phosphodiesterase inhibition by measuring lung cAMP levels and assessed pulmonary and circulating TNF-α (Table 1). Lung cAMP levels significantly increased twofold after CBDL compared with control animals. However, levels were not further modulated by PTX treatment. Circulating TNF-α levels were dramatically elevated in 3-wk CBDL animals relative to control and were significantly decreased by PTX. However, levels remained markedly elevated at >400 pg/ml following PTX treatment, despite the improvement in HPS. In contrast, lung TNF-α mRNA levels measured by RT-PCR were not increased in 3-wk CBDL animals relative to control and were not altered by PTX treatment (data not shown).
Effects of PTX on mediators of pulmonary alterations after CBDL
We assessed the effects of PTX on the pulmonary endothelium (plasma ET-1, lung ETB receptor/eNOS) and on pulmonary intravascular macrophages (lung ED1, HO-1, and iNOS) after CBDL (Fig. 1, Table 2). As previously described, the development of HPS in 3-wk CBDL animals was accompanied by a significant increase in plasma ET-1 levels, pulmonary endothelial ETB-receptor and eNOS levels, and eNOS activation reflected by an increase in peNOSser1177 levels. After PTX treatment, circulating ET-1 levels were not diminished, but pulmonary ETB-receptor and eNOS levels were significantly decreased, and eNOS activation was abolished, supporting a direct effect of PTX in endothelial cells. The decrease in eNOS activation after PTX treatment was also associated with normalization of pulmonary NOS activity. Pulmonary intravascular macrophage accumulation and activation were also observed after CBDL, as previously established (18), reflected in an increase in ED1 (specific macrophage marker), iNOS, and HO-1 levels and in an increase in pulmonary NOS and HO activity. After PTX treatment, macrophage accumulation diminished, reflected by lower ED1 levels, although levels remained significantly higher (threefold) than that for control animals. Activation of macrophages was not diminished after PTX, reflected by the persistent increase in HO-1 and iNOS levels and in line with persistently increased circulating TNF-α levels and HO activity in the lung.
Fig. 1.

Effects of chronic pentoxifylline (PTX) administration on pulmonary endothelin B (ETB) receptor, endothelial nitric oxide synthase (eNOS), heme oxygenase (HO)-1, and inducible nitric oxide synthase (iNOS) protein expression and intravascular macrophage accumulation (reflected by ED1 expression) after common bile duct ligation (CBDL). Protein levels are quantitated by densitometry. All values are expressed as means ± SE (n = 5–8 each group). *P < 0.05 compared with control animals; †P < 0.05 compared with CBDL animals.
Table 2.
Effects of PTX on eNOS phosphorylation at Ser1177, NOS and HO activities in lung, and ET-1 levels after CBDL
| Control | 3-wk CBDL | 3-wk CBDL+PTX | |
|---|---|---|---|
| peNOS, fold control | 1 | 2.4±0.3* | 1.1±0.4† |
| NOS activity, pmol·mg protein−1·h−1 | 4.4±1.1 | 12.7±0.6* | 4.1±1.0† |
| HO activity, pmol·mg protein−1·h−1 | 17.6±3.3 | 27.6±6.8* | 25.8±3.9* |
| Plasma ET-1, pg/ml | 8.1±0.2 | 26.7±3.1* | 35.8±3.4* |
Values are expressed as means ± SE (n = 5–8 for each group). NOS, nitric oxide synthase; peNOS, phosphorylation of endothelial NOS at Ser1177; HO, heme oxygenase; ET-1, endothelin-1.
P<0.05 compared with control animals,
P<0.05 compared with CBDL animals.
