

Abstract
Apoptosis, a programmed cell death mechanism, is a fundamental process during the normal development and somatic maintenance of all multicellular organisms and thus is highly conserved and tightly regulated through numerous signaling pathways. Apoptosis is of particular clinical importance as its dysregulation contributes significantly to numerous human diseases, primarily through changes in the expression and activation of key apoptotic regulators. Each of the four families of heterotrimeric G proteins (Gs, Gi/o, Gq/11 and G12/13) has been implicated in numerous cellular signaling processes, including proliferation, transformation, migration, differentiation, and apoptosis. Heterotrimeric G protein signaling is an important but not widely studied mechanism regulating apoptosis. G protein Signaling and Apoptosis broadly cover two large bodies of literature and share numerous signaling pathways. Examination of the intersection between these two areas is the focus of this review. Several studies have implicated signaling through each of the four heterotrimeric G protein families to regulate apoptosis within numerous disease contexts, but the mechanism(s) are not well defined. Each G protein family has been shown to stimulate and/or inhibit apoptosis in a context-dependent fashion through regulating numerous downstream effectors including the Bcl-2 family, NF-κB, PI3 Kinase, MAP Kinases, and small GTPases. These cell-type specific and G protein coupled receptor dependent effects have led to a complex body of literature of G protein regulation of apoptosis. Here, we review the literature and summarize apoptotic signaling through each of the four heterotrimeric G protein families (and the relevant G protein coupled receptors), and discuss limitations and future directions for research on regulating apoptosis through G protein coupled mechanisms. Continued investigation in this field is essential for the identification of important targets for pharmacological intervention in numerous diseases.
Keywords: G proteins, cell death, apoptosis, signal transduction, cancer, ischemia, autoimmunity
I. OVERVIEW OF CELL DEATH AND APOPTOSIS
The regulation of cell death is highly conserved throughout evolution and is a critical process in nearly all human diseases. Cell death is also a part of normal development and maintenance of somatic tissues. Cell death can occur through two distinct mechanisms: apoptosis, an ordered and programmed process, and necrosis, an accidental and fairly unordered process [1]. Necrosis is usually a disorderly process characterized by inflammation, release of cytoplasmic content to the extracellular space, and subsequent widespread tissue damage. Necrosis typically occurs in response to extreme or extensive cellular or tissue damage. On the other hand, apoptosis (summarized in Fig. 1) is a highly conserved programmed cell death mechanism that morphologically consists of nuclear membrane fragmentation, chromatin compaction and fragmentation, and cell disintegration through membrane blebbing; these apoptotic bodies are then quickly ingested and degraded by professional phagocytes and neighboring cells, preventing inflammation and tissue scarring [2-4]. Apoptosis is of major clinical relevance because dysregulated apoptosis caused by the suppression, overexpression, or mutation of key apoptotic regulators contributes significantly to numerous human diseases [5]. Major diseases involving dysregulated apoptosis include polycystic kidney disease, ischemic heart disease, stroke, hepatitis B and C, and type 1 diabetes mellitus, among many others. While cell death has been classically divided into these two mechanisms, this dichotomy is excessively restrictive and fails to recognize the heterogeneity in mechanisms and morphological responses in cell death. Appreciation of the complex nature of these processes is evident by the evolution of new nomenclature such as necroptosis [6], a process comprising cell death mechanisms that overlap both mechanistically and morphologically with apoptosis and necrosis. As cell death research progresses, it will be important to recognize the heterogeneity of cell death mechanisms.
Fig. (1). Apoptosis Signaling Pathways.
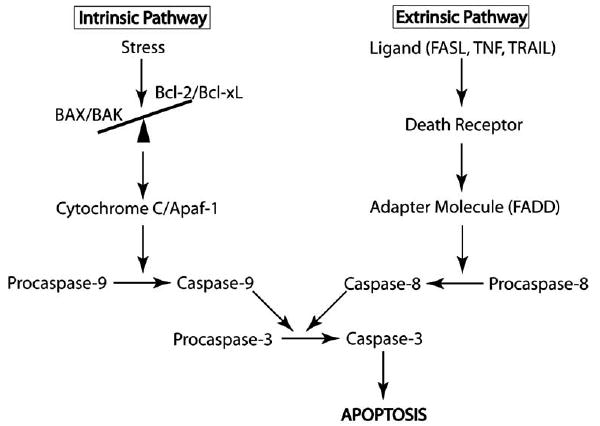
The Intrinsic pathway is triggered by various stresses including hypoxia/ischemia, growth factor deprivation, and cell damage. These stimuli cause a disruption in the ratio of pro-apoptotic (BAX, BAK) to anti-apoptotic (Bcl-2, Bcl-xL) Bcl-2 family proteins at the mitochondria. Relative increase in the levels of the pro-apoptotic proteins allows for their homodimerization, and subsequent Cytochrome C efflux into the cytoplasm activates Apaf-1. Apaf-1 triggers Caspase-9 aggregation and activation and the subsequent caspase cascade. The extrinsic pathway is triggered by ligand binding to the death receptors, which causes receptor trimerization and activation. Activated receptors recruit adapter proteins, such as FADD, leading ultimately to the activation of Caspase-8 and the subsequent caspase cascade. Both pathways converge on Caspase-3, leading to apoptosis.
Apoptosis is classically stimulated through two major pathways (see Fig. 1), the intrinsic pathway (mitochondrial pathway) and the extrinsic pathway (death-receptor pathway), and each is associated with unique stimuli and independent signal transduction cascades. Both pathways converge downstream on the activation of Caspase-3 (Fig. 1), the cysteine aspartyl protease that initiates most of the aforementioned morphological changes associated with apoptosis. Activated Caspase-3 has a number of substrates, including Caspase Activated DNAse (CAD), the nuclease responsible for apoptosis-associated DNA fragmentation. The extrinsic pathway results from stimulation of death receptors, primarily consisting of the tumor necrosis factor (TNF) receptor superfamily, and is mediated by Caspase-8 [7, 8] upstream of Caspase-3. The TNF receptor superfamily comprises more than 20 proteins with various functions, including the regulation of proliferation, apoptosis, and differentiation. These receptors are highly homologous, with cysteine-rich extracellular domains and 80 amino acid intracellular “death domains” that are critical in the transduction of extracellular signals [9]. The major apoptosis regulating receptors of this family include CD95 (FAS) and TNF-related apoptosis-inducing ligand (TRAIL) receptor. Binding of ligand to the receptor leads to receptor trimerization and recruitment of adaptor molecules, including FAS associated Death Domain (FADD). Binding of FADD recruits Procaspase-8 directly to the activated receptor complex. Procaspase-8 is activated to Caspase-8 at the receptor through self-cleavage and in turn activates Procaspase-3 to Caspase-3, leading to downstream apoptosis [10]. The extrinsic pathway is essential for normal development and modulation of the immune system and has been shown to be prominently dysregulated in various cancers. Many resistant forms of tumors show either downregulation or complete suppression of death receptor expression, suggesting that this may be a major mechanism of cancer cell immortalization [9].
The intrinsic apoptosis pathway (Fig. 1), mediated by the Bcl-2 family of proteins and Caspase-9, is a general response to cell damage, improper proliferation signals, and growth factor deprivation. The Bcl-2 family (reviewed in [11, 12]), consisting of both pro-apoptotic and anti-apoptotic proteins, is an important checkpoint of the intrinsic apoptosis pathway. The relative activity of the pro-apoptotic and anti-apoptotic Bcl-2 family members is a major determinant of whether apoptosis will be initiated. The major anti-apoptotic proteins of this family include the constitutively expressed Bcl-2 and the inducible Bcl-xL. The pro-apoptotic members of the family are typically divided into transmembrane proteins, which include BAX and BAK, and membrane associated and cytosolic effectors, including BID, BIM, BAD, and others. Bcl-2 is an important cell survival protein: its over-expression increases the survival of cells in various adverse circumstances including loss of cell adhesion [13] and even γ-irradiation [14], and it has been implicated in numerous proliferative and degenerative disorders, including various lymphomas [15], juvenile parkinsonism [16], Alzheimer’s dementia [17] and cystic kidney disease [18]. Deletion of Bcl-2 results in numerous disorders, including polycystic kidney disease with death of renal epithelial progenitors, melanocyte progenitors, and lymphocyte progenitors [12]. Bcl-2 is a transmembrane protein localized in the outer mitochondrial, endoplasmic reticulum and perinuclear membranes that under proliferative and anti-apoptotic (pro-survival) conditions heterodimerizes with pro-apoptotic BAX or BAK to form a stable, non-conductive complex [19]. Under pro-apoptotic conditions, Bcl-2 is inactivated and BAX or BAK homodimerize and catalyze mitochondrial outer membrane permeabilization and the formation of the mitochondrial apoptosis-induced channel. This leads to the efflux of Cytochrome C, which activates Apaf-1 [11, 19]. Apaf-1 catalyzes the conversion of Procaspase-9 to active Caspase-9, triggering the caspase cascade (reviewed in [20]), leading eventually to Caspase-3 activation and apoptosis. Bcl-2 function is regulated by phosphorylation through several kinases and phosphatases; phosphorylation of Bcl-2 prevents dimerization with BAX and targets Bcl-2 for degradation through the proteasome pathway, and additional mechanisms regulate Bcl-2 transcription and translation [21]. Pro-apoptotic Bcl-2 family proteins include the BH3-only proteins, such as BAD and BID, which regulate Bcl-2 functionality through direct interactions. Regulation of the intrinsic pathway by such factors as the inhibitors of apoptosis (IAP), which associate with and inhibit activated cytosolic caspases, is an important additional level of control. During pro-apoptotic signaling, inhibitors of IAPs, including Smac/Diablo and Omi/HtrA2 promote the maintenance of caspase activity, leading to cell death (reviewed in [22]).
It is now recognized that there is significant overlap between the intrinsic and extrinsic pathways. Bcl-2 family members have been shown to participate in extrinsic pathway mechanisms, and concordant activation of both the intrinsic and extrinsic pathways has also been observed. As the complexities of these pathways are slowly being unraveled, new complexities continue to arise in this original paradigm.
II. OVERVIEW OF G PROTEIN SIGNALING
Among the many important regulators of the apoptotic cascades are the guanine nucleotide binding proteins (G proteins). G proteins are important cellular signal transduction molecules that consist of two major groups: the small monomeric GTPases and the heterotrimeric G proteins. Both classes of G proteins regulate numerous cellular processes including proliferation, differentiation, junctional assembly, apoptosis, and motility [23]. G proteins are unified by a common activation mechanism involving conformational changes with GTP/GDP exchange, and there are many overlapping signal transduction pathways. To some extent, the functions of monomeric and heterotrimeric G proteins are divergent: monomeric GTPases are typically downstream of receptor mediated events, while heterotrimeric G proteins primarily transduce extracellular signals from cell surface receptors. As such, we focus this review on heterotrimeric G proteins which are readily targeted through their highly specific cell surface receptors and are thus primary therapeutic targets.
Because the interaction of heterotrimeric G proteins (henceforth referred to as G proteins) with plasma membrane receptors places them at the earliest step in cellular signal transduction pathways, there is great potential for modulating down-stream signaling through drugs that bind to the cell surface receptors. G protein coupled receptors (GPCRs) are a diverse family of proteins important for cellular responses to hormones, neurotransmitters and sensory stimuli and are essential for nearly all aspects of cellular function. Furthermore, GPCRs are highly cell type specific and thus present a selective therapeutic target. G proteins consist of Gα and Gβγ subunits, and are grouped into four major families. The families are named for the Gα subunit, and in humans, there are 21 Gα subunits encoded by 16 genes, 6 Gβ subunits encoded by 5 genes and 12 Gγ subunits. The four main G protein families are Gs, Gi, Gq, and G12 (reviewed in [24]). GαS is ubiquitously expressed and activates adenylylcyclase and some calcium channels. The second family of G proteins comprises Gαi, Gαo, Gαt (transducin) and Gαz. Gαi is found in all tissues, and Gαo is found in largest amounts in the brain and heart. The Gαq family primarily stimulates the activity of polyphosphoinositide-specific phospholipase C β (PLCβ). The Gαq family is important in regulation of intracellular calcium through the generation of IP3 and diacylglycerol and is especially important in cardiac and neuronal signal transduction. The Gα12/13 protein family (reviewed in [25, 26]) consists of the ubiquitously expressed members Gα12 and Gα13, and they regulate a variety of cellular responses including proliferation [27], transformation [28], tight junction assembly [29-31], directed cell migration [32], regulation of the actin cytoskeleton and stress fiber formation [33]. Gα12/13 activation of Rho signaling has been directly demonstrated through RhoGEF proteins (p115-RhoGEF, PDZ-RhoGEF, and others) that also contain RGS domains [25, 34]. Gα12/13 are potent oncogenes in some cell types [35, 36], and are up-regulated in breast and prostate cancer cells with high invasion potential [37, 38].
