

Summary
Poxviruses encode their own type IB topoisomerases (TopIBs) which release superhelical tension generated by replication and transcription of their genomes. To investigate the reaction catalyzed viral TopIBs, we have determined the structure of a variola virus topoisomerase-DNA complex trapped as a vanadate transition state mimic. The structure reveals how the viral TopIB enzymes are likely to position the DNA duplex for ligation following relaxation of supercoils and identifies the sources of friction observed in single molecule experiments that argue against free rotation. The structure also identifies a conformational change in the leaving group sugar that must occur prior to cleavage and reveals a mechanism for promoting ligation following relaxation of supercoils that involves a novel Asp-minor groove interaction. Overall, the new structural data support a common catalytic mechanism for the TopIB superfamily but indicate distinct methods for controlling duplex rotation in the small vs. large enzyme subfamilies.
Keywords: topoisomerase, poxvirus, smallpox, mechanism, structure
Introduction
Topoisomerases release super-helical tension and decatenate DNA in order to facilitate essential processes such as replication, transcription, and recombination (Champoux, 2001; Wang, 1996). The type IB class of topoisomerases uses a nicking-joining mechanism to release DNA supercoils that does not utilize high energy cofactors such as ATP. During this reaction, a conserved tyrosine nucleophile attacks a DNA phosphodiester to form a covalent 3′-phosphotyrosine linkage and release a free 5′-hydroxyl group. Supercoils are released in this covalent reaction intermediate as the flanking DNA duplexes rotate about the uncleaved strand at the site of the nick. Following rotation, the cleavage reaction is run in reverse to ligate the broken strand where the 5′-hydroxyl group serves as the nucleophile and the enzyme's tyrosine hydroxyl group is released. This trans-esterification chemistry is identical to that used by the tyrosine recombinase family of site-specific recombination enzymes (Grindley et al., 2006), which shares a similar catalytic domain architecture with the type IB topoisomerases (Cheng et al., 1998).
Human topoisomerase I is a well studied example of a type IB enzyme and is also the target of the anti-cancer drug camptothecin, which binds tightly to the covalent intermediate of the topoisomerase reaction and inhibits re-ligation of the nicked genome (Leppard and Champoux, 2005). Poxviruses such as the variola (smallpox) and vaccinia viruses also produce a type IB topoisomerase. At ∼33 kDa in size, the poxvirus enzymes are much smaller than their 70 kDa cellular counterparts, making them attractive model systems for studying the topoisomerase reaction as well as potential drug targets to treat or prevent poxvirus infections (Shuman, 1998). The poxvirus topoisomerases are also unique for requiring a conserved core sequence 5′-(T/C)CCTT-3′ for activity, where specificity is used both in substrate binding and in a sequence-dependent activation step that promotes cleavage (Hwang et al., 1999; Hwang et al., 1998; Shuman and Prescott, 1990).
The vaccinia virus topoisomerase has been the target of extensive mutagenesis, biochemical, and biophysical studies (Shuman, 1998). The variola virus topoisomerase (which differs by only three amino acids from the vaccinia virus protein) has also been well-studied, including a detailed analysis of sequence preferences in the regions flanking the core pentanucleotide motif (Minkah et al., 2007). An analysis of TopIB cleavage sites has revealed a distribution throughout the poxvirus genomes, with a central clustering of sites observed only for the orthopox viruses (Minkah et al., 2007). The poxvirus topoisomerases are part a broader family of small type IB enzymes which includes the topoisomerases from mimi virus and a number of eubacteria (Benarroch et al., 2006; Krogh and Shuman, 2002a).
We recently reported the crystal structures of covalent and non-covalent variola virus topoisomerase-DNA (referred to here as vTopIB-DNA) complexes (Perry et al., 2006). These structures were able to explain a great deal of the biochemical data that are available for the poxvirus enzymes. In particular, the structural models revealed the basis for sequence-specific binding and a mechanism for activation of catalysis. However, one important aspect of the poxvirus topoisomerase reaction that is not yet well-understood concerns the nature of TopIB-DNA contacts downstream of the cleavage site (colored in yellow in Fig. 1) that occur during the reaction pathway. Single molecule experiments for both the human and viral enzyme have indicated that relaxation of supercoils does not occur by free rotation of the nicked DNA duplex, but instead the enzyme provides a source of friction that restricts rotation (Koster et al., 2005). These experiments also indicated that relaxation of supercoils involves multiple cleavage-rotation-ligation cycles, where ∼20 supercoils are released on average for each poxvirus TopIB reaction cycle. Human TopIB has an elaborate protein architecture with structural elements poised to partially enclose the DNA duplex downstream of the cleavage site, providing a basis for understanding how “controlled rotation” could occur within the enzyme (Stewart et al., 1998). In contrast, the poxvirus enzyme has very few structural elements in this region and it is less clear how the downstream DNA duplex is restricted from rotating freely and is guided into a position that promotes re-ligation.
Figure 1.
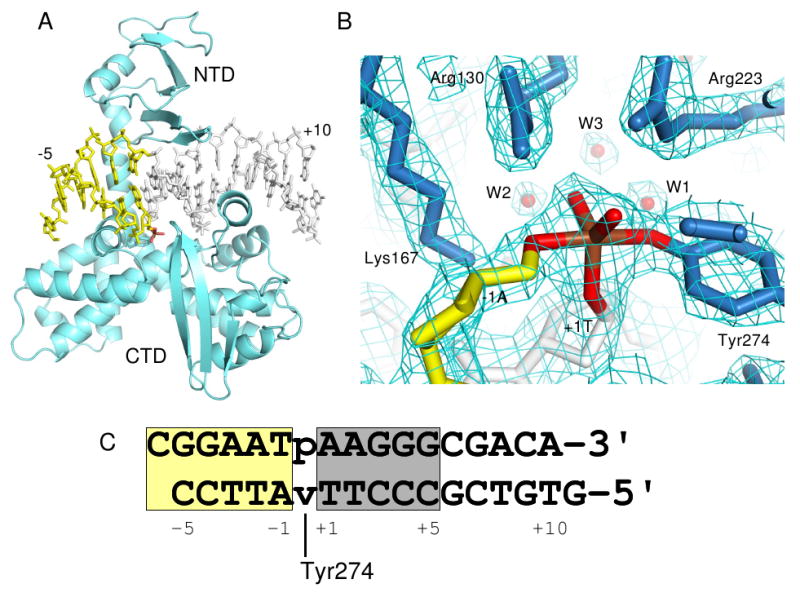
Variola virus TopIB-DNA-vandate complex. (A) Overall structure of the complex. NTD=N-terminal domain; CTD=C-terminal domain. (B) Electron density within the active site following refinement. The density is from a σA-weighted 2Fo-Fc map contoured at 1.2σ. (C) Sequence of the DNA duplex used to form the vTopIB-DNA-vanadate complex with the core recognition/activation sequence shaded gray. The vanadate linkage is indicated by ‘v’, with covalent attachment of Tyr274 indicated. The numbering indicated is used throughout the text. In all panels, DNA downstream of the cleavage site is colored yellow and the vanadium linkage is highlighted in brown and red. Figures 1a,b, 2a, 3, 5, & 6 were produced using Pymol (Delano, 2002).
To address these questions, we have determined the 2.1 Å crystal structure of a variola virus TopIB-DNA complex with intact duplex DNA segments flanking the active site of the enzyme. Replacement of the scissile phosphate by a penta-coordinated vanadium transition state mimic (Davies and Hol, 2004) was crucial to forming well-diffracting crystals. The structure provides a number of insights into the viral topoisomerase reaction, including the nature and extent of downstream contacts that are unique to the poxvirus enzymes, a network of water molecules in the active site that links the catalytic tyrosine to the O5′ leaving group, an unusual sugar geometry that requires a conformational change coupled to DNA cleavage, and a novel aspartic acid-minor groove interaction that is unique to the type IB topoisomerases and plays a role in positioning the cleaved nucleoside for ligation.
