

Abstract
The tyrosine kinase Pyk2 plays a unique role in intracellular signal transduction by linking Ca2+ influx to tyrosine phosphorylation, but the molecular mechanism of Pyk2 activation is unknown. We report that Pyk2 oligomerization by antibodies in vitro or overexpression of PSD-95 in PC6-3 cells induces trans-autophosphorylation of Tyr402, the first step in Pyk2 activation. In neurons, Ca2+ influx through NMDA-type glutamate receptors causes postsynaptic clustering and autophosphorylation of endogenous Pyk2 via Ca2+- and calmodulin-stimulated binding to PSD-95. Accordingly, Ca2+ influx promotes oligomerization and thereby autoactivation of Pyk2 by stimulating its interaction with PSD-95. We show that this mechanism of Pyk2 activation is critical for long-term potentiation in the hippocampus CA1 region, which is thought to underlie learning and memory.
Introduction
Pyk2 consists of a FERM (band 4.1/ezrin/radixin/moesin homology), kinase, proline-rich, and focal adhesion targeting (FAT) domain (targets the homologous focal adhesion kinase FAK to focal adhesions) (see Fig. 1A) (Avraham et al., 1995; Lev et al., 1995; Sasaki et al., 1995; Yu et al., 1996). Tyr402 between the FERM and kinase domain becomes autophosphorylated during Pyk2 activation (Dikic et al., 1996; Salter and Kalia, 2004). Pyk2 is stimulated by depolarization-induced Ca2+ influx (Earp et al., 1995; Lev et al., 1995; Della Rocca et al., 1997; Girault et al., 1999) or PKC (Lev et al., 1995; Dikic et al., 1996; Girault et al., 1999; Huang et al., 2001; Salter and Kalia, 2004). However, it is unclear how Ca2+ or PKC activates Pyk2.
Figure 1.

Pyk2 dimerization by antibodies or by PSD-95 induces autophosphorylation. A, Schematic of Pyk2 depicting the FERM, tyrosine kinase, and FAT domains. Proline-rich segments are orange. Src phosphorylation sites are denoted as orange circles, and the autophosphorylation site (Tyr402) is red. The magenta bar on top represents the PSD-95 binding region (residues 671-875). B, Pyk2 was immunoprecipitated (αN Pyk2 antibody) from rat brain cytosol and incubated for 1 h at 4°C in the absence or presence of Mg-ATP, Ca2+ (10 mm), calmodulin (10 μm), or a mixture of PKC inhibitors (200 nm bisindolylmaleimide, 5 μm chelerythrine chloride, and 1 μm calphostin C) before immunoblotting with anti-phospho-Tyr402 (pY402), followed by reprobing with the monoclonal Pyk2 antibody. Autophosphorylation occurred independent of Ca2+/calmodulin (compare lanes 3, 4, 7, 8) or PKC activity (lanes 10, 11). C, Rat brain cytosol was incubated with αN, αC, monoclonal Pyk2 antibodies, or nonspecific rabbit or mouse IgG in the absence or presence of Mg-ATP as indicated and directly analyzed by immunoblotting with anti-phospho-Tyr402 and subsequently the monoclonal Pyk2 antibody. D, Increasing amounts of αN Pyk2 antibody were immobilized on protein A Sepharose, washed, incubated with identical amounts of rat brain cytosol, washed, incubated with Mg2+-ATP for 1 h at 4°C, and processed for immunoblotting with anti-phospho-Tyr402 and subsequently monoclonal Pyk2 antibody and an antibody against the light chain of the immunoprecipitating antibody. E, Immunoprecipitation from brain lysate with antibodies against Pyk2 (αN) or control rabbit IgG was followed by SDS-PAGE and silver staining. The thin band near the top of the blot corresponding to the molecular size of Pyk2 is labeled with an arrow and absent in control precipitates. The bands at 55 kDa are the heavy chains of the antibodies used in the immunoprecipitations. F, PC6-3 cells were transfected with PSD-95 24 h before immunoblotting. Immunosignals were quantified by film densitometry and phospho-Tyr402 values (top of inset) per total Pyk2 signal (bottom of inset) normalized to the no-transfection control. Shown are averages ± SEM of n = 3. *p < 0.05, statistically different from GFP-transfected cells by t test. G, E. coli was transformed with pET28a carrying His-tagged Pyk2 and expression induced with IPTG before extraction and immunoblotting with anti-phospho-Tyr402, followed by probing for total Pyk2. The lower band in the phospho-probing is likely a slightly truncated degradation product and was not visible in the reprobing for total Pyk2 because the reprobing was less sensitive than the original phospho-probing. A–G, All blots are representative of three independent experiments. IB, Immunoblots; IP, immunoprecipitation.
Phosphorylated Tyr402 stimulates Src by binding its Src homology 2 domain. Src in turn phosphorylates Tyr579 and Tyr580 in the activation loop of Pyk2 to enhance its catalytic activity (Lev et al., 1995; Dikic et al., 1996; Barsacchi et al., 1999; Avraham et al., 2000). Pyk2-activated Src also phosphorylates NR2 subunits to increase NMDA receptor (NMDAR) peak currents, which is important for induction of long-term potentiation (LTP) (Wang and Salter, 1994; Chen and Leonard, 1996; Köhr and Seeburg, 1996; Lu et al., 1998; Xiong et al., 1999; Huang et al., 2001; Salter and Kalia, 2004).
The FERM domain of FAK binds to its kinase domain causing autoinhibition (Lietha et al., 2007). An additional interaction of the FERM domain with the region containing Tyr397, which is homologous to Tyr402 in Pyk2, fosters the autoinhibited FAK conformation. How these structural features relate to Pyk2 activation is unclear, but the FERM domain of Pyk2 does play a role in regulating catalytic activity and localization to focal adhesions (Dunty and Schaller, 2002). Ca2+/calmodulin from cell extracts can directly or indirectly induce Pyk2 activation (Kohno et al., 2008). This activation was inhibited by mutations in the FERM domain, which might mediate Ca2+/calmodulin binding (Kohno et al., 2008). However, the homologous site is primarily buried in the FAK structure (Lietha et al., 2007) and unlikely to bind calmodulin. The above findings do not explain how FAK and Pyk2 are generally activated or specifically by Ca2+ influx or PKC. In contrast to FAK Tyr397 autophosphorylation, which depends on Src, autophosphorylation of Tyr402 as the initial step in Pyk2 activation occurs often independent of Src (Park et al., 2004; Corvol et al., 2005; Wu et al., 2006) (but see Discussion and Sorokin et al., 2001; Cheng et al., 2002). Also, deletion of the FERM domains leads to autophosphorylation of FAK on Tyr397 but not of Pyk2 on Tyr 402 (Wu et al., 2006).
PSD-95 links Pyk2 to NMDARs (Seabold et al., 2003). PSD-95 binds with its Src homology 3 (SH3) domain to the proline-rich region of Pyk2 (see Fig. 1A) (Seabold et al., 2003) and via PDZ domains to NMDAR C termini (Kornau et al., 1995; Kim et al., 1996). We found that NMDAR-mediated Ca2+ influx induces postsynaptic clustering and trans-autophosphorylation of Pyk2 on Tyr402 by PSD-95 binding. This event is stimulated by Ca2+/calmodulin and critical for LTP.
Materials and Methods
Ethical approval.
All animal procedures had been approved by the University of Iowa and followed National Institutes of Health guidelines or were performed in agreement with the guidelines of the policies on the Use of Animals at the University of Toronto.
Antibodies and other reagents.
Rabbit polyclonal antibodies against the N- and C-terminal portions of Pyk2 (residues 1-80 and 680-860, respectively) were provided by Drs. L. M. Graves and H. S. Earp (University of North Carolina at Chapel Hill, Chapel Hill, NC). These antibodies have been characterized and used extensively for immunofluorescence and immunoprecipitation (Yu et al., 1996; Li et al., 1999; Seabold et al., 2003). The mouse monoclonal anti-Pyk2 antibody, recognizing residues 833-899, was purchased from BD Transduction Laboratories. Rabbit polyclonal phosphospecific antibody against Pyk2 pY402 was purchased from Biosource International. Rabbit polyclonal anti-PSD-95 antibody was produced against residues 494-510 and purified as described previously (Sans et al., 2000). The mouse monoclonal antibodies anti-PSD-95, anti-MAP2B, and anti-green fluorescent protein (GFP) were obtained from Affinity Bioreagents, BD Transduction Laboratories, and Covance Research Products, respectively. Rabbit polyclonal anti-glutathione S-transferase (GST) antibody was as described previously (Leonard et al., 1998). Rabbit polyclonal anti-synapsin antibody was provided by Dr. P. De Camilli (Yale University, New Haven, CT) (De Camilli et al., 1983). Chromatographically purified nonspecific rabbit and mouse IgG antibodies were purchased from Zymed. Horseradish peroxidase (HRP)-conjugated goat anti-mouse and goat anti-rabbit secondary antibodies were purchased from Bio-Rad, and protein A HRP was purchased from GE Healthcare. Alexa Fluor-conjugated secondary antibodies, including goat anti-rabbit 568, goat anti-mouse 488, and goat anti-mouse 660, were obtained from Invitrogen. Purified recombinant calmodulin was provided by Dr. M. A. Shea (University of Iowa, Iowa City, IA) (Akyol et al., 2004). MK801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate], EGTA, HEPES, glutamate, NMDA, ionomycin, phorbol 12-myristate 13-acetate (PMA), trifluoperazine (TFP), and W7 [N-(6-aminohexyl)-5-chloro-1-naphthalenesulfonamide] were purchased from Sigma, calmidazolium and calphostin C from Calbiochem, and bisindolylmaleimide and chelerythrine chloride from LC Laboratory. Pervanadate was prepared from 1 m sodium orthovanadate (Sigma) dissolved in H2O, pH 10 (the solution was heated at 60°C until the solution became clear), by addition of H2O2 at a 1:1 ratio no more than 15 min before use.
Expression constructs.
Bacterial expression vectors for GST fused to the N terminus of full-length PSD-95 and the SH3 domain of PSD-95 (residues 431-500) were provided by Dr. C. C. Garner (Stanford University, Palo Alto, CA) and Dr. B. Kay (University of Illinois at Chicago, Chicago, IL), respectively. GST was expressed using the pGEX4T-1 vector (Pfizer). His-tagged Pyk2(1-1009) was produced by ligating the Pyk2 cDNA encoding residues 1-1009 into the BamHI and EcoRI sites of pET28a. N-terminally GST-tagged Pyk2(671-875) was constructed using PCR amplification of the fragment and subsequent ligation into the BamHI and EcoRI sites of pGEX4T-1. PCR primers were the 5′ primer 5′-CAT CAT GGA TCC CGC TTT ACG GAG CTT GTG TGC AGT CTC-3′ and 3′ primer 5′-CAC GAG GAA TTC TCA CCT GTC CAA GTT GGC TGT AGG CTG GAT-3′. For expression in mammalian cell lines, PSD-95 pCMV vector was provided by Dr. C. C. Garner. GFP expression was achieved with pEGFP–C3 (Clontech). For production of enhanced GFP (EGFP) constructs, the pEGFP–C3 multiple cloning site was modified by annealing and ligating corresponding primers into the XhoI and XbaI sites of pEGFP–C3. The 5′ primer was 5′-phos-TCG AGG GCG GCC GCG GAT CCG ATA TCA AGC TTG CTA GCG AAT TCG TCG ACT-3′, and the 3′ primer was 5′-phos-CTA GAG TCG ACG AAT TCG CTA GCA AGC TTG ATA TCG GAT CCG CGG CCG CCC-3′. The new vector (pEGFP–C3G) was used to transfer constructs directly from pGEX4T-1 vectors. Pyk2(1-1009)–GFP was constructed by BamHI/EcoRI-mediated excision of the fragment and subsequent ligation into the pEGFP–C3G vector. PSD-95 SH3–GFP (residues 431-500) was constructed by cutting the SH3 domain from PSD-95 SH3–GST with EcoRI. It was then ligated into the pEGFP–C3G vector. All sequences were verified before use of the construct.
Lentiviral constructs.
The University of Iowa Center for Gene Therapy for Cystic Fibrosis provided the feline immunodeficiency virus (FIV) parent vector (pVETL) and produced the final viral vectors (Johnston et al., 1999; Stein and Davidson, 2002). PSD-95 SH3–GFP pVETL (residues 431-500) was constructed using AgeI and SalI to excise PSD-95 SH3–GFP from the pEGFP–C3G construct, followed by Klenow-mediated filling of the overhangs and blunt-end ligation into the EcoRV site of pVETL. The sequences were verified before submitting them to the University of Iowa Center for Gene Therapy for Cystic Fibrosis for production of the virus. The viral titers varied between 1 × 106 and 1 × 108.
Expression of fusion proteins.
GST- and poly-His-tagged fusion constructs were transformed into Escherichia coli by electroporation. Cells were grown in Lauria broth and induced at an optical density of 0.6–0.8 with 0.2 mm isopropyl β-d-thiogalactoside (IPTG). Cells were then collected by centrifugation and frozen for storage. For purification, cell pellets were thawed, resuspended, and incubated for 30 min in ice-cold TBS (150 mm NaCl and 15 mm Tris-Cl, pH 7.4) containing 100 μg/ml lysozyme and a low concentration of protease inhibitors [200 μm phenylmethylsulphonylfluoride (PMSF), 1 μg/ml pepstatin A, 2 μg/ml aprotinin, and 1 μg/ml leupeptin]. Sarkosyl (1.5%) and β-mercaptoethanol (10 mm) were then added for 15 min on ice. Once the incubation was complete, lysates were centrifuged for 45 min at 250,000 × g. The supernatants were removed and neutralized with 2% Triton X-100.
Transient transfection of PC6-3 cells.
