

Abstract
BACKGROUND
Brugada syndrome (BrS) is a common heritable channelopathy. Mutations in the SCN5A-encoded sodium channel (BrS1) culminate in the most common genotype.
OBJECTIVE
This study sought to perform a retrospective analysis of BrS databases from 9 centers that have each genotyped >100 unrelated cases of suspected BrS.
METHODS
Mutational analysis of all 27 translated exons in SCN5A was performed. Mutation frequency, type, and localization were compared among cases and 1,300 ostensibly healthy volunteers including 649 white subjects and 651 nonwhite subjects (blacks, Asians, Hispanics, and others) that were genotyped previously.
RESULTS
A total of 2,111 unrelated patients (78% male, mean age 39 ± 15 years) were referred for BrS genetic testing. Rare mutations/variants were more common among BrS cases than control subjects (438/2,111, 21% vs. 11/649, 1.7% white subjects and 31/651, 4.8% nonwhite subjects, respectively, P <10−53). The yield of BrS1 genetic testing ranged from 11% to 28% (P = .0017). Overall, 293 distinct mutations were identified in SCN5A: 193 missense, 32 nonsense, 38 frameshift, 21 splice-site, and 9 in-frame deletions/insertions. The 4 most frequent BrS1-associated mutations were E1784K (14×), F861WfsX90 (11×), D356N (8×), and G1408R (7×). Most mutations localized to the transmembrane-spanning regions.
CONCLUSION
This international consortium of BrS genetic testing centers has added 200 new BrS1-associated mutations to the public domain. Overall, 21% of BrS probands have mutations in SCN5A compared to the 2% to 5% background rate of rare variants reported in healthy control subjects. Additional studies drawing on the data presented here may help further distinguish pathogenic mutations from similarly rare but otherwise innocuous ones found in cases.
Keywords: Brugada syndrome, Genetic testing, Ion channels, Sodium channel, Sudden death
Introduction
Brugada syndrome (BrS) is a rare heritable arrhythmia syndrome characterized by an electrocardiographic (ECG) pattern consisting of coved-type ST-segment elevation in the right precordial leads V1 through V3 (often referred to as a type-1 Brugada ECG pattern) and an increased risk for sudden cardiac death (SCD).1,2 The penetrance and expressivity of this autosomal-dominant disorder is highly variable, ranging from a lifelong asymptomatic course to SCD during the first year of life. The syndrome is thought to account for up to 4% of all SCDs and 20% of unexplained sudden death in the setting of a structurally normal heart;3 however, some patients display a more benign course. BrS is generally considered a disorder involving young male adults, with arrhythmogenic manifestation first occurring at an average age of 40 years, with sudden death typically occurring during sleep.4 However, BrS has also been demonstrated in children and infants as young as 2 days old and may serve as a pathogenic basis for some cases of sudden infant death syndrome.3
Since the disorder’s sentinel clinical and ECG description in 1992 by Drs. Pedro and Josep Brugada,5 SCN5A-encoded cardiac sodium channel loss-of-function mutations have been shown to confer the pathogenic basis for an estimated 15% to 30% of BrS, currently representing the most common BrS genotype and classified as Brugada syndrome type 1 (BrS1).6–8 Loss-of-function mutations in SCN5A reduce the overall available sodium current (INa) through either impaired intracellular trafficking of the ion channel to the plasma membrane, thereby reducing membrane surface channel expression, or through altered gating properties of the channel. Gain-of-function SCN5A mutations cause a clinically and mechanistically distinct arrhythmia syndrome, long-QT syndrome type 3 (LQT3). Interestingly, some identical SCN5A mutations may provide either a loss-of-function BrS1-phenotype or a gain-of-function LQT3-phenotype, depending on the individual host. In fact, LQT3/BrS/conduction-disorder SCN5A overlap syndromes do exist within single large families.9,10
After a decade of genetic testing by research laboratories worldwide, BrS genetic testing has made the transition from discovery to translation to clinical implementation with the availability of clinical BrS1 genetic testing (since 2004 in North America and even earlier in Europe), which provides comprehensive open-reading frame and canonical splice site mutational analysis of SCN5A. However, it must be recognized that nearly 2% of healthy Caucasians and 5% of healthy nonwhite subjects also host rare missense SCN5A variants, leading to a potential conundrum in the interpretation of the genetic test results.11 Distinguishing pathogenic mutations from rare harmless genetic variants is of critical importance in the interpretation of genetic testing and the management of genotype-positive BrS patients.
Presently, there are over 100 BrS1-associated mutations publicly available (http://www.fsm.it/cardmoc). We sought to assemble an international compendium of putative BrS1-associated mutations through a retrospective analysis of BrS genomic databases from 9 reference centers throughout the world (5 Europe, 3 United States, 1 Japan) that have each genotyped >100 unrelated cases of clinically suspected BrS. Such a compendium may illuminate further key structure–function properties and provide a foundational building block for the development of algorithms to assist in distinguishing pathogenic mutations from similarly rare but otherwise innocuous ones.
Methods
Study population
A retrospective analysis of BrS databases from 9 centers throughout the world that have each genotyped >100 unrelated cases of clinically suspected BrS was performed. In total, 2,111 unrelated patients (78% male, mean age 39 ± 15 years) were referred for SCN5A genetic testing (Table 1). For the purpose of this compendium of identified mutations, only minimal demographic information for each center’s cohort, such as the average age and range of age at diagnosis and the number of male and female subjects was provided. The specific age and gender were collected for mutation-positive patients. A sample was accepted for genetic testing if the referring physician had made a clinical diagnosis of either possible or definite BrS. An ECG was not always available for each patient. Although DNA samples were accepted for analysis based on a referral diagnosis of BrS, several of the international centers did collect and examine 12-lead ECGs to confirm the presence of an ECG pattern consistent with BrS.
Table 1.
Demographics and mutation yield for 9 Brugada syndrome genetic testing centers
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|---|
| Total | 451 | 365 | 311 | 237 | 195 | 158 | 153 | 130 | 111 |
| Positive | 92 | 88 | 69 | 50 | 44 | 26 | 43 | 14 | 12 |
| Age (yrs) | 47 ± 14 | 40 ± 11 | 45 ± 18 | 43 ± 14 | 35 ± 18 | 36 ± 21 | 45 ± 15 | 48 ± 10 | 37 ± 21 |
| Range (yrs) | 3 to 70 | 0 to 77 | 0 to 82 | 8 to 80 | 0 to 76 | 0 to 69 | 5 to 65 | 28 to 64 | 7 to 78 |
| Yield (%) | 20.4 | 24.1 | 22.2 | 21.1 | 22.6 | 16.5 | 28.1 | 10.8 | 10.8 |
| Male | 357 | 277 | 239 | 193 | 140 | 117 | 106 | 124 | 72 |
| Positive | 68 | 67 | 55 | 40 | 31 | 17 | 30 | 13 | 7 |
| Yield (%) | 21.6 | 24.2 | 22.2 | 20.7 | 23.4 | 14.5 | 28.3 | 10.5 | 9.7 |
| Female | 94 | 88 | 72 | 44 | 55 | 41 | 47 | 6 | 39 |
| Positive | 23 | 19 | 14 | 10 | 13 | 9 | 13 | 1 | 5 |
| Yield (%) | 25.5 | 21.6 | 19.4 | 22.7 | 23.6 | 22.0 | 27.7 | 16.7 | 12.8 |
Center: 1 = Nantes, 2 = Brugada, 3 = AMC, 4 = Paris, 5 = PGxHealth, 6 = MMRL, 7 = UKM, 8 = NCVC, 9 = BCM.
Mutational analysis
Patient genomic DNA was analyzed for mutations in all 27 translated exons, including splice sites and adjacent regions, of the SCN5A-encoded cardiac sodium channel NaV1.5 using a combination of polymerase chain reaction (PCR), either denaturing high-performance liquid chromatography or single-stranded conformation polymorphism and DNA sequencing.12 In addition, frequency, location, and mutation type of SCN5A genetic variation found among 1,300 ostensibly healthy volunteers,11,13 including 649 white subjects and 651 nonwhite subjects (black, Asian, and Hispanic), was analyzed and compared with the possible BrS1-associated mutations. To be reported in this compendium as a possible BrS1-associated mutation, the case mutation must have been absent among all 1,300 control subjects who underwent comprehensive mutation scanning. Further, each reference center examined a local set of control samples from usually 200 to 400 additional unrelated, healthy individuals to determine the presence or absence of each possible case mutation observed in patients from their respective geographical region.