Results
Structure of a vTopIB-DNA transition state mimic
The DNA duplex used for crystallization was originally designed as a 15-mer with 5′-overhanging dC and dG residues on the top and bottom strands, respectively (Fig. 1) in order to promote formation of an extended DNA duplex in the crystal lattice. Although crystals of vTopIB-DNA complexes utilizing inactive protein mutants (e.g., Y274F) were readily obtained with this DNA, diffraction was limited to ∼3 Å and the regions of interest in the C-terminal subdomain of the protein were poorly ordered. To form a vanadate transition state mimic where the active site tyrosine is covalently tethered to the DNA, a similar duplex was assembled from three oligonucleotides where a nick is located at the scissile phosphate position. After complex formation with variola virus TopIB and addition of activated potassium metavanadate, VO2+ was incorporated into the site of the nick and well-diffracting crystals could be grown with properties shown in Table I.
Table I. Crystallographic data.
| Diffraction Data | |
|---|---|
| Resolution (Å) | 2.1 Å |
| Space Group | P42212 |
| Unit Cell | a=b= 104.0 Å, c = 93.0 Å |
| Mosaicity (°) | 0.4 |
| Completeness (%) | 96.3 (98.5) |
| Rmerge | 0.072 (0.523) |
| Total Reflections | 258965 |
| Unique Reflections | 27692 |
| I/σ | 21.5 (3.3) |
| Redundancy | 9.4 (8.8) |
| Refinement | |
| Rfree | 0.244 (0.28) |
| Rwork | 0.177(0.20) |
| Number of atoms | |
| Protein | 2587 |
| DNA | 656 |
| Water | 426 |
| A verage B factors (Å2) | |
| Protein | 47 |
| DNA | 46 |
| Water | 27 |
| Rmsd | |
| Bond lengths (Å) | 0.02 |
| Bond angles (°) | 1.74 |
The structure was determined by molecular replacement using a truncated vTopIB-DNA model (Perry et al., 2006) lacking active site residues, residues 250-280, and all downstream DNA, and refined to 2.1 Å resolution. A view of the overall structure is shown in Fig. 1a and electron density within the enzyme active site following refinement is shown in Fig. 1b. All protein and DNA residues are well defined in the structure. In the region upstream of the cleavage site (base pairs +1 through +10 in Fig. 1c), the structure is nearly identical to the previously reported non-covalent vTopIB-DNA complex, with an r.m.s. deviation of 1.0 Å for protein and DNA atoms. The differences present in the current structure are located in the region immediately surrounding the active site of the enzyme and in the region downstream of the cleavage site.
Downstream contacts in the vTopIB-DNA complex
The poxvirus topoisomerase contacts DNA downstream of the cleavage site via four different structural motifs, shown in an overall view in Fig. 2a and schematically in Fig. 2b. The N-terminal domain (NTD) interacts with the backbone of the uncleaved strand through hydrogen bond and van der Waals contacts using Lys35 and His39 side chains. These interactions were also observed in the covalent and non-covalent vTopIB-DNA complexes (Perry et al., 2006). A second source of contacts involves Asp-168, located in the conserved β-turn that also delivers Lys167 to the active site. This interaction will be discussed in detail in a later section. The most extensive downstream contacts are provided by the α9 and α10a helices.
Figure 2.
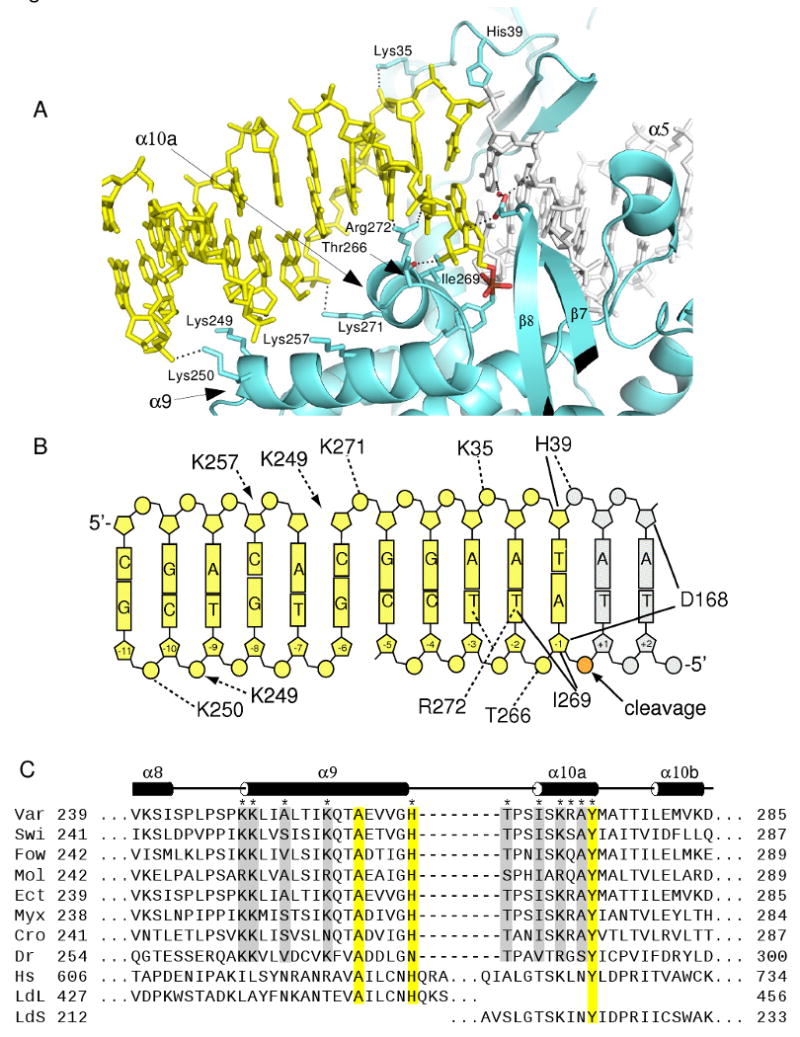
Downstream DNA contacts. (A) View of the DNA downstream of the cleavage site with interactions illustrated for α10a, α9, the amino-terminal domain, and the β7- β8 loop. The α5 and α10a helices are bound in adjacent major grooves flanking the cleavage site. (B) Schematic of vTopIB-DNA contacts downstream of the cleavage site. Dashed lines indicate hydrogen bonds and solid lines indicate van der Waals contacts. The downstream DNA in panels A and B is colored yellow and includes part of an adjacent duplex related by crystallographic symmetry (see Figure S1). (C) Sequence alignment of seven poxvirus TopIBs, one bacterial TopIB, human TopIB, and the two subunits of trypanosomal TopIB in the α8-α10 region. Secondary structure assignments are for variola virus TopIB. Protein sequence accession numbers are Var: variola (NP_042133), Swi: swinepox (NP_570234), Fow: fowlpox (NP_039106), Mol: mollescum contagiosum(NP_044038), Ect: Ectromelia (NP_671606), Myx: Myxoma (NP_051788), Cro: crocodilepox (YP_784296), Dr: Deinococcus radiodurans (NP_294413), Hs: human (NP_003277), LdL (AAF73185)/LdS (ABW86320): large and small subunits of Leishmania donovani, respectively. Conserved TopIB residues are shaded yellow and residues likely to be involved in downstream contacts in the small TopIB enzymes are shaded gray.
The α10a helix is docked in the major groove, contacting nucleotides -3 through -1 on the cleaved strand. Thr266 hydrogen bonds to the -1/-2 phosphate using its amide nitrogen and side chain hydroxyl groups, Ile269 makes van der Waals contact with the -2T base and the -1A sugar, and Arg272 makes a pair of hydrogen bonds, bridging between the O4 groups of -2T and -3T bases. In addition, Lys271 reaches across the major groove to interact with the -5/-6 phosphate on the non-cleaved strand. Most of these interactions are likely to be present for all poxvirus TopIB enzymes, based on strong conservation of the residues involved (Fig. 2c). The exception is Arg272, where amino acids other than arginine are also found in the viral topoisomerases. Interestingly, human TopIB becomes resistant to camptothecin when the corresponding residue Leu721 is substituted by arginine (Knab et al., 1993).