PC6-3 cells (supplied by Dr. S. Strack, University of Iowa, Iowa City, IA) were seeded at 2.5 × 106 cells per 100 mm dish in RPMI medium (RPMI 1640 supplemented with 5% horse serum, 5% fetal bovine serum, 5% calf serum, 0.5% penicillin/streptomycin, 1% glutamine, and 1 mm sodium pyruvate). Cells were transfected with Lipofectamine 2000 when 80–90% confluent. Briefly, 30 μg of DNA was added to serum-free Opti-MEM. An 8% Lipofectamine 2000 solution was made simultaneously in serum-free Opti-MEM. After 5 min at room temperature, the DNA mix was added to the Lipofectamine mix, followed by a 20 min incubation at room temperature. The medium on the cells was then replaced with Opti-MEM, followed by addition of the DNA/Lipofectamine solution. The dishes were gently mixed and incubated for 6 h. The medium was then replaced with RPMI-containing serum. The cells were harvested 48 h after transfection using a cell scraper and Triton X-100 homogenization buffer (1% Triton X-100, 150 mm NaCl, 10 mm Tris-Cl, 20 mm EDTA, and 10 mm EGTA, pH 7.4) containing protease inhibitors (here 200 μm PMSF, 1 μg/ml pepstatin A, 20 μg/ml aprotinin, 10 μg/ml leupeptin, and 8 μg/ml calpain inhibitor I/II) and phosphatase inhibitors (1 mm pervanadate, 25 μm NaF, and 25 mm NaPPi). The cells were then homogenized with a dounce homogenizer, followed by centrifugation at 250,000 × g for 15 min. Supernatant was removed, and the total protein was quantified with a BCA assay. An equal amount of protein (25 μg) was extracted with SDS sample buffer and loaded for SDS-PAGE and subsequent immunoblotting with phosphospecific pY402 Pyk2 antibody. The immunoblots were then stripped and reprobed for total Pyk2 using the monoclonal anti-Pyk2 antibody.
Primary hippocampal culture production and maintenance.
Primary hippocampal cultures were prepared as described previously (Lim et al., 2003; Chen et al., 2008). Briefly, hippocampi from embryonic day 18 Harlan Sprague Dawley rats were removed and incubated in HBSS (Invitrogen) with trypsin (0.03%) for 15 min at 37°C. The cells were then washed three times with HBSS, followed by trituration to dissociate cells. Dissociated cells were counted and plated for immunofluorescence on glass coverslips (60,000 cells per 35 mm dish) for microscopic analysis or in 100 mm culture dishes (800,000 cells per 100 mm dish) for biochemical analysis. The cells were incubated in Neurobasal medium (Invitrogen) containing custom-made NS21 supplement (Chen et al., 2008), 0.6 mm glutamine, and 5% fetal bovine serum (Brewer et al., 1993). After 3–4 h, the incubation medium was replaced with serum-free medium, and cells were maintained at 37°C in humidified air composed of 95% air and 5% CO2. One-third of the medium was exchanged weekly.
Transient transfection of primary hippocampal cultures.
Primary hippocampal cultures [15 d in vitro (DIV)] were transfected using an adapted calcium phosphate protocol. The medium was replaced with freshly prepared Neurobasal medium containing NS21 30 min before transfection. The removed conditioned medium was then retained for use later in the procedure. DNA (5 μg) was added to CaCl2 (200 mm). An equal volume of 2× BBS (final concentrations in mm: 140 NaCl, 0.75 Na2HPO4, and 25 N,N-bishydroxyethyl-2-aminoethane-sufonic acid, pH 7.1) was added dropwise, followed by immediate vortexing. The DNA/BBS mixture was then added dropwise to the neurons, followed by gentle mixing. After a 3.5 h incubation, the medium was removed and the cultures were washed once with HBSS (in mm: 135 NaCl, 4 KCl, 1 Na2HPO4, 2 CaCl2, 1 MgCl2, 10 glucose, and 20 HEPES, pH 7.35). Immediately after the wash, the conditioned medium was added. The cultures were allowed to express protein for 72 h at 37°C in humidified air composed of 95% air and 5% CO2, followed by use for immunofluorescence.
Infection of primary hippocampal cultures.
Primary hippocampal cultures (15 DIV) were infected using FIV carrying GFP-tagged PSD-95 SH3. Cultures (100 mm dishes) received virus at a multiplicity of infection of 10:1. The virus was added, followed by gentle mixing. After 24 h, half of the medium was replaced with fresh Neurobasal medium supplemented with NS21. Immunofluorescence and biochemical studies were initiated 72 h after infection.
Stimulation of hippocampal cultures for biochemical analysis and use of various agonists and antagonists.
Cultures (18 DIV, 100 mm plates) were pretreated with calmodulin inhibitors (W7, TFP, and calmidazolium) for 30 min, with tetrodotoxin (TTX) for 15 min, and with pervanadate for 5 min if indicated before stimulation with vehicle, NMDA, ionomycin, or PMA for 15 min. The three different calmodulin inhibitors block Ca2+-dependent binding of calmodulin to substrates and were used to evaluate the role of calmodulin in the interaction between Pyk2 and PSD-95, as well as Pyk2 activation, at the following concentrations: W7 at 10 μm, TFP at 20 μm, and calmidazolium at 30 μm (Schweitzer, 1987; Wakamori et al., 1993; Della Rocca et al., 1997; Liu et al., 1999; Heidinger et al., 2002). W7 is a member of the naphthalene-sulfonamide class of drugs, which also appears to block NADPH oxidase activation by arachidonate (Sakata et al., 1987). TFP, a member of the phenothiazine family, is the best characterized calmodulin antagonist because crystal structures have been solved illustrating its interaction with calmodulin (Cook et al., 1994; Vandonselaar et al., 1994). However, similar to W7, TFP can inhibit NADPH oxidase activation. Calmidazolium can inhibit adenylyl cyclase activity at concentrations ranging between 20 and 50 μm (Haunsø et al., 2003). However, using each of the three inhibitors individually ensures that the observed inhibition of Pyk2 binding to PSD-95 and the ensuing Pyk2 activation is attributable to their common inhibition of calmodulin instead of idiosyncratic side effects. Bath application of the specific NMDAR agonist NMDA in the presence of TTX is an established method for analyzing signaling pathways involving the NMDAR (Hell et al., 1996; Siciliano et al., 1996; Leonard et al., 1999; Ahmed et al., 2006). Of specific relevance to our studies, Src signaling downstream of NMDAR activation and the ensuing NMDAR potentiation have been monitored in intact neurons with NMDA treatments lasting 15 min (Lu et al., 1999; Crossthwaite et al., 2004). Similarly, the Ca2+ ionophore ionomycin is commonly used in studies on activation of Pyk2 by Ca2+ influx at 1 μm for 10–30 min (Sabri et al., 1998; Byron and Lucchesi, 2002; Cheng et al., 2002). PMA is a PKC activator that is often used to activate Pyk2 or test the role of PKC in Pyk2 signaling at 100 nm over 10–15 min and longer (Siciliano et al., 1996; Sabri et al., 1998; Lu et al., 1999, 2000; Huang et al., 2001; Frank et al., 2002).
Neurons were harvested immediately after treatments in 1 ml of ice-cold deoxycholate homogenization buffer (1% deoxycholate, 137 mm NaCl, 50 mm Tris-Cl, 10 mm EDTA, and 10 mm EGTA, pH 8.5) containing protease and phosphatase inhibitors as before, followed by homogenization with a dounce homogenizer and high-speed centrifugation (250,000 × g for 15 min). Supernatant was then collected for either immunoprecipitation or direct immunoblotting.
Preparation and treatment of acute cortical slices.
Transverse cortical slices (350 μm) were prepared from 21-d-old male Harlan Sprague Dawley rats (Hell et al., 1995; Leonard et al., 1999; Lim et al., 2003; Lu et al., 2007). Slices were sectioned in artificial CSF (ACSF) (127 mm NaCl, 26 mm NaHCO3, 1.9 mm KCl, 1.2 mm KH2PO4, 1 mm CaCl2, 2 mm MgSO4, and 10 mm dextrose, 290–300 mOsm/kg, equilibrated in 95%O2/5%CO2). Each slice was then incubated in ACSF for 30 min at 34°C, followed by 30 min at 22°C with continuous aeration in a submersion chamber. Slices were then transferred to modified ACSF containing 2.2 mm CaCl2 and 1 mm MgSO4 for 15 min before pretreatment with calmodulin inhibitors (W7, TFP, and calmidazolium; 30 min) and TTX (15 min) if indicated and subsequent incubation with vehicle, NMDA, ionomycin, or PMA for 15 min. Slices were immediately homogenized with a dounce homogenizer in 1 ml of deoxycholate homogenization buffer (1% deoxycholate, 137 mm NaCl, 50 mm Tris-Cl, 10 mm EDTA, and 10 mm EGTA, pH 8.5) containing protease and phosphatase inhibitors as before. Lysates were then centrifuged (250,000 × g for 15 min), and supernatants were collected for immunoprecipitation or direct immunoblotting.
Preparation of cytosolic brain extracts.
Male Harlan Sprague Dawley rats (8 weeks old) were anesthetized with halothane and decapitated. Brains were immediately placed in ice-cold sucrose buffer (in mm: 300 sucrose, 10 EDTA, 10 EGTA, 10 Tris-Cl, and 1 pervanadate, pH7.4) containing protease inhibitors as before, homogenized in a dounce homogenizer, and centrifuged at low speed (5,000 × g) to clear larger cell fragments and then at high speed (200,000 × g) to remove the crude membrane fraction. Supernatant was collected for use in experiments requiring the brain cytosol. Triton X-100 (1%) was added to the isolated cytosol before use to reduce nonspecific interactions.
Immunoprecipitation.
Triton X-100 extracts from hippocampal culture (one 100 mm dish), cortical slices (one slice), or brain (cytosol; 3 mg of total protein unless otherwise noted) were added to protein A Sepharose (rabbit antibodies) or protein L Sepharose (mouse antibodies) along with 1 μg of the desired antibody. Samples were incubated for 3 h at 4°C, followed by washes with 0.1% Triton X-100/TBS. Experiments involving hippocampal culture or cortical slice lysates, when material was limited, used sequential immunoprecipitation with proper nonspecific and then specific antibody. Resins were washed with 0.1% Triton X-100/TBS before SDS-PAGE and immunoblotting.
Pyk2 autophosphorylation.
Autophosphorylation in cytosolic extract, or after immunoprecipitation using 3 mg of total protein as starting material (unless otherwise noted), was initiated with the addition of 2.5 mm ATP and 25 mm MgCl2 or TBS as indicated. Incubation buffers also contained 50 μm EGTA to remove free Ca2+ contaminations for nominally Ca2+-free conditions. For incubations in the presence of free Ca2+, 10 mm CaCl2 was added to this buffer. After 1 h at 4°C, the reaction was terminated by addition of 50 mm EDTA or SDS sample buffer.
Pulldown assays.
For pulldown of calmodulin by the bacterially expressed His-tagged SH3–GK module of PSD-95, this polypeptide was immobilized on Ni2+ chelate agarose (GE Healthcare), washed with 0.1% Triton X-100/TBS, and incubated with calmodulin in 0.1% Triton X-100/TBS containing 50 μm EGTA. If indicated, 10 mm CaCl2 was added to the incubation buffer. Resins were washed again with the same buffer used for incubation except calmodulin was omitted. For pulldown of PSD-95–GST by bacterially expressed Pyk2, Pyk2 was immunoprecipitated with the αN Pyk2 antibody before incubation with GST or PSD-95–GST and washing analogous to the pulldown experiments with His-tagged SH3–GK.
Immunoblotting.
Samples were extracted with SDS sample buffer (3% SDS, 10% sucrose, 90 mm Tris-Cl, 20 mm DTT, and 60 μm bromophenol blue) at 90°C for 10 min, resolved by SDS-PAGE, and transferred to polyvinylidene fluoride membranes. Membranes were blocked with 7% milk/TBS for 2 h, incubated with primary antibody for 2 h at room temperature or overnight at 4°C, washed, incubated with HRP-conjugated secondary antibodies for 1 h at room temperature, washed extensively, and developed with ECL detection kit (GE Healthcare). When indicated, membranes were stripped (2% SDS, 20 mm DTT, and 62.5 mm Tris-Cl, pH 6.7), washed with water, and reprobed (Hell et al., 1993). Immunosignals were in the linear range and digitalized and quantified with Photoshop (Adobe Systems) as detailed previously (Davare and Hell, 2003; Merrill et al., 2007).
Immunofluorescence microscopy.
Primary hippocampal neurons (18 DIV) were cultured on coverslips in 35 mm dishes, preincubated with W7 (10 μm), EGTA (2 mm), or MK801 [10 μm (Lu et al., 1998; Fong et al., 2002; Gardoni et al., 2002)] for 30 min and with TTX for 15 min, and treated with vehicle, glutamate, NMDA, ionomycin, or PMA for indicated time periods at 37°C. Coverslips were rapidly washed with PBS (in mm: 137 NaCl, 3 KCl, 10 Na2HPO4, and 1 KH2PO4, pH 7.4), fixed in 4% paraformaldehyde/4% sucrose/PBS for 15 min, washed (three times for 5 min in PBS), permeabilized with 0.5% Triton X-100/PBS for 20 min, washed (two times for 5 min in PBS), and incubated with blocking buffer (2% glycerol, 5% FBS, 2% goat serum, and 50 mm NH4Cl in PBS) for 2 h at room temperature and then with primary antibody in blocking buffer overnight at 4°C. Antibodies included the N-terminal anti-Pyk2 antibody (1:500), anti-PSD-95 (1:500), anti-bassoon (1:1,000), anti-MAP2B (1:1,000), and anti-synapsin (1:1,000). Coverslips were washed (three times for 5 min in PBS) and incubated with blocking buffer for 30 min and with secondary antibody (1:500) in blocking buffer for 1 h at 4°C. Coverslips were washed with PBS (four times for 5 min) and water (three times for 5 min), mounted on slides using Prolong Antifade Gold Reagent (Invitrogen), and incubated for 24 h at 4°C. Images were collected using an epifluorescence Olympus microscope with a 100× objective. Bandpass filter sets were 490 nm/528 nm, 555 nm/617 nm, and 635 nm/685 nm. Negative controls included omission of primary antibody to exclude nonspecific signals from secondary antibodies and bleed through from other channels. Coverslips were coded and randomized for image collection by an individual, who was blind to the treatments. Identical exposure time and contrast settings were used for each field in a given experiment. The dendritic length and total number of puncta in each image were also determined in a blind manner. Dendritic length was calculated using NIH ImageJ, whereas puncta were quantified using a customized MATLAB package (MathWorks). The MATLAB package removed smoothly distributed staining like that found in dendritic shafts and the soma with the top-hat transform. A threshold value was determined and held constant for all images such that pixels with intensities below that level were disregarded. It then counted the remaining puncta, defined as a series of 5 or more contiguous pixels above threshold. Colocalization of Pyk2 and PSD-95 puncta was determined by designating the 10 pixels in each punctum with the highest intensity as the region of interest (ROI). Overlap of four pixels in two ROIs was required for colocalization. This provided two variables for comparison between various treatment groups. The first is the total number of puncta under different stimulation conditions, and the second is the colocalization of Pyk2 puncta with a synaptic marker such as PSD-95.