The mechanisms of signal transduction through G proteins have been partially defined by elegant crystallographic studies of G proteins in different conformations (reviewed in [24]). Gα subunits are composed of a GTPase domain (shared with monomeric G proteins) and a helical domain. The GTPase domain binds guanine nucleotides and contains the conformationally sensitive regions necessary for GTP hydrolysis. In addition, the GTPase domain contains the interaction sites for the Gβγ dimer. The helical domain is unique to heterotrimeric Gα subunits and its functions are not well-characterized. All Gα subunits (except transducin) are palmitoylated at the N-terminus and several are also myristoylated. These modifications are important for interactions with the Gβγ subunit and the plasma membrane. Exogenous toxins are also capable of modifying specific Gα subunits. Gαs is ADP-ribosylated by cholera toxin, resulting in its constitutive activation. Conversely, Gαi, Gαo, and Gαt are ADP-ribosylated by pertussis toxin, which uncouples it from its receptor and prevents GTP/GDP exchange, leading to inactivation. The Gβ subunit is a protein composed of seven WD40 sequence repeats and forms a seven-bladed propeller. The Gβ N-terminus interacts tightly with the Gγ subunit through a coiled-coil interaction and Gγ cannot be dissociated from Gβ except under denaturing conditions. Membrane interactions of Gβγ are facilitated by isoprenylation of Gγ on its C-terminus.
In the resting state, G protein coupled receptors form a stable complex with GDP-liganded Gαβγ (Fig. 2). When agonists bind to GPCRs, there are conformational changes in the cytoplasmic domains of the receptor that alter Gα conformation and lead to GDP release from Gα. Cellular GTP concentration exceeds GDP by several-fold favoring the binding of GTP to Gα. GTP binding induces conformational changes in Gα, leading to changes in the orientation of Gα with the receptor and dissociation from Gβγ subunits. Thus, new interactions of Gα and Gβγ with down-stream effectors are initiated, leading to signal transduction. The duration of activation varies and is determined by rate of GTP hydrolysis on Gα.
Fig. (2). The G Protein Cycle.
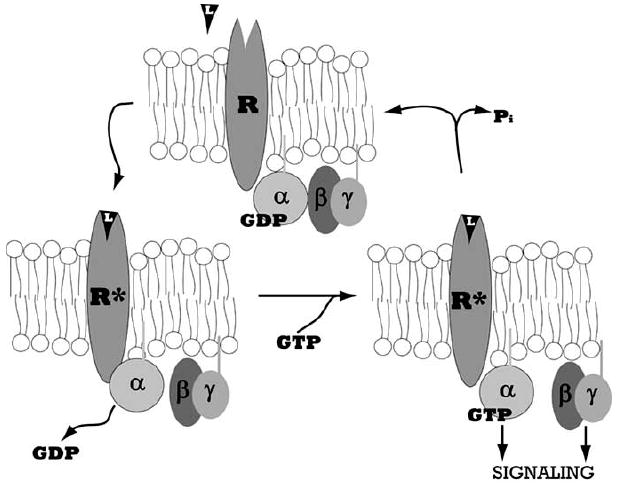
Heterotrimeric G proteins comprise three subunits, α, β, and γ, which associate in the inactive Gα-GDP bound state. The seven transmembrane domain G protein coupled receptor (GPCR), denoted by R, is activated by ligand binding, L. The activated receptor, denoted by R*, associates with Gα and catalyzes the dissociation of GDP. The subsequent binding of GTP causes Gα to dissociate from Gβγ, exposing interaction sites on Gα and Gβγ through which these proteins can activate downstream effectors and transduce signals. Subsequent hydrolysis of GTP to GDP by the intrinsic GTPase activity of Gα causes inactivation and reassociation of the Gαβγ complex.
The major issue of signaling specificity from a vast number of receptors through a limited number of G proteins remains only partially understood. Some specificity resides in the interactions of receptors with specific G proteins, but there is significant overlap and many other mechanisms are likely to account for the unique cellular responses initiated by agonist binding to a GPCR. Evidence now suggests that subcellular localization of signaling molecules into discrete microdomains, differences in the stoichiometry of receptors and G proteins, post-translational modifications of G proteins, and interactions with other proteins including scaffolding or regulatory proteins are all important for establishing signaling specificity. In the past several years, additional families of proteins that regulate the canonical G protein cycle have been identified. These include the large family of RGS proteins (Regulators of G protein Signaling) that interact with GTP-liganded Gα subunits and stimulate the intrinsic GTPase activity of Gα (GAP function) (reviewed in [39, 40]). Another family of regulatory proteins is the GPR (G protein Regulator) family consisting of proteins that share a conserved motif (Goloco). These proteins bind to GDP-liganded Gα subunits to form a stable complex and inhibit GDP release (reviewed in [41]). Finally, non-receptor proteins can also activate G protein signaling (activators of G protein signaling – AGS) (reviewed in [42]) through non-classical mechanisms. G proteins have also been found to couple directly to a variety of single transmembrane domain receptors, including receptor tyrosine kinases, integrins, insulin and insulin-like growth factor receptors, and others (reviewed in [43]). There is now recognition for broad cellular roles for G proteins in many cellular processes beyond the initial GPCR/Gα interface at the plasma membrane and include vesicular trafficking, protein processing through the Golgi, cytokinesis [44, 45], as well as cell growth and other fundamental cellular properties including apoptosis.
Signaling through G proteins is an important mechanism regulating apoptosis. Numerous studies have implicated signaling through each of the four G protein families to regulate apoptosis, but the mechanism(s) are not well defined. Most results in the literature are cell type and stimulus specific, so clear themes have been slow to emerge. This review will summarize the available data on the role of each G protein family in pro-apoptotic and anti-apoptotic pathways, discuss limitations of the available literature and highlight some emerging themes. With further elucidation of these pathways, modulation of G protein signaling by targeting specific GPCRs or Gα subunits may prove to be an important and effective therapeutic strategy in the treatment of numerous human diseases.
III. REVIEW OF LITERATURE ON G PROTEIN REGULATION OF APOPTOSIS
A. Gαs and Apoptosis
Gαs has been implicated in numerous pro-apoptotic and anti-apoptotic pathways and this literature is summarized in Table 1. The most prominent paradigm of Gαs signaling involves the cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA) pathways. In many cell types, activation of Gαs leads to induction of adenylylcyclase, increasing intracellular cAMP levels and leading to activation of PKA. Downstream signaling through PKA leads to the eventual physiological effect of Gαs activation. While most studies of apoptosis involving Gαs have implicated this pathway, several involve cAMP-independent induction of apoptosis as well.
Table 1.
Cell Culture and Animal Studies Examining the Role of Gαs in Apoptosis
| No. | G Protein | Model System | Cell Type | Apoptotic Stimulus | Phenotype | Pathway | Comments | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | Gαs | Overexpression | Neurons (Human SH-SY5Y neuroblastoma cells) | Serum starvation H2O2 tunicamycin |
Stimulated | Independent of cAMP/PKA Caspase dependent |
May be physiologically relevant to bipolar disorders | [46] |
| 2 | Q227L-Gαs | Overexpression | Neurons (Human SH-SY5Y neuroblastoma cells) | H2O2 | Inhibited | cAMP/PKA dependent repression of BAK induction of Bcl-xL inhibition of NF-κB, NFAT, AP1 |
[47] | |
| 3 | Gαs | Endogenous Prostaglandin E2 – activation of Gαs |
Neurons (Human SH-SY5Y neuroblastoma cells) | H2O2 | Inhibited | cAMP/PKA dependent repression of BAK inhibition of NF-κB, NFAT, AP1 |
[47] | |
| 4 | Gαs | Endogenous β1-adrenergic receptor activation |
Cardiomyocytes (murine primary cultures) | NONE | Stimulated | cAMP/PKA dependent | β2-knockout | [48] |
| 5 | Gαs | Endogenous β2-adrenergic receptor activation |
Cardiomyocytes (murine primary cultures) | NONE | Stimulated | cAMP/PKA dependent | β1-knockout Also anti-apoptotic through the actions of Gαi |
[48] |
| 6 | Gαs | Overexpression | Cardiomyocytes (murine transgenic) | NONE | Stimulated | cAMP/PKA signaling | Also amplifies β-adrenergic simulation of cardiac contractility | [49] |
| 7 | Gαs | Knockout | Pancreatic β cells (murine transgenic) | NONE | Stimulated | cAMP/PKA signaling with unestablished downstream mediators | May be related to downstream IRS2 activation | [50] |
| 8 | Gαs | Endogenous Ghrelin – activation of Gαs |
Pancreatic β cells (Hamster HIT-T15 cells) | Serum starvation Interferon-γ TNF-α |
Inhibited | cAMP/PKA signaling and downstream ERK1/2 and PI3K/Akt activation | Also stimulated proliferation | [51,52] |
| 9 | Gαs | Endogenous Ghrelin – activation of Gαs |
Pancreatic β cells (Human primary culture) | Serum starvation Interferon-γ TNF-α |
Inhibited | cAMP/PKA signaling and downstream ERK1/2 and PI3K/Akt activation | Also stimulated proliferation | [51,52] |
| 10 | Gαolf | Overexpression | Pancreatic β Cells (Murine βTC-3 cells) | Serum starvation | Inhibited | cAMP/PKA signaling | Adenylylcyclase I and VIII dependent | [53] |
| 11 | Gαs | Endogenous Isoproterenol (β-adrenergic agonist) |
T Lymphocytes (Murine S49 cells) | NONE | Stimulated | cAMP/PKA signaling | Inhibited by Bcl-2 overexpression | [54] |
| 12 | Gαs | Endogenous Isoproterenol (β-adrenergic agonist) |
T Lymphocytes (Murine S49 cells) | NONE | Stimulated | Lck signaling | PKA independent | [55] |
| 13 | Gαs | Endogenous Isoproterenol (β-adrenergic agonist) |
T Lymphocytes (Murine primary CD4+/CD8+ thymocytes) | NONE | Stimulated | Lck signaling | [55] | |
| 14 | Q227L-Gαs | overexpression | Breast carcinoma (Human MCF7 cells) | Serum starvation | Stimulated | cAMP/PKA dependent inhibition of ERK1/2 |
[64] |
Studies of Gαs in neuronal cells have led to conflicting findings, and these are summarized in Table 1, Rows 1-3. Gαs overexpression in neuronal cells increased sensitivity to apoptotic stimuli independent of cAMP and PKA activity. Specifically, overexpression of Gαs in the SH-SY5Y neuroblastoma cell line augmented apoptosis induced by serum starvation, hydrogen peroxide and tunicamycin. However, in this same SH-SY5Y neuroblastoma cell line, the transgenic expression of a constitutively activated Gαs inhibited hydrogen peroxide induced apoptosis by preventing the transcriptional upregulation of BAK and downregulation of Bcl-xL [46]. Similar effects were seen upon treatment of non-transfected SH-SY5Y cells with prostaglandin E2, a Gαs agonist, and these effects were mediated by cAMP production and PKA activation. PKA activation in turn led to the inhibition of the transcription factors AP-1, Nf-κB, and NFAT [47]. In interpreting these results, the physiological relevance of overexpression systems must be carefully considered. It is possible that overexpression of Gαs induces non-physiological apoptosis, but activation of endogenous Gαs through receptor (such as prostaglandin E2) mediated mechanisms may indeed be anti-apoptotic. Further studies will be necessary to resolve these issues.
In cardiomyocytes (Table 1, Rows 4-6), Gαs activation by both the β1-adrenergic receptor and β2-adrenergic receptor led to a pro-apoptotic state. These effects were modulated by downstream activation of adenylylcyclase and PKA. However, activation of the β2-adrenergic receptor also induced dominant anti-apoptotic effects through Gαi signaling (discussed subsequently), suggesting the possibility of reciprocal regulation of apoptosis through different Gα families in cardiomyocytes. Selective inhibition of Gαi with pertussis toxin in myocytes allowed for the examination of Gαs-specific pro-apoptotic signaling [48]. Similarly, transgenic mice overexpressing Gαs in cardiomyocytes using the α-myosin heavy chain promoter showed a distinct cardiomyopathy characterized by myocytes with chromatin condensation, DNA laddering and cellular vacuolization characteristic of apoptosis. cAMP levels and PKA activity were increased. These mice also showed increased cardiomyocyte contractility in response to β-adrenergic stimulation, suggesting that overexpressing Gαs amplifies β-adrenergic signaling in the heart. This may be a mechanism by which apoptosis is induced [49]. Taken together, these findings in cardiac myocytes are consistent with Gαs stimulated apoptosis, although the physiologic relevance of overexpressing Gαs is not clear. Studying the activation of endogenous Gαs will present a clearer physiological picture.