The sequence of the -3 to -1 region of the vTopIB target site has been shown to have minimal effects on non-covalent binding of the enzyme, but strongly influences the rate of DNA cleavage (Hwang et al., 1999; Hwang et al., 1998; Minkah et al., 2007; Shuman and Prescott, 1990). These sequence preferences can be readily explained by the observed α10a contacts. In the cleaved strand -1 position, for example, T is strongly disfavored, but A, C and G are tolerated (Minkah et al., 2007). The 5-methyl group of a -1T substitution would produce a severe steric clash with the side chain of Ile269, whereas -1G would be expected to be similar to -1A as observed in the current structure and steric affects introduced by -1C would be expected to be minimal. In the -2 position, G is favored, A and T are tolerated, and C is somewhat disfavored. Arg272 could interact strongly with -2G (via hydrogen-bonding to N7 and/or O6), productively with -2A (via N7) and as observed in the current structure for -2T. A C in the -2 position would not be expected to interact directly with Arg272. Only small sequence preferences have been observed in the -3 position, where a number of possibilities may exist for bridging interactions with the -2 base.
Unfavorable major groove contacts in the -3 to -1 region would most likely result in a shift in the position of the α10a helix. Since Tyr274 is located at the C-terminus of α10a, proper positioning of this essential catalytic residue is clearly dependent on proper docking of the helix in the adjacent major groove. Thus, the sequence flanking the cleavage site could modulate the efficiency of cleavage by altering the mode of interaction with α10a. This sequence-dependent positioning is similar to that observed for the α5 helix, whose docking in the upstream major groove is essential for proper delivery of catalytic Arg130 to the active site (Perry et al., 2006). An additional role for α10a may involve positioning of the downstream DNA helix for ligation in the final step of the TopIB reaction cycle (discussed later).
As shown in Fig. 2, the α9 helix contacts the DNA substrate ∼10 bp downstream of the cleavage site. These contacts could not be reliably inferred from previous structural models of vTopIB-DNA complexes, where there was no downstream DNA duplex present. In order to visualize the α9-DNA contacts shown in Fig. 2, we examined the continuous DNA duplex in the crystal lattice that results from Watson-Crick base pairing of the complementary 5′-overhangs on the ends of the 15-mer duplex shown in Fig. 1c. The duplex in Fig. 1c includes all of the vTopIB-DNA contact sites upstream of the cleavage site (bp +1 to +9), but contains only five base pairs (-1 to -5) on the dowstream side. However, the downstream flanking DNA forms a continuous helix with a neighboring DNA molecule in the crystal, effectively extending the amount of downstream DNA that is available for interaction with the TopIB protein (see Fig. S1).
The closest α9-DNA contact is made by Lys250, which hydrogen bonds to the -10/-11 phosphate. Lys249 is ideally positioned to interact with the -6/-7 phosphate on the non-cleaved strand, which is missing in our structural model. This residue instead is directed towards the -9/-10 phosphate as shown in Figs. 2a-b. Lys257 is located in a position where it could make close contact with the -7/-8 phosphate on the non-cleaved strand, but in the current structure this side chain adopts a position midway between the -7/-8 and the -8/-9 phosphates. Inspection of sequence alignments in this region reveals that Lys249 and Lys250 are strongly conserved in the poxvirus TopIB family and Lys257 is most often lysine or arginine. Since Lys249 and Lys250 play no obvious role in stabilizing the protein structure, we suggest that the observed and inferred α9-DNA interactions shown in Fig. 2 are conserved among the poxvirus TopIBs and that the interactions are likely to be functionally important. Although Lys257 could interact with downstream DNA, this residue may also have a role in stabilizing helix α9 via a salt bridge, since a conserved Asp/Glu is located one helical turn away in the small TopIB proteins (Fig. 2c).
The structure and sequence alignment further suggest that Ala253 may lie in an important position in the α9 helix in variola virus TopIB. The DNA backbone closely approaches this residue, with a 4.3 Å distance to the -7 sugar on the uncleaved strand. Residues in this position for the poxvirus TopIBs are strongly biased for short chained amino acids (Ala, Ser, Thr, Val) that are well suited to make hydrogen bonding and/or van der Waals contact with the sugar-phosphate backbone. The lack of long-chain residues in this position (which would most likely prevent a close DNA contact) further supports the idea that α9-DNA interactions are functionally relevant. The full contact surface of the vTopIB-DNA interface therefore extends from 10 bp upstream of the cleavage site (α7-major groove interaction) to 11 base-pairs downstream of the cleavage site (α9-minor groove interaction), in good agreement with early DNaseI footprinting studies (Shuman, 1991).
A comparison of the downstream contacts shown in Fig. 2 with those observed in the TopIB-DNA complexes that have been reported for human (hTopIB) and Leishmania donovani (ldTopIB) topoisomerases (Davies et al., 2006; Redinbo et al., 1998; Stewart et al., 1998) reveals some major differences in the way that the enzymes interact with DNA downstream of the cleavage site. The vTopIB and ldTopIB complexes with DNA are compared in similar orientations in Fig. 3. One difference between the two complexes is that the α10a helix (viral TopIB labeling scheme) does not contact bases in the major groove in the larger topoisomerases. This helix is instead more loosely associated with the backbone of the cleaved strand.
Figure 3.
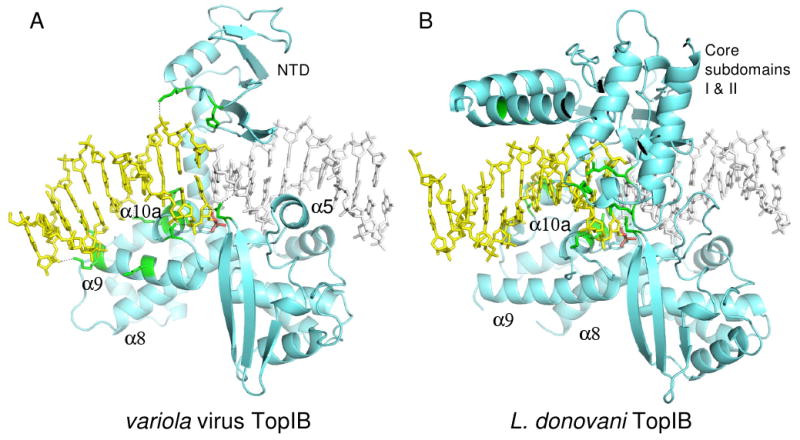
Comparison of (A) variola virus TopIB and (B) L. donovani TopIB (PDB code 2B9S) transition state mimic complexes. The view orientations of the two complexes are identical with respect to the conserved active site residues. Protein regions that contact DNA downstream of the cleavage site are colored green. In (A), the DNA is bent by 35-40°, resulting in a trajectory that facilitates interaction with the N-terminus of α9. Note the different orientations of the α9 helix in the two complexes. The downstream DNA in panels A and B is colored yellow and in panel A the downstream DNA includes part of an adjacent duplex related by crystallographic symmetry (see Figure S1).
The second major difference involves the relationship between α9 and the downstream DNA duplex. In the ldTopIB (and hTopIB) complexes, there is a considerable distance between α9 and the downstream DNA, whereas the two interact in the vTopIB complex. One reason for this difference is that the α9 helices have slightly different orientations in the two complexes. The second reason is evident from a comparison of the panels in Fig. 3. The DNA is slightly bent in the vTopIB complex, but not in the ldTopIB complex. Normal vector analysis in FREEHELIX (Dickerson, 1998) indicates a 15-20° bend between the starting and ending segments of the duplex shown in Fig. 1c and a 35-40° bend between the start and end of the extended duplex shown in Fig. 3a. Together, DNA-bending and an alternative α9 orientation lead to a scenario where the downstream duplex is able to dock via the minor groove onto the N-terminus of α9 in the vTopIB system.
The primary sources of DNA-bending in the vTopIB-DNA complex are an opening of the major groove within the core recognition sequence and compression of the major groove at the junction between symmetry-related duplexes in the downstream segment. This results in a negative roll angle of 8-10° for the +8/+9 and +9/+10 base pair steps within the core sequence, and a downward trajectory for the helix in the view shown in Fig. 3a. A number of other small distortions in base-pair parameters within the core sequence may also contribute to the observed bend.