Cell isolation and whole-cell recording.
CA1 neurons were isolated from hippocampal slices of postnatal day 14–21 Wistar rats as described previously (Wang and MacDonald, 1995). Whole-cell patch recordings were performed under perfusion at room temperature. The extracellular solution was composed of 140 mm NaCl, 1.3 mm CaCl2, 25 mm HEPES, 33 mm glucose, 5.4 mm KCl, 0.5 μm tetrodotoxin, and 0.5 μm glycine, pH 7.3–7.4, with osmolarity ranging from 320 to 330 mOsm. No Mg2+ was added to record NMDA-evoked currents. The intracellular solution consisted of the following (in mm): 11 EGTA, 10 HEPES, 2 MgCl2, 2 tetraethylammonium chloride, 1 CaCl2, 140 CsF, and 4 K2ATP. NMDA currents were evoked by rapid application of NMDA (50 μm) delivered from a multi-barreled fast perfusion system. NMDA was applied once every 60 s for a period of 3 s. Glycine at 1 μm was present throughout the recordings in the bath. The membrane potential was held at −60 mV. A voltage step of 10 mV was applied before NMDA application to monitor series resistance.
Whole-cell patch recordings from hippocampal slices.
Transverse hippocampal slices were prepared from 4- to 6-week-old male Sprague Dawley rats and placed in a holding chamber for at least 1 h before recording. A single slice was transferred to the recording chamber and superfused with ACSF (2 ml/min) composed of the following (in mm):124 NaCl, 3 KCl, 1.25 NaH2PO4, 1.3 MgCl2, 2.6 CaCl2, 26 NaHCO3, 10 glucose, and 0.01 bicuculline methiodide (saturated with 95% O2–5%CO2 at 28–30°C). Synaptic responses were evoked with a bipolar tungsten electrode located ∼50 μm from the cell body layer in CA1. Test stimuli were evoked at 0.05 Hz with the stimulus intensity set to 25% of maximal synaptic response. Tetanic stimulations consisted of two trains of 100 Hz stimuli lasting 500 ms at an intertrain interval of 10 s and were performed under current clamp. The average of EPSC amplitudes from the 5 min period immediately before tetanus was defined as baseline. For voltage-clamp recordings, the patch pipette (4–6 MΩ) solution contained the following (in mm):132.5 K-gluconate, 17.5 KCl, 10 HEPES, 0.2 EGTA, 2 Mg-ATP, 0.3 GTP, and 5 QX-314 [2(triethylamino)-N-(2,6-dimethylphenyl) acetamine], pH 7.25 (290 mOsm). The intracellular solution was supplemented with GST, GST–SH3, or GST–Pyk2(671-875) polypeptides. The peptides were numbered, and the individual performing the recordings was unaware of which was applied in experiments. Patch recordings in slice were performed using the “blind” patch method. Cells were voltage clamped at −65 mV. Data were used only after the access resistance had stabilized. Signals were amplified using an Axopatch 1-D, sampled at 5 kHz, and analyzed with pClamp6 software (Molecular Devices). Series resistance ranged from 10 to 20 MΩ, as estimated from series resistance compensation of current responses to voltage steps of 5 mV, and cells were discarded if the resistance changed by >15%.
To test whether the GST polypeptides affected basal synaptic NMDAR activity, NMDAR-mediated EPSCs were recorded in the presence of CNQX (10 μm) to block AMPA-type glutamate receptors (AMPARs). To increase the amplitude of these currents, the holding potential of the neurons was transiently raised from −65 to −40 mV for 2.5 s whenever a test stimulus was applied.
Statistical analysis.
Statistical analysis was performed in Prism (GraphPad Software). Statistical significance was determined using a paired two-tailed t test with a 95% confidence interval. Three independent experiments were performed separately on different days for all quantifications. For each immunofluorescence experiment, 5–10 images were taken from any single coverslip, and values were averaged before statistical analysis for the means from the three independent experiments.
Results
Pyk2 dimerization induces its autophosphorylation
As the unique central intermediary between Ca2+ influx and tyrosine phosphorylation, Pyk2 has been the subject of numerous studies. We attempted to stimulate Pyk2 after immunoprecipitation from brain lysate by addition of Ca2+ and calmodulin. Surprisingly, addition of Mg-ATP alone to Pyk2 immunocomplexes was sufficient to induce strong Tyr402 phosphorylation (Fig. 1B, compare lanes 2, 4), an established and reliable readout for Pyk2 activation (Dikic et al., 1996; Park et al., 2004). Neither Ca2+ alone nor Ca2+/calmodulin enhanced this autophosphorylation. In agreement with previous findings (Lev et al., 1995), these data indicate that Ca2+ and calmodulin alone are not sufficient to stimulate autoactivation of Pyk2. Furthermore, autophosphorylation was unaffected by inclusion of a mixture of three structurally and functionally different PKC inhibitors (Fig. 1B, compare lanes 10, 11). We also evaluated whether Ca2+/calmodulin or calcium/calmodulin-dependent kinase II (CaMKII) were present in Pyk2 immunoprecipitates from rat brain with negative results (data not shown), yet Pyk2 effectively autophosphorylated in such immunocomplexes (Fig. 1B). Accordingly, antibody-induced autophosphorylation bypasses the Ca2+ and PKC dependence of Pyk2 activation.
The effective autophosphorylation of Pyk2 in the immunocomplexes, which was not observed if Pyk2 is in solution (Fig. 1C, lanes 1, 8), suggests that dimerization of Pyk2 itself can induce the phosphorylation reaction. Such a mechanism would be analogous to trans-autophosphorylate by several receptor-type tyrosine kinases during ligand-induced dimerization (Ullrich and Schlessinger, 1990; Hubbard and Miller, 2007), which induces structural changes in their activation loops to disinhibit their catalytic domains (Zhang et al., 2006). In fact, overexpression of Pyk2 leads to its activation via trans-autophosphorylation of Tyr402 (Park et al., 2004). Although stable Pyk2 oligomers were not observed in this study, an attractive explanation is that high enough concentrations of Pyk2 foster transient or semi-stable formation of protein complexes that aligns two Pyk2 polypeptides for trans-autophosphorylation.
To determine whether dimerization is sufficient to induce autophosphorylation of Pyk2 in brain cytosol, three different Pyk2 antibodies targeting various regions of Pyk2 were used: a polyclonal antibody directed against the N-terminal 80 residues of Pyk2 (αN), a polyclonal antibody recognizing residues 680-860 of Pyk2 (αC), and a monoclonal antibody targeting residues 833-899 of Pyk2 (mono). The recognition sites for each of the antibodies selected for these experiments are distant from the catalytic domain and from Tyr402 to allow the structural flexibility that might be necessary for trans-autophosphorylation. Each of the three antibodies induced Pyk2 autophosphorylation in brain cytosol in the presence of Mg-ATP (Fig. 1C, lanes 3, 5, 7). Nonspecific antibodies did not lead to substantial autophosphorylation (lanes 1, 8).
To further scrutinize this point, Pyk2 was immunoprecipitated with increasing amounts of αN immobilized on protein A Sepharose before addition of aliquots of brain cytosolic extracts. Each aliquot contained 500 μg of protein (Bradford protein assay). After washing, samples were incubated with Mg-ATP and analyzed by immunoblotting (Fig. 1D). Probing for total Pyk2 shows that the amount of immunoprecipitated Pyk2 saturated at ∼0.25 μg of αN (middle panel), which completely removed Pyk2 from the cytosol samples (data not shown). Probing for the antibody light chain in the immunoprecipitates illustrates the increase in antibody (bottom panel). Probing for Tyr402 phosphorylation indicates that the total amount of phosphorylated Tyr402 initially increased in parallel with the amount of Pyk2 protein. However, beyond an optimal concentration at which Pyk2 immunoprecipitation was saturated (∼0.25 μg of αN), Tyr402 phosphorylation rapidly decreased. This decrease must be attributable to a change in the Pyk2:antibody stoichiometry, being closest to the optimal 2:1 ratio at ∼0.25 μg of αN and reaching nearly 1:1 during additional increases of αN because an excess of αN increasingly promoted binding of one rather than two Pyk2 molecules per antibody complex. Accordingly, trans-autophosphorylation occurred at intermediate antibody concentrations when the amount of IgG-dimerized Pyk2 was highest. cis-autophosphorylation was not detectable as illustrated by the lack of autophosphorylation at higher αN concentrations when individual Pyk2 molecules were on separate antibody complexes too far apart for trans-autophosphorylation. Finally, monoclonal anti-Pyk2 was treated with papain to produce Fab fragments, which are no longer linked together via the heavy chain disulfide bridge but rather constitute monomeric epitope binding sites. The Fab fragments were no longer capable of inducing Tyr402 phosphorylation in cytosolic solution when the same amount of untreated monoclonal antibody effectively induced Tyr402 phosphorylation as originally seen in Figure 1C (data not illustrated).
It is theoretically possible that another kinase is stoichiometrically associated with Pyk2 in such a steric arrangement that enables such a hypothetical kinase to phosphorylate a second Pyk2 complex in trans but not the Pyk2 directly bound to it. However, silver staining for total protein in the Pyk2 immunoprecipitates only identified one main single band of the expected size of Pyk2 itself near the top of this relatively high-percentage gel (Fig. 1E) except for the heavy chain of the immunoprecipitating antibody. This band was absent in control immunoprecipitates, confirming its identity as being Pyk2. No other band was detectable, indicating that there is no other major protein precipitating with Pyk2 with a near stoichiometric relationship under basal conditions.
Ectopic PSD-95 expression leads to Pyk2 activation
Pyk2 binds to PSD-95 (Seabold et al., 2003), which is one of the most prominent scaffolding proteins at postsynaptic sites. PSD-95 can oligomerize via two mechanisms. One involves palmitoylation of the cysteine residues in position 3 and 5 near the N terminus of PSD-95 (Craven et al., 1999; El-Husseini Ael et al., 2002), although precisely how palmitoylation leads to PSD-95 oligomerize is unclear. The other is mediated by binding of the SH3 domain of one PSD-95 molecule to the GK domain of another PSD-95 molecule (Masuko et al., 1999; McGee and Bredt, 1999; Shin et al., 2000; McGee et al., 2001). We hypothesized that ectopic expression of PSD-95 fosters Pyk2 dimerization and thereby trans-autophosphorylation. We used PC6-3 cells, a subclone of PC12 cells (Pittman et al., 1993). PC12 cells are widely used for studying Pyk2 because of their high levels of endogenous Pyk2 and their similarity to neurons after treatment with nerve growth factor (Lev et al., 1995; Barsacchi et al., 1999; Ivankovic-Dikic et al., 2000; Park et al., 2000; Haglund et al., 2004; Park et al., 2004; Banno et al., 2005). However, PC6-3 cells do not possess detectable levels of endogenous PSD-95 (data not shown). PC6-3 cells were transfected with either GFP or GFP and PSD-95. After 48 h, immunoblotting of cell extracts showed that ectopic expression of PSD-95, but not GFP alone, induced Pyk2 Tyr402 phosphorylation without altering total Pyk2 levels (Fig. 1F). The most obvious explanation for this effect is that increased PSD-95 levels shift the equilibrium from free to PSD-95-bound Pyk2 under basal activity levels of the PC6-3 cells. In neurons, Pyk2 binding to PSD-95 and thereby Pyk2 autoactivation is triggered by influx of Ca2+, which acts in conjunction with calmodulin, as detailed below. However, PSD-95 overexpression in PC6-3 cells can lead to Pyk2 autoactivation without Ca2+ influx because Pyk2 binds even without Ca2+/calmodulin to PSD-95, although this interaction is much weaker than if Ca2+/calmodulin are present (see Fig. 3B, lanes 3–6). These findings thus indicate that Pyk2 binding to PSD-95 induces Pyk2 trans-autophosphorylation and thereby activation.
Figure 3.

Ca2+/calmodulin stimulates PSD-95 binding to Pyk2. A, Ni2+-agarose was incubated with or without the bacterially expressed His-tagged SH3–GK module of PSD-95 and subsequently with calmodulin (10 μm) in the presence and absence of Ca2+ (10 mm) before immunoblotting (IB). Ponceau S staining indicates immobilization of SH3–GK (lanes 1, 2). Probing with anti-calmodulin antibodies shows specific calmodulin binding to the SH3–GK domain when Ca2+ is present. B, Bacterially expressed Pyk2 was immunoprecipitated (IP) using αN Pyk2 (lanes 1–6) or, as negative control, nonspecific rabbit IgG (lanes 7, 8) before incubation with GST or full-length PSD-95–GST with and without Ca2+ (10 mm) and calmodulin (10 μm) as indicated. Samples were washed with and without Ca2+ corresponding to the binding incubation conditions. Samples underwent immunoblotting with monoclonal Pyk2 and subsequently GST antibodies. GST–PSD-95 (lanes 3–6) but not GST alone (lanes 1, 2) bound to Pyk2. This interaction was increased in the presence of Ca2+ and calmodulin (lane 6). Lanes 9 and 10 show immunoblots with anti-GST of input material. GST–PSD-95 was partially degraded at the C terminus (GST resided at the N terminus). The lack of binding of the truncated polypeptide in the middle region but not the top one-third of the gel (compare lane 10 with lane 6) is consistent with proteolytic removal of SH3 in the more extensively truncated form of GST–PSD-95.
Notably, we also observed massive Tyr402 phosphorylation when Pyk2 was expressed in E. coli (Fig. 1G). Such an induction of tyrosine phosphorylation by ectopic expression of a tyrosine kinase in E. coli appears unusual. It is once more best explained by our model of trans-autophosphorylation of Pyk2 at elevated Pyk2 concentrations, which can lead to trans-autophosphorylation during Pyk2 overexpression in PC12 cells attributable to weak or transient direct or indirect interactions between individual Pyk2 molecules (Park et al., 2004).
