

Abstract
Glycogen is degraded during brain activation but its role and contribution to functional energetics in normal activated brain have not been established. In the present study, glycogen utilization in brain of normal conscious rats during sensory stimulation was assessed by three approaches, change in concentration, release of 14C from pre-labeled glycogen and compensatory increase in utilization of blood glucose (CMRglc) evoked by treatment with a glycogen phosphorylase inhibitor. Glycogen level fell in cortex, 14C release increased in three structures and inhibitor treatment caused regionally selective compensatory increases in CMRglc over and above the activation-induced rise in vehicle-treated rats. The compensatory rise in CMRglc was highest in sensory-parietal cortex where it corresponded to about half of the stimulus-induced rise in CMRglc in vehicle-treated rats; this response did not correlate with metabolic rate, stimulus-induced rise in CMRglc or sequential station in sensory pathway. Thus, glycogen is an active fuel for specific structures in normal activated brain, not simply an emergency fuel depot and flux-generated pyruvate greatly exceeded net accumulation of lactate or net consumption of glycogen during activation. The metabolic fate of glycogen is unknown, but adding glycogen to the fuel consumed during activation would contribute to a fall in CMRO2/CMRglc ratio.
Keywords: brain activation, brain imaging, energetics, glucose utilization, glycogenolysis, sensory stimulation
The importance of quantifying metabolic fluxes induced by brain activation at a cellular level is emphasized by positron emission tomographic, magnetic resonance spectroscopic (MRS) and optical (infrared or fluorescence) studies of brain function in health and disease that rely on measurement of signals generated from endogenous or exogenous tracers metabolized by energy-producing pathways. Astrocytes are increasingly recognized as having essential roles in both signaling and energetics during brain activation, including modulation of neurotransmission via gliotransmitters, synthesis and cycling of amino acid neurotransmitters, regulation of extracellular glutamate and K+ levels and blood flow control (Hertz and Peng 1992; Walz 2000; Newman 2003; Hertz and Zielke 2004; Volterra and Meldolesi 2005; Zhang and Haydon 2005; Takano et al. 2006; Hertz et al. 2007). Linkage of excitatory neurotransmission to energy production has focused attention on astrocytic glycolysis and the possibility of lactate trafficking from activated astrocytes to neurons (Pellerin and Magistretti 2004; Hyder et al. 2006), but quantitative aspects of potential lactate shuttling coupled to lactate oxidation in nearby cells compared with lactate release from activated tissue remains to be experimentally established (Chih and Roberts 2003; Dienel and Cruz 2004, 2006). However, fluxes in pathways preferentially or exclusively localized in astrocytes do increase during activation in vivo, including glycogenolysis (Swanson et al. 1992; Cruz and Dienel 2002), CO2 fixation for de novo amino acid biosynthesis via oxidative pathways (Öz et al. 2004) and acetate oxidation (Cruz et al. 2005). Thus, working astrocytes increase glycolytic and oxidative fluxes, and their contribution to overall energetics is higher than recognized (Hertz et al. 2007). Glycogen is the major glucose store in brain, but its functional role and contribution to brain energetics are not well understood (Swanson 1992; Wiesinger et al. 1997; Gruetter 2003; Brown 2004; Dienel and Cruz 2006).
Brain glycogen (Cataldo and Broadwell 1986a,b) and glycogen phosphorylase (Pfeiffer et al. 1990, 1992; Pfeiffer-Guglielmi et al. 2003) are predominantly localized in astrocytes, and immunoelectron microscopic analysis showed that glycogen phosphorylase is located throughout astrocytes, including their processes near synapses and endfeet surrounding capillaries; ependymal cells were also immunoreactive, but endothelial cells, pericytes and neurons (except in the trigeminal nucleus) were not labeled (Richter et al. 1996). Astrocytes have a complex architecture with a large fraction of their cellular volume and surface area composed of highly specialized thin processes that surround and interact with neuronal elements. These processes are too narrow to accommodate mitochondria and are, therefore, likely to be dependent upon glycolytic metabolism of glucose and glycogen to satisfy energy demands, although diffusion of high-energy metabolites from the mitochondria-containing larger processes and cell soma may supply ATP during sustained activation (Hertz et al. 2007). Many reports of low-brain glycogen levels, slow turnover under resting conditions, and rapid mobilization during an energy crisis have lead to the notion of glycogen as mainly an emergency fuel reserve, but much higher-brain glycogen levels in carefully handled rats, glycogen phosphorylase activation by behavioral activity, glycogenolysis regulation by neuronal signals and glycogen utilization during sensory stimulation contradict this concept (see Discussion). Furthermore, the complexity of local and diffuse regulatory mechanisms that govern glycogen turnover suggests that glycogen has an important role in astrocytic energetics, but functions of glycogen are poorly understood, in part, due to technical difficulties of assay of its level and turnover in vivo and because the mechanisms that integrate energy metabolism with neurotransmitter and intracellular signaling also confer high sensitivity of glycogen to experimental and analytical conditions.
In vivo assays of brain glycogen have employed two general approaches, determination of glycogen concentration or glycogen labeling using biochemical, radiochemical and MRS methods. Glycogen level reflects the net balance between synthesis and degradation and glycogen is generally assayed in dissected tissue samples that may contain activated and unstimulated structures; changes in both synthesis and degradation can stabilize its level and local concentration shifts can be masked by averaging in mixed tissue samples. Labeling studies can target either incorporation of label from glucose into glycogen or release of label from previously labeled glycogen. At steady state, the rate of label accumulation via biosynthesis is inversely related to the half-life of a molecule, and incorporation rates are low for compounds that turn over slowly. Label release is a very sensitive indicator of glycogenolysis because changes during short intervals are readily detected by radiochemical assays; interpretation of release after short-term labeling of outer tiers of glycogen has the limitation that the actual quantity of glycogen degraded is not known. In vivo MRS assays can quantitatively track the time courses of labeling and release in longitudinal assays in the same subject, but they generally have less temporal and spatial resolution and lower sensitivity than the radiochemical and biochemical approaches. The present study employed a new approach to evaluate glycogen and glucose utilization at a local level by testing the hypothesis that inhibition of glycogen phosphorylase causes compensatory increases in utilization of blood-borne glucose in activated brain structures. Firstly, changes in glycogen content and label release from pre-labeled glycogen were assessed to ensure that glycogen utilization was enhanced by the stimulation paradigm, and then local rates of glucose utilization were assayed during rest and stimulation in vehicle and inhibitor-treated rats.
Materials and methods
Chemicals
d-[1-14C]glucose and 2-deoxy-d-[1-14C]glucose (DG) (specific activities 54–55 mCi/mmol) were purchased from Perkin-Elmer Life Sciences (Boston, MA, USA). Amylo-α-1,4-α-1,6, glucosidase (Aspergillus niger) and glycogen were obtained from Roche Molecular Biochemicals (Indianapolis, IN, USA); polyethylene glycol 200 was from Sigma-Aldrich (St Louis, MO, USA). CP-316,819 ([R-R*,S*]-5-chloro-N-[2-hydroxy-3-(methoxymethylamino)-3-oxo-1-(phenylmethyl)propyl]-1H-indole-2-carboxamide) was obtained from Pfizer (Groton, CT, USA).
Experimental procedures
Two types of metabolic studies were carried out, measurement of levels of unlabeled and labeled carbohydrates in extracts of dissected samples from brain frozen in situ and in vivo assays of local rates of glucose utilization (CMRglc) in brain during rest and activation after treatment with vehicle or an inhibitor of glycogen phosphorylase. Male Hanover-Wistar rats (250–400 g, Taconic Farms, Germantown, NY, USA) were housed under a standard 12-h light/dark cycle and given free access to food and water; the rats were not fasted overnight prior to the experiment. On the morning of the experiment, the rats were surgically prepared for metabolic and in situ-freezing assays as described (Cruz and Dienel 2002; Dienel et al. 2002; Cruz et al. 2005), restrained by plaster casts around the hind limbs and sequestered in a shelter-box to minimize sensory stimulation; body temperature was maintained at 37°C. Physiological variables (mean arterial blood pressure, arterial blood PCO2, PO2 and pH, hematocrit and arterial plasma glucose and lactate levels were determined prior to and at intervals during the experiment; all were within the normal range (data not shown). All animal use procedures were in strict accordance with the NIH Guide for Care and Use of Laboratory Animals, and were reviewed and approved by the local animal care and use committee.
Glycogen labeling
After 3 h recovery from surgical procedures, brain glycogen was pre-labeled for 30 min with [1-14C]glucose (100 μCi/kg, intravenously) in 18 rats while they remained in the shelter. Timed samples of arterial blood were drawn throughout the 30-min labeling period interval for determination of plasma glucose and lactate levels and total 14C contents. The integrated glucose specific activity was calculated for each rat and used to normalize the concentration of labeled glycogen to the time-activity integral in that animal.