Mutation nomenclature
All possible BrS1-associated mutations were denoted using the accepted Human Genome Variation Society’s guidelines for nomenclature.14 The nucleotide and amino acid designations were based on the SCN5A transcript NM_198056.2. For example, the missense mutation E1784K would indicate the wild-type amino acid (E = glutamic acid) at position 1784 is replaced by lysine (K). Frameshift mutations resulting from nucleotide insertions or deletions were annotated using the F861WfsX90 format, which indicates that the wild-type phenylalanine (F) at position 861 is altered to a tryptophan (W) followed by 89 miscoded amino acids prior to a termination codon (X) 90 residues from the beginning of the altered reading frame.
A substitution of either the first or the last 2 nucleotides of a particular exon has the capacity to alter proper mRNA splicing, regardless of whether the nucleotide substitution codes for a different amino acid (missense mutation) produces a stop codon (nonsense mutation) or does not alter the open reading frame at all (i.e., a synonymous or silent single-nucleotide substitution).15–17 As such, mutations involving this exonic portion of the splice site were considered as possible splicing mutations in this study and annotated as either missense/splicesite, nonsense/splice-site, or silent/splice-site mutations to distinguish them from intronic mutations predicted to disrupt splicing.
Topological placement of the mutations was assigned using a combination of Swissprot (http://ca.expasy.org/uniprot/) and recent studies of the linear topologies for the sodium channel pore-forming alpha subunit.18–20 The Swissprot database provides generally accepted residue ranges corresponding with each ion-channel region and specialized subregions. For Nav1.5, mutations were localized to either the N-terminus (amino acids 1 to 126), interdomain linker (IDL I-II, aa 416-711, IDL II-III, aa 940-1200, and IDL III-IV, aa 1471-1523), transmembrane/linker (Domain I, aa 127-415, Domain II, aa 712-939, Domain III, aa 1201-1470, Domain IV, aa 1524-1772), or C-terminus (aa 1773-2016). The transmembrane-spanning region was further subdivided into S1 through S4 (DI S1-S4/S5, aa 127-252, DII S1-S4/S5, aa 712-841, DIII S1-S4/S5, aa 1201-1336, and DIV S1-S4/S5, aa 1524-1659) and S5 through S6, the pore region and selectivity filter of the channel (DI S5-S6, aa 253-415, DII S5-S6, aa 842-939, DIII S5-S6, aa 1337-1470, DIV S5-S6, aa 1660-1772).
Defining terminology: variant versus mutation
For the purposes of this compendium, a variant will be defined as any change to the wild-type sequence, whether it is in case or control subjects. Mutations will be identified as rare, caseonly (absent in the 1,300+ healthy volunteers) variants that are possibly pathogenic. Variants identified with a minor allele frequency (MAF) >0.5% among the 1,300 healthy control subjects will be termed common polymorphisms. If the MAF is <0.5%, these variants will be termed uncommon/rare polymorphisms.
Defining a variant as a possible BrS1-causative mutation
To be considered as a possible BrS1-causing mutation, the variant must disrupt either the open reading frame (i.e., missense, nonsense, insertion/deletion, or frameshift mutations) or the splice site (polypyrimidine tract, splice acceptor, or splice donor recognition sequences). In addition to the exonic splice sites described above, the acceptor splice site was defined as the 3 intronic nucleotides preceding an exon (designated as IVS−1, −2, or −3) and the donor splice site as the first 5 intronic nucleotides after an exon (designated as IVS+1, +2, +3, +4, or +5).17 Additionally, single-nucleotide substitutions (namely, a purine [A or G] for a pyrimidine [C or T]) within the polypyrimidine tract immediately preceding the acceptor splice-site may be causative.17 As such, some pyrimidine-to-purine substitutions in this region of the intron have been included as potentially pathogenic. For example, an IVS-5 cytosine (C) that falls within the polypyrimidine tract and is substituted by an adenine (A) that would predictably disrupt the polypyrimidine tract and consequently result in aberrant splicing would be included as a possible pathogenic mutation. Hence, single-nucleotide substitutions that obviously did not change the open reading frame (i.e., synonymous single-nucleotide polymorphisms) or those outside of the splice site recognition sequence were not included in either case or control subjects for this study.
Additionally, to be considered as a possible BrS1-causing mutation, the nonsynonymous variant must have been absent in all published databases listing the SCN5A channel common polymorphisms and previously published reports or compendia of rare control variants, e.g., those found in over 2,600 reference alleles (Table 2) derived from over 1,300 ostensibly healthy adult volunteers.11,13 As such, the sole or concomitant presence of a common polymorphism such as H558R-SCN5A or a rarer polymorphism such as A572D-SCN5A would not by definition warrant the annotation of possible BrS1-associated mutation and would not be counted toward the assignment of compound or multiple mutation status to an individual in this compendium. This does not imply that common and rare polymorphisms may not possibly modulate the BrS1 phenotype.
Table 2.
Control variants found in 2,600 reference alleles
| Exon | Nucleotide change | Variant | Mutation type | Location | Number | Ethnicity | Status |
|---|---|---|---|---|---|---|---|
| 2 | 52 C>T | R18W | Missense | N-terminal | 1 | O | Rare control |
| 2 | 100 C>T | R34C | Missense | N-terminal | 44 | B>H=O>W>A | Polymorphism |
| 2 | 101 G>A | R34H | Missense | N-terminal | 1 | B | Rare control |
| 6 | 647 C>T | S216L | Missense | DI-S3/S4 | 4 | W | Polymorphism |
| 7 | 856 G>T | A286S | Missense | DI-S5/S6 | 1 | B | Rare control |
| 7 | 872 A>G | N291S | Missense | DI-S5/S6 | 1 | O | Rare control |
| 7 | 895 T>A | L299M | Missense | DI-S5/S6 | 1 | B | Rare control |
| 9 | 1126 C>T | R376C | Missense | DI-S5/S6 | 1 | W | Rare control |
| 11 | 1340 C>G | A447G | Missense | DI/DII | 1 | B | Rare control |
| 11 | 1345 A>G | T449A | Missense | DI/DII | 1 | O | Rare control |
| 11 | 1381 T>G | L461V | Missense | DI/DII | 2 | B | Polymorphism |
| 11 | 1425 A>C | R475S | Missense | DI/DII | 1 | B | Rare control |
| 11 | 1441 C>T | R481W | Missense | DI/DII | 6 | B>H=O | Polymorphism |