Ca2+ influx and stimulation of PKC induce Pyk2 binding to PSD-95
Based on the above results (Fig. 1), we hypothesized that treatment with pharmacological agents known to activate Pyk2 would do so by inducing its clustering. In neurons, such clustering could be accomplished by binding to PSD-95. After a 5 min pretreatment with the Na+ channel blocker TTX to prevent action potentials and thereby overexcitation (Hell et al., 1996; Leonard et al., 1999), acute cortical slices and primary hippocampal cultures were incubated with vehicle, the NMDAR agonist NMDA (to selectively induce Ca2+ influx through NMDARs), the Ca2+ ionophore ionomycin, or the PKC activator PMA (for more details and discussion of the use of these drugs see, Materials and Methods). Slices and cultures were then extracted with 1% deoxycholate before sequential immunoprecipitation with nonspecific control IgG and anti-PSD-95 and immunoblotting with anti-Pyk2 and subsequently anti-PSD-95. NMDA, ionomycin, and PMA increased coimmunoprecipitation of Pyk2 with PSD-95 several fold in both systems compared with vehicle control (Fig. 2A,B). None of the treatments affected the total amount of Pyk2 present in tissue or culture lysates (Fig. 2A,B). Immunoprecipitation with nonspecific IgG resulted in no signal, ensuring that immunoprecipitation of PSD-95 and the associated Pyk2 was specific.
Figure 2.
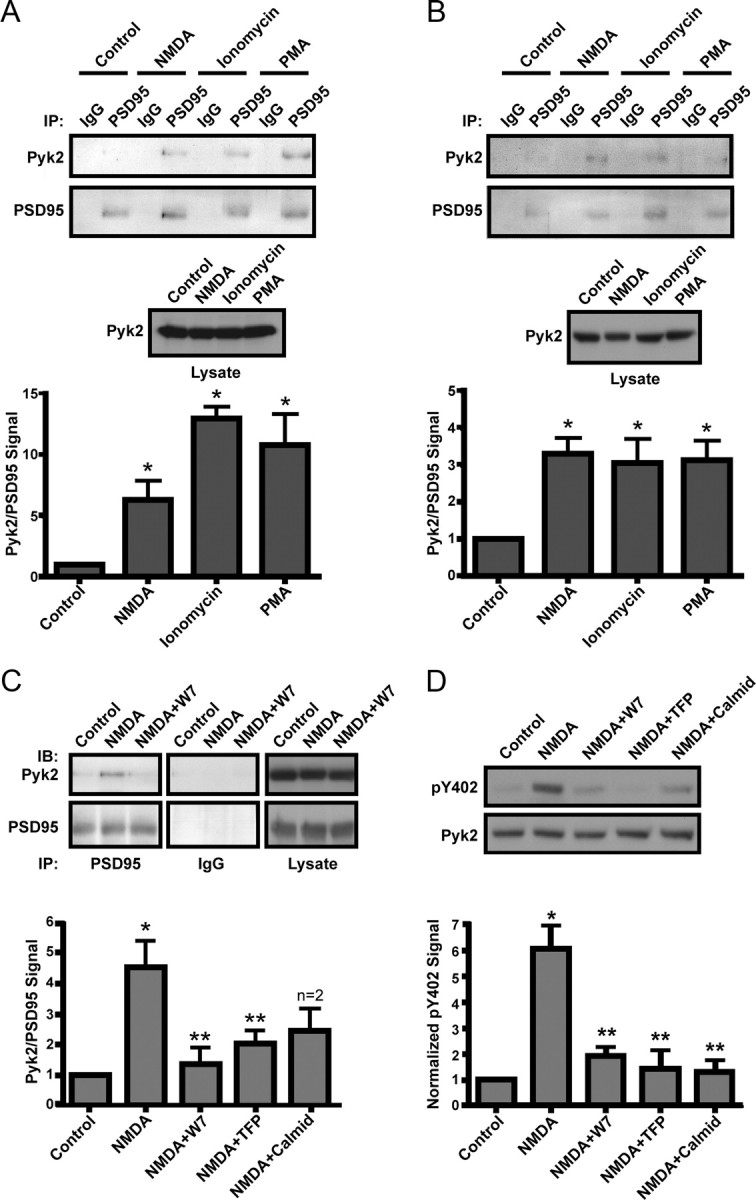
Stimulation of Pyk2 binding to PSD-95 and Pyk2 autophosphorylation in intact neurons. A, B, Acute cortical slices (A) or primary hippocampal cultures (B; 18 DIV) were incubated with TTX (1 μm) plus vehicle (Control), NMDA (50 μm), ionomycin (1 μm), or PMA (100 nm) for 15 min and extracted (1% deoxycholate) before immunoprecipitation (IP) with control IgG and then anti-PSD-95. Immunoblots (IB) were probed with anti-PSD-95 and reprobed with monoclonal Pyk2 antibody (top). Immunosignals were quantified by film densitometry (bottom). Although the variability in PSD-95 precipitation was low, Pyk2 signals were corrected by dividing each Pyk2 signal by the corresponding PSD-95 signal. The Pyk2/PSD-95 ratio was normalized to control equaling 1. Immunoblotting of lysates illustrates that the same amounts of Pyk2 were present in all samples (middle). Stimulation with NMDA, ionomycin, or PMA induced a several fold increase in Pyk2 coimmunoprecipitation with PSD-95 in slices and cultures. C, Acute cortical slices were preincubated with calmodulin inhibitors (10 μm W7, 20 μm TFP, and 30 μm calmidazolium) for 15 min before incubation with TTX plus vehicle or NMDA, processing, and analysis as in A. Control IgG precipitations (middle column) were from the same blots and exposures as PSD-95 precipitations (left column). All three calmodulin inhibitors prevented the NMDA-induced increase in coprecipitation of Pyk2 with PSD-95 (bottom row) without changing total Pyk2 in lysates (right column). D, Primary hippocampal cultures (18 DIV) were pretreated with calmodulin inhibitors before incubation with TTX plus vehicle or NMDA. Immunoblotting was performed on lysates with the phosphospecific antibody against Pyk2 Tyr402 (pY402) and, after stripping, monoclonal Pyk2 antibody. Signals were quantified by densitometry. The pY402/total Pyk2 ratio was normalized to control equaling 1. NMDA induced a several fold increase in Pyk2 Tyr402 phosphorylation, which was blocked by all three calmodulin antagonists. A–D, Asterisks indicate statistical significance compared with control (*) or NMDA (**) treatment (t test, p < 0.05; n = 4 ± SEM for all experiments).
NMDA-induced Pyk2 association with PSD-95 and Pyk2 autophosphorylation requires calmodulin
There is good evidence that Ca2+/calmodulin mediates activation of Pyk2 in vitro (Kohno et al., 2008). Furthermore, calmodulin has been implicated previously in Pyk2 signaling. For instance, α1b and α2a adrenergic receptor-triggered ERK1/2 phosphorylation was antagonized by dominant-negative Pyk2 and Src and by three different calmodulin inhibitors (Della Rocca et al., 1997). Similarly, upregulation of NMDAR currents and the correlated tyrosine phosphorylation of the NMDAR, Pyk2, and Src required Ca2+/calmodulin when induced by stimulation of the metabotropic glutamate receptor mGluR1 (Heidinger et al., 2002). Interestingly, PKC inhibitors had no effect in these studies, pointing toward release of Ca2+ from intracellular stores as the main signaling mechanism in these two cases. We hypothesized that inhibition of calmodulin would reduce NMDAR-mediated Pyk2 binding to PSD-95. Acute cortical slices were pretreated with three different calmodulin inhibitors (W7, TFP, or calmidazolium) for 15 min before TTX was added for an additional 15 min and ultimately NMDA or vehicle for the final 15 min treatment. All three calmodulin inhibitors antagonized the NMDA-induced Pyk2 coimmunoprecipitation with PSD-95 (Fig. 2C) without affecting the cellular levels of Pyk2 (for more details on the calmodulin inhibitors, see Materials and Methods).
Pyk2 activation was monitored by measuring phosphorylation of Tyr402. This phosphorylation is induced by Ca2+ influx through NMDARs in response to ischemia (Huang et al., 2001; Guo et al., 2004). Primary hippocampal cultures (18 DIV) were pretreated with vehicle or calmodulin antagonists before incubation with vehicle or NMDA. NMDA induced a sixfold increase in Tyr402 autophosphorylation (Fig. 2D). All three calmodulin antagonists blocked this increase in Pyk2 autophosphorylation (Fig. 2D) similar to their blockade of NMDA-stimulated Pyk2 coprecipitation with PSD-95 (Fig. 2C).
Ca2+/calmodulin increases binding between Pyk2 and PSD-95 in vitro
The SH3 domain of PSD-95 binds to Pyk2 residues 671-875, which include two proline-rich regions (Seabold et al., 2003). Calmodulin interacts in a Ca2+-dependent manner with the HOOK region of PSD-95 and its homologues SAP97 and SAP102 immediately adjacent to the SH3 domain (Masuko et al., 1999; Paarmann et al., 2002; Fukunaga et al., 2005). Ca2+/calmodulin induces binding of the SH3 domain of SAP102 with the GK domain of PSD-95 (Masuko et al., 1999), possibly by loosening the intramolecular interaction between the SH3 and GK domains of these two proteins (McGee and Bredt, 1999; McGee et al., 2001). We found that Ca2+/calmodulin but not calmodulin alone associates with the SH3–GK region of PSD-95 (Fig. 3A). We then tested whether Ca2+/calmodulin can stimulate the interaction between PSD-95 and Pyk2. For this purpose, His-tagged full-length Pyk2(1-1009) was expressed in E. coli. After immunoprecipitation from the bacterial extracts with the αN Pyk2 antibody or nonspecific IgG as negative control, the resin samples with the Pyk2 immunocomplexes were incubated with bacterially expressed GST–PSD-95 or GST alone in the presence or absence of Ca2+ and calmodulin. GST–PSD-95 bound specifically to Pyk2 after its immunoprecipitation with αN (Fig. 3B, lanes 3–6). No binding was observed between GST–PSD-95 and control IgG precipitates (lane 8) or between αN precipitates and GST (lanes 1,2), indicating the specificity of the GST–PSD-95 binding to immunoisolated Pyk2. Ca2+/calmodulin in combination, but not individually, strongly increased PSD-95 binding to Pyk2 (compare lane 6 with lanes 3–5). Importantly, both calmodulin binding to PSD-95 and the calmodulin-induced stimulation of Pyk2 binding to PSD-95 was Ca2+ dependent.
Ca2+ influx through the NMDAR induces Pyk2 clustering at synapses
Biochemical data described above raise the possibility that an increase in intracellular Ca2+ will induce Pyk2 translocation to postsynaptic sites in which PSD-95 is concentrated (Hunt et al., 1996; Rao et al., 1998; Valtschanoff and Weinberg, 2001; Sans et al., 2005). Mature (18 DIV) primary hippocampal cultures were pretreated with TTX, followed by stimulation for 15 min with vehicle or glutamate or for 5 min with NMDA. In subsequent studies, cultures received 30 min pretreatment with vehicle, EGTA to chelate extracellular Ca2+, MK801 to block NMDARs, or W7 followed by treatment with vehicle, glutamate, NMDA, PMA, or ionomycin as described above. Cultures were fixed immediately after stimulation and double labeled with αN Pyk2 antibody and one of the following: anti-PSD-95, anti-bassoon, or anti-MAP2B antibodies (Fig. 4).
Figure 4.

Stimulation-induced synaptic clustering of Pyk2 in hippocampal neurons. A, Primary hippocampal cultures (18 DIV) were treated with TTX (100 nm) plus vehicle or glutamate (100 μm) for 15 min or NMDA (50 μm) for 5 min, fixed, and stained with αN Pyk2 (red in overlay at higher magnification at bottom) plus PSD-95, bassoon, or MAP2B antibodies (green). Scale bars at bottom right represent 10 μm for the corresponding magnifications. Arrows indicate Pyk2 puncta that colocalize with PSD-95 or bassoon puncta after stimulation. B, Density of Pyk2 and PSD-95 immunoreactive puncta during treatment with TTX plus vehicle, glutamate (100 μm), PMA (100 nm), ionomycin (1 μm), NMDA (50 μm), glutamate plus EGTA (2 mm), glutamate plus MK801 (10 μm), or glutamate plus W7 (10 μm) for 15 min (5 min for NMDA). C, Colocalization of Pyk2 puncta with PSD-95 puncta indicates that NMDA and glutamate stimulated Pyk2 clustering preferentially at synaptic PSD-95 clusters. A–C, For each experiment, three independent repetitions were performed except for the costaining of Pyk2 with bassoon and with MAP2B after glutamate treatment, which were only performed twice (but see Fig. 5). For quantifications shown in B and C, 5–12 different fields were analyzed for each experiment, and values were averaged before statistical analysis for the means from the three independent experiments (4 experiments for control, glutamate, and NMDA treatments). Asterisks indicate significant differences compared with control (*) or glutamate (**) treatment (t test, p < 0.05; error bars indicate SEM). Both scale bars are 10 μm.
Under control conditions, Pyk2 was predominantly smoothly distributed throughout the somata and dendrites (Fig. 4A, top left panel). Glutamate and NMDA induced a dramatic redistribution of Pyk2 to a mainly punctate appearance as expected if synaptically targeted. Quantification of Pyk2 puncta per dendritic length indicates a near sevenfold increase in Pyk2 cluster density during glutamate and NMDA application (per 10 μm dendritic length from 0.4 ± 0.11 in control to 2.8 ± 0.37 and 3.1 ± 0.37 in glutamate- and NMDA-treated neurons, respectively) (Fig. 4B). These puncta were chiefly colocalized with PSD-95 (Fig. 4A,C) and bassoon (Fig. 4A) particularly after glutamate and NMDA application. Bassoon is a well established specific marker for presynaptic active zones at mature synapses (Friedman et al., 2000; Zhai et al., 2001; Altrock et al., 2003; Shapira et al., 2003). Staining of parallel cultures for the microtubule-associated protein MAP2B, a dendritic marker (Hirokawa et al., 1988; Goedert et al., 1991), shows that the integrity of the dendrites was not affected by glutamate (Fig. 4A, right panels) or NMDA (Fig. 5, middle) application. GFP-transfected neurons also showed no change in dendritic morphology when stimulated with NMDA (Fig. 6A).
Figure 5.