Sequence alignments indicate that the conserved basic residues at the N-terminus of α9 in the viral TopIB enzymes are not present in the human or Leishmania enzymes (Fig. 2c). Indeed, the residue corresponding to Ala253 in vTopIB (which is a short-chain amino acid for the small TopIBs) is asparagine in both of the larger TopIB sequences. Interestingly, the Deinococcus radiodurans topoisomerase (drTopIB) shares the conserved sequences discussed above for the viral enzymes in α9 and in α10a. Like the viral enzymes, the bacterial TopIBs are minimal topoisomerases in terms of sequence length and therefore lack the additional structural motifs found in the large eukaryotic enzymes (Krogh and Shuman, 2002a). The structure of 70 kDa hTopIB bound non-covalently to DNA reveals that the helical “nose-cone” and “linker” elements are responsible for most of the downstream contacts in that system (Stewart et al., 1998). Neither of these elements is present in the poxvirus TopIB enzymes.
Poxvirus topoisomerase active site
Our original motivation for constructing a vanadate transition state mimic was to obtain a vTopIB-DNA complex structure containing a downstream DNA sequence with intact strands and with well-ordered C-terminal domain structural elements. However, the vTopIB-DNA complex described here is one of the few structural examples of a penta-coordinate vanadium linkage in the context of an intact DNA strand. The first reported was the ldTopIB-DNA complex (Davies et al., 2006), which, as described above, differs in some important ways with viral TopIB in the region downstream of the cleavage site. We therefore compared the two structures in the active site of the enzyme to determine if there are any significant differences.
Overall, the vTopIB and ldTopIB-vanadate active sites are quite similar. Both contain a trigonal bipyramidal vanadium complex that mimics the expected transition state for a concerted phosphoryl transfer mechanism. As shown in Fig. 4, Tyr274-OH and O5′ of the -1A residue occupy axial coordination positions around vanadium, with O3′ of +1T and the two non-bridging oxygens in equatorial positions. Arg130 and Arg223 are symmetrically arranged around the vanadium complex, with each side chain forming hydrogen bonds via their Nε and Nη2 hydrogens to coordinated oxygen atoms. Arg130 interacts with O5′ and O2 (corresponding to OP2, the pro-Rp non-bridging oxygen), whereas Arg223 interacts with the Tyr274 oxygen and O1. Lys167 hydrogen-bonds to both O5′ and to O2 of the +1T residue. Each of these active site residues is further anchored by a series of additional hydrogen bonds involving solvent, protein, and DNA atoms, shown schematically in Fig. 4b.
Figure 4.
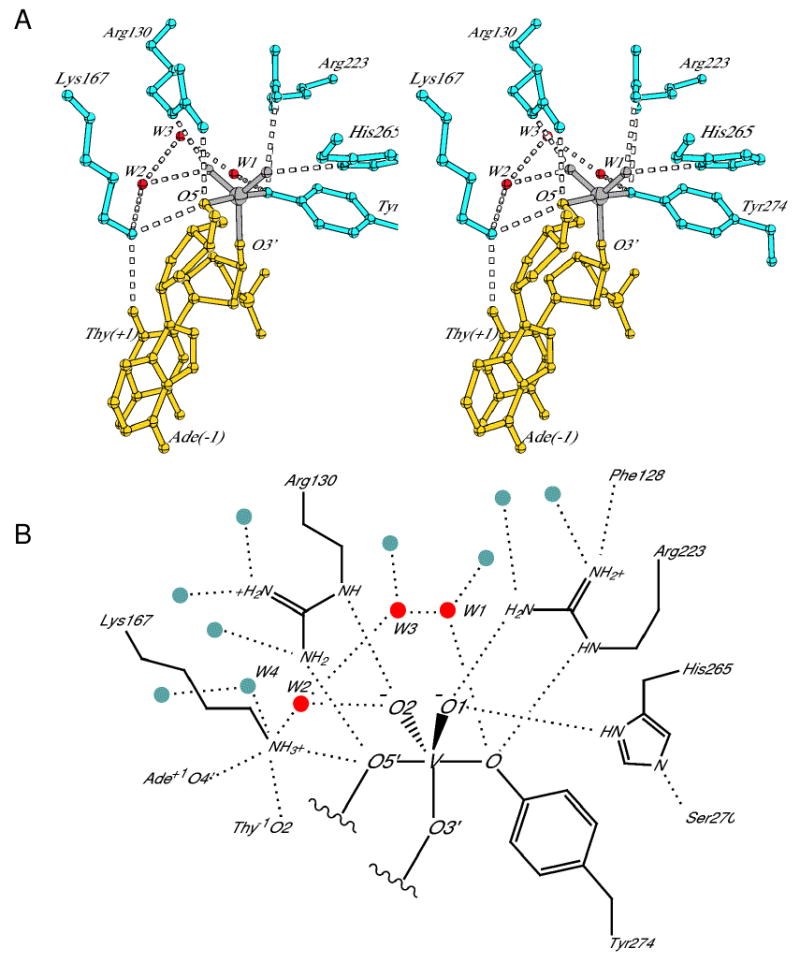
Active site of the vTopIB-DNA transition state mimic. (A) Stereo diagram showing the five conserved active site residues and three water molecules most closely associated with the reaction center. The three water molecules and Lys167 form a chain linking Tyr274 and O5′ of the -1Ade residue. (B) Schematic of the active site showing all of the hydrogen-bonding interactions made by the residues in (A). Solvent molecules are indicated by red and gray circles. See Fig. S2 for a comparison of active site hydrogen-bonding distances for the non-covalent, covalent, and transition state mimic structures.
The striking correspondence between the positioning of active site residues in the LdTopIB-DNA transition state mimic and the interactions inferred from extensive biochemical studies of vaccinia virus TopIB has been recently summarized by Yakovleva et al. (2008). For example, the contacts to O1 and O2 observed in the structure agree nicely with the results from biochemical experiments involving non-bridging phosphorothiolate and methylphosphonate substitutions (Stivers et al., 2000; Tian et al., 2005). Most of the active site residues form identical hydrogen-bonding interactions in the vTopIB-DNA complex described here, further supporting the earlier biochemical studies. Most of the hydrogen-bonding interactions to oxygen atoms in coordination around vanadium have nearly ideal hydrogen-bonding distances, as sumarized in Fig. S2. The exception is the Arg223 to Tyr274 hydrogen bond, where the Nη2-O distance is slightly longer, at 3.3 Å.
One important difference between the vTopIB and ldTopIB transition state active sites is the conformation of His265. In the vTopIB-DNA complex, the δ-nitrogen of His265 hydrogen bonds to O1 and the ε-nitrogen accepts a hydrogen bond from Ser270. Thus, the α10a helix (which contains Ser270) plays a role in positioning two catalytic residues in the active site: His265 and Tyr274. In the ldTopIB-DNA active site, the corresponding active site histidine adopts an alternative conformation where Nε hydrogen bonds to O1 and Nδ forms a water-mediated hydrogen bond to the conserved serine (not shown). This configuration involves more than simply a rotation of the imidazole ring; the entire side chain adopts a distinct conformation in order to make the appropriate interactions.
We were particularly interested in examining the solvent structure in the vTopIB active site. An active site water has been implicated as the functional group responsible for accepting a proton from the catalytic tyrosine during cleavage in hTopIB (Redinbo et al., 1998) and various networks of solvent molecules have been observed in the active sites of other TopIB-DNA complexes (Davies et al., 2006; Perry et al., 2006). As expected, we observe a water molecule (W1) hydrogen-bonded to Tyr274-OH whose position corresponds to the water molecule previously reported in hTopIB-DNA complexes (Fig. 4). We also observe two well-ordered water molecules (W2 and W4) within somewhat longer hydrogen bonding distance of Lys167, where W2 also hydrogen bonds to non-bridging oxygen O2 and W4 hydrogen bonds to O2 of +2T.