In pancreatic islet β-cells (Table 3, Rows 7-10), Gαs mediates glucagon-like peptide 1 and incretin hormone signaling, which promote β-cell survival and insulin release. β-cell specific Gαs knockout mice showed significantly increased β-cell apoptosis and symptoms of diabetes mellitus, including severe hyperglycemia and glucose intolerance. The pro-survival effects of Gαs in β-cells may be mediated by downstream cAMP/PKA stimulation of insulin receptor substrate 2 (IRS2), although this study found primarily IRS2-independent effects [50]. Additional studies have also implicated ghrelin, a hormone produced by the gastric P/D1 cells and hypothalamic arcuate nucleus that stimulates insulin secretion and glucose metabolism. Specifically, ghrelin has been shown to activate cAMP/PKA mediated resistance to serum starvation and interferon-γ/tumor necrosis factor-α induced apoptosis in β-cells [51, 52]. These phenotypes were associated with elevated intracellular cAMP levels, and inhibition of adenylylcyclase with MDL12330A and KT5720 abrogated these anti-apoptotic effects. Notably, ghrelin also stimulated β-cell proliferation through these same pathways. Similarly, the overexpression of constitutively activated Gαolf (a Gαs family member) in murine βTC-3 cells, a β-cell line derived from SV40-T transgenic mice, protected these cells from serum withdrawal induced apoptosis. These effects were mediated by increased cAMP specifically through the activation of adenylylcyclase I and VIII and presumably downstream PKA signaling, although the precise mechanisms have not been established [53]. Taken together, these findings suggest that in pancreatic β-cells, Gαs may be necessary to prevent apoptosis and its activation or overexpression may lead to inhibition of stimulated apoptosis.
Table 3.
Cell Culture and Animal Studies Examining the Role of Gαq/11 in Apoptosis
| No. | G Protein | Model System | Cell Type | Apoptotic Stimulus | Phenotype | Pathway | Comments | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | Gαq/11 | Endogenous Carbachol activation (M3) |
Chinese Hamster Ovary (CHO cells) | etoposide | Inhibited | PLC-independent Increased Bcl-2 expression Activation of ERK1/2, JNK |
M3 receptor truncation caused loss of anti-apoptotic activity | [93,94] |
| 2 | Gαq/11 | Endogenous Carbachol activation (M3) |
CHO Cells | NONE | Stimulated | Downstream Rac-1 activation | Cells overexpressing Rac1 | [95] |
| 3 | Gαq/11 | Endogenous Adenosine activation (P2Y1) |
Human Astrocytoma (1231N1 cells) | Serum starvation | Stimulated | PKC activation PI3K, ERK1/2 activation |
[96] | |
| 4 | Gα11 | Overexpression | HeLa Cells | NONE | Stimulated | Activation of ROCK | Blocked by overexpression of Bcl-2 | [97,98] |
| 5 | Gα11 | Endogenous M1 activation |
HeLa Cells | NONE | Stimulated | Activation of ROCK | Blocked by overexpression of Bcl-2 | [97,98] |
| 6 | R183C-Gαq | Overexpression | COS-7 Cells, CHO Cells | NONE | Stimulated | PKC activation | Blocked by overexpression of Bcl-2 | [99] |
| 7 | Gαq | Overexpression | Rat Ventricular Myocytes | Low-glucose | Inhibited | Activation of PI3K, EGFR, and Src kinases | [100] | |
| 8 | Gαq | Endogenous P. multocida toxin activation |
Rat Ventricular Myocytes | H2O2 | Stimulated | Activation of PLC Inhibition of Akt |
[101] | |
| 9 | Q209L-Gαq | Overexpression | Rat Ventricular Myocytes | NONE | Stimulated | Depletion of PIP2 Reduced Akt phosphorylation |
Inhibited by overexpression of myristoylated Akt | [102-104] |
In mouse S49 lymphoma cells, a model of T lymphocytes (Table 1, Rows 11-13), activation of Gαs by isoproterenol induced PKA-dependent apoptosis. Isoproterenol is a non-selective β-adrenergic receptor agonist. Forskolin, an adenylylcyclase activator, also stimulated apoptosis, suggesting that the effects of Gαs activation are indeed predominantly mediated by cAMP. These pro-apoptotic effects were completely blocked by concomitant overexpression of Bcl-2 [54], implicating a predominantly intrinsic apoptosis cascade in this pathway. Gαs may also induce a non-PKA dependent apoptosis in these lymphocytes. Specifically, in another study, isoproterenol induction of apoptosis was blocked in S49 lymphoma cells with Gαs knocked out, but was not abrogated in PKA knockout S49 cells, suggesting PKA-independent mechanisms of apoptosis induced by Gαs. These effects were mediated by the Src family kinase, Lck which is directly activated by Gαs. Similar pathways of Gαs mediated apoptosis were also established in double positive (CD4+/CD8+) primary murine thymocytes grown in culture [55]. Collectively, these studies suggest that Gαs activation stimulates apoptosis, although the signaling pathways may be both cAMP/PKA dependent and independent.
Finally, in human cancers, Gαs polymorphisms have been shown to correlate with clinical outcome of various cancers. For example, the GNAS1 T393C mutation, which increases the transcription of Gαs, has been associated with more favorable clinical outcomes in breast carcinoma [56], renal carcinoma [57], bladder carcinoma [58], colorectal adenocarcinoma [59], chronic lymphocytic leukemia [60], and intrahepatic cholangiocarcinoma [61], although the precise mechanisms remain to be established. It is possible that the increase in Gαs expression affects apoptosis. However, many activating Gαs mutations have also been found in various cancers, most of which activate Gαs by inhibiting its intrinsic GTPase activity and extending its active GTP-bound half-life. Such mutations have been found in various renal carcinomas [62] and Gαs activating mutations were found in 30% of juvenile ovarian granulosa cell tumors [63]. When a constitutively activated Gαs was expressed in MCF7 breast carcinoma cells, tumorigenesis was inhibited, possibly through its pro-apoptotic effects. These effects were mediated by downstream production of cAMP, activation of PKA and inhibition of ERK1/2 signaling [64]. Thus, the precise role(s) of Gαs in malignancy remain unclear at this time, and it remains to be determined whether or not Gαs regulation of apoptosis is an important process in these cancers.
The experimental approaches to study Gαs regulated apoptosis include Gαs activation through endogenous receptors, overexpression of wild type or constitutively active Gαs protein and Gαs knockout in some animals/cell types. Each approach has particular advantages and limitations, necessitating the use of multiple methods to conclusively define these phenotypes. Because many early studies relied exclusively on single methods, it is difficult to draw definitive conclusions. However, some clear patterns of Gαs regulation of apoptosis begin to emerge. In cardiomyocytes, the studies to date have been consistent and reveal a pro-apoptotic effect of Gαs activation through the β-adrenergic receptors. Similarly, in lymphocytes, activation of Gαs also has clear pro-apoptotic effects. On the other hand, in pancreatic β-cells, expression and activation of Gαs appears to be primarily anti-apoptotic. However, in neuronal cell lines the limited studies have conflicting results and will require additional study. At this time, it is also not possible to reach any conclusions about the potential role of Gαs regulated apoptosis in human cancers. Using multiple models will help clarify the exact roles of Gαs in these cell types, as will the use of other cell lines, primary cultures, and animal models.
B. Gαi/o and Apoptosis
A large number of studies have examined the role of the Gαi family in regulating apoptosis (Table 2). Unlike the studies with Gαs, the results with Gαi are highly consistent. With few exceptions, agonists and stimuli can be neatly categorized as pro-apoptotic (Table 2, Rows 1-9) or anti-apoptotic (Table 2, Rows 10-23). Although some members of the Gαi/o family inhibit adenylylcyclase, lower cAMP levels and might be expected to decrease apoptosis, the regulation of adenylylcyclase is complex (reviewed in [65]). Therefore, it is not surprising that the effects of this family of G proteins on regulating apoptosis are also diverse (see Table 2). Where adenylylcyclase was studied (Table 2, Rows 1,2 and 6), inhibition was associated with increased apoptosis. There are also several examples where activation of Gαs and Gαi have similar effects on apoptosis, suggesting that adenylylcyclase independent pathways are important in the regulation of apoptosis by these G proteins.
Table 2.
Cell Culture and Animal Studies Examining the Role of Gαi/o in Apoptosis
| No. | G Protein | Model System | Cell Type | Apoptotic Stimulus | Phenotype | Pathway | Comments | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | Gαi | Endogenous CD47-stimulation |
T Lymphocytes (Human CD4+/CD8- Jurkat T cells) | NONE | Stimulated | Inhibition of cAMP/PKA | CD47 is also known as Integrin-associated Protein | [66] |
| 2 | Gαi | Endogenous CD47-stimulation |
Breast carcinoma cells | NONE | Stimulated | Inhibition of cAMP/PKA | CD47 is also known as Integrin-associated Protein | [67] |
| 3 | Gαi | Endogenous- Mastoparam activation |
Macrophages (murine bone marrow derived) | NONE | Stimulated | Induced PI3K, PBK, and acid Sphingomyelinase pathways Induced NF-κB nuclear translocation |
[68] | |
| 4 | Q205LGαi2 | Overexpression | Neurons (Human SH-SY5Y neuroblastoma cells | H2O2 | Stimulated | Induction of BAK Activation of PARP |
[47] | |
| 5 | Gαi2 | Endogenous Adenosine analog activation |
Neurons (Human SH-SY5Y neuroblastoma cells | H2O2 | Stimulated | Induction of BAK Activation of PARP |
[47] | |
| 6 | Gαi | Endogenous Clonidine, Norepinephrine (α2-adrenergic agonists) |
Melanocytes (Oryzias latipes skin derived) | NONE | Stimulated | Inhibition of cAMP/PKA | Inhibited by Yohimbine, an α2-adrenergic receptor antagonist | [69] |
| 7 | Gαi | Endogenous LHRH (GnRH) activation |
endometrial and ovarian adenocarcinomas | NONE | Stimulated | activation of phosphotyrosine phosphatase | [70] | |
| 8 | Gαi | Endogenous GnRH (LHRH) and antagonist activation |
Hormone Dependent Cancers (JEG-3, BPH-1) | NONE | Stimulated | Activation of JNK and p38 | Specific to GnRH receptor type I; both agonists and antagonists activate Gαi | [71] |
| 9 | Gαi | Endogenous LPA-stimulated |
Enterocytes (rat IEC-6 cells) | serum withdrawal campothecin TNF-α γ-irradiation |
Inhibited | Induced ERK1/2 and Akt phosphorylation increased Bcl-2 expression |
[72,73] | |
| 10 | Gαi | Endogenous LPA-stimulated |
T Lymphocytes (Human CD4+/CD8+/CD3low Tsup-1 cells) | Stimulation of FAS, CD2, CD3, and CD28 | Inhibited | Suppression of BAX expression | models apoptosis induced by thymic selection | [75] |
| 11 | Gαi | Endogenous LPA-stimulated |
Fibroblasts | Inhibited | MAPK, PI3K and Akt activation inhibition of downstream Caspase-9 activation |
[74,75] | ||
| 12 | Gαi | Endogenous LPA-stimulated |
Cardiomyocytes | Ischemia-reperfusion injury | Inhibited | Possibly IGF and calpain mediated | Ischemia induces necrosis; reperfusion induces apoptosis | [76] |
| 13 | Gαi | Endogenous LPA-stimulated |
Hepatocytes (murine AML12 cells) |
Clostridium difficile toxin TNF-α D-galactosamine |
Inhibited | ERK1/2, PI3K, and Akt activation | [78] | |
| 14 | Gαi | Endogenous LPA-stimulated |
Cancer-derived (HeLa, DLD-1, HOS, Mcf-7 cell lines) | TRAILactivation | Inhibited | PI3K and Akt activation Induction of cFLIP Phosphorylation of BAD |
[77] | |
| 15 | Gαi | Endogenous S1P-stimulated |
T Lymphocytes (Human CD4+/CD8+/CD3low Tsup-1 cells) | C6 ceramide | Inhibited | Suppression of BAX expression | models apoptosis induced by thymic selection | [75] |
| 16 | Gαi | Endogenous S1P-stimulated |
Hepatocytes (rat hepatoma HTC4 cells) | Serum starvation | Inhibited | ERK1/2 activation | Possibly mediated by Rho activation | [79,80] |
| 17 | Gαi | Endogenous C-peptide stimulated |
Renal Proximal Tubule Cells (opossum OK cells) | TNF-α | Inhibited | PI3K activation Activation of NF-κB |
A Potential Model for Diabetic Nephropathy | [81] |
| 18 | Gαi | Endogenous T. Gondii infection stimulated |
Macrophages (murine) | Staurosporine | Inhibited | PI3K activation ERK1/2 and Akt activation Downregulation of PARP |
[82] | |
| 19 | Gαi | Endogenous DHEA stimulated |
Endothelial Cells (Bovine BAEC cells) | Serum starvation | Inhibited | PI3K/Akt activation Upregulation of Bcl-2 |
A Potential Atherosclerosis Model | [85] |
| 20 | Gαi | NGF UK14304 |
Neurons (rat PC12 cells) | None | Inhibited | PI3K/Akt activation Phorphorylation of BAD |
May also be Gβγ mediated | [86] |
| 21 | Gαi | Endogenous β2-adrenergic receptor activation |
Cardiomyocytes (murine primary cultures) | Inhibited | PI3K/Akt activation | β1-knockout Also pro-apoptotic through the actions of Gαs |
[48] | |
| 22 | Gαi | Endogenous β2-adrenergic receptor activation |
Cardiomyocytes (rat primary cultures) | Hypoxia | Inhibited | PI3K/Akt activation | β1-blockade using CGP 20712A | [87,88] |
| 23 | Gαo | Endogenous | Neurons (mouse-rat FC11 cells) | Expression of mutant γ-secretase | Stimulated | Caspase-3 activation | Alzheimer’s Disease model | [90] |
| 24 | Gαo | Endogenous | Neurons (rat PC12 cells) | Serum withdrawal β-amyloid |
Stimulated | Alzheimer’s Disease model | [91] |
For example, in T lymphocytes, activation of Gαs (discussed above) and Gαi both lead to increased apoptosis in a highly contextual manner. CD47, also known as Integrin-associated Protein, induces activation-dependent cell death in Jurkat T cells (a lymphoma derived cell line), a process essential for immunological tolerance and contraction. The use of pertussis toxin, a potent inhibitor of Gαi, completely abrogated this phenotype implicating Gαi in this pathway. The inhibitory effects of pertussis toxin have been shown to be mediated by cAMP and PKA [66] (Table 2, Rows 1,2). Thus, in lymphocytes, activation and inhibition of adenylylcyclase through Gαs and Gαi respectively can both lead to increased apoptosis in a context dependent manner. Specifically, immature double positive thymocytes undergo apoptosis upon activation of Gαs through β-adrenergic receptors, while mature CD3+/CD4+/CD8- T lymphocytes (modeled by Jurkat cells) undergo apoptosis upon activation of Gαi. This trend needs to be corroborated in future studies and may prove useful in immunomodulation.