NMDA-induced Pyk2–GFP clustering occurs postsynaptically and independently of Pyk2 kinase activity. Primary hippocampal cultures (15 DIV) were transfected with Pyk2–GFP or GFP-tagged kinase-dead Pyk2 (KD-GFP) (72 h; red in all overlays at higher magnification at bottom), treated with TTX plus either vehicle or NMDA (50 μm, 5 min), fixed, and stained with antibodies against synapsin (green in all dual overlays at the very bottom labeled “1 + 3”) and either MAP2B or PSD-95 (green in the respective overlays in the row labeled “1 + 2”). Scale bars, 10 μm for the corresponding magnifications. Pyk2–GFP underwent a dramatic relocation from a fairly smooth distribution under control conditions in dendritic shafts that was primarily colocalized with MAP2B to a pronounced punctate synaptic distribution away from MAP2B staining in dendritic shafts. Arrows illustrate examples of colocalization of Pyk2 and PSD-95 puncta that are juxtaposed to synapsin puncta. Kinase-dead Pyk2 also clusters during NMDA stimulation (right column). All pictures are representatives from three independent experiments.
Figure 6.
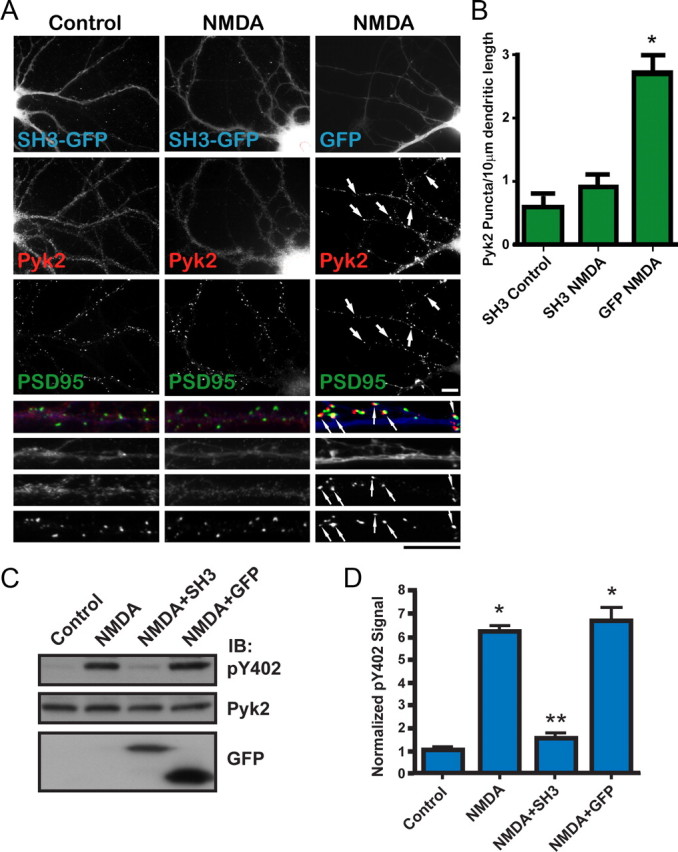
Overexpression of the PSD–95 SH3 domain inhibits NMDA-induced Pyk2 clustering and Pyk2 autophosphorylation. A, Primary hippocampal cultures (15 DIV) were transfected with the GFP-tagged SH3 domain of PSD-95 (SH3-GFP) or GFP alone (72 h; blue in overlay at higher magnification at bottom), treated with TTX plus vehicle or NMDA (50 μm, 5 min), fixed, and stained with antibodies against endogenous Pyk2 (αN; red in overlay) and PSD-95 (green in overlay). Scale bars, 10 μm for the corresponding magnifications. B, Density of Pyk2 immunoreactive puncta. SH3–GFP blocked NMDA-induced Pyk2 clustering seen in the presence of GFP alone. Five to 10 different fields were analyzed for each experiment, and values were averaged before statistical analysis for the means from the three independent experiments. The asterisk indicates statistical significance in Pyk2 clustering in neurons expressing GFP alone versus SH3–GFP (t test, p < 0.05; error bars indicate SEM). C, Primary hippocampal cultures (15 DIV) were infected with packaged FIV pVETL vectors encoding PSD-95 SH3–GFP or GFP alone (72 h) and treated with TTX plus pervanadate (1 mm) plus vehicle or NMDA (50 μm, 15 min) before immunoblotting (IB) with anti-phospho-Tyr402, monoclonal Pyk2, and GFP antibodies. D, Immunosignals were quantified by film densitometry. The pY402/Pyk2 ratios were normalization to control equaling 1. Asterisks denote statistical significance compared with control (*) or NMDA plus GFP (**) (t test, p < 0.05; n = 3 ± SEM).
Chelating extracellular Ca2+ with EGTA, blocking NMDAR pores with MK801, or inhibiting calmodulin with W7 blocked Pyk2 clustering without affecting PSD-95 distribution (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) (Fig. 4B). Ionomycin induced clustering similar to glutamate and NMDA (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) (Fig. 4B). These results indicate that increasing intracellular Ca2+, either with a general Ca2+ ionophore, which presumably acts along the whole dendritic shaft, or specifically via NMDARs, is necessary and sufficient for stimulating Pyk2 clustering at synapses. The PKC activating PMA also induced Pyk2 clustering (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) (Fig. 4B), in agreement with the above finding that PMA, like NMDA and ionomycin, stimulates Pyk2 binding to PSD-95. However, PMA-induced Pyk2 clustering and binding to PSD-95 might depend on a mechanism different from that underlying Ca2+ influx-induced Pyk2 clustering and PSD-95 binding and is therefore not within the scope of this work.
NMDA induces postsynaptic Pyk2 clustering
Pyk2 is present not only in dendrites but also in axons (Seabold et al., 2003). GFP-tagged Pyk2 was expressed in 15 DIV hippocampal cultures by low-efficiency calcium phosphate transfection so that only a few widely dispersed neurons expressed Pyk2–GFP. Cultures were at low density, with the average distance between neighboring somata being 200 μm. After 3 d, neurons were pretreated with TTX, treated with vehicle or NMDA, fixed, and stained with anti-synapsin and either anti-MAP2B or anti-PSD-95 antibodies. Synapsin is a synaptic vesicle-associated protein that is concentrated presynaptically and serves, like bassoon, as a synaptic marker (De Camilli et al., 1983; Chin et al., 1995). Under control conditions, Pyk2–GFP was smoothly distributed throughout dendritic areas of transfected neurons as identified by MAP2B labeling (Fig. 5, left column). Stimulation with NMDA induced clustering of Pyk2–GFP similar to that observed with endogenous Pyk2. This clustering was within the dendritic areas of transfected neurons that were identified as a result of Pyk2–GFP signals in somata from which the dendrites originated (Fig. 5). NMDA-induced Pyk2–GFP clusters primarily colocalized with synapsin and PSD-95. Because in all analyzed samples no other neurons close enough to form axonal contacts with one of the Pyk2–GFP-positive neurons were transfected with Pyk2–GFP, this clustering is attributable to Pyk2–GFP accumulation at postsynaptic sites. Transfected GFP alone did not cluster when stimulated with NMDA (Fig. 6A).
Mutating Lys457 to Ala in GFP–Pyk2 creates a kinase-dead isoform. Like wild type, GFP–Pyk2(K457A) also clustered at postsynaptic sites (Fig. 5). Accordingly, postsynaptic Pyk2 clustering is independent of its activity, consistent with our hypothesis that clustering precedes its activation.
NMDA-induced Pyk2 clustering depends on the SH3 domain of PSD-95
To address the role of PSD-95 in Pyk2 clustering, the GFP-tagged SH3 domain of PSD-95 was ectopically expressed in primary hippocampal cultures. Because Pyk2 interacts with the PSD-95 SH3 domain (Seabold et al., 2003), overexpression of the SH3 domain was hypothesized to disrupt the stimulation-dependent interaction between Pyk2 and synaptically localized PSD-95. In fact, NMDA stimulation was unable to initiate clustering of endogenous Pyk2 in the presence of SH3–GFP, whereas Pyk2 clustered readily in GFP-expressing neurons (Fig. 6A). The Pyk2 puncta density was threefold higher in GFP than in GFP–SH3-transfected neurons when treated with NMDA (2.7 ± 0.26 compared with 0.9 ± 0.20) (Fig. 6B). The Pyk2 puncta density of GFP-transfected neurons stimulated with NMDA was comparable with that of untransfected NMDA-stimulated neurons (Fig. 4A,B). Endogenous PSD-95 was unaffected by expression of GFP or SH3 domain (Fig. 6A).
NMDA-induced Pyk2 autophosphorylation depends on the SH3 domain of PSD-95
Induction of Pyk2 autoactivation and binding to PSD-95 in intact neurons show the same pharmacological profiles. We investigated whether Pyk2 binding to PSD-95 is required for NMDA-induced Pyk2 activation with the hypothesis that inhibition of the Pyk2–PSD-95 interaction would eliminate Pyk2 activation by NMDA. Primary hippocampal cultures (15 DIV) were infected with the GFP-tagged PSD-95 SH3 domain similar to that used above, except a lentiviral vector based on a minimal FIV was used. This vector provided >90% infection efficiency in hippocampal cultures without detectably affecting the health of the neurons. Control infections were performed with GFP alone expressed from an analogous FIV vector. At 18 DIV, the neurons were stimulated with NMDA in the presence of TTX and pervanadate. Lysates were analyzed by immunoblotting with phospho-Tyr402, monoclonal Pyk2, and GFP antibodies as before. The results clearly show that GFP–SH3 but not GFP alone suppresses the NMDA-induced upregulation of Pyk2 autophosphorylation (Fig. 6C,D).
Binding of Pyk2 to PSD-95 is necessary for LTP
We showed previously that the sequential activation of Pyk2 and Src plays a critical role in the induction of LTP at CA3–CA1 synapses (Lu et al., 1998; Xiong et al., 1999; Huang et al., 2001; Salter and Kalia, 2004). To test the relevance of the interaction between Pyk2 and PSD-95 in LTP induction, whole-cell patch recordings were obtained from pyramidal neurons in the hippocampal CA1 region. Recording electrodes contained GST alone as control, the GST-tagged SH3 domain of PSD-95, or the GST-tagged Pyk2 residues 671-875, which mediate Pyk2 binding to the SH3 domain of PSD-95 (Seabold et al., 2003). None of the polypeptides affected basal NMDAR EPSCs, as measured with CNQX to block AMPARs (Fig. 7A,B), or NMDA-evoked whole-cell currents (Fig. 7C) under basal conditions over a time course of 25–30 min. This is consistent with our recent finding that, contrasting cultured neurons, in acute slices Src-mediated upregulation of NMDAR activity is constitutively suppressed under basal conditions by the C-terminal Src kinase CSK (Xu et al., 2008). However, the induction of LTP was entirely blocked by including either the SH3 domain of PSD-95 or the SH3 binding site on Pyk2 in the pipette, whereas GST by itself failed to block LTP (Fig. 7D–F). Currents were averaged 5 min before and 30 min after tetani. Potentiation under control conditions (GST) was 161 ± 12% (p < 0.001 vs baseline) compared with 92 ± 0.06% for GST–SH3 (p < 0.001 vs control LTP) and 104 ± 0.05% for GST–Pyk2(671-875) (p = 0.004 vs control LTP).
Figure 7.

Interfering with the PSD-95–Pyk2 interaction prevents induction of LTP. A, B, Peak currents of NMDAR-mediated EPSCs in CA1 isolated with CNQX are not affected during infusion of GST (n = 5; CTL; black circles), GST–SH3 (n = 3; green circles), or GST–Pyk2(671-875) (n = 5; red squares). C, NMDA-induced whole-cell patch peak currents in acutely isolated CA1 neurons are not affected during infusion of GST (n = 7; CTL; black circles), GST–SH3 (n = 7; green circles), or GST–Pyk2(671-875) (n = 6; red squares). D–F, LTP induced by two tetani of 100 Hz/500 ms, 10 s apart, was blocked by infusion of GST–SH3 (n = 6; green circles) and GST–Pyk2(671-875) (n = 6; red squares) but not GST alone (n = 7; CLT; black circles). Shown are sample traces (D) before (a) and 30 min after (b) tetani, exemplary time courses for each condition (E), and averaged EPSCs that were normalized to baseline for each experiment from all time courses for each condition ±SEM (F). Arrows indicate time of tetani.
Discussion
Role of PSD-95 in postsynaptic clustering and activation of Pyk2
Under basal conditions, PSD-95 is concentrated at postsynaptic sites, whereas Pyk2 is diffuse throughout dendrites. We hypothesize that the interaction between the SH3 and GK domain in the basal state of PSD-95 is responsible for inhibiting Pyk2 binding. Ca2+ induces calmodulin binding to the HOOK regions of PSD-95 and its homologs SAP97 and SAP102, which connect their SH3 and GK domains (Masuko et al., 1999; Paarmann et al., 2002; Fukunaga et al., 2005). Ca2+/calmodulin binding results in a rearrangement, which likely includes displacement of the GK domain from the SH3 domain promoting Pyk2 binding to the SH3 domain. Pyk2 could consequently accumulate at postsynaptic sites, in which binding to PSD-95 clusters would mediate its oligomerization. This model is supported by the finding that PSD-95 and its homologs form homomultimers and heteromultimers (Tomita et al., 2001; Christopherson et al., 2003; Kim and Sheng, 2004). The proximity of Pyk2 molecules could promote trans-autophosphorylation, thereby leading to Pyk2 activation. SAP97, SAP102, and the related CHAPSYN110/PSD-93 are present at postsynaptic sites and might also contribute to Pyk2 clustering and activation. However, PSD-95 is the functionally prevailing postsynaptic representative of this protein family in hippocampal neurons (Schlüter et al., 2006).
Precise localization of kinases is important for effective and specific phosphorylation of targets (Pawson and Scott, 1997; Wong and Scott, 2004). CaMKII translocates to postsynaptic sites during Ca2+ influx through NMDAR binding (Leonard et al., 1999; Shen and Meyer, 1999; Bayer et al., 2001; Merrill et al., 2005) for postsynaptic signaling (Barria and Malinow, 2005). We now demonstrate the postsynaptic translocation of Pyk2 during NMDAR-mediated Ca2+ influx (Figs. 4, 5). Ectopic expression of PSD-95 SH3–GFP prevented postsynaptic Pyk2 clustering, indicating that this clustering is via Pyk2 binding to PSD-95 or homologs (Fig. 6).