An additional molecule (W3) bridges between W1 and W2, completing a chain of water molecules that connects Tyr274-OH and -1A-O5′ through Lys167. This water network provides a plausible pathway for movement of protons between the nucleophile and leaving group during catalysis of cleavage and ligation. A water in a similar position as W2 is present in the deposited ldTopIB-DNA complex, but there is no water corresponding to W3. The B-factors of all three waters in the vTopIB-DNA complex (W1, W2, and W3) are less than 25 A2, and omit maps following random parameter shifts and refinement show clear density for all three atoms in weighted Fo-Fc and 2Fo-Fc electron density maps. These water molecules are therefore likely to be integral to the active site and may play a role in catalysis. Each water is further contacted by solvent molecules in a second layer, which in turn interact with DNA, protein, and/or additional solvent molecules (Fig. 4b).
A final observation in the vTopIB-DNA active site concerns the unusual coordination of the Lys167 Nζ amino group. In addition to the three hydrogen-bonded groups shown in Fig. 4a, there is an additional water molecule (W4) and a 3 Å contact with O4′ of the -1A sugar (Fig. 4b). Of the five hydrogen-bonded groups, two are strict hydrogen bond acceptors (Ade-O4′ and Thy-O2), two are donors or acceptors (waters), and the fifth is O5′. The hydrogen bonding geometries are not ideal, with only the O2-Nζ-O5′ angle close to tetrahedral and the two Nζ-water distances ∼ 3.4 Å. This arrangement presumably plays a role in tuning the pKa of Lys167 for efficient acid and base catalysis. A summary of hydrogen-bonding distances observed in the vTopIB-DNA transition state mimic is compared to those observed in the structures of the pre-cleavage complex and the covalent intermediate structures in Figure S2.
Conformation of the -1 sugar
The -1A sugar is the leaving group during cleavage of the DNA and is the attacking group during ligation of the DNA following relaxation of supercoils. The conformation of this sugar is unusual in the vTopIB and ldTopIB vanadate transition state model structures. In DNA duplexes, the backbone torsion angles tend to cluster around a set of standard values (Schneider et al., 1997). For the α (P-O5′), β (O5′-C5′), and γ (C5′-C4′) angles, these values are (-60, 180, and +60°), respectively, or (g-, t, g+). As shown in Fig. 5a, the -1A sugar in the vTopIB-DNA complex has an atypical γ angle of 180°, resulting in both O5′ and O3′ being oriented towards the minor groove. This conformation is compared to the corresponding phosphodiester linkage in a hTopIB-DNA pre-cleavage complex in Fig 5b, where γ ∼ 60° and O5′ points towards the major groove.
Figure 5.
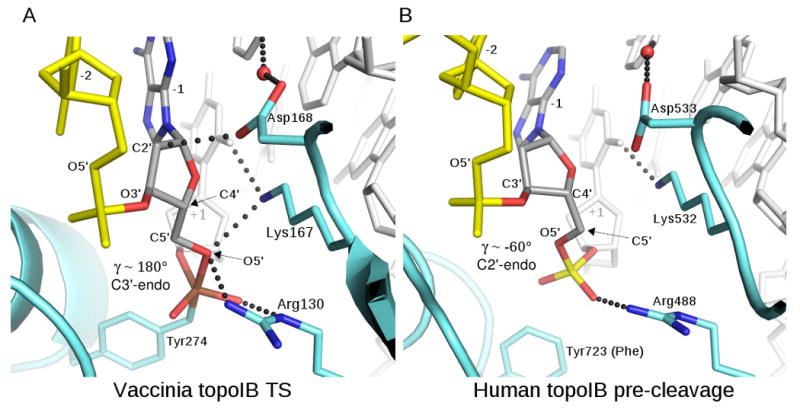
Conformation of the -1 sugar during TopIB catalysis. (A) The -1Ade sugar in the vTopIB-DNA-vanadate complex. The backbone torsion angle γ (the C3′-C4′-C5′-O5′ dihedral angle) adopts a trans configuration, placing O5′ in position for axial coordination of vanadium and in position for hydrogen-bonding by Lys167 and Arg130. The sugar also adopts an RNA-like C3′-endo pucker. (B) The corresponding sugar in a pre-cleavage complex of human TopIB (Y723F mutant) bound to DNA (PDB code 1A35). Here, the γ angle is close to the standard value of -60°, and the sugar is C2′-endo. The phosphate has not yet rotated and O5′ is not in position for interaction with Lys532 and Arg488.
In addition to the unusual backbone torsion angle, the -1A ribose ring adopts a C3′-endo sugar pucker in the vTopIB-DNA transition state model, rather than the more commonly observed C2′-endo conformation. Thus, a conformational change involving the scissile phosphate and the -1 sugar accompanies attack of the phosphate by tyrosine during the cleavage reaction. It is important to note that this conformational change is not only a geometric requirement for formation of a penta-coordinate transition state structure. The flipping of O5′ to the minor groove side of the DNA backbone is required in order to form hydrogen-bonding interactions with Lys167 and Arg130, the two residues responsible for leaving group activation during cleavage and for nucleophile activation during ligation (Krogh and Shuman, 2000; Krogh and Shuman, 2002b).
The conformational changes described above could occur in a concerted fashion upon attack by the tyrosine nucleophile. Alternatively, the DNA substrate could be reconfigured prior to cleavage, either as a consequence of enzyme binding or as an activation step that occurs subsequent to binding. For the TopIB enzymes, the only structures of DNA complexes containing an intact phosphodiester at the cleavage site are pre-cleavage complexes of hTopIB where a Y723F mutant was used to prevent cleavage of the substrate (Redinbo et al., 2000; Redinbo et al., 1998). In these structures, the sugar geometry is close to that found in standard B-DNA, with standard (α,β,γ) angles of (g-,t,g+) and C2′-endo pucker for the -1 sugar (e.g., as shown in Fig. 5b). These structures imply that the ground state of the enzyme-substrate complex is not pre-configured for cleavage, assuming that the absence of Tyr723 does not influence the conformation of this region in the structure. Interestingly, pre-cleavage complexes of a Cre recombinase K201A mutant (Lys201 in Cre corresponds to vTopIB Lys167) bound to loxP site DNA do appear to be pre-configured for cleavage, with similar geometry for the -1 sugar as that shown in Fig. 5a for the vTopIB-DNA transition state mimic (Ghosh et al., 2007).
The stereochemistry of the -1 sugar shown in Fig. 5A helps to explain why DNA-RNA hybrid substrates where the downstream strand is composed of ribonucleotides is efficiently cleaved and ligated by vaccinia virus TopIB (Sekiguchi and Shuman, 1997). The -1 sugar adopts an RNA-like pucker during catalysis and there appears to be room for O2′ if Asp168 is rotated to an alternative configuration.
A role for Asp168
Asp168 is conserved in the topoisomerase IB family but its role in the topoisomerase reaction has previously been uncertain. In all structures of TopIB-DNA complexes that have been determined, this residue is inserted into the minor groove adjacent to the cleavage site, where it contacts the -1 sugar (Fig. 6). The variola virus TopIB D168A mutant has an unusual phenotype (Hwang et al., 2006; Perry et al., 2006). The enzyme is defective in relaxation of supercoiled plasmid substrates under conditions where substrate turnover is required, but is ∼13-fold faster in cleavage of suicide substrates and ∼5-fold faster in ligation compared to the unsubstituted enzyme. The loss of Asp168 results in an increase in DNA-binding affinity, which inhibits product release and is responsible for the low activity observed in plasmid relaxation assays. The vTopIB D168A mutant also generates a threefold greater fraction of covalent intermediate in equilibrium assays than does the wild-type enzyme, which may explain why the mutant is also toxic to E. coli (Hwang et al., 2006). Asp168 therefore has a negative effect on DNA-binding affinity and a negative effect on the absolute rates of cleavage and ligation in the TopIB reaction, but shifts the balance between cleavage and ligation so as to favor the ligated state.
Figure 6.