Sensory stimulation paradigm and metabolite assays
Procedures for sensory stimulation by brushing, in situ freezing, dissection of frozen tissue, powdering and weighing, ethanol extraction of metabolites, analysis of glucose, lactate, and labeled and unlabeled glycogen were previously described in detail (Madsen et al. 1999; Cruz and Dienel 2002; Dienel et al. 2002; Cruz et al. 2005). In brief, at 30 min after the pulse of [1-14C]glucose, the ‘resting’ group of nine rats remained undisturbed in a sound-insulated box to minimize any exposure to external stimuli. The activated group of nine rats was given a 10-min mixed-modal stimulation paradigm consisting of generalized sensory, acoustic and visual components to produce widespread changes in neural activity and metabolism. Somatosensory stimulation consisted of continuous gentle bilateral brushing of the head, whiskers, face, back, paws and tail with soft paintbrushes. The acoustic stimulus was a 40 Hz–8 kHz broadband click-tone (Grass S10CTCM Module; Astro-Med, West Warwick, RI, USA) with an intensity setting of 95 dB, corresponding to a measured level of ~88 dB (Digital Sound Meter; Extech Instruments, Waltham, MA, USA); the ambient (‘resting’) sound level in the box was ~55 dB. The photic stimulus was a 16 Hz on–off flash (Grass PS33 Plus photic stimulator at an intensity setting of one, attenuated by three sheets of white paper). Ambient light in the room was reduced and particular care was taken to minimize all noise and other stimuli throughout the preparative and experimental procedures. At stimulus onset, the rats were highly alert due to the abrupt, novel paradigm; they watched the brushes and moved around, sometimes grasping the brush bristles, but rarely vocalized. After 10–15 min of continuous brushing, rats seemed to adapt and they stopped moving and became much more passive. At the end of the experimental interval, rats were anesthetized with fast-acting thiopental (25 mg/kg, i.v.), the brains quickly frozen in situ and stored at −80°C. Tissue from three brain regions, entire dorsal cerebral cortex, inferior colliculus and superior colliculus was dissected out, extracted and analyzed for labeled and unlabeled metabolites. Any influence of anesthesia on metabolite levels is minimal due to the brief time under thiopental (<30 s) prior to rapid cortical tissue freezing.
Glycogen phosphorylase inhibition
Twelve rats were pre-treated with CP-316,819, a glycogen phosphorylase inhibitor using a protocol provided by Dr. R. Swanson (personal communication) and another 12 were given equivalent volumes of vehicle according to the same schedule. The solution containing CP-316,819 was freshly prepared daily by suspension of CP-316,819 in 10% polyethylene glycol 200 in 0.9% saline, pH 7.4 with heating (40–50°C). The total CP-316,819 dose was 250 mg/kg, given in divided doses via three intraperitoneal injections starting the day before the metabolic assay; the initial dose was 150 mg/kg, followed 6 and 24 h later by 50 mg/kg booster injections. Rats were given the last injection 3 h prior to metabolic assays, and each group was sub-divided for assays in resting and stimulated rats (n = 6/ group).
Glucose utilization assays
Local rates of glucose utilization (CMRglc) were determined with the autoradiographic [14C]deoxyglucose ([14C]DG) method using a 30-min assay interval, which is fully quantitative but shorter than the routine 45 min experimental interval (Sokoloff et al. 1977). The stimulation paradigm was carried out exactly as described above, except that photic stimulation was not included. Acoustic plus sensory stimulation onset coincided with the intravenous pulse of [14C]DG (100 μCi/kg) and was continued throughout the metabolic labeling interval. Timed samples of arterial blood were drawn for determination of plasma glucose and lactate levels and total 14C contents in plasma. At the end of the experimental period the rats were killed and their brains processed for determination of local CMRglc.
Results
Brief sensory stimulation alters metabolite levels and glycogen turnover
Normal conscious rats were given 10 min of acoustic, visual, plus somatosensory (brushing) stimulation that was initiated immediately after abrupt removal of a shelter in which the rats were sequestered from environmental cues; ‘resting’ rats remained in the shelter and were not given a specific stimulus. The concentrations of the major carbohydrate stores in brain and release of 14C from glycogen pre-labeled with [1-14C]glucose were assessed in three dissected brain regions, entire dorsal cerebral cortex, inferior colliculus (a structure in the auditory pathway) and superior colliculus (the dorsal region receives input from the retina, whereas the ventral region integrates input from various pathways). Sudden arousal and physical movement of the rat in response to somatosensory stimulation with the brushes caused arterial plasma glucose levels to rise about 20% (Fig 1a). Glucose levels in all three brain structures increased in proportion to the rise in plasma glucose (Fig 1a), as reflected by brain-to-plasma distribution ratios for glucose; these ratios were equivalent (0.21–0.24) during rest and after 10-min stimulation. Thus, brain glucose level equilibrated with that in plasma and glucose supply to brain from blood matched the stimulus-induced rise in demand. Plasma lactate levels also rose, presumably due to muscular activity and exceeded those in brain during rest and activation (Fig. 1b). As lactate transport across the blood brain barrier is passive and concentration-driven, influx into brain and altered metabolism within brain may have contributed to the small net increases in regional lactate levels during stimulation, ranging from 0.1 to 0.5 μmol/g wet weight. Net glycogen consumption was evident only in cerebral cortex where its concentration fell by 14% (Fig. 1c), but glycogen degradation increased in all three regions, reflected by 11–24% lower retention of 14C in pre-labeled glycogen (Fig. 1d). Thus, glycogenolysis increased in all dissected regions even though blood and brain glucose levels were in the normal range (but slightly increased by the stimulus), with regional differences in the balance between glycogen synthesis and degradation.
Fig. 1.
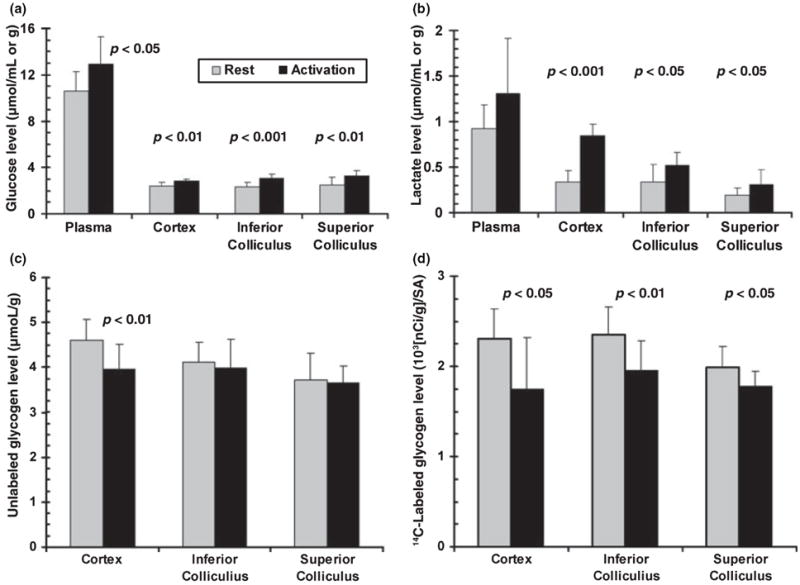
Sensory stimulation activates glycogenolysis and increases regional glucose and lactate levels in brain. Stimulus-induced changes in plasma (μmol/mL) and brain (μmol/g wet wt.) glucose (a) and lactate (b) level, brain glycogen content (c) and 14C-labeled brain glycogen (d) were determined at 10 min after onset of visual (16 Hz on–off flashing light), acoustic (95 dB broadband tone) and generalized sensory stimulation (gentle brushing of the whiskers and body) of conscious rats. Brain metabolite levels were assayed in tissue extracts. Glycogen turnover was determined as 14C retained in glycogen after pre-labeling with [1-14C]glucose for 30 min prior to stimulation; the [14C]glycogen level in each rat was normalized by dividing by the time-activity integral of arterial plasma glucose for that animal (ISA = integrated specific activity); see Materials and methods. Values are means; vertical bars = 1 SD (n = 9/group). Statistically significant differences between activation and rest were identified with the t-test.