Effects of PTX administration on relevant signaling pathways after CBDL
Since both TNF-α signaling and Akt activation may contribute to pulmonary alterations in experimental HPS and may be influenced by PTX, we studied these pathways after CBDL. Specifically, we evaluated TNF-α signaling by assessing lung IκB-α activation (pIκB-αser32/36) and Akt activation by measuring phosphorylated Akt (pAktser473) (Fig. 2). As expected, activation of both Akt and TNF-α signaling pathways was evident in 3-wk CBDL animals. After PTX treatment, there was incomplete inhibition of TNF-α activation reflected in persistent IκB-α phosphorylation, consistent with the persistent marked elevation in circulating TNF-α levels and with evidence of persistent macrophage activation. In contrast, PTX completely blocked Akt activation in vivo, supporting a potential effect on shear mediated events in the microvasculature of the lung.
Fig. 2.
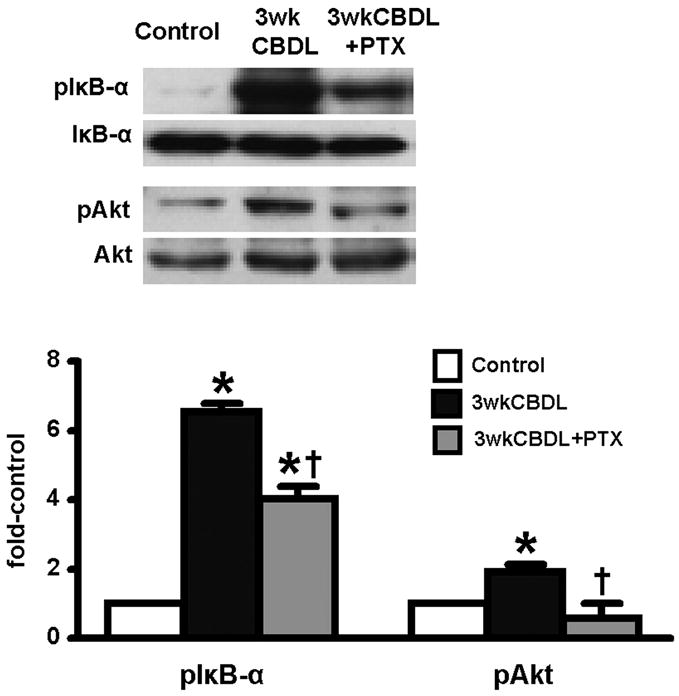
Effects of chronic PTX administration on phosphorylation of IκB-α and Akt in lung after CBDL. Protein levels are quantitated by densitometry. pIκB-α, phosphorylation of IκB-αser32/36; pAkt, phosphorylation of Aktser473. All values are expressed as means ± SE (n = 5–8 each group). *P < 0.05 compared with control animals; †P < 0.05 compared with CBDL animals.
Shear stress and TNF-α effects on ETB-receptor and eNOS levels in RPMVECs
We explored if shear stress or TNF-α modulate pulmonary endothelial alterations in vitro and how PTX modulates these events (Fig. 3). Compared with static conditions, exposure of RPMVECs to a level of laminar shear stress intermediate between venous and arterial values (15 dyn/cm2) for 24 h resulted in a 3.5-fold increase in ETB-receptor protein levels, a 1.4-fold increase in eNOS levels, as well as marked activation of Akt. Pretreatment of the endothelial monolayers for 30 min with PTX blocked the shear stress-associated increase in ETB receptor and eNOS levels and also decreased shear stress-induced Akt activation. In contrast, TNF-α had no effect on ETB-receptor or eNOS levels, despite IκB-α phosphorylation (pIκB-αser32/36), which was reduced by PTX pretreatment.
Fig. 3.

Effects of PTX on shear stress (A) or TNF-α-induced (B) ETB-receptor and eNOS expression and phosphorylation of Akt and IκB-α in rat pulmonary microvascular endothelial cells (RPMVECs) at 24 h. Protein levels are quantitated by densitometry. All values are expressed as means ± SE (n = 4–5 each group). *P < 0.05 compared with control; †P < 0.05 compared with shear stress or TNF-α treatment.