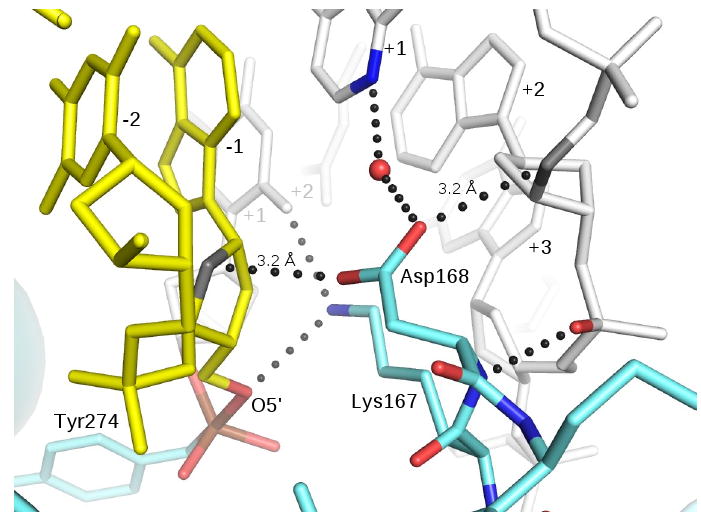
A role for Asp168 in strand ligation. Asp168 makes short van der Waals or CH-O hydrogen bonding contacts to the −1 and +2 sugars located on opposite faces of the minor groove. The β7-β8 loop is anchored to the upstream DNA via four distinct interactions involving the backbone and side chain of Asp168 and the side chain of Lys167. Lys167 and Asp168 in turn provide a docking surface for the -1 sugar, where interactions with the ribose ring and O5′ may promote a favorable conformation for efficient ligation. Asp168 contacts several ribose ring atoms with distances < 3.5 Å; the closest contacts are shown.
Asp168 occupies a nearly identical position in most TopIB-DNA complexes, with only small changes in the orientation of the carboxylate group. The ionization state of this side chain in the TopIB-DNA structures is not known. We refer to it here as a carboxylate, although it is possible that its local environment stabilizes the carboxyl form. In each structure, the Asp side chain makes a water-mediated hydrogen bond with N3 of +1A on the uncleaved strand. In the vTopIB and ldTopIB transition state structures, however, Asp168 also forms bridging interactions between sugars on opposite faces of the minor groove. On one face, there are close contacts between a carboxylate oxygen and carbon atoms in the -1A sugar. On the other face of the minor groove, the second carboxylate oxygen contacts carbon atoms in the +1 sugar of the uncleaved strand. These interactions are shown for the vTopIB-DNA transition state mimic in Fig. 6.
The oxygen-carbon distances involved in the Asp168 bridging interaction are closer than one might expect for van der Waal's contacts. For example, the distances to C1′ and C2′ of the -1 sugar shown in Fig. 6 are 3.25 and 3.15 Å, respectively. Similarly, the distances to C4′ and C5′ of the +1 sugar are 3.4 and 3.2 Å. A similar set of contacts exists in the deposited ldTopIB-DNA transition state complex (Davies et al., 2006), although the C4′ and C5′ distances are somewhat longer (3.4-3.5 Å). We assume that these close oxygen-carbon contacts are weak CH-O hydrogen bonds, which are thought to make small, but important energetic contributions to a variety of molecular interfaces (Mandel-Gutfreund et al., 1998; Wahl and Sundaralingam, 1997).
To determine if this type of Asp-DNA interaction has been previously observed, we searched the Protein Data Bank for protein-dsDNA complexes that contain Asp carboxylate-DNA contacts closer than 3.5 Å. We found that Asp contacts to the sugar-phosphate backbone are relatively common, although most complexes have at most 1-2 such interactions. Of the 1176 complexes examined, 243 entries (20%) contain Asp carboxylate-sugar contacts. Of these, 83 make contacts using both carboxylate oxygen atoms, but most of these are made to the same or adjacent sugars. Only three entries contain a bridging interaction of the type shown in Fig. 6; two of these are hTopIB/DNA complexes (PDB codes 1EJ9 and 1K4S) and the third is the ldTopIB/DNA transition state mimic (PDB code 2B9S). In each case, conserved Asp533 (human) or Asp168 (poxvirus) is responsible. Thus, the bridging Asp minor groove interaction shown in Fig. 6 appears to be unique to the type IB topoisomerases.
The precise function of Asp168 in the topoisomerase reaction is not yet understood in detail, but the properties of the vTopIB D168A mutant strongly imply a role in promoting ligation. The views presented in Figs. 5 & 6 suggest a model for how Asp168 might assist in ligation of the broken DNA strand. A crucial step in this process is the proper positioning of the -1 residue, which requires docking the downstream duplex into the correct position and orientation to restore base-stacking and helical phase with respect to the upstream duplex. In addition, however, the -1 sugar must adopt the correct geometry for nucleophilic attack on the 3′-phosphotyrosine linkage. In the absence of Asp168 and Lys167, it is likely that a larger number of alternative, unproductive conformations of this sugar would be readily accessible. As shown in Fig. 6, the close CH-O contacts made by Asp168 strongly influence the sugar ring position and Lys167 influences the torsion angle about the C4′-C5′ bond. Together, these residues are likely to play important structural roles in positioning O5′ for nucleophilic activation and attack.
It is also interesting to note that the β-turn that provides Lys167 and Asp168 is anchored to the upstream DNA duplex (base-pairs +1 and up) via a backbone amide-phosphate interaction, hydrogen-bonding interactions to the +1 base (via Lys167), and both water-mediated hydrogen bonds to the +1 base and van der Waals interactions with the +1 sugar (via Asp168). All of these interactions are shown in Fig. 6. We suggest that together, the upstream duplex and this conserved β-turn form a docking surface that promotes the precise re-engagement of the downstream duplex that is required to efficiently seal the broken strand during topoisomerase activity.
Discussion
The mechanism of supercoil relaxation used by the type IB topoisomerases differs in an important way from that used by the type IA and type II enzymes (Champoux, 2001; Wang, 1996). For the latter, the cleavage and ligation steps of the reaction are tightly coupled to strand passage so that a fixed change in topology occurs for each catalytic event. In contrast, the type IB enzymes have a finite probability of ligation occurring following each supercoil release as the upstream and downstream duplexes undergo rotation relative to one another about the site of the nick. Single molecule experiments indicate that vaccinia virus TopIB ligates highly supercoiled DNA after ∼20 rotations on average (Koster et al., 2005), whereas the results from ensemble measurements with less supercoiled substrates in solution suggest a value of ∼5 supercoils released per cycle (Stivers et al., 1997). Single molecule experiments also indicate that the rate of rotation is slower for human TopIB than for viral TopIB (Koster et al., 2005). This result can be rationalized by simply comparing the structures; hTopIB has a far more extensive protein framework that partially encloses both the upstream and downstream DNA, providing a larger impediment to free rotation. Whereas the notion of DNA rotation within an enzyme cavity captures the essence of structural and biochemical data for the human TopIB reaction, viral TopIB makes do with a far less elaborate framework.
Based on initial structural models of the vTopIB-DNA complex, it was difficult to explain how the viral TopIB enzymes inhibit free rotation of the DNA and are able to reposition the rotating duplex for efficient ligation. The vTopIB-DNA transition state mimic structure described in this report contains intact dsDNA downstream of the cleavage site and provides a framework for considering how controlled rotation of the DNA is likely to occur in the viral TopIB systems. A series of docking points composed of α9, α10a, the NTD, and the β-turn docking surface comprised of Lys167 and Asp168 appear to be responsible for capturing the rotating downstream duplex and positioning it for ligation. Many of the downstream DNA contacts observed for viral TopIB are distinct from what has been observed for the human and Leishmania TopIB enzymes. Thus, the small TopIBs have unique strategies for controlling strand rotation that are not simply a subset of the mechanisms used by the larger and more complex enzymes.
One of these strategies involves a set of distal protein-DNA contacts that requires DNA-bending in order to make optimal interactions. Since the DNA bend observed in the vTopIB-DNA complex described here is partly a result of flexibility in the DNA-DNA junction formed between symmetry-related duplexes (i.e., base-pairs -5 to -7 in Fig. 2b), the detailed interactions we observe are likely to differ somewhat in the context of an extended DNA substrate that lacks a flexible junction. Indeed, an optimal α9-DNA interaction may only form when the DNA substrate has been cleaved and the downstream duplex is less restrained. This would be consistent with the idea that the downstream contacts are more important during strand rotation and ligation rather than in the pre-cleavage state.