Heterogeneous responses of CMRglc to glycogen phosphorylase blockade during stimulation
Rationale for experimental design
Hexokinase is the first irreversible step of glucose utilization and it generates glucose-6-phosphate (glc-6-P) from glucose delivered to brain by blood. Hexokinase is regulated by feedback inhibition by glc-6-P, a ‘branch-point’ metabolite that can also be generated from glycogenolysis and metabolized by the glycolytic, pentose phosphate shunt or glycogen synthesis pathways (Fig. 2). The downstream fate glycogen in brain in vivo is not known, but in cultured astrocytes, glycogen is converted mainly to lactate and released to the medium (Dringen et al. 1993). Functional activation of brain increases metabolic demand, consumes ATP and activates phosphofructokinase, causing glc-6-P levels to fall, thereby dis-inhibiting hexokinase. We hypothesize that glycogen turnover has an important role in functional energetics of normal, activated astrocytes and, if true, inhibition of glycogen phosphorylase to render blood glucose the sole source of glc-6-P would cause a compensatory increase in utilization of blood-borne glucose by hexokinase during brain activation (Fig. 2), but not during rest when glycogen turnover is slow (Watanabe and Passonneau 1973). Experimental design optimized the influence on measured CMRglc of prolonged metabolic responses to glycogenolysis blockade and minimized any potential effects of transient hypoglycemia at stimulus onset that might trigger only brief glucose demand. The hexokinase reaction was directly and simultaneously measured throughout brains of conscious rats with the autoradiographic [14C]DG method, which employs pulse labeling and a routine 30-to 45-min experimental interval; it registers [14C]DG-6-P accumulation throughout the experimental period and is relatively insensitive to brief changes in metabolic rate at onset of the labeling period due to the lag before precursor enters brain and is metabolized (Sokoloff et al. 1977). The glycogen phosphorylase inhibitor Pfizer compound CP-316,819 binds to the inactive form of glycogen phosphorylase and impairs conversion to the active form; this inhibition is enhanced by high-glucose concentration and relieved by low-glucose level (Hoover et al. 1998; Martin et al. 1998; Treadway et al. 2001). Thus, if brief depletion of local glucose level occurs at stimulus onset inhibion by CP-316,819 would be released, attenuating any compensatory rise in CMRglc that may arise from transient hypoglycemia. The CP-316,819 dosing paradigm was generously provided by Dr. R. Swanson (personal communication), who observed an 88% increase in rat brain glycogen content after a 24-h dosing regimen. To identify brain regions with a compensatory response to glycogenolysis inhibition, CMRglc was assayed during rest and sensory stimulation in groups of rats pre-treated for 24 h with CP-316,819 or vehicle.
Fig. 2.
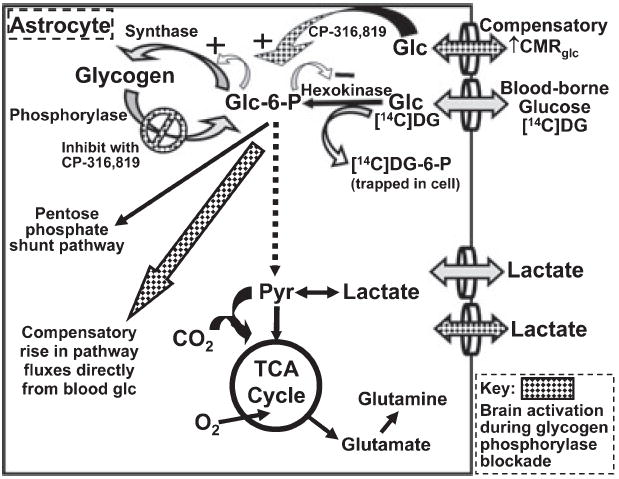
Compensatory increase in glucose utilization in astrocytes during sensory stimulation with blockade of glycogen phosphorylase. Schematic diagram of glucose metabolic pathways illustrating the three major pathways for utilization of glucose-6-phosphate (glc-6-P) in astrocytes: the glycolytic pathway to form pyruvate, the pentose phosphate shunt pathway to generate NADPH and pentose phosphates (most of which re-enter the glycolytic pathway after transformation by the transketolase/transaldolase reactions) and the glycogen pathway (for more details, see Hertz et al. 2007). Note that glc-6-P feedback inhibits hexokinase (negative sign) and activates the glycogen synthesis pathway (plus sign). Inhibition of glycogen phosphorylase with CP-316,819 during brain activation renders blood glucose the sole source for glc-6-P via the hexokinase reaction, which is specifically measured with the [14C]deoxyglucose (DG) method (see text). CP-316,819-induced compensatory changes in utilization of blood-borne glucose (hatched symbols) at the hexokinase step are quantified by means of changes in the rate of [14C]DG phosphorylation, from which rates of glucose utilization are calculated.
Effects of vehicle and CP-316,819 on CMRglc during rest and sensory stimulation
CMRglc was similar in unstimulated vehicle and CP-treated rats in the 21 regions of interest (Fig. 3). The arithmetic mean rate of glucose utilization (i.e. an average not weighted by the mass of each structure) was 1.00 ± 0.27 μmol/(g min) for resting vehicle-treated rats and 0.94 ± 0.26 μmol/(g min) for resting CP-treated rats. A lack of measurable effect by glycogen phosphorylase inhibition is consistent with slow glycogen turnover under unstimulated conditions (Watanabe and Passonneau 1973) even though the brain is metabolically active. In contrast, during the 30 min of continuous stimulation during the [14C]DG assay interval, there were robust, heterogeneous increases in CMRglc throughout the brain in both treatment groups, with a higher overall increase in the CP-treated rats (Fig. 3). The arithmetic mean percent increase for all structures assayed during activation compared with respective control value in vehicle-treated rats was 28.7 ± 20.8%, compared with 46.8 ± 33.0% in CP-treated rats (p < 0.05). Only the corpus callosum, superior colliculus and auditory cortex did not respond to the stimulus paradigm with increased glucose utilization (Fig. 3). Baseline plasma glucose levels were similar in all groups (means were 9–10 mmol/L) and were stable over time during the [14C]DG assay in resting vehicle- and CP-treated rats (<3% change), whereas after stimulus onset plasma glucose levels rose in all rats (~10% at 5 min and ~25% at 20 min) and tended to be higher at 5 min in vehicle-treated rats (33%).
Fig. 3.
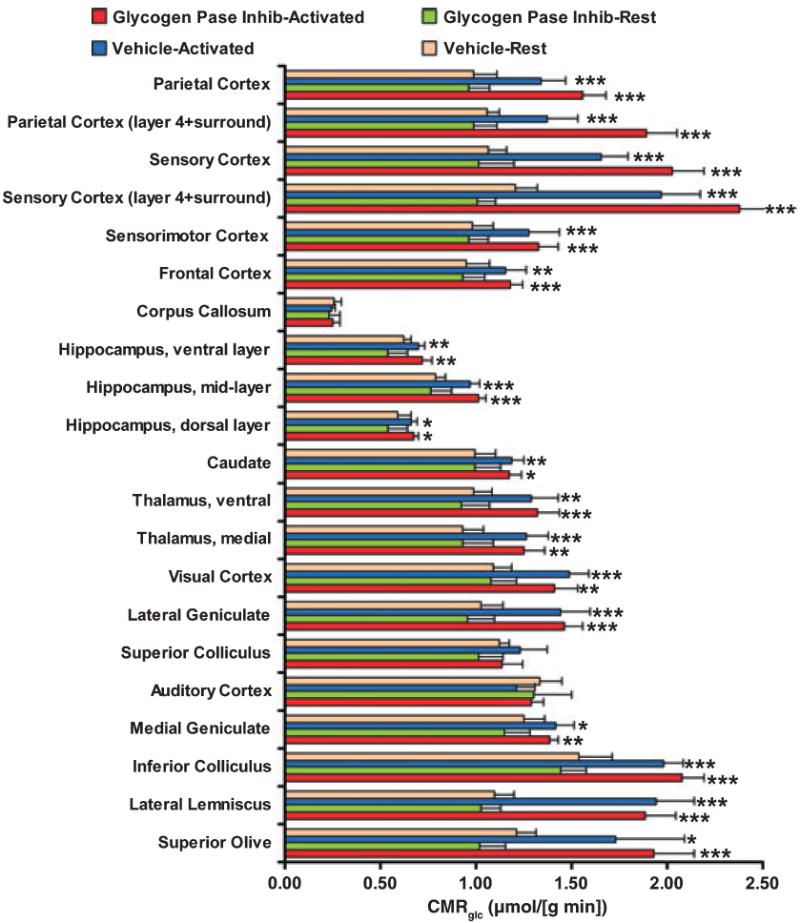
Sensory stimulation increases local rates of glucose utilization (CMRglc) in many brain structures. Rats were pre-treated with the glycogen phosphorylase inhibitor, CP-316,819 (the abbreviation Pase Inhib denotes phosphorylase inhibitor), or an equal volume of vehicle and CMRglc assayed during a 30-min interval of rest or activation. Acoustic plus generalized sensory stimulation commenced with the pulse intravenous injection of [14C]deoxyglucose and continued throughout the 30-min experimental interval. CMRglc in regions of cerebral cortex corresponds to all laminae; in parietal and sensory cortex, layer 4 plus the immediately adjacent tissue was also assayed (Zilles and Wree 1995). The dorsal hippocampal layer includes the alveus, oriens, pyramidal and radiatum layers of dorsal CA1 hippocampus; the mid-layer includes the lacunosum-molecular layer of CA1 sector plus the molecular layer of the external limb of dentate gyrus; and the ventral layer includes the multiform layers of the external limb plus granular and molecular layers of the internal limb (Beck et al. 1990). Values are means; bars = 1 SD (n = 6/group). Statistically significant differences between activated and resting groups: *p < 0.05; **p < 0.01; ***p < 0.001; t-test.