CD47 has also proved important in the immunological regulation of some cancers. Activation of CD47 by monoclonal antibodies or thrombospondins, native ligands of CD47, in various breast cancer cell lines led to a Gαi mediated increase in apoptosis [67] (Table 2, Row 2). Activation of Gαi by mastoparan, a wasp venom peptide, has been recently shown to induce apoptosis in murine bone marrow derived macrophages. The mechanism involved increased ceramide levels through the induction of acid sphingomyelinase, an important sphingomyelin degrading enzyme whose deficiency is associated with Niemann-Pick Disease (Table 2, Row 3). Activation of PI3K and PKB pathways, which causes NF-κB nuclear translocation, was also associated with this phenotype, and these effects were abrogated by pertussis toxin [68]. Importantly and in contrast, sphingosine-1-phosphate (S1P) has been shown to activate Gαi signaling in T lymphocytes to inhibit C6 ceramide induced apoptosis (discussed later) (Table 2, Row 14).
In SH-SY5Y neuroblastoma cells, overexpression of activated Gαi2 led to an augmentation of hydrogen peroxide induced apoptosis by inducing the expression of pro-apoptotic BAK, promoting activation of Caspase-3 and Poly ADP-ribose polymerase (PARP). Similar effects were seen with the activation of endogenous Gαi2 with 2-chloro-N(6)-cyclopentyl-adenosine, an adenosine A1 receptor agonist [47] (Table 2, Row 4-5). In melanocytes from Oryzias latipes, the Japanese killifish, clonidine, an α2-adrenergic receptor agonist, stimulated apoptosis through Gαi by inhibiting cAMP and PKA signaling, while yohimbine, an α2-adrenergic receptor antagonist, prevented these effects. Norepinephrine, a natural α2 receptor agonist, had similar effects which were also abrogated by yohimbine [69] (Table 2, Row 6). Additionally, anti-proliferative and pro-apoptotic signaling in endometrial and ovarian adenocarcinomas through leuteinizing hormone-releasing hormone (LHRH), also known as gonadotropin-releasing hormone (GnRH), is mediated by Gαi induced activation of phosphotyrosine phosphatase [70]. Similarly, in several hormone dependent cancers, dysregulated GnRH receptor signaling increased apoptosis through direct effects on Gαi pathways, that were independent of Gαq effects [71] (Table 2, Row 7-8). Gαi signaling through the GnRH receptor is unusual in that both known receptor agonists (GnRH I and GnRH II) and antagonists (Ac-D-Nal(2)-D-4-ClPhe-D-Pal-Ser-1-MePal-D-Isopropyl-Lys-Leu-IsopropylLys-Pro-D-AlaNH2, also known as 135-25) seem to activate Gαi mediated pathways, while only agonists activate Gαq pathways. In these models, activation of Gαi downstream of the GnRH receptor type I induced JNK and p38 activation in a pertussis toxin dependent manner and stimulated apoptosis.
While the aforementioned examples represent numerous pro-apoptotic activities of Gαi, the majority of Gαi family signaling has been primarily implicated in anti-apoptotic mechanisms in various cell lines (Table 2, Row 9-23). Gαi signaling through the lysophosphatidic acid (LPA; 1-acyl-2-lyso-sn-glycero-3-phosphate) pathway has been shown in numerous systems to inhibit apoptosis. For example, in intestinal epithelial (IEC-6) cells, LPA was shown to reduce apoptosis induced by serum withdrawal, campothecin, TNF-α, and γ-irradiation through a Gαi/o pathway (Table 2, Row 9). While LPA is known to activate three families of G proteins, Gi, Gq, and G12/13, through receptors EDG2 (LPA1), EDG4 (LPA2), and EDG7 (LPA3) respectively, the use of pertussis toxin to inhibit Gαi pathways[72] indicates that this effect is most likely mediated by Gαi/o [73]. In this system, LPA induced the phosphorylation of ERK1/2 and Akt and increased the expression of Bcl-2 [72]. Similarly, LPA mediated cytoprotective effects in fibroblast cell lines are abrogated by the addition of pertussis toxin. These pathways seem to be mediated by downstream MAPK, PI3K and Akt activation and inhibition of downstream Caspase-9 activation (Table 2, Row 11)[72,74,75]. Additional reports have also shown an anti-apoptotic effect of LPA in other cell lines as well (Table 2, Row 11-13). In human CD4+/CD8+/ CD3low Tsup-1 cells, LPA shows protective effects against T-cell maturation-induced death and immunological contraction by reducing the expression of pro-apoptotic BAX [75]. Specifically, antibody activation of FAS, CD2, CD3, or CD28 led to an apoptosis that was suppressed by LPA; this phenotype was abrogated by antisense suppression of the LPA receptors. LPA also prevents death from ischemia-reperfusion injury in cardiomyocytes [76]. Notably, in this study, ischemia alone induced necrotic myocyte death, but reperfusion induced characteristic apoptosis that was inhibited by IGF and calpain inhibitors. Thus, it appears that reperfusion specifically induces apoptosis in a Gαi-dependent manner, perhaps through the generation of reactive oxygen species, calcium influx, or other inflammatory mediators. The precise downstream G protein signaling mechanisms have not been determined in these models, [76]. In several cancer derived cell lines, including HeLa (cervical carcinoma), DLD-1 (colon adenocarcinoma), HOS (prostate carcinoma), and MCF-7 (breast adenocarcinoma), LPA has been shown to inhibit TRAIL-induced apoptosis by preventing the activation of Caspase-8 and Caspase-3, and this signaling may be important in immunological evasion by cancers, especially ovarian carcinomas where serum levels of LPA are significantly higher than baseline [77] (Table 2, Row 13). In this model, LPA induced PI3K dependent Akt activation, expression of the anti-apoptotic cellular FLICE-inhibitory protein (cFLIP), and induced phosphorylation of pro-apoptotic BAD, which prevents downstream Caspase-8 activation. LPA also protected murine hepatocytes (murine AML12 cells) against Clostridium difficile toxin and TNF-α/D-galactosamine induced apoptosis by activating ERK1/2, PI3K, and Akt [78].
Sphingosine-1-Phosphate (S1P), an analog of LPA, has also been shown to have anti-apoptotic effects in various models (Table 2, Row 14-15). S1P was shown to have anti-apoptotic effects in Tsup-1 double positive (CD4+/CD8+/CD3low) lymphoblastoma cells [75] by suppressing cellular expression of BAX when the cells were stimulated with C6 ceramide, a potent stimulator of apoptosis. Activation of S1P receptors Egd3 and Egd5 protected rat hepatoma (HTC4) cells from serum starvation induced apoptosis by activating of Gαi and downstream signaling through ERK1/2. This S1P effect was not only pertussis toxin sensitive but was also inhibited by C3 exoenzyme, a Rho inhibitor, indicating that Rho dependent pathways may also be important in S1P-mediated cytoprotection [79, 80].
Gαi has also been shown to inhibit apoptosis independent of LPA and S1P signaling (Table 2, Row 17-22). In renal tubular epithelium, TNF-α induced apoptosis was prevented by the activation of NF-κB and PI3K through a pertussis toxin sensitive G protein pathway that was mediated by C-peptide, a cleavage product of pro-insulin. Additionally, in studying renal apoptosis associated with such syndromes as diabetes mellitus, insulin and C-peptide protected proximal tubular epithelial cells against TNF-α induced cytotoxicity by activating NF-κB and inducing the expression of the pro-survival protein TRAF2 [81]. Notably, this is one of the first studies to implicate the C-peptide in insulin-independent signaling and may be a model for diabetic nephropathy associated apoptosis [81] (Table 2, Row 17). Resistance to staurosporine, a potent inducer of apoptosis, in macrophages infected by Toxoplasma gondii infection is also mediated by Gαi dependent activation of PI3K, ERK1/2 and Akt [82] (Table 2, Row 18). This study suggests that T. gondii may appropriate cellular Gαi signaling pathways to prevent induced death of infected cells, and this may be a potential pharmacological target. Downstream effects include upregulation of various anti-apoptotic genes, reduced caspase activation, and downregulated poly ADP-ribose polymerase (PARP) expression [83]. These anti-apoptotic effects were also abrogated by pertussis toxin [82]. Gαi dependent PI3K activation has also been implicated in inflammatory pathways in neutrophils and macrophages [84], and it may be these pathways that are upregulated by T. gondii infection. The adrenal steroid dehydroepiandrosterone (DHEA), a precursor of androstenedione, estradiol, and testosterone, has also been shown to protect cells from apoptosis through the upregulation of Bcl-2 transcription through the PI3K/Akt Pathway and Gαi [85] (Table 2, Row 19). These effects were independent of estrogen receptor signaling and were neither stimulated by es-tradiol nor inhibited by estrogen receptor antagonists. This may be an important model of atherosclerosis, as DHEA levels have been shown to fall with aging.
Additionally, Gαi1, Gαi3, GαoA, and GαoB activation by nerve growth factor (NGF) protected rat pheochromocytoma (PC12) cells against apoptosis by inducing BAD phosphorylation and NF-κB nuclear translocation, again in a pertussis toxin sensitive manner [86] (Table 2, Row 20). In this model, stimulation of α2-adrenergic receptor by UK14304 resulted in a similar phenotype that was blocked by pertussis toxin. Importantly, the use of Gαz as a Gβγ scavenger resulted in diminished Akt activation, suggesting that Gβγ may be the relevant signal transducers in this system. In rat neonatal and adult cardiomyocytes, activation of Gαi through the β2-adrenergic receptor protected cells against hypoxia induced apoptosis through the activation of PI3K and Akt, which may have therapeutic implications in various ischemic heart diseases [87,88] (Table 2, Row 21-22). In this study, specific activation of the β2 receptor was achieved using a specific β1-blocker, CGP 20712A. In the same cell type, however, Gαs activation by the β2-adrenergic receptor led to a pro-apoptotic state [48], as discussed above. The specific conditions under which Gαs signaling dominates over Gαi signaling through these β-adrenergic receptors and vice verse are still unclear and await further study. Studies have suggested that initial activation of the Gαs cascade leads to receptor phosphorylation, which then causes a shift in the receptor affinity and initiation of predominantly Gαi mediated signaling [89]. Specifically examining acute stimulation versus chronic stimulation, as would be the case with congestive heart failure, could refine our understanding of these pathways. While the overall effect of β2-adrenergic stimulation was anti-apoptotic, this dual effect suggests that the decision between survival and apoptosis may be mediated by other simultaneous signaling mechanisms (second signals). Examination of the controls that determine pro-apoptotic versus anti-apoptotic states may yield important pharmacologically targetable pathways in the treatment of acute coronary syndromes and the associated cell death.
Gαo is ubiquitously expressed throughout the central nervous system, but many of its functions remain to be defined. It has been implicated in numerous apoptotic pathways associated with neurodegenerative disorders (Table 2, Row 23-24). While NGF induces neuro-protection mediated by Gαo as mentioned above, Gαo has been shown to mediate apoptosis in neurodegenerative disorders such as Alzheimer’s Disease (AD). AD is associated with the accumulation of the amyloid precursor protein (APP) cleavage product Aβ42, which forms the amyloid plaques characteristic of the disease. In F11 cells, a cell line created from the fusion of mouse neuroblastoma N18TG-2 cells with embryonic rat dorsal root ganglion neurons, EcD-induced expression of transfected mutant presenilin-2 (M239V presenilin-2), the γ-secretase implicated in cleavage of APP to Aβ42, induces apoptosis through Gαo [90]. These effects were abrogated by inhibition of Caspase-3 and by pertussis toxin. Similarly, overexpression of presenilin-2 increases apoptosis induced by serum withdrawal or the addition of β-amyloid in PC12 cells in a pertussis toxin dependent manner that is most likely mediated by Gαo [91].