Shear stress and TNF-α effects on eNOS activation in RPMVECs
Since shear stress and TNF-α have both been reported to modulate activation of eNOS in endothelial cells (3, 6), we assessed the effects of shear stress and TNF-α on eNOS activation (peNOSser1177 levels) in RPMVECs (Fig. 4). Application of shear stress (15 dyn/cm2) for 30 min increased peNOSser1177 levels approximately fourfold over control. The addition of PTX completely blocked the shear stress modulation of peNOSser1177 levels. Shear-mediated activation of eNOS was associated with Akt phosphorylation (pAktser473), which was blocked by the addition of PTX. In contrast, TNF-α incubation inhibited basal peNOSser1177 by 50% and was associated with evidence of IκB-α phosphorylation (pIκB-αser32/36). The addition of PTX decreased TNF-α-induced IκB-α phosphorylation.
Fig. 4.

Effects of PTX on shear stress (A) or TNF-α-induced (B) eNOS phosphorylation and activation of Akt and IκB-α in RPMVECs at 30 min. Protein levels are quantitated by densitometry. All values are expressed as means ± SE (n = 4–5 each group). *P < 0.05 compared with control; †P < 0.05 compared with shear stress or TNF-α treatment. peNOS, phosphorylation of eNOSSer1177.
Effects of Akt signaling inhibition on ETB-receptor expression and eNOS activation in RPMVECs
To further explore the involvement of Akt pathway in shear stress-mediated events, endothelial cell monolayers were pretreated with a specific Akt inhibitor, wortmannin, before undergoing shear stress (15 dyn/cm2). ETB-receptor expression was examined after application of shear stress for 24 h (Fig. 5A). eNOS phosphorylation (peNOSser1177 levels) was measured after exposure of shear stress for 30 min (Fig. 5B). Akt activation (pAktser473) was also assessed at each time point. Similar to the effects of PTX, wortmannin blocked shear stress-induced Akt activation, ETB-receptor upregulation, and eNOS phosphorylation, suggesting a direct role for the Akt signaling pathway in shear stress-mediated endothelial alterations.
Fig. 5.

Effects of wortmanin on shear stress-induced ETB receptor expression, eNOS phosphorylation, and Akt activation in RPMVECs. A: 24-h exposure to shear stress. B: 30-min exposure to shear stress. All values are expressed as means ± SE (n = 4–5 each group). *P < 0.05 compared with control; †P < 0.05 compared with shear stress.
DISCUSSION
In this study, we have assessed the effects of PTX on the initiation of experimental HPS. We find that PTX administration, initiated 1 wk after CBDL and continued for 2 wk, markedly improved intrapulmonary vasodilatation and gas exchange abnormalities without altering systemic or portal pressures. The improvement in HPS was accompanied by a decrease in lung NOS activity, downregulation of pulmonary endothelial ETB-receptor and eNOS levels, decreased eNOS activation, and reduced pulmonary intravascular macrophage accumulation. These changes were associated with complete reversal of pulmonary Akt activation in vivo. In contrast, PTX only partially reversed the increase in circulating TNF-α levels and pulmonary activation of NF-κB. In RPMVECs, shear stress, but not TNF-α, increased ETB receptor and eNOS levels and eNOS activation, effects associated with Akt activation and inhibited by PTX. Finally, the effects of PTX on shear stress-mediated increases in ETB-receptor levels and eNOS activation in PRMVECs were reproduced by the specific Akt inhibitor wortmannin. Taken together, these findings demonstrate that PTX administration significantly improves experimental HPS and support that direct effects on Akt activation in the pulmonary microvascular endothelium contribute to this effect.