| 12 | 1571 C>A | S524Y | Missense | DI/DII | 18 | B>W=H | Polymorphism |
| 12 | 1673 A>G | H558R | Missense | DI/DII | 408 | W>B>H>O>A | Polymorphism |
| 12 | 1703 G>A | R568H | Missense | DI/DII | 1 | W | Rare control |
| 12 | 1735 G>A | G579R | Missense | DI/DII | 1 | W | Rare control |
| 12 | 1776 C>A | N592K | Missense | DI/DII | 1 | W | Rare control |
| 12 | 1787 A>G | D596G | Missense | DI/DII | 1 | B | Rare control |
| 12 | 1802 T>C | V601A | Missense | DI/DII | 1 | W | Rare control |
| 12 | 1852 C>T | L618F | Missense | DI/DII | 1 | O | Rare control |
| 13 | 1913 G>A | G638D | Missense | DI/DII | 1 | A | Rare control |
| 13 | 1967 C>T | P656L | Missense | DI/DII | 1 | B | Rare control |
| 13 | 2014 G>A | A672T | Missense | DI/DII | 1 | A | Rare control |
| 14 | 2066 G>A | R689H | Missense | DI/DII | 1 | H | Rare control |
| 14 | 2074 C>A | Q692K | Missense | DI/DII | 3 | W | Polymorphism |
| 14 | 2114 C>T | S705F | Missense | DI/DII | 1 | A | Rare control |
| 16 | 2770 G>A | V924I | Missense | DII-S6 | 2 | B=O | Polymorphism |
| 17 | 2924 C>T | R975W | Missense | DII/DIII | 1 | W | Rare control |
| 17 | 2957 G>A | R986Q | Missense | DII/DIII | 1 | B | Rare control |
| 17 | 3047 C>T | T1016M | Missense | DII/DIII | 1 | A | Rare control |
| 17 | 3118 G>A | G1040R | Missense | DII/DIII | 1 | B | Rare control |
| 18 | 3245 T>C | V1082A | Missense | DII/DIII | 1 | B | Rare control |
| 18 | 3269 C>T | P1090L | Missense | DII/DIII | 5 | A>W | Polymorphism |
| 18 | 3292 G>T | V1098L | Missense | DII/DIII | 1 | A | Rare control |
| 18 | 3308 C>A | S1103Y | Missense | DII/DIII | 31 | B>O>H | Polymorphism |
| 18 | 3319 G>A | E1107K | Missense | DII/DIII | 1 | A | Rare control |
| 18 | 3346 C>T | R1116W | Missense | DII/DIII | 2 | A | Polymorphism |
| 20 | 3578 G>A | R1193Q | Missense | DII/DIII | 12 | A>W | Polymorphism |
| 21 | 3751 G>A | V1251M | Missense | DIII-S2 | 1 | B | Rare control |
| 22 | 3878 T>C | F1293S | Missense | DIII-S3/S4 | 2 | W | Polymorphism |
| 22 | 3922 C>T | L1308F | Missense | DIII-S4 | 3 | O>B | Polymorphism |
| Intron 24 | 4299 +2 T>A | 1433sp | Splice site | DIII-S5/S6 | 1 | W | Rare control |
| 26 | 4534 C>T | R1512W | Missense | DIII/DIV | 1 | H | Rare control |
| 28 | 5360 G>A | S1787N | Missense | C-terminal | 3 | W | Polymorphism |
| 28 | 5507 T>C | I1836T | Missense | C-terminal | 1 | B | Rare control |
| 28 | 5701 G>A | E1901K | Missense | C-terminal | 1 | W | Rare control |
| 28 | 5755 C>T | R1919C | Missense | C-terminal | 1 | B | Rare control |
| 28 | 5851 G>T | V1951L | Missense | C-terminal | 16 | H>A>B=O | Polymorphism |
| 28 | 5873 G>A | R1958Q | Missense | C-terminal | 1 | B | Rare control |
| 28 | 5885 C>T | P1962L | Missense | C-terminal | 1 | B | Rare control |
| 28 | 5904 C>G | I1968M | Missense | C-terminal | 1 | B | Rare control |
| 28 | 5972 G>A | R1991Q | Missense | C-terminal | 1 | B | Rare control |
| 28 | 6010 T>C | F2004L | Missense | C-terminal | 7 | W>H | Polymorphism |
| 28 | 6016 C>G | P2006A | Missense | C-terminal | 3 | W>B | Polymorphism |
The > and = symbols represent the relative prevalence of the variant of interest in each corresponding ethnicity.
A = Asian; B = black; H = Hispanic; O = other; and W = white.
Results
Overall, 2,111 unrelated patients (78% male, average age at testing 39 ± 15 years) were referred for BrS genetic testing across 9 testing centers (Table 1). As expected, rare SCN5A missense mutations were far more common among BrS cases (438/2,111, 21%) than similarly rare genetic variants were among control subjects (43/1,300 [11/649, 1.7% white subjects and 31/651, 4.8% nonwhite subjects], P <10−55). The yield differed significantly across centers (P = .0017; chi2 = 24.7, degrees of freedom (df) = 8), ranging from 11% (centers 8 [14/130] and 9 [12/111]) to 28% (center 7 [43/153], P = .0017 for the 9 centers) (Table 1, Figure 1). There was no significant difference in yield between male (324/1,520, 21.3%) and female (112/439, 25.5%, P = .07) subjects. Of the 438 SCN5A mutation-positive cases, 13 (3%) harbored multiple mutations (Table 3). All 13 were male and trended toward younger age at diagnosis (29.7 ± 16.2 years) than male subjects with a single mutation (39.2 ± 14.4 years, P = .07).
Figure 1.
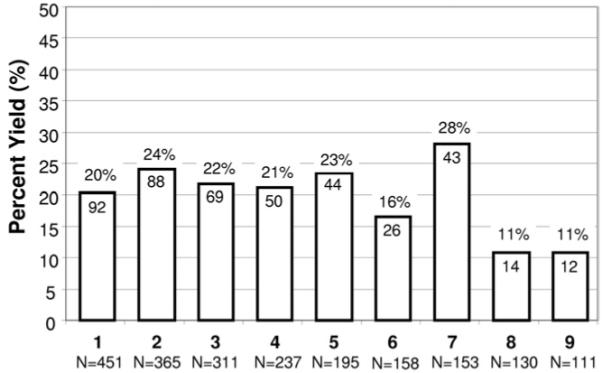
Mutation detection yield by genetic testing center. Depicted here is a comparison of Brugada syndrome genetic testing for each of the 9 centers ordered according to the total number (N = X) of unrelated patients tested. The number within each column represents the number of genotype positive patients for the respective center. For example, Center 1 analyzed 451 unrelated cases and identified a putative pathogenic mutation in 92 (20%). Center: 1 = Nantes, 2 = Brugada, 3 = AMC, 4 = Paris, 5 = PGxHealth, 6 = MMRL, 7 = UKM, 8 = NCVC, 9 = BCM.
Table 3.
Brugada syndrome patients with multiple SCN5A mutations
| Gender | Age at diagnosis (yrs) |
Mutation 1 | Location | Mutation 2 | Location |
|---|---|---|---|---|---|
| M | 35 | N109K | N-terminal | V240M | DI-S4/S5 |
| M | 16 | A185V | DI-S2/S3 | A226V | DI-S4 |
| M | 44 | 611+1 G>A |
DI-S3 | V300I | DI-S5/S6 |
| M | 26 | T220I | DI-S4 | E439K | DI-DII |
| M | 21 | 934+4 C>T |
DI-S5/S6 | G1642E | DIV-S4 |
| M | 51 | P336L | DI-S5/S6 | I1660V | DIV-S5 |
| M | 40 | L619F | DI-DII | Q1383X | DII-S5/S6 |
| M | 49 | T632M | DI-DII | M764R | DII-S2 |
| M | 2 | Q646RfsX5 | DI-DII | D1243N | DIII-S2 |
| M | 24 | A647D | DI-DII | P1332L | DIII-S4/S5 |
| M | 7 | G752R | DII-S2 | K1872N | C-terminal |
| M | 41 | E1053K | DII-DIII | R1583C | DIV-S2/S3 |
| M | 23 | R1232W | DIII-S1/S2 | T1620M | DIV-S3/S4 |
M = male.
Overall, 293 distinct, possible BrS1-associated mutations (200, 68% novel to this cohort), absent in 2,600 reference alleles, were identified in the 438 genotype-positive cases, 225 (77%) of which were identified only once (Table 4, Figure 2). Only 68 mutations were found in multiple unrelated patients. The 4 most frequent BrS1-associated mutations were E1784K (14 patients), F861WfsX90 (11 patients), D356N (8 patients), and G1408R (7 patients) (Table 4, Figure 2). Two-thirds of the unique mutations were missense mutations (193), whereas the remaining third (100) involved radical mutations (38 frameshift, 32 nonsense, 21 splice-site, and 9 in-frame insertions or deletions) (Table 4, Figure 3).
Table 4.