In summary, most Gαi/o pathways have not been examined for specific effects of the various subtypes, Gαi1, Gαi2, and Gαi3, GαoA, and GαoB. These investigations are particularly difficult without the use of dominant negative subunits or subunit specific siRNA based knockdown as small molecule inhibitors are relatively non-specific. Future studies investigating the roles of individual subunits would allow a much finer understanding of the precise signaling pathways in each of these apoptosis models and give more feasible pharmacological targets. As discussed above, these studies are heterogeneous and limited by the approaches utilized. Nevertheless, as with Gαs, some general patterns of Gαi/o regulation of apoptosis appear to be relatively consistent in the literature. LPA and similar agonists appear to consistently inhibit apoptosis in a wide variety of cell types and in response to various apoptotic stimuli. The consistency of these findings is particularly noteworthy as most studies with other receptors and ligands often have largely conflicting and context dependent phenotypes. In some studies, these effects were shown to be pertussis toxin sensitive, but one cannot exclude co-activation of G12/13 or Gq pathways. There are a wide variety of Gαi activating ligands that are pro-apoptotic. One trend that is particularly noteworthy is the specificity of response to Gαi activation in T lymphocytes. The differentiation state of the T lymphocytes is particularly important in determining the consequences of Gαi activation. Trends in cardiomyocytes suggest interesting context dependent activation of Gαs or Gαi through the same β-adrenergic receptors lead to either pro-apoptotic or anti-apoptotic effects respectively. It is highly probable, based on the current understanding of the switch between Gαs and Gαi that signaling through the β2 receptor may be controlled by the strength and duration of receptor activation. Very few studies have been done with Gαo, but these initial studies do suggest a predominantly pro-apoptotic role in neurons.
C. Gαq/11 and Apoptosis
As with the other G protein families, Gαq has been implicated in both pro-apoptotic and anti-apoptotic mechanisms, often through the same signaling pathways and downstream effectors, in a cell type and stimulus specific manner (see Table 3). In this regard, the muscarinic acid receptor family has most been widely studied. While the muscarinic acid receptors activate both Gαq/11 and Gαi/o pathways, the M1, M3, and M5 signal through Gαq/11 while M2 and M4 signal through Gαi/o. Studies of these receptors in the context of apoptosis have primarily involved the isolation of particular receptors and examination of downstream signaling. In the heart, muscarinic receptor activation of Gαq pathways protected cardiomyocytes from apoptosis [92]. In this study, deletion of the carboxy-terminus of these receptors led to a decoupling of anti-apoptotic signaling but preserved Gαq/11 dependent activation of PLC and MAPK pathways (Table 3, Row 1). Similarly, in chinese hamster ovary cells transgenically expressing M3 receptors, carbachol activation of these receptors caused resistance to etoposide-induced apoptosis through a PLC-independent pathway that was associated with the upregulation of Bcl-2 expression [93] (Table 3, Row 1). There is some evidence that some of the anti-apoptotic effects of muscarinic acid receptor activation may be mediated by G protein independent effects. For example, carbachol activation of overexpressed M3 receptors in Chinese hamster ovary cells led to the inhibition of apoptosis induced by the topoisomerase inhibitor etoposide. Another transfected M3 receptor truncation mutant, Δ565-M3, that preserved the G protein activation capacity did not have this anti-apoptotic effect, although Gαq signaling through PLC remained intact [92]. In chinese hamster ovary cells, this effect was accompanied by activation of ERK1/2 and JNK [94]. In contrast, in chinese hamster ovary cells overexpressing the small GTPase Rac1 or constitutively activated Rac1, activation of Gαq through M3 receptors induced apoptosis. In cells expressing normal levels of Rac1, activation of Gαq only led to PCK mediated inhibition of cell proliferation. While the physiological relevance of this Rac1 overexpression system has not been established, it is clear that various cellular factors can completely shift the M3-Gαq signaling system from pro-survival to pro-apoptotic [95] (Table 3, Row 2). Further elucidation of these mechanisms may provide valuable therapeutic targets in disorders such as Alzheimer’s disease, cardiac failure, and gastroduodenal ulcers, where muscarinic acid receptor signaling is dysregulated.
Gαq has also been shown to regulate apoptosis through non-muscarinic acid receptor pathways. In human astrocytoma (1231N1) cells, adenosine-binding to the Gαq/11 coupled purinergic receptor P2Y1 induced apoptosis through PKC signaling and activation of ERK1/2 (Table 3, Row 3). In the same study, however, activation of the muscarinic acid receptors in the astrocytoma cells by carbachol induced cell proliferation, indicating again that at least in this system, there is exquisite receptor-specificity to the G protein mediated apoptosis signaling pathways [96]. This is another example of potential second signals modulating the downstream effects of G protein regulation of apoptosis. In HeLa cells, constitutively activated Gα11 and activation of the M1 receptor stimulated apoptosis by decreasing Akt activation utilizing a Rho-dependent pathway [97,98] (Table 3, Row 5). Specifically, activation of Gαq/11 led to proteolytic activation of Rho-associated kinase (ROCK), and this was blocked by overexpression of Bcl-2, suggesting that this is an intrinsic pathway mediated apoptosis [98]. Dominant negative mutants of RhoA inhibited this phenotype. In Cos-7 cells and CHO cells (Table 3, Row 6), expression of constitutively activated Gαq (R183C Gαq) also induced apoptosis through a PKC pathway that was inhibited by the overexpression of Bcl-2 [99]. The use of the pancaspase inhibitor z-VAD-fmk abrogated this phenotype.
Gαq/11 regulation of cell survival and apoptosis has been particularly well studied in cardiomyocytes (Table 3, Row 7-9). Overexpression of Gαq protected neonatal rat ventricular cardiomyocytes from low-glucose induced apoptosis through the activation of PI3K, EGFR, and Src kinases, and phosphorylation of Akt. These effects were shown to be independent of Gαq-mediated cardiac hypertrophy [100]. However, activation of Gαq in non-transfected neonatal rat ventricular cardiomyocytes with Pasteurella multocida toxin enhanced H2O2 stimulated apoptosis through the activation of PLC and possible downstream inhibition of Akt [101]. The effects of Pasteurella multocida toxin appear to be the result of direct stimulation of Gαq although the precise mechanism remains to be defined. Other studies in the same model have shown that overexpression of wild type Gαq induces cardiac hypertrophy accompanied by increased Akt phosphorylation, while overexpression of a constitutively activated mutant or activation of the overexpressed wild type Gαq induces apoptosis [102,103]. This may contribute to the cell death that is observed in cardiomyopathy associated with eccentric and concentric hypertrophy. These findings were also demonstrated in mice overexpressing (eight times normal expression) Gαq in cardiomyocytes, which showed lethal peripartum cardiomyopathy. The constitutively activated Gαq induced apoptosis in cardiomyocytes was accompanied by depletion of phosphatidylinositol 4,5-bisphosphate (PIP2) and reduced Akt phosphorylation. Further, the introduction of myristoylated Akt into these cells expressing constitutively activated Gαq significantly reduced apoptosis [104]. Myristoylation of Akt specifically targets it to the membrane and has been reported to prevent serum deprivation-induced apoptosis in cultured cardiomyocytes and to reduce apoptosis caused by ischemia-reperfusion in vivo or hypoxia in vitro [104]. Thus, it is clear that Gαq is probably mediating survival and apoptotic signaling in both the inactive and active conformation and activation may provide an important switch for cellular fate. Again, strength and duration of signaling may be important determinants of cellular fate.
In summary, the studies on Gαq and apoptosis suffer from the same limitations of studies with other Gα families. Nevertheless, like with other Gα families, some patterns of regulation are beginning to emerge. In cardiomyocytes, activation of Gαq seems to predominantly induce apoptosis, and even cardiac-specific overexpression of wild type Gαq in mice seems to produce a similar apoptotic phenotype. However, under certain circumstances, Gαq may actually promote cardiomyocyte survival. Thus, further analysis of differences in Gαq signaling may help distinguish the pro-hypertrophy and pro-survival signaling pathways from the proapoptotic pathways. In other cell types, Gαq seems to predominantly stimulate apoptosis, although there are several examples of Gαq -mediated suppression of apoptosis (Table 3).
D. Gα12/13 and Apoptosis
As with other cellular signaling pathways, the role of the Gα12/13 family in the regulation of cell survival and apoptosis is least understood among the four major G protein families. While Gα12 and Gα13 have been implicated in many cellular processes, most of the signaling pathways remain only partially characterized and defined. Remarkably, there have been few studies on apoptosis regulated by the Gα12/13 family in any context. While several studies have implicated Gα12 in proliferation and neoplastic transformation [105,106], some recent studies have suggested that Gα12 does not stimulate proliferation in the context of tumorigenesis but does promote cell migration. Expression of Gα12 was significantly elevated in breast and prostate cancer, and thrombin stimulation resulted in increased cell invasion but not proliferation [37,38]. However, the effects of up-regulated Gα12 levels on apoptosis were not addressed in these studies. It is possible that apoptosis may be playing a role in these tumor phenotypes. The few studies of Gα12/13 regulation of apoptosis have indicated that in various contexts both Gα12 and Gα13 can be primarily promoters of apoptosis (summarized in Table 4). In transiently transfected COS cells, constitutively activated Gα12 (Q229L Gα12) and Gα13 (Q226L Gα13) induced apoptosis through activation of MEKK1 and Ask1 (Table 4, Row 1,2). In this system, wild type Gα12 and Gα13 also induced apoptosis compared to control, although the constitutively activated isoforms showed a much stronger phenotype. Co-transfection of Bcl-2 abrogated apoptosis induced by transfected Gα12 and Gα13 proteins, suggesting a Bcl-2 dependent intrinsic apoptosis pathway [107]. In a human adenocarcinoma cell line, a naturally occurring truncated Gα13 that is missing its C-terminus stimulated apoptosis through Ask1 and JNK (Table 4, Row 3). The pro-proliferative and transformative properties of Gα13 were lost in this model, however. Crystal structure analysis suggested that this truncated mutant has impaired GTP binding and hydrolysis capacity. The apoptosis in this model was blocked by the co-transfection of dominant negative Ask1. Recent evidence suggests that activated Gα13 may also increase Ask1 expression by reducing its ubiquitination and subsequent degradation [108]. Additionally, overexpression of this truncated Gα13 stimulated apoptosis in Rat 1A fibroblasts through JNK signaling (Table 4, Row 4). Overexpression of a Q226L truncated Gα13 stimulated apoptosis to similar levels, suggesting that Gα13 may have unique pro-apoptotic properties that are independent of its activation state [109]. Transfected constitutively activated Gα13 (Q226L Gα13) also stimulated apoptosis in Cos-7 cells and chinese hamster ovary (CHO) cells through a RhoA dependent pathway that was inhibited by the overexpression of Bcl-2 [99]. The use of the pancaspase inhibitor z-VAD-fmk abrogated this phenotype. However, constitutively activated Gα13 rescued mouse F9 teratocarcinoma cells in which apoptosis was induced by the depletion of membrane-organizing extension spike protein (moesin) using shRNA or morpholino, suggesting again an added level of complexity to this picture [110]. In this study, it appears that constitutively activated Gα13 acts through Rho, Rac and/or Cdc42 on ezrin and radixin, and this may be a model for embryonic differentiation.
Table 4.