In addition to supporting early DNA-binding and DNaseI footprinting results (Shuman, 1991), the distal contacts observed in the vTopIB-DNA structure may provide additional insight into recent biochemical studies where an energetic coupling has been observed between major groove interactions within the core recognition site and interactions involving the downstream DNA duplex (Nagarajan and Stivers, 2007). Single-atom changes in the core sequence (e.g., replacement of N7 by carbon in the +3 or +4 guanine) can cause changes in the rate of ligation without significantly changing the rate of cleavage. These effects have an unexpected dependence on the length of the scissile DNA strand downstream of the cleavage site, suggesting that downstream interactions are somehow coupled to core site interactions in an anticooperative manner.
Although it is difficult to explain individual observations in detail based on a snapshot of a single reaction intermediate, the energetic coupling of upstream and downstream vTopIB-DNA interactions can in general terms be understood based on the work described here since downstream interactions are clearly dependent on DNA-bending that is influenced to some extent by core site binding. In particular, a model in which strain that prevents simultaneous optimal binding to core-site and downstream elements is relieved upon cleavage at the scissile phosphate would be nicely explained by the current structure and the models discussed above. We may have artificially relieved this strain to some extent by use of a flexible DNA-DNA junction that presumably allows closer α9-DNA contacts than would normally be made prior to cleavage. The notion of a competing set of downstream interactions that would be strengthened in the cleaved state also explains how the viral TopIB enzyme could extend the lifetime of the covalent intermediate in order to allow for topoisomerase activity.
The vTopIB-DNA structure described here has also provided an opportunity to reinforce and extend the findings of Davies et al., who recently described the structure of a ldTopIB transition state mimic (Davies et al., 2006). Both structures contain a penta-coordinated vanadium (V) species representing an approximation of the expected transition state structure in the TopIB phosphoryl transfer reaction. Although the actual transition state may differ somewhat from that represented by the vanadate complex, the differences are likely to be subtle and we would argue that the protein-DNA interactions represented by the LdTopIB and vTopIB structures are strongly indicative of the contacts responsible for promoting catalysis. For example, both the LdTopIB and vTopIB-DNA structures strongly support a crucial role for Arg130 in stabilization of negative charge in the transition state. Indeed, this is the only protein residue that contacts the non-bridging OP2 atom during catalysis. In contrast, the non-bridging OP1 oxygen is hydrogen bonded by both Arg223 and His265. The vTopIB structure also confirms hydrogen bonding between Arg223 and the Tyr274 hydroxyl group, an interaction that was also observed in the covalent intermediate structure (Perry et al., 2006).
The nature of the interactions involving O5′ have been of particular interest in the TopIB active site. Both transition state mimic structures support a role for Lys167 as proton donor/acceptor for O5′ and the vTopIB-DNA complex described here further reveals a continuous network of three water molecules that link nucleophile and leaving group via Lys167. This observaton is particularly interesting in the context of the tyrosine recombinase active site, which differs primarily by the presence of a conserved active-site histidine that is not present in the TopIB enzymes. This histidine is located in the same space that is occupied by waters W1 and W3 in the vTopIB structure described here, suggesting that the two alternative active site components may play similar functional roles.
Materials and Methods
Crystallization of vTopIB-DNA-Vanadate
Variola virus topoisomerase IB was expressed in E. coli and purified as described (Perry et al., 2006). Three 2′-deoxyoligonucleotides with sequences 5′-CGGAATAAGGGCGACA, 5′-GTGTCGCCCTT, and 5′-ATTCC were synthesized and the two longer oligos were purified by anion exchange chromatography at pH 12. The 5-mer was used without purification. The three oligonucleotides were annealed to form a vTopIB-binding substrate, where the scissile phosphodiester linkage is missing. Sodium orthovanadate (Sigma) was activated at pH 10 as previously described (Gordon, 1991) and added to a solution containing vTopIB and DNA. A mixture containing 127 μM vTopIB, 200 μM annealed DNA duplex, 20mM sodium HEPES (pH 7.5), 125mM NaCl, and 200 μM activated sodium orthovanadate was incubated at 4 °C for 30 minutes.
Immediately prior to crystallization, spermidine was added to a final concentration of 16.6 mM. vTopIB-DNA-vanadate crystals were formed by hanging drop vapor diffusion by mixing 1:1 with and equilibrating against a well reservoir of 5% polyethylene glycol (PEG) 400, 5 mM MgCl2, 5 mM CaCl2, 200 mM NaCl, 50 mM Tris-HCl (pH 7.4). Crystals were cryoprotected by transfer into a solution containing 5% PEG 400, 5 mM MgCl2, 5 mM CaCl2, 200 mM NaCl, 50 mM Tris-HCl (pH 7.4), 2.5% glucose, 63.5 μM topoisomerase, 100 μM DNA, 100 μM activated sodium orthovanadate and equilibrated against a reservoir containing 5% PEG 400, 5 mM MgCl2, 5 mM CaCl2, 200 mM NaCl, 50 mM Tris-HCl (pH 7.4), 25% glucose. Crystals were flash frozen in liquid nitrogen prior to data collection.
Structure Solution and Refinement
Diffraction data were measured at the Advanced Light Source beamline 8.2.1 and processed using HKL2000 (Otwinowski and Minor, 1997). Initial phases were determined by molecular replacement using a search model composed of the non-covalent vTopIB-DNA complex (PDB code 2H7G), excluding all DNA downstream of the cleavage site and amino acid residues 240-314. Clear electron density for the missing amino acid residues, downstream DNA, and active site VO2 were observed in electron density maps based on initial phases. The structure was refined using REFMAC (Murshudov et al., 1997), model building was performed with O (Jones et al., 1991). We were not able to identify high resolution structures of penta-coordinate vanadium (V) model compounds with similar ligands as observed in the vanadate-DNA linkage. We therefore used the structure of potassium metavanadate mononhydrate (Christ et al., 1954) as the basis for structural restraints of the vanadate linkage during refinement. In that structure, vanadium is coordinated by five oxygen atoms in a distorted trigonal bipyramid. V-O bond lengths range from 1.63 to 1.99 A, with trigonal geometry for the equatorial oxygens. A series of restraints for the vTopIB-DNA complex were examined, where target values for V-O bond lengths were varied from 1.6 A to 2.0 Å, with a large σ value of 0.2 Å. The values of the V-O bond lengths varied only slightly following these trial refinements, indicating that the refined structure is not strongly influenced by the values of the restraints employed in the refinement. The final structure was obtained following refinement with V-O bond distances restrained to 1.8 Å (the average in potassium metavanadate), with σ=0.2 Å. Coordinates for the refined structure have been deposited with the Protein Data Bank with accession code 3IGC.
Database searches
Structure searches of the Protein Data Bank were performed using a local copy of the PDB containing released entries up to May, 2009 and locally written searching software. Only entries containing protein bound to double-stranded DNA were examined.