In both vehicle- and CP-treated rats, the largest increases in CMRglc during activation compared with the corresponding resting structure in the same treatment group, 1.6 to 2.4-fold, were in the superior olive and layer 4 (plus some immediately adjacent tissue) of parietal and sensory cortex in CP-treated rats (Fig. 3). Cortical layer 4 is the location of thalamo-cortical input known to have high-functional activity and metabolic rate (Zilles and Wree 1995), and layer 4 is highly activated in the whisker-to-barrel cortex by brushing the whiskers (Kossut et al. 1988). The acoustic component of the stimulus increased CMRglc in three auditory structures (superior olive, lateral lemniscus and inferior colliculus) to similar levels, about 1.7–2 μmol/(g min), whereas more distal structures in this pathway had a smaller change (medial geniculate) or no response (auditory cortex). This group of rats was not given an on–off flash visual stimulus (in contrast to the paradigm in Fig. 1), but they actively watched the brushes during the first 10–15 min of stimulation, and CMRglc increased in visual structures, the lateral geniculate and visual cortex.
Selective increases in CMRglc evoked by glycogen phosphorylase inhibition
Structures that responded to restricted use of glycogen with compensatory increases in the rate of utilization of blood-borne glucose during stimulation were identified by means of activation/rest ratios calculated for each structure in CP- and vehicle-treated rats (Fig. 4). Because CMRglc was assayed in separate groups of rats during rest and activation, the individual values for each activated rat were normalized to the mean value in resting rats for that structure in the same treatment group, then used to calculate mean ratio ± SD for that structure in the vehicle- and CP-treated groups.
Fig. 4.
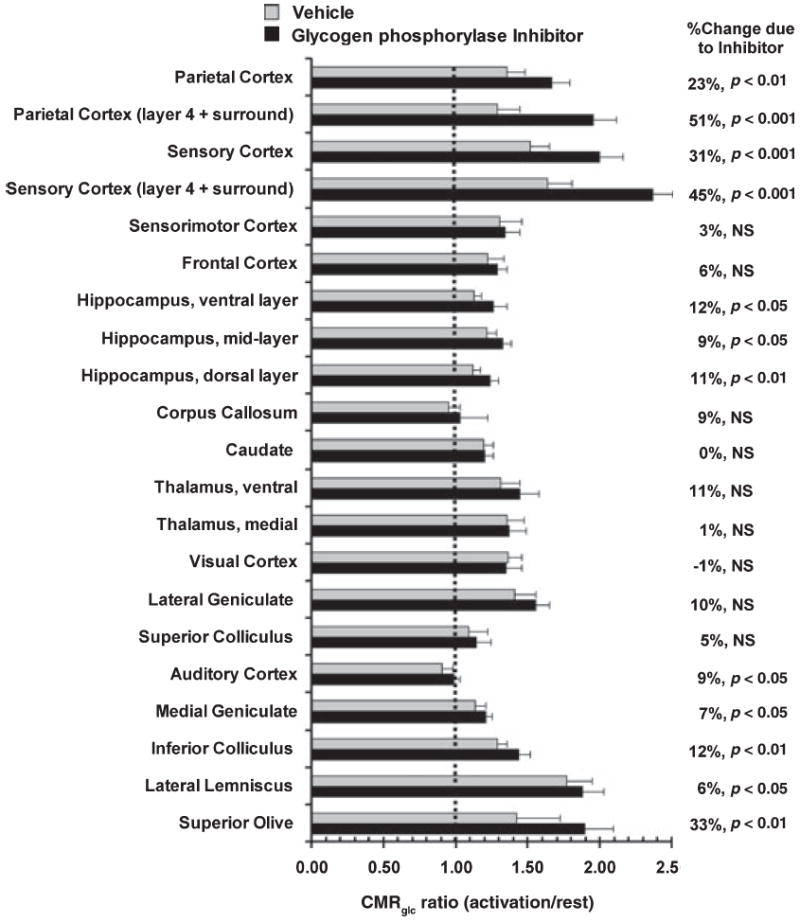
Differential activation of CMRglc in functional pathways during glycogenolysis blockade. The relative change in CMRglc during stimulation compared with rest in the same treatment group (vehicle- and CP-316,819-treated) was calculated from data shown in Fig. 3 by dividing each value for stimulated rats by the respective mean value for resting CMRglc for that structure in that group. Values are mean ratios (bars = 1 SD); ratios > 1.0 indicate activation due to sensory stimulation. Statistically significant differences between ratios for each structure in CP-316,819- compared with vehicle-treated rats were identified with the t-test after logarithic transformation of the ratio data (Zar 1974).
Glycogen phosphorylase inhibitor treatment significantly enhanced the stimulus-induced relative rise in CMRglc in 12 of 21 regions over and above the respective relative rise in vehicle-treated rats, and an unexpected finding was that regional responses to CP-treatment were not in proportion to increased metabolic demand during activation (Fig. 4). For example, the largest increases in the CMRglc activation/rest ratio in CP-treated rats compared with same ratio in vehicle-controls were in layer 4 of sensory and parietal cortex (45–51%), superior olive (33%) and the overall sensory and parietal cortical tissue (23–31%). However, this selective response was not related to the percent rise in CMRglc during activation. For example, CP-treatment had little, if any, differential effect on the CMRglc activation/rest ratio in many structures (lateral geniculate, visual cortex, medial and ventral thalamus and sensorimotor cortex; Fig. 4), even though these structures exhibited significant, 20–40% increases in CMRglc during activation compared with rest in both CP- and vehicle-treated rats (Fig. 3). As a contrasting example, parietal cortex in vehicle-treated rats responded to activation with a similar, 35%, rise above rest (Fig. 3), but had an even larger response during glycogen phosphorylase inhibition (Fig. 4). Notably, the inferior colliculus, the brain structure with the highest resting metabolic rate in resting brain and a strong (30%) response to acoustic stimulation compared with rest (Fig. 3) showed only a 12% rise in the CMRglc activation/rest ratio when use of endogenous stored fuel was blocked (Fig. 4), suggesting low-net glycogen utilization during activation. This conclusion is consistent with data for inferior colliculus (Fig. 1) that showed increased 14C release during stimulation but stable glycogen concentration. In the auditory pathway, four (superior olive, lateral lemniscus, inferior colliculus and medial geniculate) of the five structures assayed had significant increases in CMRglc during activation in vehicle-treated rats, with the lateral lemniscus having the greatest percent increase above rest (77%) (Fig. 3) but only the superior olive had a large response to glycogenolysis blockade in CP- versus vehicle-treated rats (33%; Fig. 4). Among the cortical structures, only sensory and parietal cortex consumed substantially more blood-borne glucose when glycogenolysis was inhibited (Figs 3 and 4). In CA1 hippocampus, the fiber-rich lacunosum and molecular layers (denoted as ‘mid-layer’ in Figs 3 and 4) have a higher-metabolic rate than the more dorsal and ventral laminae (Beck et al. 1990), but this sector was not activated by CP-316,819 treatment to a greater extent than other hippocampal regions during this stimulation paradigm, contrasting the differential laminar responses to the inhibitor in sensory-parietal cortex (Fig. 4). Note that the ‘compensatory increase’ in CMRglc arising from glycogen phosphorylase inhibition (calculated as the mean CMRglc in activated CP-treated rats minus the mean CMRglc in the same structure in activated vehicle-treated rats) was about half the magnitude of the activation-induced rise in CMRglc in vehicle treated rats (Figs 3 and 4); for the overall sensory and parietal cortex, this net increase over and above the CMRglc in the activated vehicle-treated rats was 0.2–0.5 μmol/ (g min).
To summarize, neither the sequential order of the processing stations in ascending sensory pathways nor resting CMRglc are predictive of a requirement for compensatory CMRglc or the magnitude of increased CMRglc during activation with glycogenolysis blockade.
Fuel consumption and metabolite generation
Increased pathway fluxes during brain activation can deplete or fill metabolic pools, and changes in pool size are sometimes used to infer shifts in pathway rates. For example, the brain-to-plasma distribution ratio is similar during rest and after 10-min stimulation (Fig. 1) even though CMRglc increased markedly (Fig. 3), indicating that overall glucose supply matched local demand, but some glycogen was consumed to help satisfy demand and small amounts of lactate accumulated (Fig 1). Because increased lactate level in activated brain is often interpreted in terms of a predominance of glycolytic metabolism, it is important to evaluate whether concentration changes are predictive of the magnitude of flux changes. Net global changes in levels of glucose, lactate and glycogen in cerebral cortex during the 10-min stimulation interval (data from Fig. 1) were, therefore, compared with the amount of flux-generated pyruvate that could be formed from glucose during the same 10-min interval, calculated from measured overall cortical glucose utilization rates (Fig. 3) using the conversion, one molecule glucose or glucosyl unit from glycogen forms two pyruvate molecules. As shown in Fig. 5, metabolite levels after activation interval do not appropriately reflect the direction or magnitude of changes in pathway fluxes. For example, in the absence of additional data, one might infer from the increase in brain glucose content that its rate of utilization slowed, whereas the converse was actually true; plasma glucose rose and brain glucose equilibrated with that in plasma (Fig. 1). Stability of brain glucose during activation was also observed in our parallel microdialysis studies to assay extracellular metabolites in the inferior colliculus of conscious rats given only an acoustic stimulus; dialysate glucose levels were similar in samples collected during 10-min intervals before, during and after acoustic stimulation, whereas lactate levels transiently doubled during the first 10 min of a 20-min stimulation interval, then fell between 10 and 20 min (Cruz et al. 2007).