Compendium of Brugada syndrome-associated SCN5A mutations
| Region | Nucleotide change | Coding effect | Mutation type | Location | No. of unrelated individuals |
Testing center |
|---|---|---|---|---|---|---|
| Exon 2 | 3 G>A | M1I* | Missense | N-terminal | 1 | 1 |
| Exon 2 | 53 G>A | R18Q* | Missense | N-terminal | 1 | 3 |
| Exon 2 | 191_193delTGC | L64del* | In-frame del | N-terminal | 1 | 5 |
| Exon 2 | 210 T>G | N70K* | Missense | N-terminal | 1 | 5 |
| Exon 2 | 217 C>T | Q73X* | Nonsense | N-terminal | 1 | 2 |
| Exon 2 | 250 G>A | D84N* | Missense | N-terminal | 2 | 1 |
| Exon 3 | 278 T>C | F93S* | Missense | N-terminal | 1 | 1 |
| Exon 3 | 281 T>G | I94S* | Missense | N-terminal | 1 | 7 |
| Exon 3 | 310 C>T | R104W* | Missense | N-terminal | 2 | 1, 2 |
| Exon 3 | 311 G>A | R104Q | Missense | N-terminal | 3 | 1, 7, 8 |
| Exon 3 | 327 C>A | N109K* | Missense | N-terminal | 1 | 3 |
| Exon 3 | 361 C>T | R121W* | Missense | N-terminal | 1 | 8 |
| Exon 3 | 362 G>A | R121Q* | Missense | N-terminal | 2 | 2, 6 |
| Exon 3 | 376 A>G | K126E | Missense | N-terminal | 1 | 9 |
| Exon 3 | 381dupT | L128SfsX44* | Frame shift | DI-S1 | 1 | 7 |
| Intron 3 | 393 −5 C>A* | Splice site | DI-S1 | 1 | 1 | |
| Exon 4 | 407 T>C | L136P | Missense | DI-S1 | 2 | 8 |
| Exon 4 | 410_418dupTCATGTGCA | I137_C139dup* | In-frame ins | DI-S1 | 1 | 3 |
| Exon 4 | 436 G>A | V146M* | Missense | DI-S1 | 1 | 7 |
| Exon 4 | 468 G>A | W156X | Nonsense | DI-S1/S2 | 1 | 3 |
| Exon 4 | 477 T>A | Y159X* | Nonsense | DI-S2 | 1 | 5 |
| Exon 4 | 481 G>C | E161Q* | Missense | DI-S2 | 1 | 6 |
| Exon 4 | 481 G>A | E161K | Missense | DI-S2 | 3 | 3, 4 |
| Exon 5 | 486delC | Y162XfsX1* | Frame shift | DI-S2 | 1 | 5 |
| Exon 5 | 525 G>C | K175N* | Missense | DI-S2 | 1 | 1 |
| Exon 5 | 533 C>G | A178G* | Missense | DI-S2 | 1 | 9 |
| Exon 5 | 535 C>T | R179X | Nonsense | DI-S2/S3 | 1 | 2 |
| Exon 5 | 544 T>C | C182R* | Missense | DI-S2/S3 | 1 | 4 |
| Exon 5 | 554 C>T | A185V* | Missense | DI-S2/S3 | 1 | 2 |
| Exon 5 | 579 G>A | W193X* | Nonsense | DI-S3 | 1 | 4 |
| Exon 5 | 611 C>T | A204V* | Missense | DI-S3 | 1 | 4 |
| Intron 5 | 611 +1 G>A* | Splice site | DI-S3 | 1 | 5 | |
| Intron 5 | 611 +3_611+4dupAA | Splice site | DI-S3 | 1 | 9 | |
| Intron 5 | 612 −2 A>G* | Splice site | DI-S3 | 1 | 4 | |
| Exon 6 | 635 T>A | L212Q* | Missense | DI-S3/S4 | 1 | 5 |
| Exon 6 | 656_657insATTCA | T220FfsX10* | Frame shift | DI-S4 | 1 | 2 |
| Exon 6 | 659 C>T | T220I | Missense | DI-S4 | 2 | 2, 3 |
| Exon 6 | 664 C>T | R222X | Nonsense | DI-S4 | 4 | 2, 4 |
| Exon 6 | 665 G>A | R222Q | Missense | DI-S4 | 1 | 4 |
| Exon 6 | 667 G>C | V223L* | Missense | DI-S4 | 2 | 2 |
| Exon 6 | 673 C>T | R225W | Missense | DI-S4 | 3 | 1, 6 |
| Exon 6 | 677 C>T | A226V | Missense | DI-S4 | 2 | 2 |
| Exon 6 | 694 G>A | V232I | Missense | DI-S4 | 2 | 2, 6 |
| Exon 7 | 718 G>A | V240M | Missense | DI-S4/S5 | 1 | 3 |
| Exon 7 | 745 A>T | K249X* | Nonsense | DI-S4/S5 | 1 | 6 |
| Exon 7 | 808 C>A | Q270K* | Missense | DI-S5 | 1 | 6 |
| Exon 7 | 827 T>A | L276Q | Missense | DI-S5 | 1 | 8 |
| Exon 7 | 832 C>G | H278D* | Missense | DI-S5/S6 | 1 | 4 |
| Exon 7 | 844 C>T | R282C* | Missense | DI-S5/S6 | 1 | 2 |
| Exon 7 | 898 G>A | V300I* | Missense | DI-S5/S6 | 1 | 5 |
| Intron 7 | 934 +1 G>A* | Splice site | DI-S5/S6 | 1 | 3 | |
| Intron 7 | 934 +4 C>T* | Splice site | DI-S5/S6 | 1 | 5 | |
| Exon 8 | 944 T>C | L315P* | Missense | DI-S5/S6 | 1 | 6 |
| Exon 8 | 959 C>A | T320N* | Missense | DI-S5/S6 | 1 | 5 |
| Exon 8 | 974 T>G | L325R | Missense | DI-S5/S6 | 1 | 4 |
| Intron 8 | 998 +1 G>A* | Splice site | DI-S5/S6 | 1 | 4 | |
| Exon 9 | 1007 C>T | P336L | Missense | DI-S5/S6 | 2 | 2, 6 |
| Exon 9 | 1036 G>T | E346X | Nonsense | DI-S5/S6 | 1 | 1 |
| Exon 9 | 1052 G>T | G351V | Missense | DI-S5/S6 | 1 | 9 |
| Exon 9 | 1052 G>A | G351D* | Missense | DI-S5/S6 | 1 | 2 |
| Exon 9 | 1066 G>A | D356N | Missense | DI-S5/S6 | 8 | 1, 2, 4, 6, 7 |
| Exon 9 | 1099 C>T | R367C | Missense | DI-S5/S6 | 2 | 3 |
| Exon 9 | 1100 G>T | R367L* | Missense | DI-S5/S6 | 1 | 1 |
| Exon 9 | 1100 G>A | R367H | Missense | DI-S5/S6 | 6 | 1, 2, 8, 9 |
| Exon 9 | 1106 T>A | M369K | Missense | DI-S5/S6 | 1 | 1 |
| Exon 9 | 1120 T>G | W374G* | Missense | DI-S5/S6 | 1 | 1 |
| Exon 9 | 1127 G>A | R376H | Missense | DI-S5/S6 | 4 | 3, 4, 8 |
| Exon 10 | 1156 G>A | G386R* | Missense | DI-S5/S6 | 1 | 1 |
| Exon 10 | 1157 G>A | G386E* | Missense | DI-S5/S6 | 2 | 2 |
| Exon 10 | 1186 G>C | V396L* | Missense | DI-S6 | 1 | 1 |
| Exon 10 | 1187 T>C | V396A* | Missense | DI-S6 | 1 | 2 |
| Exon 10 | 1255 C>T | Q419X* | Nonsense | DI/DII | 1 | 7 |
| Exon 10 | 1315 G>A | E439K* | Missense | DI/DII | 1 | 3 |
| Intron 10 | 1338 +2 T>A* | Splice site | DI/DII | 1 | 6 | |
| Exon 11 | 1428_1431delCAAG | S476RfsX30* | Frame shift | DI/DII | 1 | 3 |
| Exon 11 | 1502 A>G | D501G | Missense | DI/DII | 1 | 3 |