Dimerization by antibodies in vitro and overexpression of PSD-95 in PC6-3 cells induce trans-autophosphorylation of Pyk2, indicating that PSD-95 binding promotes Pyk2 activation by oligomerization (Fig. 1). Ca2+ influx through NMDAR stimulates coimmunoprecipitation of Pyk2 with PSD-95 and concomitant Pyk2 Tyr402 autophosphorylation (Figs. 2, 3, 6). GFP–SH3 completely blocked activity-induced Pyk2 autophosphorylation, indicating that the autoactivation depends on access to the SH3 domain of PSD-95 (Fig. 6).
Role of Ca2+/calmodulin and PKC
Ca2+/calmodulin stimulates Pyk2 binding to PSD-95 (Figs. 2C, 3) and Pyk2 trans-autophosphorylation (Fig. 2D), consistent with the finding that calmodulin only interacts with PSD-95 and SAP102 when Ca2+ is present (Masuko et al., 1999; Paarmann et al., 2002; Fukunaga et al., 2005). Ca2+/calmodulin is also critical for postsynaptic Pyk2 clustering during Ca2+ influx through the NMDAR (Fig. 4B). PKC inhibitors did not inhibit glutamate- or NMDA-induced Pyk2 clustering (data not shown), suggesting that PKC is not necessary for postsynaptic Pyk2 clustering and activation induced by Ca2+ influx.
However, stimulation of PKC also led to PSD-95 binding (Fig. 2) and postsynaptic Pyk2 clustering (Fig. 4). PSD-95 binding might thus be a general mechanism for Pyk2 activation in neurons. One isoform of PKC (PKCα) binds PSD-95 (Lim et al., 2002). NMDAR-mediated Ca2+ influx can activate the Ca2+-sensitive phospholipase Cδ (Rebecchi and Pentyala, 2000) for production of PKC-activating diacylglycerol compounds in neurons (Codazzi et al., 2006). Nevertheless, additional characterization of this PKC-mediated mechanism is beyond the scope of this article.
We do not know at this point with certainty how Ca2+/calmodulin induces Pyk2/PSD-95 association. Because Ca2+/calmodulin can regulate the SH3–GK domain interaction, we hypothesize that Ca2+/calmodulin is promoting Pyk2 binding to PSD-95 by relaxing the intramolecular interaction between SH3 and GK of PSD-95 (Masuko et al., 1999; McGee and Bredt, 1999; McGee et al., 2001). Once bound to PSD-95, which forms dimers (Craven et al., 1999; Masuko et al., 1999; Shin et al., 2000; McGee et al., 2001; El-Husseini Ael et al., 2002) and perhaps oligomers at postsynaptic sites, Pyk2 proteins within a complex could trans-autophosphorylate, thereby conferring activation (Fig. 1F).
Ca2+/calmodulin might also directly bind to Pyk2 for its activation (Kohno et al., 2008). However, the involvement of intermediary proteins, possibly PSD-95 homologs that are present in non-neuronal cells such as SAP97, has not been excluded in this work. Based on point mutations, the authors propose that Ca2+/calmodulin binds to a segment in the middle of the FERM domain. However, this segment is essentially buried in the crystal structure of FAK (Lietha et al., 2007) and likely not accessible to Ca2+/calmodulin (Schaller, 2008).
Ca2+/calmodulin is also important for CaMKII binding to the NMDAR NR2B subunit (Bayer et al., 2001; Leonard et al., 2002). This Ca2+/calmodulin-induced binding promotes autophosphorylation between subunits of the dodecameric CaMKII holoenzyme (Bayer et al., 2001; Merrill et al., 2005) (see also Sun et al., 2004). Calmodulin thus emerges as a key mediator of recruiting and regulating postsynaptic signaling elements at activated postsynaptic sites. KN93 (2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)amino-N-(4-chlorocinnamyl)-N-methylbenzylamine), which inhibits Ca2+/calmodulin-dependent kinases including CaMKII, failed to affect glutamate-induced Pyk2 clustering, indicating that the effects of Ca2+/calmodulin were independent of CaMKII (data not shown).
Role of Src in Pyk2 signaling
Pyk2 stimulates the NMDAR activity through Src, which acts downstream of Pyk2 at postsynaptic sites (Yu et al., 1997; Lu et al., 1998; Huang et al., 2001; Salter and Kalia, 2004) (see also Corvol et al., 2005). However, in some non-neuronal cells, the initial Ca2+-dependent Pyk2 activation through Tyr402 autophosphorylation is inhibited, although not fully blocked, by Src antagonists (Sorokin et al., 2001; Cheng et al., 2002). These results suggest that interdependence between Pyk2 and Src activation can exist. Autophosphorylation of Pyk2 on Tyr402 recruits and activates Src for additional phosphorylation and additional activation of Pyk2 (see Introduction). It is obvious that Src binding to phosphorylated Tyr402 would protect Tyr402 from dephosphorylation, although this possibility is untested because the relevant phosphatase is unknown. Furthermore, the Src-mediated increase in Pyk2 activity would allow more effective rephosphorylation of Tyr402 should a Tyr402 residue in an oligomeric complex become dephosphorylated. These properties provide a positive feedback loop that prolongs the activity of Pyk2/Src complexes. Such complexes thus might constitute a molecular memory, which might be important for the establishment of LTP.
Physiological relevance of Pyk2 clustering in LTP
PKC increases NMDAR currents by stimulating Pyk2. Pyk2 then activates Src, which then phosphorylates NMDARs (Yu et al., 1997; Lu et al., 1998; Huang et al., 2001; Salter and Kalia, 2004). The PKC effect is blocked by injection of kinase-dead Pyk2 and various Src inhibitors. Src inhibitors but not PKC inhibitors block upregulation of NMDAR currents after injection of active Pyk2. Inhibition of PKC or Pyk2 activity does not affect upregulation of NMDAR activity by Src activators, placing Pyk2 downstream of PKC and upstream of Src. The PKC–Pyk2–Src signaling cascade is required for LTP in the hippocampal CA1 region (Yu et al., 1997; Lu et al., 1998; Huang et al., 2001; Salter and Kalia, 2004). We find that two different constructs that interfere with Pyk2 binding to PSD-95, GST–SH3 and Pyk2 residues 671-875, blocked LTP without directly affecting NMDAR function (Fig. 7). Because an increasing body of evidence indicates that LTP is the physiological correlate of learning and memory (Martin et al., 2000; Bredt and Nicoll, 2003; Collingridge et al., 2004; Malenka and Bear, 2004; Pastalkova et al., 2006; Whitlock et al., 2006), Pyk2 anchoring by PSD-95 is likely important for these higher brain functions.
The concomitant postsynaptic recruitment and activation of Pyk2 during NMDAR-mediated Ca2+ influx is an economical and effective mechanism to elevate Pyk2 activity, specifically at those synapses experiencing LTP. This targeting mechanism will ensure that Pyk2 is mainly active at those synapses undergoing potentiation. By providing for accumulation of Pyk2 specifically at activated postsynaptic sites, this mechanism likely contributes to the critical synapse specificity of LTP, which is crucial for preventing saturation of neuronal input.
LTP at mossy fiber synapses in the hippocampal CA3 area is NMDAR independent and attributable to increased presynaptic glutamate release. However, postsynaptic NMDARs at CA2 mossy fiber synapses can undergo LTP themselves independent of increased glutamate release (Kwon and Castillo, 2008; Rebola et al., 2008). This NMDAR LTP requires coactivation of NMDAR and mGluR5 and the ensuing stimulation of PKC and Src, suggesting a PKC–Pyk2–Src–NMDAR cascade analogous to that established for CA1 synapses.
Clinical aspects of Pyk2 clustering in ischemia
Ischemia causes reversal of glutamate transporters and overstimulation of NMDAR-mediated Ca2+ influx triggering neuronal damage (Rothman and Olney, 1987; Lee et al., 1999). Pyk2 translocates to postsynaptic densities after ischemia, in which it interacts with NMDARs via PSD-95 (Liu et al., 2001; Cheung et al., 2003; Seabold et al., 2003; Hou et al., 2005). NMDAR antagonists inhibit ischemia-induced stimulation of Pyk2, Src, and tyrosine phosphorylation of NR2A and NR2B (Liu et al., 2001; Guo et al., 2004). Hence, ischemia likely activates Pyk2 through PSD-95, which could contribute to consequent neuropathologies.
In summary, Pyk2 oligomerization in general and specifically by Ca2+/calmodulin-induced PSD-95 binding in neurons induces its activation by trans-autophosphorylation. The concomitant postsynaptic Pyk2 accumulation at synapses undergoing potentiation allows for synapse-selective targeting and activation Pyk2, which contributes to the synapse-selective upregulation of NMDAR activity and thereby induction of LTP.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants R01 NS046450 and R01 NS035563 (J.W.H.), Canadian Institutes of Health Research (CIHR) Grant 44008 (J.F.M.), NIH Predoctoral Training Grant T32 GM008365 (J.A.B., J.D.U.), American Heart Association Predoctoral Fellowship 0515567Z (J.A.B.), NIH National Research Service Award F31 MH081420 (J.D.U.), and a CIHR postdoctoral award (M.A.B.). Parental viral vectors were provided and FIV viruses produced by the University of Iowa Center for Gene Therapy for Cystic Fibrosis, which is supported by NIH Grant P30 DK54759. We thank Drs. L. M. Graves and H. S. Earp (University of North Carolina at Chapel Hill, Chapel Hill, NC) for the N- and C-terminal Pyk2 antibodies, Dr. P. DeCamilli (Yale University, New Haven, CT) for the anti-synapsin antibody, Dr. M. A. Shea (University of Iowa, Iowa City, IA) for purified calmodulin, Dr. S. Strack for the PC6-3 cell line, and Drs. J. C. Houtman (University of Iowa, Iowa City, IA) and Lee M. Graves (University of North Carolina at Chapel Hill, Chapel Hill, NC) for critically reading this manuscript.
References
- Ahmed R, Zha XM, Green SH, Dailey ME. Synaptic activity and F-actin coordinately regulate CaMKIIalpha localization to dendritic postsynaptic sites in developing hippocampal slices. Mol Cell Neurosci. 2006;31:37–51. doi: 10.1016/j.mcn.2005.08.020. [DOI] [PubMed] [Google Scholar]
- Akyol Z, Bartos JA, Merrill MA, Faga LA, Jaren OR, Shea MA, Hell JW. Apo-calmodulin binds with its C-terminal domain to the N-methyl-D-aspartate receptor NR1 C0 region. J Biol Chem. 2004;279:2166–2175. doi: 10.1074/jbc.M302542200. [DOI] [PubMed] [Google Scholar]
- Altrock WD, tom Dieck S, Sokolov M, Meyer AC, Sigler A, Brakebusch C, Fässler R, Richter K, Boeckers TM, Potschka H, Brandt C, Löscher W, Grimberg D, Dresbach T, Hempelmann A, Hassan H, Balschun D, Frey JU, Brandstätter JH, Garner CC, Rosenmund C, Gundelfinger ED. Functional inactivation of a fraction of excitatory synapses in mice deficient for the active zone protein bassoon. Neuron. 2003;37:787–800. doi: 10.1016/s0896-6273(03)00088-6. [DOI] [PubMed] [Google Scholar]
- Avraham H, Park SY, Schinkmann K, Avraham S. RAFTK/Pyk2-mediated cellular signalling. Cell Signal. 2000;12:123–133. doi: 10.1016/s0898-6568(99)00076-5. [DOI] [PubMed] [Google Scholar]
- Avraham S, London R, Fu Y, Ota S, Hiregowdara D, Li J, Jiang S, Pasztor LM, White RA, Groopman JE, Avraham H. Identification and characterization of a novel related adhesion focal tyrosine kinase (RAFTK) from megakaryocytes and brain. J Biol Chem. 1995;270:27742–27751. doi: 10.1074/jbc.270.46.27742. [DOI] [PubMed] [Google Scholar]
- Banno Y, Ohguchi K, Matsumoto N, Koda M, Ueda M, Hara A, Dikic I, Nozawa Y. Implication of phospholipase D2 in oxidant-induced phosphoinositide 3-kinase signaling via Pyk2 activation in PC12 cells. J Biol Chem. 2005;280:16319–16324. doi: 10.1074/jbc.M410903200. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Barsacchi R, Heider H, Girault J, Meldolesi J. Requirement of pyk2 for the activation of the MAP kinase cascade induced by Ca2+ (but not by PKC or G protein) in PC12 cells. FEBS Lett. 1999;461:273–276. doi: 10.1016/s0014-5793(99)01468-4. [DOI] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–805. doi: 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Byron KL, Lucchesi PA. Signal transduction of physiological concentrations of vasopressin in A7r5 vascular smooth muscle cells. A role for PYK2 and tyrosine phosphorylation of K+ channels in the stimulation of Ca2+ spiking. J Biol Chem. 2002;277:7298–7307. doi: 10.1074/jbc.M104726200. [DOI] [PubMed] [Google Scholar]
- Chen C, Leonard JP. Protein tyrosine kinase-mediated potentiation of currents from cloned NMDA receptors. J Neurochem. 1996;67:194–200. doi: 10.1046/j.1471-4159.1996.67010194.x. [DOI] [PubMed] [Google Scholar]
- Chen Y, Stevens B, Chang J, Milbrandt J, Barres BA, Hell JW. NS21: re-defined and modified supplement B27 for neuronal cultures. J Neurosci Methods. 2008;171:239–247. doi: 10.1016/j.jneumeth.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JJ, Chao YJ, Wang DL. Cyclic strain activates redox-sensitive proline-rich tyrosine kinase 2 (PYK2) in endothelial cells. J Biol Chem. 2002;277:48152–48157. doi: 10.1074/jbc.M110937200. [DOI] [PubMed] [Google Scholar]
- Cheung HH, Teves L, Wallace MC, Gurd JW. Inhibition of protein kinase C reduces ischemia-induced tyrosine phosphorylation of the N-methyl-d-aspartate receptor. J Neurochem. 2003;86:1441–1449. doi: 10.1046/j.1471-4159.2003.01951.x. [DOI] [PubMed] [Google Scholar]