Increased pulmonary NO production appears to play a major contributory role in microvascular changes in both human and experimental HPS (5). Our finding that pulmonary NOS activity is significantly increased after CBDL and normalizes with improvement of intrapulmonary vasodilatation after PTX administration is consistent with this concept. Prior studies in CBDL animals support that liver-derived ET-1 stimulation of pulmonary microvascular eNOS through the ETB receptor contributes to NO production and the onset of vasodilatation (8, 10, 19). Our finding that PTX downregulates ETB and eNOS levels and eNOS activation in CBDL lung in vivo without altering systemic or portal hemodynamics and inhibits shear stress-mediated increases in these proteins in RPMVECs in vitro are in line with these prior studies and are consistent with direct PTX effects in the pulmonary microvascular endothelium. The observation that exposure of RPMVECs to shear stress recapitulates ETB and eNOS alterations found as the hyperdynamic state and increased pulmonary shear stress develops after CBDL in vivo suggests that shear stress may contribute to endothelial susceptibility to HPS and that PTX may influence this process.
We have also explored the potential mechanisms for the observed PTX effects in the pulmonary endothelium in experimental HPS. Although PTX is established to inhibit TNF-α-mediated events and to increase cAMP levels through phosphodiesterase inhibition, our in vivo findings support that the modulation of pulmonary endothelial molecular changes and improvement of HPS occur, despite persistent lung NF-κB activation and without altering lung cAMP levels. These results point toward a potential distinct effect in the endothelium. Based on the observations that increased vascular shear stress is associated with both endothelial Akt activation (3) and the onset of HPS (10) and the recent finding that PTX can inhibit Akt activation in mesangial cells (7), we assessed PTX effects on lung Akt activation after CBDL in vivo and in response to exposure to shear stress in vitro. We found that lung Akt activation occurred following CBDL and that PTX administration abolished this effect over the same time frame that molecular and physiological alterations of HPS improved. In RPMVECs, exposure to levels of shear stress anticipated to be found in the pulmonary vasculature in portal hypertension produced molecular changes similar to those after CBDL and were associated with Akt activation. PTX and the specific Akt inhibitor wortmannin blocked these shear-mediated effects. Although the precise role of lung Akt activation in HPS in vivo and the mechanisms for the effect of PTX on Akt in RPMVECs remain to be defined, these results are consistent with the concept that shear stress-mediated Akt activation may contribute to pulmonary microvascular endothelial alterations in experimental HPS.
Our observations are in line with prior results showing that increased circulating TNF-α levels derived from bacterial translocation modulate pulmonary intravascular macrophage accumulation and iNOS and HO-1 expression and activity after CBDL (12, 18). In prior studies when antibiotics were used to inhibit bacterial translocation (12) or PTX was given to antagonize TNF-α (16) beginning before CBDL, circulating TNF-α levels did not rise, a hyperdynamic state did not develop, intrapulmonary vasodilatation was attenuated, and macrophage accumulation and iNOS expression were decreased in 4- to 5-wk CBDL animals. In contrast, in the present study, PTX administered after the onset of hepatic injury at a dose similar to that used in prior work significantly improved HPS without influencing systemic or portal hemodynamics and without normalizing TNF-α levels, NF-κB activation, or lung iNOS or HO-1 levels. One hypothesis for the differing effects of PTX in experimental HPS when administered before rather than after the establishment of liver injury is that PTX pretreatment blocks local endotoxin and TNF-α release, thereby preventing splanchnic and systemic hemodynamic changes, including increased vascular shear stress that may mediate the molecular changes in the ETB receptor and eNOS that result in endothelial susceptibility to HPS. Our in vitro studies support that TNF-α does not directly influence these molecular events in RPMVECs. We found a persistent increase in lung HO-1 and iNOS levels, despite a reduction in intravascular macrophage accumulation in the present work. This observation is not fully explained but could reflect production of these proteins in another cell type in the lung, incomplete TNF-α inhibition, or an insufficient decrease in pulmonary intravascular macrophage numbers to significantly lower protein levels. The persistent increase in HO-1 after PTX treatment is accompanied by elevated lung HO activity, an event that contributes to maintenance of intrapulmonary vasodilatation after CBDL (18), and may explain why intrapulmonary vasodilatation does not completely resolve with PTX treatment here. The observation that lung NOS activity normalizes after PTX, despite a persistent increase in iNOS levels, is unusual, as iNOS activity is classically regulated by changes in expression. Although this is an important area for further investigation, recent work showing that carbon monoxide may inhibit iNOS activity without reducing expression provides one explanation for this finding (14).