| Exon 12 | 1537delC | R513VfsX8* | Frame shift | DI/DII | 1 | 8 |
| Exon 12 | 1562delA | K521SfsX102* | Frame shift | DI/DII | 1 | 2 |
| Exon 12 | 1577 G>A | R526H* | Missense | DI/DII | 2 | 1, 5 |
| Exon 12 | 1595 T>G | F532C | Missense | DI/DII | 1 | 2 |
| Exon 12 | 1603 C>T | R535X | Nonsense | DI/DII | 4 | 1, 2, 4, 5 |
| Exon 12 | 1629 T>A | F543L* | Missense | DI/DII | 1 | 2 |
| Exon 12 | 1654 G>A | G552R* | Missense | DI/DII | 1 | 9 |
| Exon 12 | 1717 C>T | Q573X* | Nonsense | DI/DII | 1 | 2 |
| Exon 12 | 1721delG | G574DfsX49* | Frame shift | DI/DII | 1 | 2 |
| Exon 12 | 1844 G>A | G615E | Missense | DI/DII | 1 | 4 |
| Exon 12 | 1855 C>T | L619F | Missense | DI/DII | 1 | 1 |
| Exon 12 | 1858 C>T | R620C* | Missense | DI/DII | 1 | 1 |
| Exon 12 | 1890 G>A | T630T* | Silent/splice site | DI/DII | 3 | 1, 3 |
| Intron 12 | 1890 +5 G>A* | Splice site | DI/DII | 2 | 2, 5 | |
| Exon 13 | 1895 C>T | T632M | Missense | DI/DII | 2 | 2, 4 |
| Exon 13 | 1918 C>G | P640A* | Missense | DI/DII | 1 | 3 |
| Exon 13 | 1936delC | Q646RfsX5* | Frame shift | DI/DII | 3 | 2, 5, 6 |
| Exon 13 | 1940 C>A | A647D* | Missense | DI/DII | 1 | 5 |
| Exon 13 | 1943 C>T | P648L | Missense | DI/DII | 1 | 7 |
| Exon 13 | 1950_1953delAGAT | D651AfsX25* | Frame shift | DI/DII | 1 | 4 |
| Exon 13 | 1981 C>T | R661W* | Missense | DI/DII | 1 | 5 |
| Exon 13 | 1983_1993dupGGCCCTCAGCG | A665GfsX16* | Frame shift | DI/DII | 1 | 1 |
| Exon 14 | 2024_2025delAG | E675VfsX45* | Frame shift/splice | DI/DII | 1 | 2 |
| Exon 14 | 2047 T>G | C683G* | Missense | DI/DII | 1 | 7 |
| Exon 14 | 2092 G>T | E698X* | Nonsense | DI/DII | 1 | 2 |
| Exon 14 | 2102 C>T | P701L | Missense | DI/DII | 1 | 4 |
| Exon 14 | 2150 C>T | P717L* | Missense | DII-S1 | 1 | 6 |
| Exon 14 | 2201dupT | M734IfsX11* | Frame shift | DII-S1 | 1 | 7 |
| Exon 14 | 2204 C>T | A735V | Missense | DII-S1 | 4 | 2, 4, 8, 9 |
| Exon 14 | 2236 G>A | E746K | Missense | DII-S1/S2 | 3 | 1, 2, 7 |
| Exon 14 | 2254 G>A | G752R | Missense | DII-S2 | 5 | 1, 5 |
| Exon 15 | 2273 G>A | G758E* | Missense | DII-S2 | 1 | 2 |
| Exon 15 | 2274delG | I759FfsX6* | Frame shift | DII-S2 | 2 | 5 |
| Exon 15 | 2291 T>G | M764R* | Missense | DII-S2 | 1 | 4 |
| Exon 15 | 2314 G>A | D772N | Missense | DII-S2/S3 | 1 | 1 |
| Exon 15 | 2317 C>T | P773S* | Missense | DII-S2/S3 | 1 | 6 |
| Exon 15 | 2320delT | Y774TfsX28* | Frame shift | DII-S2/S3 | 2 | 3 |
| Exon 15 | 2326_2328delTAC | Y776del* | In-frame del | DII-S2/S3 | 1 | 2 |
| Exon 15 | 2365 G>A | V789I* | Missense | DII-S3 | 1 | 4 |
| Exon 15 | 2423 G>C | R808P* | Missense | DII-S4 | 1 | 1 |
| Exon 15 | 2435_2436 3delTGGTAinsCGCCT | L812P†* | Indel/splice site | DII-S4 | 1 | 5 |
| Exon 16 | 2465 G>A | W822X | Nonsense | DII-S4 | 1 | 4 |
| Exon 16 | 2516 T>C | L839P* | Missense | DII-S4/S5 | 1 | 1 |
| Exon 16 | 2533delG | V845CfsX2* | Frame shift | DII-S5 | 1 | 6 |
| Exon 16 | 2549_2550insTG | F851GfsX19* | Frame shift | DII-S5 | 1 | 2 |
| Exon 16 | 2550_2551dupGT | F851CfsX19* | Frame shift | DII-S5 | 1 | 5 |
| Exon 16 | 2553 C>A | F851L | Missense | DII-S5 | 1 | 2 |
| Exon 16 | 2582_2583delTT | F861WfsX90 | Frame shift | DII-S5 | 11 | 3, 7 |
| Exon 16 | 2599 G>C | E867Q* | Missense | DII-S5/S6 | 1 | 2 |
| Exon 16 | 2602delC | L868X | Frame shift | DII-S5/S6 | 2 | 6, 7 |
| Exon 16 | 2632 C>T | R878C | Missense | DII-S5/S6 | 1 | 2 |
| Exon 16 | 2633 G>A | R878H* | Missense | DII-S5/S6 | 5 | 1, 2, 4, 5, 7 |
| Exon 16 | 2657 A>C | H886P* | Missense | DII-S5/S6 | 1 | 2 |
| Exon 16 | 2677 C>T | R893C* | Missense | DII-S5/S6 | 2 | 4 |
| Exon 16 | 2678 G>A | R893H* | Missense | DII-S5/S6 | 3 | 1, 3, 4 |
| Exon 16 | 2701 G>A | E901K* | Missense | DII-S5/S6 | 3 | 1, 4 |
| Exon 16 | 2729 C>T | S910L | Missense | DII-S5/S6 | 1 | 1 |
| Exon 16 | 2743 T>C | C915R* | Missense | DII-S6 | 1 | 3 |
| Exon 16 | 2750 T>G | L917R* | Missense | DII-S6 | 1 | 2 |
| Exon 16 | 2780 A>G | N927S | Missense | DII-S6 | 3 | 3, 7 |
| Exon 16 | 2783 T>C | L928P* | Missense | DII-S6 | 1 | 1 |
| Exon 17 | 2804 T>C | L935P* | Missense | DII-S6 | 1 | 5 |
| Exon 17 | 2850delT | D951MfsX6* | Frame shift | DII/DIII | 1 | 4 |
| Exon 17 | 2893 C>T | R965C | Missense | DII/DIII | 3 | 2, 4, 5 |
| Exon 17 | 2894 G>A | R965H | Missense | DII/DIII | 1 | 3 |
| Exon 17 | 2914_2923delTTTGTCAAGC | F972GfsX170* | Frame shift | DII/DIII | 1 | 6 |
| Exon 17 | 2989 G>A | A997T* | Missense | DII/DIII | 1 | 5 |
| Exon 17 | 3005_3012delCCAGCTGC | P1002HfsX25* | Frame shift | DII/DIII | 1 | 7 |
| Exon 17 | 3140_3141dupTG | P1048CfsX98* | Frame shift | DII/DIII | 1 | 3 |
| Exon 17 | 3157 G>A | E1053K | Missense | DII/DIII | 3 | 1 |
| Exon 17 | 3164 A>G | D1055G* | Missense | DII/DIII | 1 | 1 |
| Exon 17 | 3171_3172delTGinsA | D1057EfsX88* | Insertion/deletion | DII/DIII | 1 | 7 |
| Intron 17 | 3228 +2delT* | Splice site | DII/DIII | 1 | 3 | |