Fig. 5.
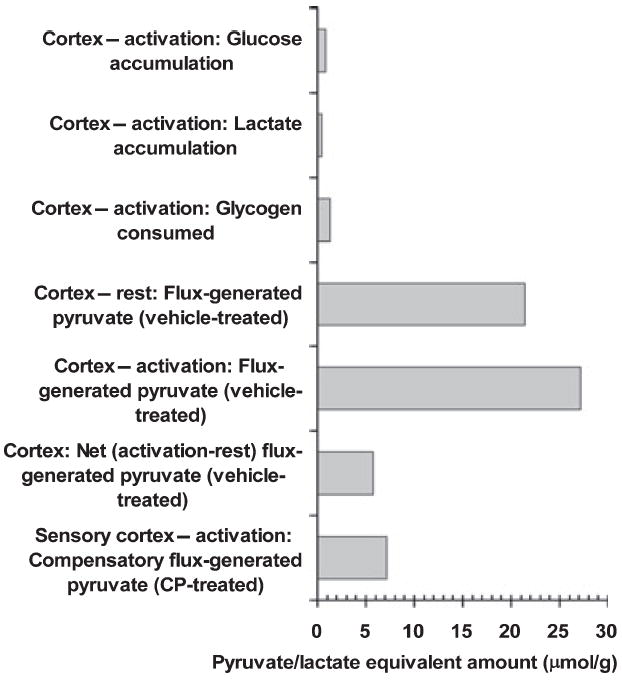
Comparison of flux-generated pyruvate/lactate with net concentration change. The amount of pyruvate generated in cerebral cortex via flux of blood-borne glucose through the glycolytic pathway during a 10-min interval was calculated from the arithmetic mean CMRglc of measured values in all six cerebral cortical structures during rest (1.07 ± 0.13 μmol glucose/[g min]) and stimulation 1.36 ± 0.19 μmol glucose/(g min) in vehicle-treated rats (Fig. 3); there was an overall 27% increase (p < 0.05) due to activation. During rest, pyruvate equivalents generated from flux of glucose = 1.07 μmol glucose/(g min) × 10 min × 2 pyruvate/glucose = 21.4. Compensatory increases in CMRglc induced by the glycogen phosphorylase inhibitor were calculated as the difference in the mean activated CMRglc in CP-316,819-treated rats minus that in activated vehicle-treated rats [i.e. 0.22, 0.52, 0.37 and 0.41 μmol/(g min) for overall parietal cortex, parietal layer 4, sensory cortex and sensory layer 4, respectively] and pyruvate equivalents were calculated by multiplying by 20, as described above. Because the value for sensory cortex approximates the mean value for all four regions, this representative structure was plotted. Net changes in brain cerebral cortical concentrations of glucose, lactate and glycogen during the 10-min stimulation (Fig. 1) are expressed as triose equivalents (1 glucose = 2 pyruvate/ lactate).
The net gains in tissue lactate content in dissected cortex, inferior colliculus and superior colliculus during the 10-min stimulation interval were large percentage increases (150%, 53%, 63%, respectively; Fig. 1) but they represented rather small quantities of lactate (0.51, 0.18, 0.12 μmol/g, respectively). The amount of pyruvate/lactate generated by cortical metabolism of glucose during a 10-min interval in resting and activated vehicle-treated rats was calculated from the arithmetic mean rates (1.07 and 1.36 μmol/[g min], respectively; Fig. 3) measured in all cortical structures (Fig. 5). The net increase in lactate and glucose concentrations in cerebral cortex corresponded to <4% of the total amount of pyruvate generated from the flux of blood glucose during rest or activation. Also, cortical lactate accumulation corresponded to (i) <10% of the net (activation-rest) amount of pyruvate generated from glucose or of the compensatory glucose utilization (i.e. the rate in activated CP-treated rats over and above that in activated vehicle-treated rats) and (ii) about 40% of the net glycogen consumed. The paucity of lactate accumulation indicates that most pyruvate/lactate generated must be metabolized or released. Finally, the decrement in mean cortical glycogen level is also small compared with the net activation-induced flux-generated pyruvate in the vehicle and CP-treated rats (Fig. 5). To summarize, fluxes through glucose, glycogen, pyruvate and lactate pools respond to shifts in energy demand with changes in functional activity, whereas metabolite concentrations reflect net difference in rates of formation and disappearance, which are influenced by many factors, including their blood levels, transport, relative fluxes of glycolytic and oxidative pathways in all cell types, malate aspartate shuttle activity, pH, cellular redox state and diffusion within brain.
Discussion
Compensatory CMRglc during activation
The major finding of this study is that pre-treatment of normal conscious rats with the glycogen phosphorylase inhibitor CP-316,819 using a protocol that causes an 88% increase in brain glycogen content (R. Swanson, personal communication) selectively stimulates local rates of utilization of blood-borne glucose in specific gray matter structures during sensory stimulation, but not during rest or in corpus callosum (Figs 3 and 4). These compensatory increases in CMRglc were not predictable on the basis of sequential stage of sensory information processing, basal metabolic rate of the structure, or percent increase in CMRglc during activation compared with rest. Because the hexokinase reaction and glycogenolysis generate glc-6-P that is used by various pathways (Fig. 2), increased reliance on blood glucose in activated CP-treated rats is linked to impaired utilization of glycogen in astrocytes. This relationship suggests preferential, active utilization of glycogen in substantial amounts compared with direct metabolism of blood glucose by the glycolytic or pentose pathways (Fig. 2) in stimulated astrocytes located in specific brain regions. High-glycogen turnover during sensory stimulation could arise if local neuronal activity triggers pulsatile glycogen degradation and re-synthesis, particularly in astrocytic fine processes that surround synaptic structures, as well as in their soma and endfeet. Increased extracellular K+ levels (Hof et al. 1988) and many neuromodulators and neurotransmitters, especially monoamines, stimulate astrocytic glycogenolysis (Hertz and Peng 1992; Hertz et al. 2007), whereas glutamate, GABA and aspartate do not (Magistretti et al. 1981), even when assayed in cultured astrocytes or brain slices using media containing 3 mmol/L glucose. Localized release of label from glycogen occurs in structures in the whisker-to-barrel pathway during whisker stimulation (Swanson et al. 1992), and the compensatory increase in CMRglc during glycogenolysis blockade was greatest (~50% of the additional glucose consumed during activation in vehicle-treated rats; Fig. 4) in sensory-parietal cortex that contains the whisker ‘barrels’. CP-316,819 was developed as a glycogen phosphorylase inhibitor to help achieve glycemic control in diabetic patients (Hoover et al. 1998; Martin et al. 1998; Treadway et al. 2001); this drug did not significantly alter plasma glucose levels during rest or activation in the present study, and the robust, compensatory shift to utilization of blood glucose should minimize any potential deleterious consequences of glycogen phosphorylase blockade or brain glycogen depletion.
The activation-dependent, CP-induced compensatory rise in CMRglc may represent, at least in part, the contribution of glycogen turnover to the overall flux into the glc-6-P pool in astrocytes in the absence of the glycogen phosphorylase inhibitor. In the CP-treated rats, the glycogen turnover-related flux could manifest itself as increased glycogen synthesis and increased downstream metabolism via the pentose phosphate shunt and glycolytic pathways. If the activation-induced compensatory rise in CMRglc in sensory-parietal cortex (~0.2–0.4 μmol/[g min]; Figs 3 and 4) reflects glycogen turnover, then during a 10-min period as much as 2–4 μmol/g of glucose is predicted to cycle through glycogen via synthesis and degradation in activated control rats, consistent with the expected smaller overall fall in glycogen level in dissected samples of cortex, ~0.6 μmol/g (Fig. 1) due to blunting by tissue averaging and re-synthesis. To summarize, the local compensatory increases in CMRglc identify brain structures in which glycogen metabolism is most likely to be linked to astrocytic functional activities evoked by the physiological stimulation paradigm. The possibility of increased glycogen turnover due to pulsatile ‘on–off’ activation of its utilization and replenishment suggests that assays of glycogen levels and of glycogen labeling during activation will underestimate or fail to detect changes in glycogenolytic activity due to re-synthesis that would stabilize levels and degradation that may remove newly incorporated label.