Cell Culture Studies Examining the Role of Gα12/13 in Apoptosis
| No. | G Protein | Model System | Cell Type | Apoptotic Stimulus | Phenotype | Pathway | Comments | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | Gα12, Q229L-Gα12 | Overexpression | COS-7 Cells | NONE | Stimulated | Activation of MEKK1 and Ask1 | Blocked by overexpression of Bcl-2 | [107] |
| 2 | Gα13, Q226L-Gα13 | Overexpression | COS-7 Cells | NONE | Stimulated | Activation of MEKK1 and Ask1 | Blocked by overexpression of Bcl-2 | [107] |
| 3 | Truncated Gα13 | Endogenous | Human Adenocarcinoma Cells | NONE | Stimulated | Activation of Ask1 and JNK | Blocked by overexpression of dominant negative Ask1 | [109] |
| 4 | Truncated Gα13 | Overexpression | Fibroblasts (RAT1A Cells) | NONE | Stimulated | Activation of Ask1 and JNK | [109] | |
| 5 | Q226L-Gα13 | Overexpression | COS-7 Cells Chinese Hamster Ovary (CHO cells) |
NONE | Stimulated | Rho activation | Blocked by overexpression of Bcl-2 | [99] |
| 6 | Q226L-Gα13 | Overexpression | Mouse teratocarcinoma (F9 Cells) | Moesin-depletion | Inhibited | Rho, Rac, Cdc42 signaling Ezrin and Radixin signaling |
[110] | |
| 7 | Q229L-Gα12 | Overexpression | Canine Kidney (MDCK Cells) | Serum Starvation | Stimulated | Activation of JNK Degradation of Bcl-2 |
Blocked by inhibition of PP2A | [111] |
| 8 | Gα12 | Endogenous Thrombin activation |
Canine Kidney (MDCK Cells); Human Kidney (HEK293 Cells) | Serum Starvation | Stimulated | Activation of JNK Degradation of Bcl-2 |
Blocked by inhibition of PP2A | [111] |
In Madin-Darby Canine Kidney (MDCK) cells, overexpression of a constitutively activated Gα12 (Q229L Gα12) induced apoptosis that was associated with the degradative loss of Bcl-2 and JNK phosphorylation. Overexpression of wild type Gα12 had no effect on apoptosis in this model, and in concordance with the studies in various tumor cell lines, constitutively activated Gα12 did not induce proliferation in confluent monolayers of MDCK cells [111]. However, there is very little known about the role of Gα12 and Gα13 in regulating apoptosis in non-transfected cells. Recently, we demonstrated for the first time that activation of an endogenous Gα12-coupled signaling pathway stimulates apoptosis in epithelial cells. Thrombin stimulation of the Gα12/13-coupled protease activated receptors augments serum-starvation induced apoptosis in MDCK and Human Embryonic Kidney 293 cells that is associated with JNK activation and downstream phosphorylation and degradation of Bcl-2. These effects were abrogated by the use of Protein Phosphatase 2A inhibitors and siRNA knockdown of the PP2A catalytic subunit, suggesting that PP2A may be acting as an intermediary in this case [111]. Gα12 has been shown to interact with the scaffolding subunit of the serine/threonine phosphatase PP2A and this interaction stimulates PP2A activity in vitro and in MDCK cells [112, 113]. Thus, PP2A may be a master regulator of Gα12 induced apoptosis. Additionally, in COS7 cells, transfection of constitutively activated Gα12 mutants that were deficient in Rho signaling also led to the loss of Bcl-2 [114]. These few studies suggest that Gα12 regulates Bcl-2 expression and mediates a Bcl-2 and JNK dependent apoptosis in various cell lines. Additionally, the mitogenic potential of Gα12 depends upon the cell-type and may be influenced by cellular confluency. Additional studies support the notion that thrombin and its protease activated receptors are important in regulating apoptosis. Recently, it was shown that intranigral injection of thrombin stimulated apoptosis by increasing JNK activity and dramatically reducing Bcl-2 expression, leading to nigral dopaminergic neurodegeneration [115], a pathology of Parkinsonism. Additionally, inhibition of protease activated receptor 1 (PAR1) in a murine model of Parkinson’s Disease by antagonist BMS-200261 reduced neuronal injury and loss [116]. However, the precise role of G protein activation was not examined in these studies.
In summary, much work remains to be done in the field of Gα12/13 mediated apoptosis. This field has been particularly limited by the extensive reliance on overexpression systems, and the transition to the use of natural agonists will be important in refining our understanding of the roles of Gα12 and Gα13 in the regulation of apoptosis. However, one interesting emerging theme is the importance of Bcl-2 signaling in both Gα12 and Gα13 mediated apoptosis signaling. Most studies to date have either shown that these G proteins lead to the downregulation of Bcl-2 expression or that overexpression of Bcl-2 leads to inhibition of apoptosis. Most of the literature points to predominantly pro-apoptotic roles for both Gα12 and Gα13 in various cell types, although many of these studies are limited by the use of overexpression systems. It is possible that examination of endogenous pathways will lead to the discovery of novel anti-apoptotic pathways as well.
Gβγ and Apoptosis
The Gβγ subunits have also been implicated in apoptosis regulation. For example, the overexpression of Gβ1γ2 in HaCaT cells was found to enhance UVB-induced apoptosis by promoting the cellular secretion of heparin-binding EGF-like growth factor (HB-EGF) and augmenting the activity of p38 and EGFR and inducing Src kinase activation. These pro-apoptotic effects mediated by Gβ1γ2 were inhibited by antibody blockade of HB-EGF and HB-EGF inhibitor CRM197, sequestration of Gβγ by the carboxyl terminus of G protein coupled receptor kinase 2 (GRK2ct), and by 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2), a potent Src inhibitor. This may be an important mechanism of apoptosis in keratinocytes and may be applicable to various dermatological disorders [117]. Additionally, in modeling Familial Alzheimer’s Disease, the expression of mutant APP, the protein implicated in the disease, induces activation of Gαo and subsequent apoptosis mediated by Gβγ in a Bcl-2 dependent manner in COS cells. The Gβγ dependency of this effect was demonstrated by the overexpression of the carboxy-terminal amino acids 495-689 of the beta-adrenergic receptor kinase-1, which blocks the specific functions Gβγ and reduced apoptosis in this model [118, 119]. The specific overexpression of Gβ2γ2 in this model also induced apoptosis. Other models have suggested that some pro-apoptotic and anti-apoptotic effects of G protein activation are actually mediated by downstream signaling by Gβγ. Analysis of similar effects in other systems for Gβγ specific effects through sequestration may yield further information and indeed may be a mechanism for added specificity of G protein regulation of apoptosis.
IV. EMERGING THEMES
In cardiomyocytes, all studies have shown a pro-apoptotic effect of Gαs activation through the β-adrenergic receptors, and activation of Gαs through either β1 or β2 receptor has similar cellular effects. Similarly, in lymphocytes, activation of Gαs through the β-adrenergic receptors also has clear pro-apoptotic effects. LPA and similar agonists appear to consistently inhibit apoptosis in a wide variety of cell types and in response to various apoptotic stimuli through Gαi signaling. The consistency of these findings is particularly noteworthy as most studies with other receptors and ligands have largely conflicting and context dependent phenotypes. In cardiomyocytes, activation of Gαq seems to predominantly induce apoptosis, and even cardiac specific overexpression of wild type Gαq in mice seems to produce a similar apoptotic phenotype. Bcl-2 signaling appears to be particularly important in both Gα12 and Gα13 mediated apoptosis signaling. Most studies to date have either shown that these G proteins lead to the downregulation of Bcl-2 expression or that overexpression of Bcl-2 leads to inhibition of apoptosis. Most of the literature points to predominantly pro-apoptotic roles for both Gα12 and Gα13 in various cell types.
V. FUTURE DIRECTIONS
In looking for new directions in this field, it is first useful to consider the limitations in both the fields of G protein signaling and apoptosis independently. Because any given cell type expresses multiple GPCRs that may couple to multiple Gα subunits and a particular ligand agonist may activate multiple Gα subunits, studying G protein signaling has been particularly challenging. This has certainly been true in the investigation of apoptosis, where downstream targets of each G protein have been used as very imprecise indicators of G protein activation. The field has relied on the use of overexpression models and constitutively activated mutant G proteins for the study of particular G protein mediated pathways, but these models may not reflect the in vivo physiology and this may be generating some of the inconsistency in the field. Clearly, overexpressing a Gα subunit may lead to engagement of pathways that would not be activated through physiologic mechanisms. Likewise, constitutively activated subunits are useful for identifying potential phenotypes in apoptosis but also need to be substantiated in more physiologic models. Direct measurement of G protein activity coupled with selective small molecular or siRNA inhibition of those subunits will be essential as the field progresses. Traditional and novel stratagies for determining Gα activity are useful adjuncts for identifying potential roles for specific Gα subunits in regulating apoptosis. Ultimately, however, the goal is to modulate an apoptotic pathway in a specific cell type that has become dysregulated as part of a disease process. The hope is that by selectively engaging a subset of Gα subunits coupled to specific receptor, it will be possible to alter signaling only in the effected cell type. To achieve these goals, it is necessary to understand the normal regulation of apoptosis through G proteins and their receptors in each cell type.
The cell death field has been hindered by the heterogeneous morphological and molecular phenotypes that have been collectively and rather loosely termed apoptosis. Indeed the field has been inundated with numerous techniques for the measurement of cell death, including ATP quantification, DNA laddering, phosphatidylserine segregation (Annexin 5), propidium iodide cell labeling, and timelapse morphological studies, among others. Many cell death phenotypes will show some but not all of these phenotypes, indicating that there is a high degree of heterogeneity in what is defined as apoptosis. Further, even with the same cell line, different stimuli elicit very heterogeneous cell death progressions. Even caspase activation, an established hallmark of apoptosis, has been called into question as a consistent marker of apoptosis, as in the case with CD47-induced apoptosis [67]. Heterogeneity of apoptosis-inducing stimuli, morphological and molecular responses, and analysis methodologies, have made interpreting the literature a challenge.
Although GPCR mediated signaling is crucial in every major cellular signaling cascade and much work has been done to uncover the cellular functions and ligand activators of each GPCR, the precise G protein functions downstream of these receptors are very poorly defined to date. This is primarily because receptors couple to multiple G proteins and studying these specific effects has been consequently difficult. Additionally, many GPCRs also signal through non-G protein mediated pathways such as the G protein independent activation of PI3K by the Gαq coupled muscarinic acid receptors. This is certainly the case in the apoptosis field, where many GPCR ligands have been found to modulate apoptosis signaling. More precise identification of the particular G protein subunits (especially Gα, but also Gβγ) involved in each cell death pathway will allow for better pharmacological targeting of these pathways. Perhaps knockdown is the method which will definitively establish the role of a particular Gα and Gβγ subunit within a particular apoptosis pathway and in a particular cell type. Small molecule and peptide inhibitors, especially for the Gα12 family, have not surfaced, and the added lack of specificity of small molecules suggests that silencing may be the best strategy. While the direct inhibition of G proteins as a therapeutic strategy is challenging because of their ubiquitous expression and diverse functionalities, it may be a route to pursue if specific small molecules are found. High sequence homology between the Gα subunits, especially within a particular family, has made this development particularly challenging, but with high-throughput cellular assays that are now well established, a screening protocol for subunit specific drugs would be immensely helpful in furthering research in the field and identifying potential pharmacological agents.
With the elucidation of these G protein mediated apoptosis pathways, the targeting of downstream signal transducers may be another potential pharmacological strategy. Because these G protein mediated apoptosis pathways show stimulus-specific and cell-type specific effects, such a strategy may improve the targetability and specificity of therapy. However, the downstream targets in these pathways are also abundant and ubiquitous molecules, so such strategies must be approached with caution.
Perhaps the most promising strategy is the targeting of GPCRs, which are indeed to a large extent differentially expressed in various cell types and thus more easily modulated therapeutically. In this regard, the identification of the particular GPCR-Gα pairs that are involved in these apoptosis pathways is also of great importance. Most of the current studies have focused on identifying either the Gα subunit or the GPCR involved in a particular apoptosis pathway, but identifying precisely which GPCRs have either a pro-apoptotic or anti-apoptotic effect and which Gα subunit mediate those effects will have much more therapeutic potential. In particular, this may lead to the identification of small molecular GPCR agonists or antagonists that are specific to the activation of desired Gα subunits coupled to that receptor for maximal control of targeting. Such compounds have been found, for example, for the separation of Gαi and Gαq signaling through the GnRH receptor and screening small molecules will likely lead to the identification of other such compounds. Developing a greater understanding of these precise GPCR-Gα pairs will be essential in developing finely tuned therapeutic strategies for the many diseases in which aberrant apoptosis is a major contributing factor.
G protein mediated apoptosis has already proved to be important in the etiology of numerous diseases, including neurodegeneration, cardiomyopathies, renal diseases, and others. The work in this field up to this point has demonstrated a clear importance for G proteins in the regulation of apoptosis. Clearly defining the physiologically relevant G protein mediated pathways with the identification of responsible GPCRs will yield novel therapies of great clinical significance.
NOTE: ADDED IN PROOF
While this article was in review, two pertinent papers appeared in the literature. Choi, et al. show that the expression of constitutively activated Gαs in the A549 and H1299 human lung cancer cell lines augments cisplatin induced apoptosis through the modulation of Bcl-2 family proteins, specifically the up regulation of BAX and BAK and the down regulation of Bcl-2 and Bcl-xL [120]. Conversely, Seo, et al. show that Gαi inhibits apoptosis induced by H2O2 in the same H1299 human lung cancer cell line through the up regulation of Bcl-2 through the NF-κB pathway [121]. This is an interesting example of the opposing nature of Gαs and Gαi signaling in the regulation of apoptosis and something we do not see consistently in other systems.
Acknowledgments
This work was supported by the Paul and Daisy Soros Fellowship, the Huntington’s Disease Society of America Donald A. King Fellowship, and the American Medical Association Seed Grant (to V. Y.) and NIH grants GM55223 and P50 DK074030, Project 1 (to B. M. D.).