| Exon 18 | 3236 C>A | S1079Y* | Missense | DII/DIII | 1 | 1 |
| Exon 18 | 3338 C>T | A1113V* | Missense | DII/DIII | 1 | 5 |
| Exon 18 | 3345 G>A | W1115X* | Nonsense | DII/DIII | 1 | 6 |
| Exon 19 | 3419 G>C | S1140T* | Missense | DII/DIII | 1 | 5 |
| Exon 20 | 3553_3554delCA | Q1185GfsX55* | Frame shift | DII/DIII | 1 | 3 |
| Exon 20 | 3576 G>A | W1192X* | Nonsense | DII/DIII | 1 | 6 |
| Exon 20 | 3622 G>T | E1208X | Nonsense | DIII-S1 | 1 | 1 |
| Exon 20 | 3634_3636delATC | I1212del* | In-frame del | DIII-S1 | 1 | 2 |
| Exon 20 | 3656 G>A | S1219N | Missense | DIII-S1 | 1 | 1 |
| Exon 20 | 3666delG | A1223PfsX7* | Frame shift/splice | DIII-S1 | 1 | 1 |
| Exon 21 | 3673 G>A | E1225K | Missense | DIII-S1/S2 | 4 | 1, 5, 6, 7 |
| Exon 21 | 3682 T>C | Y1228H | Missense | DIII-S1/S2 | 1 | 1 |
| Exon 21 | 3694 C>T | R1232W | Missense | DIII-S1/S2 | 3 | 1, 2, 9 |
| Exon 21 | 3695 G>A | R1232Q* | Missense | DIII-S1/S2 | 1 | 7 |
| Exon 21 | 3716 T>C | L1239P* | Missense | DIII-S2 | 1 | 2 |
| Exon 21 | 3727 G>A | D1243N* | Missense | DIII-S2 | 5 | 1, 2, 5 |
| Exon 21 | 3746 T>A | V1249D* | Missense | DIII-S2 | 1 | 6 |
| Exon 21 | 3758 A>G | E1253G* | Missense | DIII-S2 | 1 | 1 |
| Exon 21 | 3784 G>A | G1262S | Missense | DIII-S2 | 1 | 1 |
| Exon 21 | 3813 G>C | W1271C* | Missense | DIII-S3 | 1 | 1 |
| Exon 21 | 3823 G>A | D1275N | Missense | DIII-S3 | 3 | 1, 5 |
| Intron 21 | 3840 +1 G>A | Splice site | DIII-S3 | 6 | 1, 3, 4 | |
| Exon 22 | 3863 C>G | A1288G* | Missense | DIII-S3 | 1 | 4 |
| Exon 22 | 3894delC | I1299SfsX13* | Frame shift | DIII-S4 | 1 | 5 |
| Exon 22 | 3932 T>C | L1311P* | Missense | DIII-S4 | 1 | 7 |
| Exon 22 | 3956 G>T | G1319V | Missense | DIII-S4/S5 | 5 | 2, 3, 7 |
| Intron 22 | 3963 +4 A>G* | Splice site | DIII-S4/S5 | 1 | 5 | |
| Intron 22 | 3963 +2 T>C | Splice site | DIII-S4/S5 | 1 | 1 | |
| Exon 23 | 3968 T>G | V1323G* | Missense | DIII-S4/S5 | 1 | 7 |
| Exon 23 | 3995 C>T | P1332L | Missense | DIII-S4/S5 | 1 | 5 |
| Exon 23 | 4018 G>A | V1340I* | Missense | DIII-S5 | 1 | 9 |
| Exon 23 | 4030 T>C | F1344L* | Missense | DIII-S5 | 1 | 1 |
| Exon 23 | 4036 C>A | L1346I* | Missense | DIII-S5 | 1 | 4 |
| Exon 23 | 4037 T>C | L1346P* | Missense | DIII-S5 | 1 | 3 |
| Exon 23 | 4052 T>G | M1351R* | Missense | DIII-S5 | 1 | 2 |
| Exon 23 | 4057 G>A | V1353M* | Missense | DIII-S5 | 2 | 2 |
| Exon 23 | 4072 G>T | G1358W* | Missense | DIII-S5 | 1 | 4 |
| Exon 23 | 4077 G>T | K1359N* | Missense | DIII-S5 | 1 | 4 |
| Exon 23 | 4079 T>G | F1360C | Missense | DIII-S5/S6 | 1 | 1 |
| Exon 23 | 4088 G>A | C1363Y | Missense | DIII-S5/S6 | 1 | 3 |
| Exon 23 | 4118 T>A | L1373X* | Nonsense | DIII-S5/S6 | 1 | 2 |
| Exon 23 | 4145 G>T | S1382I | Missense | DIII-S5/S6 | 1 | 1 |
| Exon 23 | 4147 C>T | Q1383X* | Nonsense | DIII-S5/S6 | 1 | 1 |
| Exon 23 | 4178 T>A | L1393X | Nonsense | DIII-S5/S6 | 3 | 1, 3, 9 |
| Exon 23 | 4182 C>G | Y1394X* | Nonsense | DIII-S5/S6 | 1 | 5 |
| Exon 23 | 4190delA | K1397RfsX2 | Frame shift | DIII-S5/S6 | 1 | 9 |
| Exon 23 | 4213 G>A | V1405M* | Missense | DIII-S5/S6 | 2 | 1, 7 |
| Exon 23 | 4213 G>C | V1405L | Missense | DIII-S5/S6 | 2 | 3 |
| Exon 23 | 4216 G>C | G1406R | Missense | DIII-S5/S6 | 1 | 3 |
| Exon 23 | 4217 G>A | G1406E* | Missense | DIII-S5/S6 | 2 | 5 |
| Exon 23 | 4222 G>A | G1408R | Missense | DIII-S5/S6 | 7 | 1, 4, 5, 7 |
| Exon 23 | 4226 A>G | Y1409C* | Missense | DIII-S5/S6 | 1 | 1 |
| Exon 23 | 4227 C>G | Y1409X* | Nonsense | DIII-S5/S6 | 1 | 8 |
| Exon 23 | 4234 C>T | L1412F* | Missense | DIII-S5/S6 | 1 | 5 |
| Exon 24 | 4255 A>G | K1419E* | Missense | DIII-S5/S6 | 1 | 1 |
| Exon 24 | 4258 G>C | G1420R* | Missense | DIII-S5/S6 | 1 | 3 |
| Exon 24 | 4279 G>T | A1427S* | Missense | DIII-S5/S6 | 1 | 2 |
| Exon 24 | 4283 C>T | A1428V* | Missense | DIII-S5/S6 | 1 | 2 |
| Exon 24 | 4294 A>G | R1432G | Missense | DIII-S5/S6 | 1 | 4 |
| Exon 24 | 4296 G>C | R1432S* | Missense | DIII-S5/S6 | 1 | 2 |
| Exon 24 | 4298 G>T | G1433V* | Missense | DIII-S5/S6 | 1 | 3 |
| Exon 24 | 4299 G>A | G1433G* | Silent/splice site | DIII-S5/S6 | 1 | 4 |
| Intron 24 | 4299 +1 G>T* | Splice site | DIII-S5/S6 | 1 | 5 | |
| Intron 24 | 4299 +1delG* | Splice site | DIII-S5/S6 | 1 | 1 | |
| Intron 24 | 4300 −1 G>A* | Splice site | DIII-S5/S6 | 1 | 2 | |
| Exon 25 | 4302 T>G | Y1434X* | Nonsense | DIII-S5/S6 | 1 | 5 |
| Exon 25 | 4313 C>T | P1438L | Missense | DIII-S5/S6 | 1 | 4 |
| Exon 25 | 4320 G>A | W1440X | Nonsense | DIII-S5/S6 | 1 | 2 |
| Exon 25 | 4321 G>C | E1441Q* | Missense | DIII-S5/S6 | 1 | 7 |
| Exon 25 | 4342 A>C | I1448L* | Missense | DIII-S6 | 1 | 4 |
| Exon 25 | 4343 T>C | I1448T* | Missense | DIII-S6 | 1 | 2 |
| Exon 25 | 4346 A>G | Y1449C* | Missense | DIII-S6 | 1 | 1 |
| Exon 25 | 4352 T>A | V1451D* | Missense | DIII-S6 | 1 | 5 |
| Exon 25 | 4376_4379delTCTT | F1459SfsX3* | Frame shift | DIII-S6 | 1 | 6 |