The present findings have potentially important clinical and mechanistic implications for treating and understanding HPS. First, from a practical standpoint, we have shown that PTX, initiated following the establishment of liver injury in CBDL animals, can significantly decrease the subsequent severity of HPS. This result supports that trials of PTX may be reasonable in patients with established liver disease. Second, our findings extend prior in vivo observations and provide support for the concept that PTX modulates events in the pulmonary microvascular endothelium, including shear stress-mediated activation of Akt that influences ETB-receptor-mediated vasodilatation. Defining the specific signaling pathways involved in these events provides the potential to develop novel targeted therapies for human disease.
Acknowledgments
GRANTS
This study was supported by DK-56804 and a Veterans Affairs Merit Review grant to M. B. Fallon.
References
- 1.Boo YC, Sorescu G, Boyd N, Shiojima I, Walsh K, Du J, Jo H. Shear stress stimulates phosphorylation of endothelial nitric-oxide synthase at Ser1179 by Akt-independent mechanisms. Role of protein kinase A. J Biol Chem. 2002;277:3388–3396. doi: 10.1074/jbc.M108789200. [DOI] [PubMed] [Google Scholar]
- 2.Chang SW, Ohara N. Pulmonary circulatory dysfunction in rats with biliary cirrhosis. An animal model of the hepatopulmonary syndrome. Am Rev Respir Dis. 1992;145:798–805. doi: 10.1164/ajrccm/145.4_Pt_1.798. [DOI] [PubMed] [Google Scholar]
- 3.Dimmeler S, Fleming I, Fissthaler B, Hermann C, Busse R, Zeiher A. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 4.Fallon MB, Abrams GA, McGrath JW, Hou Z, Luo B. Common bile duct ligation in the rat: a model of intrapulmonary vasodilatation and hepatopulmonary syndrome. Am J Physiol Gastrointest Liver Physiol. 1997;272:G779–G784. doi: 10.1152/ajpgi.1997.272.4.G779. [DOI] [PubMed] [Google Scholar]
- 5.Gaines DI, Fallon MB. Hepatopulmonary syndrome. Liver Int. 2004;24:397–401. doi: 10.1111/j.1478-3231.2004.0944.x. [DOI] [PubMed] [Google Scholar]
- 6.Kim F, Gallis B, Corson MA. TNF-α inhibits flow and insulin signaling leading to NO production in aortic endothelial cells. Am J Physiol Cell Physiol. 2001;280:C1057–C1065. doi: 10.1152/ajpcell.2001.280.5.C1057. [DOI] [PubMed] [Google Scholar]
- 7.Lin SL, Chen RH, Chen YM, Chiang WC, Tsai TJ, Hsieh BS. Pentoxifylline inhibits platelet-derived growth factor-stimulated cyclin D1 expression in mesangial cells by blocking Akt membrane translocation. Mol Pharmacol. 2003;64:811–822. doi: 10.1124/mol.64.4.811. [DOI] [PubMed] [Google Scholar]
- 8.Ling Y, Zhang J, Luo B, Song D, Liu L, Tang L, Stockard C, Grizzle W, Ku D, Fallon MB. The role of endothelin-1 and the endothelin B receptor in the pathogenesis of experimental hepatopulmonary syndrome. Hepatology. 2004;39:1593–1602. doi: 10.1002/hep.20244. [DOI] [PubMed] [Google Scholar]
- 9.Luo B, Abrams GA, Fallon MB. Endothelin-1 in the rat bile duct ligation model of hepatopulmonary syndrome: correlation with pulmonary dysfunction. J Hepatol. 1998;29:571–578. doi: 10.1016/s0168-8278(98)80152-9. [DOI] [PubMed] [Google Scholar]