References
- 1.Kanduc D, Mittelman A, Serpico R, Sinigaglia E, Sinha AA, Natale C, Santacroce R, Di Corcia MG, Lucchese A, Dini L, Pani P, Santacroce S, Simone S, Bucci R, Farber E. Int J Oncol. 2002;21:165–170. [PubMed] [Google Scholar]
- 2.Jacobson MD, Weil M, Raff MC. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- 3.Raff MC. Nature. 1992;356:397–400. doi: 10.1038/356397a0. [DOI] [PubMed] [Google Scholar]
- 4.Cohen JJ. Immunol Today. 1993;14:126–130. doi: 10.1016/0167-5699(93)90214-6. [DOI] [PubMed] [Google Scholar]
- 5.Kam PC, Ferch NI. Anaesthesia. 2000;55:1081–1093. doi: 10.1046/j.1365-2044.2000.01554.x. [DOI] [PubMed] [Google Scholar]
- 6.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 7.Ashe PC, Berry MD. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:199–214. doi: 10.1016/S0278-5846(03)00016-2. [DOI] [PubMed] [Google Scholar]
- 8.Zimmermann KC, Bonzon C, Green DR. Pharmacol Ther. 2001;92:57–70. doi: 10.1016/s0163-7258(01)00159-0. [DOI] [PubMed] [Google Scholar]
- 9.Fulda S, Debatin KM. Oncogene. 2006;25:4798–4811. doi: 10.1038/sj.onc.1209608. [DOI] [PubMed] [Google Scholar]
- 10.Walczak H, Krammer PH. Exp Cell Res. 2000;256:58–66. doi: 10.1006/excr.2000.4840. [DOI] [PubMed] [Google Scholar]
- 11.Sorenson CM. Biochim Biophys Acta. 2004;1644:169–177. doi: 10.1016/j.bbamcr.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 12.Youle RJ, Strasser A. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 13.Li L, Backer J, Wong AS, Schwanke EL, Stewart BG, Pasdar M. J Cell Sci. 2003;116:3687–3700. doi: 10.1242/jcs.00644. [DOI] [PubMed] [Google Scholar]
- 14.Kumar P, Coltas IK, Kumar B, Chepeha DB, Bradford CR, Polverini PJ. Cancer Res. 2007;67:1193–1202. doi: 10.1158/0008-5472.CAN-06-2265. [DOI] [PubMed] [Google Scholar]
- 15.Aisenberg AC, Wilkes BM, Jacobson JO. Blood. 1988;71:969–972. [PubMed] [Google Scholar]
- 16.Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 17.Vyas S, Javoy-Agid F, Herrero MT, Strada O, Boissiere F, Hibner U, Agid Y. J Neurochem. 1997;69:223–231. doi: 10.1046/j.1471-4159.1997.69010223.x. [DOI] [PubMed] [Google Scholar]
- 18.Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. Cell. 1993;75:229–240. doi: 10.1016/0092-8674(93)80065-m. [DOI] [PubMed] [Google Scholar]
- 19.Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ. Mol Cell. 2001;8:705–711. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- 20.Cohen GM. Biochem J. 1997;326(Pt 1):1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruvolo PP, Deng X, May WS. Leukemia. 2001;15:515–522. doi: 10.1038/sj.leu.2402090. [DOI] [PubMed] [Google Scholar]
- 22.Saelens X, Festjens N, Vande Walle L, van Gurp M, van Loo G, Vandenabeele P. Oncogene. 2004;23:2861–2874. doi: 10.1038/sj.onc.1207523. [DOI] [PubMed] [Google Scholar]
- 23.Melien O. Methods Mol Biol. 2007;361:119–144. doi: 10.1385/1-59745-208-4:119. [DOI] [PubMed] [Google Scholar]
- 24.Oldham WM, Hamm HE. Nat Rev Mol Cell Biol. 2008;9:60–71. doi: 10.1038/nrm2299. [DOI] [PubMed] [Google Scholar]
- 25.Kelly P, Casey PJ, Meigs TE. Biochemistry. 2007;46:6677–6687. doi: 10.1021/bi700235f. [DOI] [PubMed] [Google Scholar]
- 26.Riobo NA, Manning DR. Trends Pharmacol Sci. 2005;26:146–154. doi: 10.1016/j.tips.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 27.Radhika V, Hee Ha J, Jayaraman M, Tsim ST, Dhanasekaran N. Oncogene. 2005;24:4597–4603. doi: 10.1038/sj.onc.1208665. [DOI] [PubMed] [Google Scholar]
- 28.Jiang H, Wu D, Simon MI. FEBS Lett. 1993;330:319–322. doi: 10.1016/0014-5793(93)80896-3. [DOI] [PubMed] [Google Scholar]
- 29.Sabath E, Negoro H, Beaudry S, Paniagua M, Angelow S, Shah J, Grammatikakis N, Yu AS, Denker BM. J Cell Sci. 2008;121:814–824. doi: 10.1242/jcs.014878. [DOI] [PubMed] [Google Scholar]
- 30.Meyer TN, Hunt J, Schwesinger C, Denker BM. Am J Physiol Cell Physiol. 2003;285:C1281–1293. doi: 10.1152/ajpcell.00548.2002. [DOI] [PubMed] [Google Scholar]
- 31.Meyer TN, Schwesinger C, Denker BM. J Biol Chem. 2002;277:24855–24858. doi: 10.1074/jbc.C200240200. [DOI] [PubMed] [Google Scholar]
- 32.Goulimari P, Kitzing TM, Knieling H, Brandt DT, Offermanns S, Grosse R. J Biol Chem. 2005;280:42242–42251. doi: 10.1074/jbc.M508690200. [DOI] [PubMed] [Google Scholar]
- 33.Buhl AM, Johnson NL, Dhanasekaran N, Johnson GL. J Biol Chem. 1995;270:24631–24634. doi: 10.1074/jbc.270.42.24631. [DOI] [PubMed] [Google Scholar]
- 34.Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, Bollag G, Sternweis PC. Science. 1998;280:2109–2111. doi: 10.1126/science.280.5372.2109. [DOI] [PubMed] [Google Scholar]
- 35.Voyno-Yasenetskaya TA, Pace AM, Bourne HR. Oncogene. 1994;9:2559–2565. [PubMed] [Google Scholar]
- 36.Xu N, Bradley L, Ambdukar I, Gutkind JS. Proc Natl Acad Sci USA. 1993;90:6741–6745. doi: 10.1073/pnas.90.14.6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kelly P, Moeller BJ, Juneja J, Booden MA, Der CJ, Daaka Y, Dewhirst MW, Fields TA, Casey PJ. Proc Natl Acad Sci USA. 2006;103:8173–8178. doi: 10.1073/pnas.0510254103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kelly P, Stemmle LN, Madden JF, Fields TA, Daaka Y, Casey PJ. J Biol Chem. 2006;281:26483–26490. doi: 10.1074/jbc.M604376200. [DOI] [PubMed] [Google Scholar]
- 39.De Vries L, Gist Farquhar M. Trends Cell Biol. 1999;9:138–144. doi: 10.1016/s0962-8924(99)01515-9. [DOI] [PubMed] [Google Scholar]
- 40.Jacoby E, Bouhelal R, Gerspacher M, Seuwen K. Chem Med Chem. 2006;1:761–782. doi: 10.1002/cmdc.200600134. [DOI] [PubMed] [Google Scholar]
- 41.Zhong H, Neubig RR. J Pharmacol Exp Ther. 2001;297:837–845. [PubMed] [Google Scholar]
- 42.Cismowski MJ. Semin Cell Dev Biol. 2006;17:334–344. doi: 10.1016/j.semcdb.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 43.Patel TB. Pharmacol Rev. 2004;56:371–385. doi: 10.1124/pr.56.3.4. [DOI] [PubMed] [Google Scholar]
- 44.Wang HX, Weerasinghe RR, Perdue TD, Cakmakci NG, Taylor JP, Marzluff WF, Jones AM. Mol Biol Cell. 2006;17:4257–4269. doi: 10.1091/mbc.E06-01-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brunk I, Holtje M, von Jagow B, Winter S, Sternberg J, Blex C, Pahner I, Ahnert-Hilger G. Handb Exp Pharmacol. 2006:305–325. doi: 10.1007/3-540-29784-7_15. [DOI] [PubMed] [Google Scholar]
- 46.Zhao C, Lai JS, Warsh JJ, Li PP. J Neurosci Res. 2006;84:389–397. doi: 10.1002/jnr.20875. [DOI] [PubMed] [Google Scholar]
- 47.Kim SY, Seo M, Kim Y, Lee YI, Oh JM, Cho EA, Kang JS, Juhnn YS. J Biol Chem. 2007 doi: 10.1074/jbc.M702344200. [DOI] [PubMed] [Google Scholar]
- 48.Zhu WZ, Zheng M, Koch WJ, Lefkowitz RJ, Kobilka BK, Xiao RP. Proc Natl Acad Sci USA. 2001;98:1607–1612. doi: 10.1073/pnas.98.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Geng YJ, Ishikawa Y, Vatner DE, Wagner TE, Bishop SP, Vatner SF, Homcy CJ. Circ Res. 1999;84:34–42. doi: 10.1161/01.res.84.1.34. [DOI] [PubMed] [Google Scholar]
- 50.Xie T, Chen M, Zhang QH, Ma Z, Weinstein LS. Proc Natl Acad Sci USA. 2007 doi: 10.1073/pnas.0704796104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Granata R, Settanni F, Biancone L, Trovato L, Nano R, Bertuzzi F, Destefanis S, Annunziata M, Martinetti M, Catapano F, Ghe C, Isgaard J, Papotti M, Ghigo E, Muccioli G. Endocrinology. 2007;148:512–529. doi: 10.1210/en.2006-0266. [DOI] [PubMed] [Google Scholar]
- 52.Granata R, Settanni F, Trovato L, Destefanis S, Gallo D, Martinetti M, Ghigo E, Muccioli G. J Endocrinol Invest. 2006;29:RC19–22. doi: 10.1007/BF03347367. [DOI] [PubMed] [Google Scholar]
- 53.Regnauld KL, Leteurtre E, Gutkind SJ, Gespach CP, Emami S. Am J Physiol Regul Integr Comp Physiol. 2002;282:R870–880. doi: 10.1152/ajpregu.00374.2001. [DOI] [PubMed] [Google Scholar]
- 54.Yan L, Herrmann V, Hofer JK, Insel PA. Am J Physiol Cell Physiol. 2000;279:C1665–1674. doi: 10.1152/ajpcell.2000.279.5.C1665. [DOI] [PubMed] [Google Scholar]
- 55.Gu C, Ma YC, Benjamin J, Littman D, Chao MV, Huang XY. J Biol Chem. 2000;275:20726–20733. doi: 10.1074/jbc.M000152200. [DOI] [PubMed] [Google Scholar]
- 56.Otterbach F, Callies R, Frey UH, Schmitz KJ, Wreczycki C, Kimmig R, Siffert W, Schmid KW. Breast Cancer Res Treat. 2007;105:311–317. doi: 10.1007/s10549-006-9462-y. [DOI] [PubMed] [Google Scholar]
- 57.Frey UH, Lummen G, Jager T, Jockel KH, Schmid KW, Rubben H, Muller N, Siffert W, Eisenhardt A. Clin Cancer Res. 2006;12:759–763. doi: 10.1158/1078-0432.CCR-05-1722. [DOI] [PubMed] [Google Scholar]
- 58.Frey UH, Eisenhardt A, Lummen G, Rubben H, Jockel KH, Schmid KW, Siffert W. Cancer Epidemiol Biomarkers Prev. 2005;14:871–877. doi: 10.1158/1055-9965.EPI-04-0720. [DOI] [PubMed] [Google Scholar]
- 59.Frey UH, Alakus H, Wohlschlaeger J, Schmitz KJ, Winde G, van Calker HG, Jockel KH, Siffert W, Schmid KW. Clin Cancer Res. 2005;11:5071–5077. doi: 10.1158/1078-0432.CCR-05-0472. [DOI] [PubMed] [Google Scholar]
- 60.Frey UH, Nuckel H, Sellmann L, Siemer D, Kuppers R, Durig J, Duhrsen U, Siffert W. Clin Cancer Res. 2006;12:5686–5692. doi: 10.1158/1078-0432.CCR-06-0288. [DOI] [PubMed] [Google Scholar]
- 61.Schmitz KJ, Lang H, Frey UH, Sotiropoulos GC, Wohlschlaeger J, Reis H, Takeda A, Siffert W, Schmid KW, Baba HA. Neoplasia. 2007;9:159–165. doi: 10.1593/neo.06796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kalfa N, Lumbroso S, Boulle N, Guiter J, Soustelle L, Costa P, Chapuis H, Baldet P, Sultan C. J Urol. 2006;176:891–895. doi: 10.1016/j.juro.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 63.Kalfa N, Ecochard A, Patte C, Duvillard P, Audran F, Pienkowski C, Thibaud E, Brauner R, Lecointre C, Plantaz D, Guedj AM, Paris F, Baldet P, Lumbroso S, Sultan C. J Clin Endocrinol Metab. 2006;91:1842–1847. doi: 10.1210/jc.2005-2710. [DOI] [PubMed] [Google Scholar]