Physiological roles of glycogen in activated astrocytes
Brain glycogen is often considered to be a slowly turning over emergency fuel reservoir, but many lines of evidence indicate that glycogen is tapped for fuel without overt or prolonged energy deficits. (i) Glycogen phosphorylase can be activated within seconds by conversion from the inactive b-form to the active a-form (Folbergrova et al. 1978), and its activation occurs during behavioral activities, including intracranial reward self-stimulation and exposure to a novel environment (Harley et al. 1995; Konkle et al. 1999; Konkle and Bielajew 2004; Walling et al. 2006). (ii) Glycogenolysis in cultured astrocytes and brain slices assayed in vitro under normoglycemic conditions is initiated by many signals arising from neuronal activity, including the wide-spread locus coeruleus noradrenergic arousal-stress-response systems, diverse neurotransmitter systems and specific electrolytes (Magistretti et al. 1981; Hof et al. 1988; Hertz and Peng 1992; Stone 1994; Dienel and Cruz 2006; Hertz et al. 2007). (iii) Glycogenolysis occurs in retina of the intact honeybee during light stimulation (Evêquoz et al. 1983), chick brain during early memory consolidation (Hertz et al. 2003; Gibbs et al. 2006) and rat whisker-to-barrel cortex in vivo during vibrissae stimulation (Swanson 1992; Swanson et al. 1992). (iv) Recent studies in carefully handled rats using improved enzyme-inactivation procedures found much higher-brain glycogen levels (9–12 μmol/g compared with the usual range of 2–3 μmol/g) that fell after brief sensory stimulation (Madsen et al. 1999; Cruz and Dienel 2002; Fig. 1). In the present study, glycogen levels in resting rats were lower and the concentration fall during stimulation was smaller than our studies cited above; the reasons for these differences are unidentified, but may include (i) less control of ambient conditions in our current laboratory resulting in higher ‘background’ sensory stimulation and lower-glycogen levels and (ii) change of rat supplier and strain from Sprague–Dawley to Wistar–Hanover rats. Rat strains differ in their response to stress (Bielajew et al. 2002, 2003), which may influence the impact of abrupt arousal and sensory stimulation on glycogen. High-brain glycogen levels have also been reported by Kong et al. (2002), who found that glycogen level fell during prolonged sleep deprivation induced by gentle handling and presentation of novel environments; conceivably, the sensory stimulation procedure itself may have induced glycogenolysis contributing to the lower levels attributed to sleep deprivation. To summarize, combination of local (e.g. neurotransmitters and K+) plus diffuse (e.g. locus coeruleus innervation) regulatory mechanisms that control glycogen turnover integrate astrocytic energy metabolism with inter- and intra-cellular signaling.
The interactive controls that regulate glycogen utilization also render glycogen level highly sensitive to the physiological state, history and ‘handling’ of experimental subjects, as well as analytical procedures. Therefore, ‘negative findings’ in studies of glycogen could be expected in previously activated or -stressed subjects due to glycogen depletion, in anesthetized (e.g. during MRS studies) or habituated subjects in which an arousal-stress-response is blunted, and in brain regions that are activated by the stimulus paradigm but do not show a strong glycogen-related CMRglc response to the specific stimulus (e.g. visual, auditory, frontal or sensorimotor cortex; Figs 3 and 4). For example, visual cortex responded to the stimulus paradigm by increasing CMRglc ~50% in vehicle-treated rats (Fig. 3), but there was no compensatory response to glycogen phosphorylase inhibition (Fig. 4) implying no glycogenolytic response. The stability of [13C]glycogen in human visual cortex when assayed by MRS during visual stimulation after prolonged pre-labeling with [13C]glucose (Öz et al. 2007) is consistent with our findings.
Metabolic fate of glycogen
Physiological roles of glycogen are tied to its metabolic functions, which can vary with cellular needs. Rapid glycogen turnover associated with tight temporal coupling of its degradation and re-synthesis would require twice as much blood glucose to obtain the same ATP yield as direct glycolytic metabolism of glucose (Shulman et al. 2001; Hertz et al. 2007), perhaps underlying the large rise in CMRglc in specific structures during activation (Figs 3 and 4), but not during rest when glycogen turnover is low (Watanabe and Passonneau 1973). A ‘tight-coupling’ hypothesis predicts that accumulation of label from glucose into glycogen should rise markedly during activation compared with rest in CP-treated rats but that labeling should be low in activated controls because rapid turnover would eliminate the label; our finding of no increase in glycogen labeling during sensory stimulation compared with rest in dissected cortical samples is consistent with this notion (Dienel et al. 2002). During CP-treatment, there may, however, be increased downstream flux directly to the glycolytic and pentose shunt pathways, not just incorporation of glucose into glycogen; direct tests of these possibilities are technically difficult, minimally requiring sensitive assays with high temporal-spatial resolution.
In cultured astrocytes, glycogen is converted to lactate and released to the medium (Dringen et al. 1993), and depending on the fate of glycogen-derived glc-6-P in brain in vivo, net glycogen consumption and increased turnover may enhance the fall in the ratio of oxygen to glucose utilization (CMRO2/CMRglc) during sensory stimulation that we previously observed with a similar stimulation paradigm (Madsen et al. 1999). For example, inclusion of the fuel consumed in addition to glucose during activation, including glycogen (Fig. 1) and acetate (Cruz et al. 2005), would further depress the CMRO2/CMRglc ratio. Because lactate does not accumulate in brain in proportion to the amount of flux-generated pyruvate in vivo (Fig. 5) pyruvate/lactate must be metabolized or quickly released from the tissue sampling volume. The fall in CMRO2/CMRglc ratio and the large underestimation of CMRglc using [1- or 6-14C]glucose as the tracer during activating conditions (Collins et al. 1987; Lear and Ackermann 1988, 1989; Ackermann and Lear 1989; Dienel and Cruz 2004; Cruz et al. 2007) suggest increased non-oxidative metabolism of glucose and rapid release of quickly labeled metabolites. An alternative scenario is lactate shuttling among cells followed by lactate oxidation; a ‘tightly coupled’ shuttle process requires that CMRO2 match CMRglc+glycogen, a stoichiometry that is not observed in most activation studies (Dienel and Cruz 2004, 2006). Putative lactate shuttling from astrocytes to neurons has been implicated under aglycemic conditions or intense electrical stimulation because glycogenolysis does support compound action potentials in excised optic nerve for a duration related to the glycogen amount at stimulus onset (Brown 2004; Brown et al. 2005; Tekkok et al. 2005). The identity of the lactate-oxidizing cells and fraction of lactate formed from glycogen actually transferred to other cells compared with that released to the perfusate have not been established, and an interesting alternative explanation is that astrocytic lactate utilization may, in fact, help maintain neuronal function. Hargittai and Lieberman (1991) concluded that glial metabolism is governed by axonal metabolic activity and accounts for most of the normal respiratory activity in isolated crayfish giant axons (also see Dienel and Hertz 2005). Even if a shuttle process exists under specific experimental conditions, it may not apply to normal brain tissue. For example, during an energy crisis provoked by severe hypoglycemia, compensatory metabolic shifts that do not reflect normal fuel usage are used in an attempt to maintain ATP levels, including oxidative consumption of endogenous amino acids, fatty acids and other compounds (Siesjö 1978; Ghajar et al. 1982). White matter has a low-metabolic rate and does not require as much energy as gray matter, and the corpus callosum, a white matter structure, did not increase CMRglc or show a compensatory response during activation (Figs 3 and 4). Isolated nerves have no blood flow so diffusion limitation of metabolite efflux may lead to greater availability for metabolism, contrasting the in vivo situation where rapid transmembrane transport compared with oxidation rate, diffusion in extracellular fluid and release to blood may be favored in gray matter (Dienel and Hertz 2001; Hertz and Dienel 2005). To summarize, analysis of the utilization and metabolic fate of glycogen can help understand its functional roles and contribution to astrocytic and overall brain energetics. The intriguing possibility of high-glycogen turnover in specific brain regions during activation complicates interpretation of standard approaches that assess concentration and labeling changes to evaluate roles or utilization of glycogen under various conditions.
Acknowledgments
We thank Pfizer Global Research and Development, Groton, CT, USA, for generously providing CP-316819 and W. Ross Tracey, PhD, Department of Cardiovascular, Metabolic and Endocrine Diseases, Pfizer, for his sponsorship of our request. We also thank Raymond Swanson, MD, Department Neurology, UC San Francisco for providing the CP-dosing regimen developed in his laboratory. This work was supported by NIH grants NS36728 and NS47546.
Abbreviations used
- CMRglc
compensatory increase in utilization of blood glucose
- CP or CP-316,819
[R-R*,S*]-5-chloro-N-[2-hydroxy-3-(methoxymethylamino)-3-oxo-1-(phenylmethyl)propyl]-1H-indole-2-carboxamide
- DG
2-deoxy-d-[1-14C]glucose
- glc-6-P
glucose-6-phosphate
- MRS
magnetic resonance spectroscopic
Footnotes
Note added in proof After this paper was accepted for publication, we became aware that work by Dr Swanson and colleagues describing the effects of CP-316,819 on glycogen accumulation, EEG activity, and neuronal survival after hypoglycemia was in press (Suh S., Bergher J.P., Anderson C.M., Treadway J.L., Fosgerau K. and Swanson R.A. (2007) Astrocyte glycogen sustains neuronal activity diring hypoglycemia: Studies with the glycogen phosphorylase inhibitor CP-316,819([R-R*,S*]-5-chloro-N-[2-hydroxy-3-(methoxymethylamino)-3-oxo-1-(phenylmethy1)propy1]-1H-indole-2-carboxamide. J. Pharmacol. Exp. Ther. 321, 45–50.