- 10.Luo B, Liu L, Tang L, Zhang J, Stockard C, Grizzle W, Fallon M. Increased pulmonary vascular endothelin B receptor expression and responsiveness to endothelin-1 in cirrhotic and portal hypertensive rats: a potential mechanism in experimental hepatopulmonary syndrome. J Hepatol. 2003;38:556–563. doi: 10.1016/s0168-8278(03)00012-6. [DOI] [PubMed] [Google Scholar]
- 11.Morawietz H, Talanow R, Szibor M, Rueckschloss U, Schubert A, Bartling B, Darmer D, Holtz J. Regulation of the endothelin system by shear stress in human endothelial cells. J Physiol. 2000;525:761–770. doi: 10.1111/j.1469-7793.2000.00761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rabiller A, Nunes H, Lebrec D, Tazi KA, Wartski M, Dulmet E, Libert JM, Mougeot C, Moreau R, Mazmanian M, Humbert M, Herve P. Prevention of gram-negative translocation reduces the severity of hepatopulmonary syndrome. Am J Respir Crit Care Med. 2002;166:514–517. doi: 10.1164/rccm.200201-027OC. [DOI] [PubMed] [Google Scholar]
- 13.Rodriguez-Roisin R, Krowka MJ, Herve P, Fallon MB ERS Task Force Pulmonary-Hepatic Vascular Disorders (PHD) Scientific Committee. Pulmonary-hepatic vascular disorders (PHD) Eur Respir J. 2004;24:861–880. doi: 10.1183/09031936.04.00010904. [DOI] [PubMed] [Google Scholar]
- 14.Sawle P, Foresti R, Mann BE, Johnson TR, Green CJ, Motterlini R. Carbon monoxide-releasing molecules (CO-RMs) attenuate the inflammatory response elicited by lipopolysaccharide in RAW264.7 murine macrophages. Br J Pharmacol. 2005;145:800–810. doi: 10.1038/sj.bjp.0706241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schenk P, Schoniger-Hekele M, Fuhrmann V, Madl C, Silberhumer G, Muller C. Prognostic significance of the hepatopulmonary syndrome in patients with cirrhosis. Gastroenterology. 2003;125:1042–1052. doi: 10.1016/s0016-5085(03)01207-1. [DOI] [PubMed] [Google Scholar]
- 16.Sztrymf B, Rabiller A, Nunes H, Savale L, Lebrec D, LePape A, de Montpreville V, Mazmanian M, Humbert M, Herve P. Prevention of hepatopulmonary syndrome by pentoxifylline in cirrhotic rats. Eur Respir J. 2004;23:752–758. doi: 10.1183/09031936.04.00080404. [DOI] [PubMed] [Google Scholar]
- 17.Wiest R, Das S, Cadelina G, Garcia-Tsao G, Milstien S, Groszmann R. Bacterial translocation in cirrhotic rats stimulates eNOS-derived NO production and impairs mesenteric vascular contractility. J Clin Invest. 1999;104:1223–1233. doi: 10.1172/JCI7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J, Ling Y, Luo B, Tang L, Stockard C, Grizzle WE, Fallon MB. Analysis of pulmonary heme oxygenase-1 and nitric oxide synthase alterations in experimental hepatopulmonary syndrome. Gastroenterology. 2003;125:1441–1451. doi: 10.1016/j.gastro.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 19.Zhang M, Luo B, Chen SJ, Abrams GA, Fallon MB. Endothelin-1 stimulation of endothelial nitric oxide synthase in the pathogenesis of hepatopulmonary syndrome. Am J Physiol Gastrointest Liver Physiol. 1999;277:G944–G952. doi: 10.1152/ajpgi.1999.277.5.G944. [DOI] [PubMed] [Google Scholar]