| Exon 25 | 4387 A>T | N1463Y* | Missense | DIII-S6 | 1 | 2 |
| Exon 25 | 4389_4396delCCTCTTTA | L1464WfsX5* | Frame shift | DIII-S6 | 1 | 8 |
| Exon 25 | 4402 G>T | V1468F* | Missense | DIII-S6 | 1 | 2 |
| Exon 25 | 4426 C>T | Q1476X* | Nonsense | DIII/DIV | 1 | 4 |
| Intron 25 | 4437 +5 G>A* | Splice site | DIII/DIV | 2 | 3, 5 | |
| Exon 26 | 4477_4479delAAG | K1493del* | In-frame del | DIII/DIV | 2 | 1, 7 |
| Exon 26 | 4477 A>T | K1493X* | Nonsense | DIII/DIV | 1 | 2 |
| Exon 26 | 4501 C>G | L1501V | Missense | DIII/DIV | 1 | 1 |
| Exon 27 | 4562 T>A | I1521K* | Missense | DIII/DIV | 1 | 5 |
| Exon 27 | 4573 G>A | V1525M* | Missense | DIV-S1 | 1 | 4 |
| Exon 27 | 4642 G>A | E1548K* | Missense | DIV-S1/S2 | 3 | 1, 4 |
| Exon 27 | 4708_4710dupATC | I1570dup* | In-frame ins | DIV-S2 | 1 | 3 |
| Exon 27 | 4712 T>G | F1571C* | Missense | DIV-S2 | 1 | 4 |
| Exon 27 | 4720 G>A | E1574K | Missense | DIV-S2 | 4 | 2, 6, 7 |
| Exon 27 | 4745 T>C | L1582P* | Missense | DIV-S2 | 1 | 3 |
| Exon 27 | 4747 C>T | R1583C* | Missense | DIV-S2/S3 | 2 | 1, 2 |
| Exon 27 | 4748 G>A | R1583H* | Missense | DIV-S2/S3 | 1 | 1 |
| Exon 27 | 4773 G>A | W1591X* | Nonsense | DIV-S3 | 1 | 2 |
| Exon 27 | 4810 G>A | V1604M* | Missense | DIV-S3 | 1 | 1 |
| Exon 27 | 4813 +2_4813+5dupTGGG | Splice site | DIV-S3 | 1 | 2 | |
| Exon 28 | 4838 A>T | Q1613L* | Missense | DIV-S3/S4 | 1 | 1 |
| Exon 28 | 4845 C>A | Y1615X* | Nonsense | DIV-S3/S4 | 1 | 2 |
| Exon 28 | 4856delC | P1619RfsX12* | Frame shift | DIV-S3/S4 | 1 | 2 |
| Exon 28 | 4859 C>T | T1620M | Missense | DIV-S3/S4 | 2 | 2, 9 |
| Exon 28 | 4867 C>T | R1623X | Nonsense | DIV-S4 | 2 | 2, 3 |
| Exon 28 | 4868 G>A | R1623Q | Missense | DIV-S4 | 1 | 5 |
| Exon 28 | 4885 C>T | R1629X* | nonsense | DIV-S4 | 1 | 3 |
| Exon 28 | 4886 G>A | R1629Q* | Missense | DIV-S4 | 1 | 3 |
| Exon 28 | 4912 C>T | R1638X | Nonsense | DIV-S4 | 3 | 2, 3 |
| Exon 28 | 4925 G>A | G1642E* | Missense | DIV-S4 | 1 | 5 |
| Exon 28 | 4978 A>G | I1660V | Missense | DIV-S5 | 5 | 2, 3, 5, 6 |
| Exon 28 | 4981 G>A | G1661R* | Missense | DIV-S5 | 2 | 1 |
| Exon 28 | 4981 G>C | G1661R* | Missense | DIV-S5 | 1 | 2 |
| Exon 28 | 4999 G>A | V1667I | Missense | DIV-S5 | 1 | 3 |
| Exon 28 | 5015 C>A | S1672Y | Missense | DIV-S5 | 2 | 1, 4 |
| Exon 28 | 5038 G>A | A1680T | Missense | DIV-S5 | 2 | 2, 6 |
| Exon 28 | 5068_5070delGA | D1690HfsX98* | Frame shift | DIV-S5/S6 | 1 | 1 |
| Exon 28 | 5083 C>T | Q1695X | Nonsense | DIV-S5/S6 | 2 | 1, 4 |
| Exon 28 | 5092 G>A | A1698T* | Missense | DIV-S5/S6 | 1 | 2 |
| Exon 28 | 5124_5126delCAC | T1709del* | In-frame del | DIV-S5/S6 | 1 | 4 |
| Exon 28 | 5126 C>T | T1709M | Missense | DIV-S5/S6 | 2 | 1, 8 |
| Exon 28 | 5126 C>G | T1709R* | Missense | DIV-S5/S6 | 1 | 4 |
| Exon 28 | 5134 G>A | G1712S* | Missense | DIV-S5/S6 | 1 | 7 |
| Exon 28 | 5141 A>G | D1714G | Missense | DIV-S5/S6 | 1 | 3 |
| Exon 28 | 5157delC | I1720SfsX67* | Frame shift | DIV-S5/S6 | 1 | 8 |
| Exon 28 | 5164 A>G | N1722D* | Missense | DIV-S5/S6 | 1 | 1 |
| Exon 28 | 5182 T>C | C1728R* | Missense | DIV-S5/S6 | 1 | 2 |
| Exon 28 | 5184 C>G | C1728W* | Missense | DIV-S5/S6 | 1 | 2 |
| Exon 28 | 5218 G>A | G1740R | Missense | DIV-S5/S6 | 1 | 3 |
| Exon 28 | 5227 G>A | G1743R | Missense | DIV-S5/S6 | 5 | 4, 5, 7, 9 |
| Exon 28 | 5228 G>A | G1743E | Missense | DIV-S5/S6 | 6 | 2, 3 |
| Exon 28 | 5290delG | V1764SfsX23* | Frame shift | DIV-S6 | 1 | 8 |
| Exon 28 | 5290 G>T | V1764F | Missense | DIV-S6 | 1 | 7 |
| Exon 28 | 5336 C>T | T1779M | Missense | C-terminal | 1 | 2 |
| Exon 28 | 5350 G>A | E1784K | Missense | C-terminal | 14 | 1, 2, 5, 6, 7 |
| Exon 28 | 5356_5357delCT | L1786EfsX2* | Frame shift | C-terminal | 2 | 1 |
| Exon 28 | 5387_5388insTGA | 1795_1796insD | In-frame ins | C-terminal | 1 | 3 |
| Exon 28 | 5420dupA | F1808IfsX3* | Frame shift | C-terminal | 1 | 7 |
| Exon 28 | 5435 C>A | S1812X | Nonsense | C-terminal | 1 | 7 |
| Exon 28 | 5464_5467delTCTG | E1823HfsX10* | Frame shift | C-terminal | 1 | 2 |
| Exon 28 | 5494 C>G | Q1832E* | Missense | C-terminal | 1 | 6 |
| Exon 28 | 5577_5578dupAA | R1860KfsX13* | Frame shift | C-terminal | 1 | 2 |
| Exon 28 | 5581 G>A | V1861I* | Missense | C-terminal | 1 | 2 |
| Exon 28 | 5616 G>C | K1872N* | Missense | C-terminal | 1 | 5 |
| Exon 28 | 5707 G>C | S1904L | Missense | C-terminal | 1 | 2 |
| Exon 28 | 5770 G>A | A1924T | Missense | C-terminal | 1 | 3 |
| Exon 28 | 5803 G>A | G1935S | Missense | C-terminal | 1 | 2 |
| Exon 28 | 5812 G>A | E1938K* | Missense | C-terminal | 1 | 2 |
| Exon 28 | 6010_6012dupTTC | F2004dup* | In-frame ins | C-terminal | 1 | 7 |
| Exon 28 | 6010 T>G | F2004V* | Missense | C-terminal | 1 | 5 |
del = deletion; dup = duplication; ins = insertion; indel = insertion/deletion; ins = insertion
novel mutation
Testing Center: 1 = Nantes; 2 = Brugada; 3 = AMC; 4 = Paris; 5 = PGxHealth; 6 = MMRL; 7 = UKM; 8 = NCVC; 9 = BCM.