- 64.Chen J, Bander JA, Santore TA, Chen Y, Ram PT, Smit MJ, Iyengar R. Proc Natl Acad Sci USA. 1998;95:2648–2652. doi: 10.1073/pnas.95.5.2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kamenetsky M, Middelhaufe S, Bank EM, Levin LR, Buck J, Steegborn C. J Mol Biol. 2006;362:623–639. doi: 10.1016/j.jmb.2006.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Manna PP, Frazier WA. J Immunol. 2003;170:3544–3553. doi: 10.4049/jimmunol.170.7.3544. [DOI] [PubMed] [Google Scholar]
- 67.Manna PP, Frazier WA. Cancer Res. 2004;64:1026–1036. doi: 10.1158/0008-5472.can-03-1708. [DOI] [PubMed] [Google Scholar]
- 68.Wang SW, Parhar K, Chiu KJ, Tran A, Gangoiti P, Kong J, Gonzalez M, Salh B, Duronio V, Steinbrecher UP, Gomez-Munoz A. Cell Signal. 2007;19:1772–1783. doi: 10.1016/j.cellsig.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 69.Uchida-Oka N, Sugimoto M. Pigment Cell Res. 2001;14:356–361. doi: 10.1034/j.1600-0749.2001.140507.x. [DOI] [PubMed] [Google Scholar]
- 70.Grundker C, Volker P, Emons G. Endocrinology. 2001;142:2369–2380. doi: 10.1210/endo.142.6.8190. [DOI] [PubMed] [Google Scholar]
- 71.Maudsley S, Davidson L, Pawson AJ, Chan R, Lopez de Maturana R, Millar RP. Cancer Res. 2004;64:7533–7544. doi: 10.1158/0008-5472.CAN-04-1360. [DOI] [PubMed] [Google Scholar]
- 72.Deng W, Wang DA, Gosmanova E, Johnson LR, Tigyi G. Am J Physiol Gastrointest Liver Physiol. 2003;284:G821–829. doi: 10.1152/ajpgi.00406.2002. [DOI] [PubMed] [Google Scholar]
- 73.Deng W, Balazs L, Wang DA, Van Middlesworth L, Tigyi G, Johnson LR. Gastroenterology. 2002;123:206–216. doi: 10.1053/gast.2002.34209. [DOI] [PubMed] [Google Scholar]
- 74.Fang X, Yu S, LaPushin R, Lu Y, Furui T, Penn LZ, Stokoe D, Erickson JR, Bast RC, Jr, Mills GB. Biochem J. 2000;352(Pt 1):135–143. [PMC free article] [PubMed] [Google Scholar]
- 75.Goetzl EJ, Kong Y, Mei B. J Immunol. 1999;162:2049–2056. [PubMed] [Google Scholar]
- 76.Umansky SR, Cuenco GM, Khutzian SS, Barr PJ, Tomei LD. Cell Death Differ. 1995;2:235–241. [PubMed] [Google Scholar]
- 77.Kang YC, Kim KM, Lee KS, Namkoong S, Lee SJ, Han JA, Jeoung D, Ha KS, Kwon YG, Kim YM. Cell Death Differ. 2004;11:1287–1298. doi: 10.1038/sj.cdd.4401489. [DOI] [PubMed] [Google Scholar]
- 78.Sautin YY, Crawford JM, Svetlov SI. Am J Physiol Cell Physiol. 2001;281:C2010–2019. doi: 10.1152/ajpcell.00077.2001. [DOI] [PubMed] [Google Scholar]
- 79.An S, Bleu T, Zheng Y. Mol Pharmacol. 1999;55:787–794. [PubMed] [Google Scholar]
- 80.An S, Zheng Y, Bleu T. J Biol Chem. 2000;275:288–296. doi: 10.1074/jbc.275.1.288. [DOI] [PubMed] [Google Scholar]
- 81.Al-Rasheed NM, Willars GB, Brunskill NJ. J Am Soc Nephrol. 2006;17:986–995. doi: 10.1681/ASN.2005080797. [DOI] [PubMed] [Google Scholar]
- 82.Kim L, Denkers EY. J Cell Sci. 2006;119:2119–2126. doi: 10.1242/jcs.02934. [DOI] [PubMed] [Google Scholar]
- 83.Goebel S, Gross U, Luder CG. J Cell Sci. 2001;114:3495–3505. doi: 10.1242/jcs.114.19.3495. [DOI] [PubMed] [Google Scholar]
- 84.Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP. Science. 2000;287:1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- 85.Liu D, Si H, Reynolds KA, Zhen W, Jia Z, Dillon JS. Endocrinology. 2007;148:3068–3076. doi: 10.1210/en.2006-1378. [DOI] [PubMed] [Google Scholar]
- 86.Wu EH, Wong YH. Cell Signal. 2005;17:881–890. doi: 10.1016/j.cellsig.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 87.Chesley A, Lundberg MS, Asai T, Xiao RP, Ohtani S, Lakatta EG, Crow MT. Circ Res. 2000;87:1172–1179. doi: 10.1161/01.res.87.12.1172. [DOI] [PubMed] [Google Scholar]
- 88.Communal C, Singh K, Sawyer DB, Colucci WS. Circulation. 1999;100:2210–2212. doi: 10.1161/01.cir.100.22.2210. [DOI] [PubMed] [Google Scholar]
- 89.Daaka Y, Luttrell LM, Lefkowitz RJ. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- 90.Abe Y, Hashimoto Y, Tomita Y, Terashita K, Aiso S, Tajima H, Niikura T, Matsuoka M, Nishimoto I. J Neurosci Res. 2004;77:583–595. doi: 10.1002/jnr.20163. [DOI] [PubMed] [Google Scholar]
- 91.Wolozin B, Iwasaki K, Vito P, Ganjei JK, Lacana E, Sunderland T, Zhao B, Kusiak JW, Wasco W, D’Adamio L. Science. 1996;274:1710–1713. doi: 10.1126/science.274.5293.1710. [DOI] [PubMed] [Google Scholar]
- 92.Tobin AB, Budd DC. Biochem Soc Trans. 2003;31:1182–1185. doi: 10.1042/bst0311182. [DOI] [PubMed] [Google Scholar]
- 93.Budd DC, Spragg EJ, Ridd K, Tobin AB. Biochem J. 2004;381:43–49. doi: 10.1042/BJ20031705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Budd DC, McDonald J, Emsley N, Cain K, Tobin AB. J Biol Chem. 2003;278:19565–19573. doi: 10.1074/jbc.M211670200. [DOI] [PubMed] [Google Scholar]
- 95.Shafer SH, Williams CL. Mol Pharmacol. 2004;65:1080–1091. doi: 10.1124/mol.65.5.1080. [DOI] [PubMed] [Google Scholar]
- 96.Sellers LA, Simon J, Lundahl TS, Cousens DJ, Humphrey PP, Barnard EA. J Biol Chem. 2001;276:16379–16390. doi: 10.1074/jbc.M006617200. [DOI] [PubMed] [Google Scholar]
- 97.Ueda H, Morishita R, Itoh H, Narumiya S, Mikoshiba K, Kato K, Asano T. J Biol Chem. 2001;276:42527–42533. doi: 10.1074/jbc.M102529200. [DOI] [PubMed] [Google Scholar]
- 98.Ueda H, Morishita R, Narumiya S, Kato K, Asano T. Exp Cell Res. 2004;298:207–217. doi: 10.1016/j.yexcr.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 99.Althoefer H, Eversole-Cire P, Simon MI. J Biol Chem. 1997;272:24380–24386. doi: 10.1074/jbc.272.39.24380. [DOI] [PubMed] [Google Scholar]
- 100.Howes AL, Miyamoto S, Adams JW, Woodcock EA, Brown JH. J Mol Cell Cardiol. 2006;40:597–604. doi: 10.1016/j.yjmcc.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 101.Sabri A, Wilson BA, Steinberg SF. Circ Res. 2002;90:850–857. doi: 10.1161/01.RES.0000016165.23795.1F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Adams JW, Pagel AL, Means CK, Oksenberg D, Armstrong RC, Brown JH. Circ Res. 2000;87:1180–1187. doi: 10.1161/01.res.87.12.1180. [DOI] [PubMed] [Google Scholar]
- 103.Adams JW, Sakata Y, Davis MG, Sah VP, Wang Y, Liggett SB, Chien KR, Brown JH, Dorn GW., 2nd Proc Natl Acad Sci USA. 1998;95:10140–10145. doi: 10.1073/pnas.95.17.10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Howes AL, Arthur JF, Zhang T, Miyamoto S, Adams JW, Dorn IG, Woodcock EA, Brown JH. J Biol Chem. 2003;278:40343–40351. doi: 10.1074/jbc.M305964200. [DOI] [PubMed] [Google Scholar]
- 105.Kumar RN, Radhika V, Audige VV, Rane SG, Dhanasekaran N. Cell Biochem Biophys. 2004;41:63–74. doi: 10.1385/CBB:41:1:063. [DOI] [PubMed] [Google Scholar]
- 106.Kumar RN, Shore SK, Dhanasekaran N. Oncogene. 2006;25:899–906. doi: 10.1038/sj.onc.1209132. [DOI] [PubMed] [Google Scholar]
- 107.Berestetskaya YV, Faure MP, Ichijo H, Voyno-Yasenetskaya TA. J Biol Chem. 1998;273:27816–27823. doi: 10.1074/jbc.273.43.27816. [DOI] [PubMed] [Google Scholar]
- 108.Kutuzov MA, Andreeva AV, Voyno-Yasenetskaya TA. FASEB J. 2007;21:3727–3736. doi: 10.1096/fj.06-8029com. [DOI] [PubMed] [Google Scholar]
- 109.Adarichev VA, Vaiskunaite R, Niu J, Balyasnikova IV, Voyno-Yasenetskaya TA. Am J Physiol Cell Physiol. 2003;285:C922–934. doi: 10.1152/ajpcell.00115.2003. [DOI] [PubMed] [Google Scholar]
- 110.Krawetz R, MacKenzie MJ, Sun Q, Walton PA, Kelly GM. Exp Cell Res. 2006;312:3224–3240. doi: 10.1016/j.yexcr.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 111.Yanamadala V, Negoro H, Gunaratnam L, Kong T, Denker BM. J Biol Chem. 2007;282:24352–24363. doi: 10.1074/jbc.M702804200. [DOI] [PubMed] [Google Scholar]
- 112.Zhu D, Kosik KS, Meigs TE, Yanamadala V, Denker BM. J Biol Chem. 2004;279:54983–54986. doi: 10.1074/jbc.C400508200. [DOI] [PubMed] [Google Scholar]
- 113.Zhu D, Tate RI, Ruediger R, Meigs TE, Denker BM. Mol Pharmacol. 2007;71:1268–1276. doi: 10.1124/mol.106.033555. [DOI] [PubMed] [Google Scholar]
- 114.Andreeva AV, Kutuzov MA, Voyno-Yasenetskaya TA. FASEB J. 2008;22:2821–2831. doi: 10.1096/fj.07-104224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Choi SH, Lee DY, Ryu JK, Kim J, Joe EH, Jin BK. Neurobiol Dis. 2003;14:181–193. doi: 10.1016/s0969-9961(03)00085-8. [DOI] [PubMed] [Google Scholar]
- 116.Hamill CE, Caudle WM, Richardson JR, Yuan H, Pennell KD, Greene JG, Miller GW, Traynelis SF. Mol Pharmacol. 2007;72:653–664. doi: 10.1124/mol.107.038158. [DOI] [PubMed] [Google Scholar]
- 117.Seo M, Lee MJ, Heo JH, Lee YI, Kim Y, Kim SY, Lee ES, Juhnn YS. J Biol Chem. 2007;282:24720–24730. doi: 10.1074/jbc.M702343200. [DOI] [PubMed] [Google Scholar]
- 118.Giambarella U, Yamatsuji T, Okamoto T, Matsui T, Ikezu T, Murayama Y, Levine MA, Katz A, Gautam N, Nishimoto I. EMBO J. 1997;16:4897–4907. doi: 10.1093/emboj/16.16.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yamatsuji T, Matsui T, Okamoto T, Komatsuzaki K, Takeda S, Fukumoto H, Iwatsubo T, Suzuki N, Asami-Odaka A, Ireland S, Kinane TB, Giambarella U, Nishimoto I. Science. 1996;272:1349–1352. doi: 10.1126/science.272.5266.1349. [DOI] [PubMed] [Google Scholar]
- 120.Choi YJ, Oh JM, Kim SY, Seo M, Juhnn YS. Cancer Sci. 2009 doi: 10.1111/j.1349-7006.2009.01136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Seo M, Nam HJ, Kim SY, Juhnn YS. Biochem Biophys Res Comm. 2009;381:153–158. doi: 10.1016/j.bbrc.2009.01.188. [DOI] [PubMed] [Google Scholar]