References
- Ackermann RF, Lear JL. Glycolysis-induced discordance between glucose metabolic rates measured with radiolabeled fluorodeoxyglucose and glucose. J Cereb Blood Flow Metab. 1989;9:774–785. doi: 10.1038/jcbfm.1989.111. [DOI] [PubMed] [Google Scholar]
- Beck T, Wree A, Schleicher A. Glucose utilization in rat hippocampus after long-term recovery from ischemia. J Cereb Blood Flow Metab. 1990;10:542–549. doi: 10.1038/jcbfm.1990.96. [DOI] [PubMed] [Google Scholar]
- sBielajew C, Konkle AT, Merali Z. The effects of chronic mild stress on male Sprague-Dawley and Long Evans rats: I. Biochemical and physiological analyses. Behav Brain Res. 2002;136:583–592. doi: 10.1016/s0166-4328(02)00222-x. [DOI] [PubMed] [Google Scholar]
- Bielajew C, Konkle AT, Kentner AC, Baker SL, Stewart A, Hutchins AA, Santa-Maria Barbagallo L, Fouriezos G. Strain and gender specific effects in the forced swim test: effects of previous stress exposure. Stress. 2003;6:269–280. doi: 10.1080/10253890310001602829. [DOI] [PubMed] [Google Scholar]
- Brown AM. Brain glycogen re-awakened. J Neurochem. 2004;89:537–552. doi: 10.1111/j.1471-4159.2004.02421.x. [DOI] [PubMed] [Google Scholar]
- Brown AM, Sickmann HM, Fosgerau K, Lund TM, Schousboe A, Waagepetersen HS, Ransom BR. Astrocyte glycogen metabolism is required for neural activity during aglycemia or intense stimulation in mouse white matter. J Neurosci Res. 2005;79:74–80. doi: 10.1002/jnr.20335. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Broadwell RD. Cytochemical identification of cerebral glycogen and glucose-6-phosphatase activity under normal and experimental conditions. I Neurons and Glia. J Elect Micro Tech. 1986a;3:413–437. [Google Scholar]
- Cataldo AM, Broadwell RD. Cytochemical identification of cerebral glycogen and glucose-6-phosphatase activity under normal and experimental conditions. II. Choroid plexus and ependymal epithelia, endothelia and pericytes. J Neurocytol. 1986b;15:511–524. doi: 10.1007/BF01611733. [DOI] [PubMed] [Google Scholar]
- Chih CP, Roberts EL., Jr Energy substrates for neurons during neural activity: a critical review of the astrocyte-neuron lactate shuttle hypothesis. J Cereb Blood Flow Metab. 2003;23:1263–1281. doi: 10.1097/01.WCB.0000081369.51727.6F. [DOI] [PubMed] [Google Scholar]
- Collins RC, McCandlesss DW, Wagman IL. Cerebral glucose utilization: comparison of [14C]deoxyglucose and [6-14C]glucose quantitative autoradiography. J Neurochem. 1987;49:1564–1570. doi: 10.1111/j.1471-4159.1987.tb01028.x. [DOI] [PubMed] [Google Scholar]
- Cruz NF, Dienel GA. High glycogen levels in brains of rats with minimal environmental stimuli: implications for metabolic contributions of working astrocytes. J Cereb Blood Flow Metab. 2002;22:1476–1489. doi: 10.1097/01.WCB.0000034362.37277.C0. [DOI] [PubMed] [Google Scholar]
- Cruz NF, Lasater A, Zielke HR, Dienel GA. Activation of astrocytes in brain of conscious rats during acoustic stimulation: acetate utilization in working brain. J Neurochem. 2005;92:934–947. doi: 10.1111/j.1471-4159.2004.02935.x. [DOI] [PubMed] [Google Scholar]
- Cruz NF, Ball KK, Dienel GA. Functional imaging of focal brain activation in conscious rats: impact of [14C]glucose metabolite spreading and release. J Neurosci Res. 2007 doi: 10.1002/jnr.21193. in press. [DOI] [PubMed] [Google Scholar]
- Dienel GA, Cruz NF. Nutrition during brain activation: does cell-to-cell lactate shuttling contribute significantly to sweet and sour food for thought? Neurochem Int. 2004;45:321–351. doi: 10.1016/j.neuint.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Dienel GA, Cruz NF. Astrocyte activation in working brain: energy supplied by minor substrates. Neurochem Int. 2006;48:586–495. doi: 10.1016/j.neuint.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Dienel GA, Hertz L. Glucose and lactate metabolism during brain activation. J Neurosci Res. 2001;66:824–838. doi: 10.1002/jnr.10079. [DOI] [PubMed] [Google Scholar]
- Dienel GA, Hertz L. Astrocytic contributions to bioenergetics of cerebral ischemia. Glia. 2005;50:362–388. doi: 10.1002/glia.20157. [DOI] [PubMed] [Google Scholar]
- Dienel GA, Wang RY, Cruz NF. Generalized sensory stimulation of conscious rats increases labeling of oxidative pathways of glucose metabolism when the brain glucose-oxygen uptake ratio rises. J Cereb Blood Flow Metab. 2002;22:1490–1502. doi: 10.1097/01.WCB.0000034363.37277.89. [DOI] [PubMed] [Google Scholar]
- Dringen R, Gebhardt R, Hamprecht B. Glycogen in astrocytes: possible function as lactate supply for neighboring cells. Brain Res. 1993;623:208–214. doi: 10.1016/0006-8993(93)91429-v. [DOI] [PubMed] [Google Scholar]
- Evêquoz V, Stadelmann A, Tsacopolous M. The effect of light on glycogen turnover in the retina of the intact honeybee drone (Apis mellifera) J Comp Physiol. 1983;150:69–75. [Google Scholar]
- Folbergrova J, Nordstrom CH, Siesjö BK. Labile metabolites and phosphorylase a in rapidly frozen rat cerebral cortex. J Neurochem. 1978;30:493–495. doi: 10.1111/j.1471-4159.1978.tb06555.x. [DOI] [PubMed] [Google Scholar]
- Ghajar JB, Plum F, Duffy TE. Cerebral oxidative metabolism and blood flow during acute hypoglycemia and recovery in unanesthetized rats. J Neurochem. 1982;38:397–409. doi: 10.1111/j.1471-4159.1982.tb08643.x. [DOI] [PubMed] [Google Scholar]
- Gibbs ME, Anderson DG, Hertz L. Inhibition of glycogenolysis in astrocytes interrupts memory consolidation in young chickens. Glia. 2006;54:214–222. doi: 10.1002/glia.20377. [DOI] [PubMed] [Google Scholar]
- Gruetter R. Glycogen: the forgotten cerebral energy store. J Neurosci Res. 2003;74:179–183. doi: 10.1002/jnr.10785. [DOI] [PubMed] [Google Scholar]
- Hargittai PT, Lieberman EM. Axon-glia interactions in the crayfish: glial cell oxygen consumption is tightly coupled to axon metabolism. Glia. 1991;4:417–423. doi: 10.1002/glia.440040410. [DOI] [PubMed] [Google Scholar]
- Harley CW, Milway JS, Fara-On M. Medial forebrain bundle stimulation in rats activates glycogen phosphorylase in layers 4, 5b and 6 of ipsilateral granular neocortex. Brain Res. 1995;685:217–223. doi: 10.1016/0006-8993(95)00481-5. [DOI] [PubMed] [Google Scholar]
- Hertz L, Dienel GA. Lactate transport and transporters: general principles and functional roles in brain cells. J Neurosci Res. 2005;79:11–18. doi: 10.1002/jnr.20294. [DOI] [PubMed] [Google Scholar]
- Hertz L, Peng L. Effects of monoamine transmitters on neurons and astrocytes: correlation between energy metabolism and intracellular messengers. Prog Brain Res. 1992;94:283–301. doi: 10.1016/s0079-6123(08)61758-6. [DOI] [PubMed] [Google Scholar]
- Hertz L, Zielke HR. Astrocytic control of glutamatergic activity: astrocytes as stars of the show. Trends Neurosci. 2004;27:735–743. doi: 10.1016/j.tins.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Hertz L, O’Dowd BS, Ng KT, Gibbs ME. Reciprocal changes in forebrain contents of glycogen and of glutamate/glutamine during early memory consolidation in the day-old chick. Brain Res. 2003;994:226–233. doi: 10.1016/j.brainres.2003.09.044. [DOI] [PubMed] [Google Scholar]
- Hertz L, Peng L, Dienel GA. Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J Cereb Blood Flow Metab. 2007;27:219–249. doi: 10.1038/sj.jcbfm.9600343. [DOI] [PubMed] [Google Scholar]
- Hof PR, Pascale E, Magistretti PJ. K+ at concentrations reached in the extracellular space during neuronal activity promotes a Ca2+-dependent glycogen hydrolysis in mouse cerebral cortex. J Neurosci. 1988;8:1922–1928. doi: 10.1523/JNEUROSCI.08-06-01922.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover DJ, Lefkowitz-Snow S, Burgess-Henry JL, Martin WH, Armento SJ, Stock IA, McPherson RK, Genereux PE, Gibbs EM, Treadway JL. Indole-2-carboxamide inhibitors of human liver glycogen phosphorylase. J Med Chem. 1998;41:2934–2938. doi: 10.1021/jm980264k. [DOI] [PubMed] [Google Scholar]
- Hyder F, Patel AB, Gjedde A, Rothman DL, Behar KL, Shulman RG. Neuronal-glial glucose oxidation and glutamatergic-GABAergic function. J Cereb Blood Flow Metab. 2006;26:865–877. doi: 10.1038/sj.jcbfm.9600263. [DOI] [PubMed] [Google Scholar]