Supplementary Material
Acknowledgments
We are grateful to Bryan Gibb for stimulating conversations and comments regarding the manuscript. This work was supported by NIH NIAID Middle Atlantic Regional Center of Excellence Grant 2 U54 AI057168 to F.D.B. G.V. is an Investigator of the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- Benarroch D, Claverie J, Raoult D, Shuman S. Characterization of mimivirus DNA topoisomerase IB suggests horizontal gene transfer between eukaryal viruses and bacteria. J Virol. 2006;80:314–321. doi: 10.1128/JVI.80.1.314-321.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champoux J. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem. 2001;70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- Cheng C, Kussie P, Pavletich N, Shuman S. Conservation of structure and mechanism between eukaryotic topoisomerase I and site-specific recombinases. Cell. 1998;92:841–850. doi: 10.1016/s0092-8674(00)81411-7. [DOI] [PubMed] [Google Scholar]
- Christ C, Clark J, Evans H. The crystal structure of potassium metavanadate monohydrate, KVO3. H2O. Acta Cryst. 1954;7:801–807. [Google Scholar]
- Davies D, Hol W. The power of vanadate in crystallographic investigations of phosphoryl transfer enzymes. FEBS Lett. 2004;577:315–321. doi: 10.1016/j.febslet.2004.10.022. [DOI] [PubMed] [Google Scholar]
- Davies DR, Mushtaq A, Interthal H, Champoux JJ, Hol WGJ. The structure of the transition state of the heterodimeric topoisomerase I of Leishmania donovani as a vanadate complex with nicked DNA. J Mol Biol. 2006;357:1202–1210. doi: 10.1016/j.jmb.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Delano W. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos: 2002. [Google Scholar]
- Dickerson RE. DNA bending: the prevalence of kinkiness and the virtues of normality. Nucleic Acids Res. 1998;26:1906–1926. doi: 10.1093/nar/26.8.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh K, Guo F, Van Duyne GD. Synapsis of loxP sites by Cre recombinase. J Biol Chem. 2007;282:24004–24016. doi: 10.1074/jbc.M703283200. [DOI] [PubMed] [Google Scholar]
- Gordon JA. Use of vanadate as protein-phosphotyrosine phosphatase inhibitor. Methods Enzymol. 1991;201:477–482. doi: 10.1016/0076-6879(91)01043-2. [DOI] [PubMed] [Google Scholar]
- Grindley N, Whiteson K, Rice P. Mechanisms of site-specific recombination. Annu Rev Biochem. 2006;75:567–605. doi: 10.1146/annurev.biochem.73.011303.073908. [DOI] [PubMed] [Google Scholar]
- Hwang Y, Burgin A, Bushman F. DNA contacts stimulate catalysis by a poxvirus topoisomerase. J Biol Chem. 1999;274:9160–9168. doi: 10.1074/jbc.274.14.9160. [DOI] [PubMed] [Google Scholar]
- Hwang Y, Minkah N, Perry K, Van Duyne GD, Bushman FD. Regulation of catalysis by the smallpox virus topoisomerase. J Biol Chem. 2006;281:38052–38060. doi: 10.1074/jbc.M608858200. [DOI] [PubMed] [Google Scholar]
- Hwang Y, Wang B, Bushman F. Molluscum contagiosum virus topoisomerase: purification, activities, and response to inhibitors. J Virol. 1998;72:3401–3406. doi: 10.1128/jvi.72.4.3401-3406.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T, Zou J, Cowan S, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Knab A, Fertala J, Bjornsti M. Mechanisms of camptothecin resistance in yeast DNA topoisomerase I mutants. J Biol Chem. 1993;268:22322–22330. [PubMed] [Google Scholar]
- Koster D, Croquette V, Dekker C, Shuman S, Dekker N. Friction and torque govern the relaxation of DNA supercoils by eukaryotic topoisomerase IB. Nature. 2005;434:671–674. doi: 10.1038/nature03395. [DOI] [PubMed] [Google Scholar]
- Krogh B, Shuman S. Catalytic mechanism of DNA topoisomerase IB. Mol Cell. 2000;5:1035–1041. doi: 10.1016/s1097-2765(00)80268-3. [DOI] [PubMed] [Google Scholar]
- Krogh B, Shuman S. A poxvirus-like type IB topoisomerase family in bacteria. Proc Natl Acad Sci U S A. 2002a;99:1853–1858. doi: 10.1073/pnas.032613199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh B, Shuman S. Proton relay mechanism of general acid catalysis by DNA topoisomerase IB. J Biol Chem. 2002b;277:5711–5714. doi: 10.1074/jbc.C100681200. [DOI] [PubMed] [Google Scholar]
- Leppard J, Champoux J. Human DNA topoisomerase I: relaxation, roles, and damage control. Chromosoma. 2005;114:75–85. doi: 10.1007/s00412-005-0345-5. [DOI] [PubMed] [Google Scholar]
- Mandel-Gutfreund Y, Margalit H, Jernigan RL, Zhurkin VB. A role for CH…O interactions in protein-DNA recognition. J Mol Biol. 1998;277:1129–1140. doi: 10.1006/jmbi.1998.1660. [DOI] [PubMed] [Google Scholar]
- Minkah N, Hwang Y, Perry K, Van Duyne G, Hendrickson R, Lefkowitz E, Hannenhalli S, Bushman F. Variola virus topoisomerase: DNA cleavage specificity and distribution of sites in Poxvirus genomes. Virology. 2007;365:60–69. doi: 10.1016/j.virol.2007.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G, Vagin A, Dodson E. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nagarajan R, Stivers J. Unmasking Anticooperative DNA-binding interactions of vaccinia DNA topoisomerase I. Biochemistry. 2007;46:192–199. doi: 10.1021/bi061706u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Perry K, Hwang Y, Bushman F, Van Duyne G. Structural basis for specificity in the poxvirus topoisomerase. Mol Cell. 2006;23:343–354. doi: 10.1016/j.molcel.2006.06.015. [DOI] [PubMed] [Google Scholar]
- Redinbo M, Champoux J, Hol W. Novel insights into catalytic mechanism from a crystal structure of human topoisomerase I in complex with DNA. Biochemistry. 2000;39:6832–6840. doi: 10.1021/bi992690t. [DOI] [PubMed] [Google Scholar]
- Redinbo M, Stewart L, Kuhn P, Champoux J, Hol W. Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science. 1998;279:1504–1513. doi: 10.1126/science.279.5356.1504. [DOI] [PubMed] [Google Scholar]
- Schneider B, Neidle S, Berman HM. Conformations of the sugar-phosphate backbone in helical DNA crystal structures. Biopolymers. 1997;42:113–124. doi: 10.1002/(sici)1097-0282(199707)42:1<113::aid-bip10>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Sekiguchi J, Shuman S. Ligation of RNA-containing duplexes by vaccinia DNA ligase. Biochemistry. 1997;36:9073–9079. doi: 10.1021/bi970705m. [DOI] [PubMed] [Google Scholar]
- Shuman S. Site-specific interaction of vaccinia virus topoisomerase I with duplex DNA. Minimal DNA substrate for strand cleavage in vitro. J Biol Chem. 1991;266:11372–11379. [PubMed] [Google Scholar]
- Shuman S. Vaccinia virus DNA topoisomerase: a model eukaryotic type IB enzyme. Biochim Biophys Acta. 1998;1400:321–337. doi: 10.1016/s0167-4781(98)00144-4. [DOI] [PubMed] [Google Scholar]
- Shuman S, Prescott J. Specific DNA cleavage and binding by vaccinia virus DNA topoisomerase I. J Biol Chem. 1990;265:17826–17836. [PubMed] [Google Scholar]
- Stewart L, Redinbo M, Qiu X, Hol W, Champoux J. A model for the mechanism of human topoisomerase I. Science. 1998;279:1534–1541. doi: 10.1126/science.279.5356.1534. see comments. [DOI] [PubMed] [Google Scholar]
- Stivers J, Jagadeesh G, Nawrot B, Stec W, Shuman S. Stereochemical outcome and kinetic effects of Rp- and Sp-phosphorothioate substitutions at the cleavage site of vaccinia type I DNA topoisomerase. Biochemistry. 2000;39:5561–5572. doi: 10.1021/bi992429c. [DOI] [PubMed] [Google Scholar]
- Stivers JT, Harris TK, Mildvan AS. Vaccinia DNA topoisomerase I: evidence supporting a free rotation mechanism for DNA supercoil relaxation. Biochemistry. 1997;36:5212–5222. doi: 10.1021/bi962880t. [DOI] [PubMed] [Google Scholar]
- Tian L, Claeboe CD, Hecht SM, Shuman S. Mechanistic plasticity of DNA topoisomerase IB: phosphate electrostatics dictate the need for a catalytic arginine. Structure. 2005;13:513–520. doi: 10.1016/j.str.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Wahl MC, Sundaralingam M. C-H…O hydrogen bonding in biology. Trends Biochem Sci. 1997;22:97–102. doi: 10.1016/s0968-0004(97)01004-9. [DOI] [PubMed] [Google Scholar]
- Wang J. DNA topoisomerases. Annu Rev Biochem. 1996;65:635–692. doi: 10.1146/annurev.bi.65.070196.003223. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.