- Chin LS, Li L, Ferreira A, Kosik KS, Greengard P. Impairment of axonal development and of synaptogenesis in hippocampal neurons of synapsin I-deficient mice. Proc Natl Acad Sci U S A. 1995;92:9230–9234. doi: 10.1073/pnas.92.20.9230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Sweeney NT, Craven SE, Kang R, El-Husseini Ael-D, Bredt DS. Lipid- and protein-mediated multimerization of PSD-95: implications for receptor clustering and assembly of synaptic protein networks. J Cell Sci. 2003;116:3213–3219. doi: 10.1242/jcs.00617. [DOI] [PubMed] [Google Scholar]
- Codazzi F, Di Cesare A, Chiulli N, Albanese A, Meyer T, Zacchetti D, Grohovaz F. Synergistic control of protein kinase Cγ activity by ionotropic and metabotropic glutamate receptor inputs in hippocampal neurons. J Neurosci. 2006;26:3404–3411. doi: 10.1523/JNEUROSCI.0478-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Isaac JT, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci. 2004;5:952–962. doi: 10.1038/nrn1556. [DOI] [PubMed] [Google Scholar]
- Cook WJ, Walter LJ, Walter MR. Drug binding by calmodulin: crystal structure of a calmodulin-trifluoperazine complex. Biochemistry. 1994;33:15259–15265. doi: 10.1021/bi00255a006. [DOI] [PubMed] [Google Scholar]
- Corvol JC, Valjent E, Toutant M, Enslen H, Irinopoulou T, Lev S, Hervé D, Girault JA. Depolarization activates ERK and proline-rich tyrosine kinase 2 (PYK2) independently in different cellular compartments in hippocampal slices. J Biol Chem. 2005;280:660–668. doi: 10.1074/jbc.M411312200. [DOI] [PubMed] [Google Scholar]
- Craven SE, El-Husseini AE, Bredt DS. Synaptic targeting of the postsynaptic density protein PSD-95 mediated by lipid and protein motifs. Neuron. 1999;22:497–509. doi: 10.1016/s0896-6273(00)80705-9. [DOI] [PubMed] [Google Scholar]
- Crossthwaite AJ, Valli H, Williams RJ. Inhibiting Src family tyrosine kinase activity blocks glutamate signalling to ERK1/2 and Akt/PKB but not JNK in cultured striatal neurones. J Neurochem. 2004;88:1127–1139. doi: 10.1046/j.1471-4159.2004.02257.x. [DOI] [PubMed] [Google Scholar]
- Davare MA, Hell JW. Increased phosphorylation of the neuronal L-type Ca2+ channel Cav1.2 during aging. Proc Natl Acad Sci U S A. 2003;100:16018–16023. doi: 10.1073/pnas.2236970100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Camilli P, Harris SM, Jr, Huttner WB, Greengard P. Synapsin I (Protein I), a nerve terminal-specific phosphoprotein. II. Its specific association with synaptic vesicles demonstrated by immunocytochemistry in agarose-embedded synaptosomes. J Cell Biol. 1983;96:1355–1373. doi: 10.1083/jcb.96.5.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della Rocca GJ, van Biesen T, Daaka Y, Luttrell DK, Luttrell LM, Lefkowitz RJ. Ras-dependent mitogen-activated protein kinase activation by G protein-coupled receptors. Convergence of Gi- and Gq-mediated pathways on calcium/calmodulin, Pyk2, and Src kinase. J Biol Chem. 1997;272:19125–19132. doi: 10.1074/jbc.272.31.19125. [DOI] [PubMed] [Google Scholar]
- Dikic I, Tokiwa G, Lev S, Courtneidge SA, Schlessinger J. A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature. 1996;383:547–550. doi: 10.1038/383547a0. [DOI] [PubMed] [Google Scholar]
- Dunty JM, Schaller MD. The N termini of focal adhesion kinase family members regulate substrate phosphorylation, localization, and cell morphology. J Biol Chem. 2002;277:45644–45654. doi: 10.1074/jbc.M201779200. [DOI] [PubMed] [Google Scholar]
- Earp HS, Huckle WR, Dawson TL, Li X, Graves LM, Dy R. Angiotensin II activates at least two tyrosine kinases in rat liver epithelial cells. J Biol Chem. 1995;270:28440–28447. doi: 10.1074/jbc.270.47.28440. [DOI] [PubMed] [Google Scholar]
- El-Husseini Ael-D, Schnell E, Dakoji S, Sweeney N, Zhou Q, Prange O, Gauthier-Campbell C, Aguilera-Moreno A, Nicoll RA, Bredt DS. Synaptic strength regulated by palmitate cycling on PSD-95. Cell. 2002;108:849–863. doi: 10.1016/s0092-8674(02)00683-9. [DOI] [PubMed] [Google Scholar]
- Fong DK, Rao A, Crump FT, Craig AM. Rapid synaptic remodeling by protein kinase C: reciprocal translocation of NMDA receptors and calcium/calmodulin-dependent kinase II. J Neurosci. 2002;22:2153–2164. doi: 10.1523/JNEUROSCI.22-06-02153.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank GD, Saito S, Motley ED, Sasaki T, Ohba M, Kuroki T, Inagami T, Eguchi S. Requirement of Ca2+ and PKCdelta for Janus kinase 2 activation by angiotensin II: involvement of PYK2. Mol Endocrinol. 2002;16:367–377. doi: 10.1210/mend.16.2.0768. [DOI] [PubMed] [Google Scholar]
- Friedman HV, Bresler T, Garner CC, Ziv NE. Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron. 2000;27:57–69. doi: 10.1016/s0896-6273(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Fukunaga Y, Matsubara M, Nagai R, Miyazawa A. The interaction between PSD-95 and Ca2+/calmodulin is enhanced by PDZ-binding proteins. J Biochem. 2005;138:177–182. doi: 10.1093/jb/mvi107. [DOI] [PubMed] [Google Scholar]
- Gardoni F, Bellone C, Viviani B, Marinovich M, Meli E, Pellegrini-Giampietro DE, Cattabeni F, Di Luca M. Lack of PSD-95 drives hippocampal neuronal cell death through activation of an alpha CaMKII transduction pathway. Eur J Neurosci. 2002;16:777–786. doi: 10.1046/j.1460-9568.2002.02141.x. [DOI] [PubMed] [Google Scholar]
- Girault JA, Costa A, Derkinderen P, Studler JM, Toutant M. FAK and PYK2/CAKbeta in the nervous system: a link between neuronal activity, plasticity and survival? Trends Neurosci. 1999;22:257–263. doi: 10.1016/s0166-2236(98)01358-7. [DOI] [PubMed] [Google Scholar]
- Goedert M, Crowther RA, Garner CC. Molecular characterization of microtubule-associated proteins tau and MAP2. Trends Neurosci. 1991;14:193–199. doi: 10.1016/0166-2236(91)90105-4. [DOI] [PubMed] [Google Scholar]
- Guo J, Meng F, Fu X, Song B, Yan X, Zhang G. N-methyl-d-aspartate receptor and L-type voltage-gated Ca2+ channel activation mediate proline-rich tyrosine kinase 2 phosphorylation during cerebral ischemia in rats. Neurosci Lett. 2004;355:177–180. doi: 10.1016/j.neulet.2003.10.076. [DOI] [PubMed] [Google Scholar]
- Haglund K, Ivankovic-Dikic I, Shimokawa N, Kruh GD, Dikic I. Recruitment of Pyk2 and Cbl to lipid rafts mediates signals important for actin reorganization in growing neurites. J Cell Sci. 2004;117:2557–2568. doi: 10.1242/jcs.01148. [DOI] [PubMed] [Google Scholar]
- Haunsø A, Simpson J, Antoni FA. Small ligands modulating the activity of mammalian adenylyl cyclases: a novel mode of inhibition by calmidazolium. Mol Pharmacol. 2003;63:624–631. doi: 10.1124/mol.63.3.624. [DOI] [PubMed] [Google Scholar]
- Heidinger V, Manzerra P, Wang XQ, Strasser U, Yu SP, Choi DW, Behrens MM. Metabotropic glutamate receptor 1-induced upregulation of NMDA receptor current: mediation through the Pyk2/Src-family kinase pathway in cortical neurons. J Neurosci. 2002;22:5452–5461. doi: 10.1523/JNEUROSCI.22-13-05452.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel a1 subunits. J Cell Biol. 1993;123:949–962. doi: 10.1083/jcb.123.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW, Yokoyama CT, Breeze LJ, Chavkin C, Catterall WA. Phosphorylation of presynaptic and postsynaptic calcium channels by cAMP-dependent protein kinase in hippocampal neurons. EMBO J. 1995;14:3036–3044. doi: 10.1002/j.1460-2075.1995.tb07306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Breeze LJ, Wang KK, Chavkin C, Catterall WA. N-methyl-d-aspartate receptor-induced proteolytic conversion of postsynaptic class C L-type calcium channels in hippocampal neurons. Proc Natl Acad Sci U S A. 1996;93:3362–3367. doi: 10.1073/pnas.93.8.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Hisanaga S, Shiomura Y. MAP2 is a component of crossbridges between microtubules and neurofilaments in the neuronal cytoskeleton: quick-freeze, deep-etch immunoelectron microscopy and reconstitution studies. J Neurosci. 1988;8:2769–2779. doi: 10.1523/JNEUROSCI.08-08-02769.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou XY, Zhang GY, Wang DG, Guan QH, Yan JZ. Suppression of postsynaptic density protein 95 by antisense oligonucleotides diminishes postischemic pyramidal cell death in rat hippocampal CA1 subfield. Neurosci Lett. 2005;385:230–233. doi: 10.1016/j.neulet.2005.05.054. [DOI] [PubMed] [Google Scholar]
- Huang Y, Lu W, Ali DW, Pelkey KA, Pitcher GM, Lu YM, Aoto H, Roder JC, Sasaki T, Salter MW, MacDonald JF. CAKbeta/Pyk2 kinase is a signaling link for induction of long-term potentiation in CA1 hippocampus. Neuron. 2001;29:485–496. doi: 10.1016/s0896-6273(01)00220-3. [DOI] [PubMed] [Google Scholar]
- Hubbard SR, Miller WT. Receptor tyrosine kinases: mechanisms of activation and signaling. Curr Opin Cell Biol. 2007;19:117–123. doi: 10.1016/j.ceb.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt CA, Schenker LJ, Kennedy MB. PSD-95 is associated with the postsynaptic density and not with the presynaptic membrane at forebrain synapses. J Neurosci. 1996;16:1380–1388. doi: 10.1523/JNEUROSCI.16-04-01380.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivankovic-Dikic I, Grönroos E, Blaukat A, Barth BU, Dikic I. Pyk2 and FAK regulate neurite outgrowth induced by growth factors and integrins. Nat Cell Biol. 2000;2:574–581. doi: 10.1038/35023515. [DOI] [PubMed] [Google Scholar]
- Johnston JC, Gasmi M, Lim LE, Elder JH, Yee JK, Jolly DJ, Campbell KP, Davidson BL, Sauter SL. Minimum requirements for efficient transduction of dividing and nondividing cells by feline immunodeficiency virus vectors. J Virol. 1999;73:4991–5000. doi: 10.1128/jvi.73.6.4991-5000.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- Kim E, Cho KO, Rothschild A, Sheng M. Heteromultimerization and NMDA receptor-clustering activity of chapsyn-110, a member of the PSD-95 family of proteins. Neuron. 1996;17:103–113. doi: 10.1016/s0896-6273(00)80284-6. [DOI] [PubMed] [Google Scholar]
- Kohno T, Matsuda E, Sasaki H, Sasaki T. Protein-tyrosine kinase CAKbeta/PYK2 is activated by binding Ca2+/calmodulin to FERM F2 alpha2 helix and thus forming its dimer. Biochem J. 2008;410:513–523. doi: 10.1042/BJ20070665. [DOI] [PubMed] [Google Scholar]
- Köhr G, Seeburg PH. Subtype-specific regulation of recombinant NMDA receptor-channels by protein tyrosine kinases of the src family. J Physiol. 1996;492:445–452. doi: 10.1113/jphysiol.1996.sp021320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- Kwon HB, Castillo PE. Long-term potentiation selectively expressed by NMDA receptors at hippocampal mossy fiber synapses. Neuron. 2008;57:108–120. doi: 10.1016/j.neuron.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399(Suppl):A7–A14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- Leonard AS, Davare MA, Horne MC, Garner CC, Hell JW. SAP97 is associated with the a-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR1 subunit. J Biol Chem. 1998;273:19518–19524. doi: 10.1074/jbc.273.31.19518. [DOI] [PubMed] [Google Scholar]
- Leonard AS, Lim IA, Hemsworth DE, Horne MC, Hell JW. Calcium/calmodulin-dependent protein kinase II is associated with the N-methyl-d-aspartate receptor. Proc Natl Acad Sci U S A. 1999;96:3239–3244. doi: 10.1073/pnas.96.6.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard AS, Bayer KU, Merrill MA, Lim IA, Shea MA, Schulman H, Hell JW. Regulation of calcium/calmodulin-dependent protein kinase II docking to N-methyl-d-aspartate receptors by calcium/calmodulin and a-actinin. J Biol Chem. 2002;277:48441–48448. doi: 10.1074/jbc.M205164200. [DOI] [PubMed] [Google Scholar]
- Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM, Plowman GD, Rudy B, Schlessinger J. Protein tyrosine kinase PYK2 involved in Ca2+-induced regulation of ion channel and MAP kinase functions. Nature. 1995;376:737–745. doi: 10.1038/376737a0. [DOI] [PubMed] [Google Scholar]
- Li X, Dy RC, Cance WG, Graves LM, Earp HS. Interactions between two cytoskeleton-associated tyrosine kinases: calcium-dependent tyrosine kinase and focal adhesion tyrosine kinase. J Biol Chem. 1999;274:8917–8924. doi: 10.1074/jbc.274.13.8917. [DOI] [PubMed] [Google Scholar]
- Lietha D, Cai X, Ceccarelli DF, Li Y, Schaller MD, Eck MJ. Structural basis for the autoinhibition of focal adhesion kinase. Cell. 2007;129:1177–1187. doi: 10.1016/j.cell.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim IA, Hall DD, Hell JW. Selectivity and promiscuity of the first and second PDZ domains of PSD-95 and synapse-associated protein 102. J Biol Chem. 2002;277:21697–21711. doi: 10.1074/jbc.M112339200. [DOI] [PubMed] [Google Scholar]
- Lim IA, Merrill MA, Chen Y, Hell JW. Disruption of the NMDA receptor-PSD-95 interaction in hippocampal neurons with no obvious physiological short-term effect. Neuropharmacology. 2003;45:738–754. doi: 10.1016/s0028-3908(03)00276-4. [DOI] [PubMed] [Google Scholar]