- Kong J, Shepel PN, Holden CP, Mackiewicz M, Pack AI, Geiger JD. Brain glycogen decreases with increased periods of wakefulness: implications for homeostatic drive to sleep. J Neurosci. 2002;22:5581–5587. doi: 10.1523/JNEUROSCI.22-13-05581.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konkle AT, Bielajew C. Tracing the neuroanatomical profiles of reward pathways with markers of neuronal activation. Rev Neurosci. 2004;15:383–414. doi: 10.1515/revneuro.2004.15.6.383. [DOI] [PubMed] [Google Scholar]
- Konkle AT, Wilson P, Bielajew C. Histochemical mapping of the substrate for brain-stimulation reward with glycogen phosphorylase. J Neurosci Methods. 1999;93:111–119. doi: 10.1016/s0165-0270(99)00136-3. [DOI] [PubMed] [Google Scholar]
- Kossut M, Hand PJ, Greenberg J, Hand CL. Single vibrissal cortical column in SI cortex of rat and its alterations in neonatal and adult vibrissa-deafferented animals: a quantitative 2DG study. J Neurophysiol. 1988;60:829–852. doi: 10.1152/jn.1988.60.2.829. [DOI] [PubMed] [Google Scholar]
- Lear JL, Ackermann RF. Comparison of cerebral glucose metabolic rates measured with fluorodeoxyglucose and glucose labeled in the 1, 2, 3-4, and 6 positions using double label quantitative digital autoradiography. J Cereb Blood Flow Metab. 1988;8:575–585. doi: 10.1038/jcbfm.1988.99. [DOI] [PubMed] [Google Scholar]
- Lear J, Ackermann RF. Why the deoxyglucose method has proven so useful in cerebral activation studies: the unappreciated prevalence of stimulation-induced glycolysis. J Cereb Blood Flow Metab. 1989;9:911–913. doi: 10.1038/jcbfm.1989.128. [DOI] [PubMed] [Google Scholar]
- Madsen PL, Cruz NF, Sokoloff L, Dienel GA. Cerebral oxygen/glucose ratio is low during sensory stimulation and rises above normal during recovery: excess glucose consumption during stimulation is not accounted for by lactate efflux from or accumulation in brain tissue. J Cereb Blood Flow Metab. 1999;19:393–400. doi: 10.1097/00004647-199904000-00005. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ, Morrison JH, Shoemaker WJ, Sapin V, Bloom FE. Vasoactive intestinal polypeptide induces glycogenolysis in mouse cortical slices: a possible regulatory mechanism for the local control of energy metabolism. Proc Natl Acad Sci USA. 1981;78:6535–6539. doi: 10.1073/pnas.78.10.6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin WH, Hoover DJ, Armento SJ, Stock IA, McPherson RK, Danley DE, Stevenson RW, Barrett EJ, Treadway JL. Discovery of a human liver glycogen phosphorylase inhibitor that lowers blood glucose in vivo. Proc Natl Acad Sci USA. 1998;95:1776–1781. doi: 10.1073/pnas.95.4.1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. New roles for astrocytes: regulation of synaptic transmission. Trends Neurosci. 2003;26:536–542. doi: 10.1016/S0166-2236(03)00237-6. [DOI] [PubMed] [Google Scholar]
- Öz G, Berkich DA, Henry PG, Xu Y, LaNoue K, Hutson SM, Gruetter R. Neuroglial metabolism in the awake rat brain:CO2 fixation increases with brain activity. J Neurosci. 2004;24:11273–11279. doi: 10.1523/JNEUROSCI.3564-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Öz G, Seaquist ER, Kumar A, Criego AB, Benedict LE, Rao JP, Henry PG, Van de Moortele PF, Gruetter R. Human brain glycogen content and metabolism: implications on its role in brain energy metabolism. Am J Physiol. 2007;292:E946–E951. doi: 10.1152/ajpendo.00424.2006. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Neuroenergetics: calling upon astrocytes to satisfy hungry neurons. Neuroscientist. 2004;10:53–62. doi: 10.1177/1073858403260159. [DOI] [PubMed] [Google Scholar]
- Pfeiffer B, Elmer K, Roggendorf W, Reinhart PH, Hamprecht B. Immunohistochemical demonstration of glycogen phosphorylase in rat brain slices. Histochemistry. 1990;94:73–80. doi: 10.1007/BF00266792. [DOI] [PubMed] [Google Scholar]
- Pfeiffer B, Meyermann R, Hamprecht B. Immunohistochemical co-localization of glycogen phosphorylase with the astroglial markers glial fibrillary acidic protein and S-100 protein in rat brain sections. Histochemistry. 1992;97:405–412. doi: 10.1007/BF00270387. [DOI] [PubMed] [Google Scholar]
- Pfeiffer-Guglielmi B, Fleckenstein B, Jung G, Hamprecht B. Immunocytochemical localization of glycogen phosphorylase isozymes in rat nervous tissues by using isozyme-specific antibodies. J Neurochem. 2003;85:73–81. doi: 10.1046/j.1471-4159.2003.01644.x. [DOI] [PubMed] [Google Scholar]
- Richter K, Hamprecht B, Scheich H. Ultrastructural localization of glycogen phosphorylase predominantly in astrocytes of the gerbil brain. Glia. 1996;17:263–273. doi: 10.1002/(SICI)1098-1136(199608)17:4<263::AID-GLIA1>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Shulman RG, Hyder F, Rothman DL. Cerebral energetics and the glycogen shunt: neurochemical basis of functional imaging. Proc Natl Acad Sci USA. 2001;98:6417–6422. doi: 10.1073/pnas.101129298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siesjö BK. Brain Energy Metabolism. Wiley; New York: 1978. [Google Scholar]
- Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, Sakurada O, Shinohara M. The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem. 1977;28:897–916. doi: 10.1111/j.1471-4159.1977.tb10649.x. [DOI] [PubMed] [Google Scholar]
- Stone EA. Glial cells as targets of the central noradrenergic system. In: Briley M, Marien M, editors. Noradrenergic Mechanisms In Parkinson’s Disease. CRC Press; Boca Raton: 1994. pp. 173–189. An update. [Google Scholar]
- Swanson RA. Physiologic coupling of glial glycogen metabolism to neuronal activity in brain. Can J Physiol Pharmacol. 1992;70(Suppl):S138–S144. doi: 10.1139/y92-255. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Morton MM, Sagar SM, Sharp FR. Sensory stimulation induces local cerebral glycogenolysis: demonstration by autoradiography. Neuroscience. 1992;51:451–461. doi: 10.1016/0306-4522(92)90329-z. [DOI] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci. 2006;9:260–267. doi: 10.1038/nn1623. [DOI] [PubMed] [Google Scholar]
- Tekkok SB, Brown AM, Westenbroek R, Pellerin L, Ransom BR. Transfer of glycogen-derived lactate from astrocytes to axons via specific monocarboxylate transporters supports mouse optic nerve activity. J Neurosci Res. 2005;81:644–652. doi: 10.1002/jnr.20573. [DOI] [PubMed] [Google Scholar]
- Treadway JL, Mendys P, Hoover DJ. Glycogen phosphorylase inhibitors for treatment of type 2 diabetes mellitus. Expert Opin Investig Drugs. 2001;10:439–454. doi: 10.1517/13543784.10.3.439. [DOI] [PubMed] [Google Scholar]
- Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci. 2005;6:626–640. doi: 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- Walling SG, Bromley K, Harley CW. Glycogen phosphorylase reactivity in the entorhinal complex in familiar and novel environments: evidence for labile glycogenolytic modules in the rat. J Chem Neuroanat. 2006;31:108–113. doi: 10.1016/j.jchemneu.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Walz W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int. 2000;36:291–300. doi: 10.1016/s0197-0186(99)00137-0. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Passonneau JV. Factors affecting the turnover of cerebral glycogen and limit dextrin in vivo. J Neurochem. 1973;20:1543–1554. doi: 10.1111/j.1471-4159.1973.tb00272.x. [DOI] [PubMed] [Google Scholar]
- Wiesinger H, Hamprecht B, Dringen R. Metabolic pathways for glucose in astrocytes. Glia. 1997;21:22–34. doi: 10.1002/(sici)1098-1136(199709)21:1<22::aid-glia3>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Zar JH. Biostatical Analysis. Prentice-Hall Inc.; Englewood Cliffs, NJ: 1974. pp. 182–185. [Google Scholar]
- Zhang Q, Haydon PG. Roles for gliotransmission in the nervous system. J Neural Transm. 2005;112:121–125. doi: 10.1007/s00702-004-0119-x. [DOI] [PubMed] [Google Scholar]
- Zilles K, Wree A. Cortex: areal and laminal structure. In: Paxinos G, editor. The Rat Nervous System. 2. Academic Press; San Diego: 1995. pp. 649–685. [Google Scholar]