- Liu J, Fukunaga K, Yamamoto H, Nishi K, Miyamoto E. Differential roles of Ca2+/calmodulin-dependent protein kinase II and mitogen-activated protein kinase activation in hippocampal long-term potentiation. J Neurosci. 1999;19:8292–8299. doi: 10.1523/JNEUROSCI.19-19-08292.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhang G, Gao C, Hou X. NMDA receptor activation results in tyrosine phosphorylation of NMDA receptor subunit 2A(NR2A) and interaction of Pyk2 and Src with NR2A after transient cerebral ischemia and reperfusion. Brain Res. 2001;909:51–58. doi: 10.1016/s0006-8993(01)02619-1. [DOI] [PubMed] [Google Scholar]
- Lu WY, Xiong ZG, Lei S, Orser BA, Dudek E, Browning MD, MacDonald JF. G-protein-coupled receptors act via protein kinase C and Src to regulate NMDA receptors. Nat Neurosci. 1999;2:331–338. doi: 10.1038/7243. [DOI] [PubMed] [Google Scholar]
- Lu WY, Jackson MF, Bai D, Orser BA, MacDonald JF. In CA1 pyramidal neurons of the hippocampus protein kinase C regulates calcium-dependent inactivation of NMDA receptors. J Neurosci. 2000;20:4452–4461. doi: 10.1523/JNEUROSCI.20-12-04452.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Allen M, Halt AR, Weisenhaus M, Dallapiazza RF, Hall DD, Usachev YM, McKnight GS, Hell JW. Age-dependent requirement of AKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO J. 2007;26:4879–4890. doi: 10.1038/sj.emboj.7601884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YM, Roder JC, Davidow J, Salter MW. Src activation in the induction of long-term potentiation in CA1 hippocampal neurons. Science. 1998;279:1363–1367. doi: 10.1126/science.279.5355.1363. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- Masuko N, Makino K, Kuwahara H, Fukunaga K, Sudo T, Araki N, Yamamoto H, Yamada Y, Miyamoto E, Saya H. Interaction of NE-dlg/SAP102, a neuronal and endocrine tissue-specific membrane-associated guanylate kinase protein, with calmodulin and PSD-95/SAP90. A possible regulatory role in molecular clustering at synaptic sites. J Biol Chem. 1999;274:5782–5790. doi: 10.1074/jbc.274.9.5782. [DOI] [PubMed] [Google Scholar]
- McGee AW, Bredt DS. Identification of an intramolecular interaction between the SH3 and guanylate kinase domains of PSD-95. J Biol Chem. 1999;274:17431–17436. doi: 10.1074/jbc.274.25.17431. [DOI] [PubMed] [Google Scholar]
- McGee AW, Dakoji SR, Olsen O, Bredt DS, Lim WA, Prehoda KE. Structure of the SH3-guanylate kinase module from PSD-95 suggests a mechanism for regulated assembly of MAGUK scaffolding proteins. Mol Cell. 2001;8:1291–1301. doi: 10.1016/s1097-2765(01)00411-7. [DOI] [PubMed] [Google Scholar]
- Merrill MA, Chen Y, Strack S, Hell JW. Activity-driven postsynaptic translocation of CaMKII. Trends Pharmacol Sci. 2005;26:645–653. doi: 10.1016/j.tips.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Merrill MA, Malik Z, Akyol Z, Bartos JA, Leonard AS, Hudmon A, Shea MA, Hell JW. Displacement of alpha-actinin from the NMDA receptor NR1 C0 domain by Ca2+/calmodulin promotes CaMKII binding. Biochemistry. 2007;46:8485–8497. doi: 10.1021/bi0623025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paarmann I, Spangenberg O, Lavie A, Konrad M. Formation of complexes between Ca2+ calmodulin and the synapse-associated protein SAP97 requires the SH3 domain-guanylate kinase domain-connecting HOOK region. J Biol Chem. 2002;277:40832–40838. doi: 10.1074/jbc.M205618200. [DOI] [PubMed] [Google Scholar]
- Park SY, Avraham H, Avraham S. Characterization of the tyrosine kinases RAFTK/Pyk2 and FAK in nerve growth factor-induced neuronal differentiation. J Biol Chem. 2000;275:19768–19777. doi: 10.1074/jbc.M909932199. [DOI] [PubMed] [Google Scholar]
- Park SY, Avraham HK, Avraham S. RAFTK/Pyk2 activation is mediated by trans-acting autophosphorylation in a Src-independent manner. J Biol Chem. 2004;279:33315–33322. doi: 10.1074/jbc.M313527200. [DOI] [PubMed] [Google Scholar]
- Pastalkova E, Serrano P, Pinkhasova D, Wallace E, Fenton AA, Sacktor TC. Storage of spatial information by the maintenance mechanism of LTP. Science. 2006;313:1141–1144. doi: 10.1126/science.1128657. [DOI] [PubMed] [Google Scholar]
- Pawson T, Scott JD. Signaling through scaffold, anchoring, and adaptor proteins. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- Pittman RN, Wang S, DiBenedetto AJ, Mills JC. A system for characterizing cellular and molecular events in programmed neuronal cell death. J Neurosci. 1993;13:3669–3680. doi: 10.1523/JNEUROSCI.13-09-03669.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Kim E, Sheng M, Craig AM. Heterogeneity in the molecular composition of excitatory postsynaptic sites during development of hippocampal neurons in culture. J Neurosci. 1998;18:1217–1229. doi: 10.1523/JNEUROSCI.18-04-01217.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebecchi MJ, Pentyala SN. Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol Rev. 2000;80:1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- Rebola N, Lujan R, Cunha RA, Mulle C. Adenosine A2A receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. Neuron. 2008;57:121–134. doi: 10.1016/j.neuron.2007.11.023. [DOI] [PubMed] [Google Scholar]
- Rothman SM, Olney JW. Excitotoxicity and the NMDA receptor. Trends Neurosci. 1987;10:299–302. doi: 10.1016/0166-2236(95)93869-y. [DOI] [PubMed] [Google Scholar]
- Sabri A, Govindarajan G, Griffin TM, Byron KL, Samarel AM, Lucchesi PA. Calcium- and protein kinase C-dependent activation of the tyrosine kinase PYK2 by angiotensin II in vascular smooth muscle. Circ Res. 1998;83:841–851. doi: 10.1161/01.res.83.8.841. [DOI] [PubMed] [Google Scholar]
- Sakata A, Ida E, Tominaga M, Onoue K. Calmodulin inhibitors, W-7 and TFP, block the calmodulin-independent activation of NADPH-oxidase by arachidonate in a cell-free system. Biochem Biophys Res Commun. 1987;148:112–119. doi: 10.1016/0006-291x(87)91083-7. [DOI] [PubMed] [Google Scholar]
- Salter MW, Kalia LV. Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci. 2004;5:317–328. doi: 10.1038/nrn1368. [DOI] [PubMed] [Google Scholar]
- Sans N, Petralia RS, Wang YX, Blahos J, 2nd, Hell JW, Wenthold RJ. A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J Neurosci. 2000;20:1260–1271. doi: 10.1523/JNEUROSCI.20-03-01260.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sans N, Wang PY, Du Q, Petralia RS, Wang YX, Nakka S, Blumer JB, Macara IG, Wenthold RJ. mPins modulates PSD-95 and SAP102 trafficking and influences NMDA receptor surface expression. Nat Cell Biol. 2005;7:1179–1190. doi: 10.1038/ncb1325. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Nagura K, Ishino M, Tobioka H, Kotani K, Sasaki T. Cloning and characterization of cell adhesion kinase beta, a novel protein-tyrosine kinase of the focal adhesion kinase subfamily. J Biol Chem. 1995;270:21206–21219. doi: 10.1074/jbc.270.36.21206. [DOI] [PubMed] [Google Scholar]
- Schaller MD. Calcium-dependent Pyk2 activation: a role for calmodulin? Biochem J. 2008;410:e3–e4. doi: 10.1042/BJ20080133. [DOI] [PubMed] [Google Scholar]
- Schlüter OM, Xu W, Malenka RC. Alternative N-terminal domains of PSD-95 and SAP97 govern activity-dependent regulation of synaptic AMPA receptor function. Neuron. 2006;51:99–111. doi: 10.1016/j.neuron.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Schweitzer E. Coordinated release of ATP and ACh from cholinergic synaptosomes and its inhibition by calmodulin antagonists. J Neurosci. 1987;7:2948–2956. doi: 10.1523/JNEUROSCI.07-09-02948.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seabold GK, Burette A, Lim IA, Weinberg RJ, Hell JW. Interaction of the tyrosine kinase Pyk2 with the N-methyl-d-aspartate receptor complex via the src homology 3 domains of PSD-95 and SAP102. J Biol Chem. 2003;278:15040–15048. doi: 10.1074/jbc.M212825200. [DOI] [PubMed] [Google Scholar]
- Shapira M, Zhai RG, Dresbach T, Bresler T, Torres VI, Gundelfinger ED, Ziv NE, Garner CC. Unitary assembly of presynaptic active zones from Piccolo-Bassoon transport vesicles. Neuron. 2003;38:237–252. doi: 10.1016/s0896-6273(03)00207-1. [DOI] [PubMed] [Google Scholar]
- Shen K, Meyer T. Dynamic control of CaMKII Translocation in hippocampal neurons by NMDA receptor stimulation. Science. 1999;284:162–166. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- Shin H, Hsueh YP, Yang FC, Kim E, Sheng M. An intramolecular interaction between Src homology 3 domain and guanylate kinase-like domain required for channel clustering by postsynaptic density-95/SAP90. J Neurosci. 2000;20:3580–3587. doi: 10.1523/JNEUROSCI.20-10-03580.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siciliano JC, Toutant M, Derkinderen P, Sasaki T, Girault JA. Differential regulation of proline-rich tyrosine kinase 2/cell adhesion kinase beta (PYK2/CAKbeta) and pp125(FAK) by glutamate and depolarization in rat hippocampus. J Biol Chem. 1996;271:28942–28946. doi: 10.1074/jbc.271.46.28942. [DOI] [PubMed] [Google Scholar]
- Sorokin A, Kozlowski P, Graves L, Philip A. Protein-tyrosine kinase Pyk2 mediates endothelin-induced p38 MAPK activation in glomerular mesangial cells. J Biol Chem. 2001;276:21521–21528. doi: 10.1074/jbc.M008869200. [DOI] [PubMed] [Google Scholar]
- Stein CS, Davidson BL. Gene transfer to the brain using feline immunodeficiency virus-based lentivirus vectors. Methods Enzymol. 2002;346:433–454. doi: 10.1016/s0076-6879(02)46070-3. [DOI] [PubMed] [Google Scholar]
- Sun XX, Hodge JJ, Zhou Y, Nguyen M, Griffith LC. The eag potassium channel binds and locally activates calcium/calmodulin-dependent protein kinase II. J Biol Chem. 2004;279:10206–10214. doi: 10.1074/jbc.M310728200. [DOI] [PubMed] [Google Scholar]
- Tomita S, Nicoll RA, Bredt DS. PDZ protein interactions regulating glutamate receptor function and plasticity. J Cell Biol. 2001;153:F19–F24. doi: 10.1083/jcb.153.5.f19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–212. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- Valtschanoff JG, Weinberg RJ. Laminar organization of the NMDA receptor complex within the postsynaptic density. J Neurosci. 2001;21:1211–1217. doi: 10.1523/JNEUROSCI.21-04-01211.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandonselaar M, Hickie RA, Quail JW, Delbaere LT. Trifluoperazine-induced conformational change in Ca2+-calmodulin. Nat Struct Biol. 1994;1:795–801. doi: 10.1038/nsb1194-795. [DOI] [PubMed] [Google Scholar]
- Wakamori M, Hidaka H, Akaike N. Hyperpolarizing muscarinic responses of freshly dissociated rat hippocampal CA1 neurones. J Physiol. 1993;463:585–604. doi: 10.1113/jphysiol.1993.sp019612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LY, MacDonald JF. Modulation by magnesium of the affinity of NMDA receptors for glycine in murine hippocampal neurones. J Physiol. 1995;486:83–95. doi: 10.1113/jphysiol.1995.sp020792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YT, Salter MW. Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature. 1994;369:233–235. doi: 10.1038/369233a0. [DOI] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- Wu SS, Jácamo RO, Vong SK, Rozengurt E. Differential regulation of Pyk2 phosphorylation at Tyr-402 and Tyr-580 in intestinal epithelial cells: roles of calcium, Src, Rho kinase, and the cytoskeleton. Cell Signal. 2006;18:1932–1940. doi: 10.1016/j.cellsig.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Xiong ZG, Pelkey KA, Lu WY, Lu YM, Roder JC, MacDonald JF, Salter MW. Src potentiation of NMDA receptors in hippocampal and spinal neurons is not mediated by reducing zinc inhibition. J Neurosci. 1999;19:RC37. doi: 10.1523/JNEUROSCI.19-21-j0003.1999. (1–6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Weerapura M, Ali MK, Jackson MF, Li H, Lei G, Xue S, Kwan CL, Manolson MF, Yang K, Macdonald JF, Yu XM. Control of excitatory synaptic transmission by C-terminal Src kinase. J Biol Chem. 2008;283:17503–17514. doi: 10.1074/jbc.M800917200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Li X, Marchetto GS, Dy R, Hunter D, Calvo B, Dawson TL, Wilm M, Anderegg RJ, Graves LM, Earp HS. Activation of a novel calcium-dependent protein-tyrosine kinase. Correlation with c-Jun N-terminal kinase but not mitogen-activated protein kinase activation. J Biol Chem. 1996;271:29993–29998. doi: 10.1074/jbc.271.47.29993. [DOI] [PubMed] [Google Scholar]
- Yu XM, Askalan R, Keil GJ, 2nd, Salter MW. NMDA channel regulation by channel-associated protein tyrosine kinase Src. Science. 1997;275:674–678. doi: 10.1126/science.275.5300.674. [DOI] [PubMed] [Google Scholar]
- Zhai RG, Vardinon-Friedman H, Cases-Langhoff C, Becker B, Gundelfinger ED, Ziv NE, Garner CC. Assembling the presynaptic active zone: a characterization of an active one precursor vesicle. Neuron. 2001;29:131–143. doi: 10.1016/s0896-6273(01)00185-4. [DOI] [PubMed] [Google Scholar]
- Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]