Figure 2.
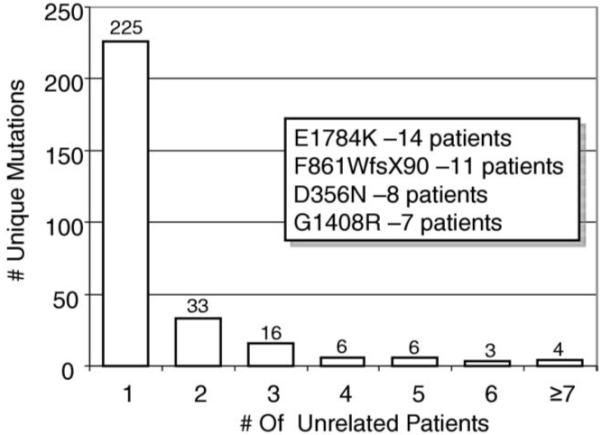
BrS1-associated mutation frequency distribution. This bar graph summarizes the distribution of specific mutations among unrelated patients. The Y-axis depicts the number of unique BrS1-associated mutations, and the X-axis represents the number of unrelated patients. For example, the first column indicates that there were 226 unique mutations each observed only once. The last column indicates that 4 different BrS1-associated mutations were each seen in ≥7 unrelated patients. The inset shows the 4 most common BrS1-associated mutations identified and the number of specified unrelated patients in whom the mutations were found.
Figure 3.
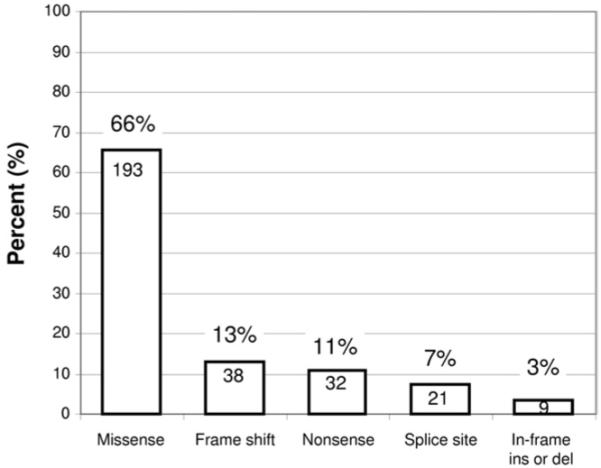
Summary of SCN5A mutation type for BrS1. The distribution of mutation type (missense, frameshift, etc.) is summarized for the possible BrS1-associated SCN5A mutations. The number within the column represents the total number of unique mutations for the respective type. For example, there were 193 unique missense mutations identified.
Of the 293 unique mutations, 208 (71%) localized to one of the 4 transmembrane-spanning regions (DI, DII, DIII, or DIV), 54 (18%) localized to an IDL (31 in IDL I-II, 17 in IDL II-III, and 6 in IDL III-IV), 17 (6%) localized to the C-terminus, and 14 (5%) localized to the N-terminus (Table 4, Figure 4). The majority of patients with a single SCN5A mutation (313/425, 74%) hosted a mutation that localized to the transmembrane region of the channel, with 31% (133/425) having a mutation that localized to either DI S1-S4, DII S1-S4, DIII S1-S4, or DIV S1-S4 and 42% (180/425) having a mutation that localized to the S5, P-loop, and S6 regions containing the pore and selectivity filter of the sodium channel (DI S5-S6, DII S5-S6, DIII S5-S6, or DIV S5-S6) (Table 4, Figure 4). In fact, compared with the topological location for the rare control variants, there was a strong predilection for a patient’s possible BrS1-associated mutation to localize to the channel’s transmembrane (S1-S4 6.3% vs. 0.2%, P <10−24) and pore-forming segments (S5-S6 8.5% vs. 0.5%, P <10−30) (Figure 5). Twenty-eight patients (1.3%) had their possible BrS1-associated missense mutation localizing to the DI-DII or DII-DIII linker domains, where the vast majority of the rare missense SCN5A variants, which were discovered among the ostensibly healthy control subjects, reside. The yield for the linker regions was 3% for cases compared with 1.7% for healthy subjects (P = NS) (Figure 5).
Figure 4.
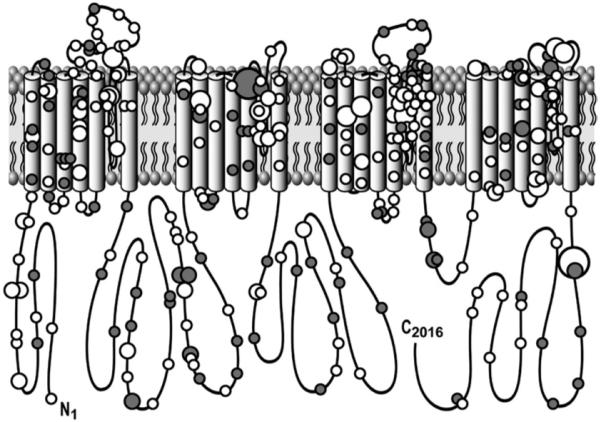
Channel topology of NaV1.5’s pore-forming alpha subunit encoded by SCN5A and location of putative BrS1-causing mutations. Missense mutations are indicated by white circles, whereas mutations other than missense (i.e., frameshift, deletions, splice-site, etc.) are depicted as gray circles. In addition, 4 different circle sizes are used, with the smallest circle indicating a mutation seen only once; a medium-sized circle for mutations observed in 2, 3, or 4 unrelated patients; a large circle for mutations observed in 5, 6, 7, 8, or 9 patients; and the largest circle indicating those mutations observed in at least 10 unrelated patients.
Figure 5.
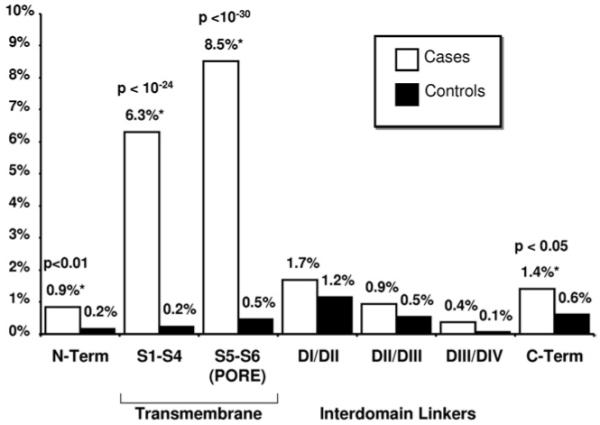
Yield of missense mutations/rare variants in cases and control subjects by location. A comparison of the yield of rare, missense case mutations/control variants in 2,111 cases versus 1,300 control subjects by protein location. * = p<0.05.
Interestingly, 10 (3.4%) of the mutations, identified in 29 patients of this BrS compendium, have been identified previously in cases of long-QT syndrome (LQTS): G615E, L619F, E1225K, P1332L, L1501V, R1623Q, I1660V, V1667I, T1779M, including the most common BrS1-associated mutation, E1784K, observed in this BrS compendium. Five of the 10 have been functionally characterized previously, with L619F,21 P1332L,22 R1623Q,23 and E1784K24 producing channel abnormalities consistent with an LQT3 phenotype and G615E25 producing a wild-type-like SCN5A channel.10
Discussion
Since the first report by Chen et al26 in 1998, a little over 100 unique SCN5A mutations have been implicated as possibly causative for BrS1. Previous small cohort studies have indicated that the prevalence of SCN5A mutations in BrS is roughly 15% to 20%, and possibly as high as 40% in cases of familial BrS. In 2000, Priori et al6 reported a 15% yield with respect to SCN5A mutations among 52 unrelated patients. In 2002, these investigators extended their analysis to 130 probands (20% with a family history of sudden unexplained death) and identified an SCN5A mutation in 22%.7 Schulze-Bahr et al8 reported a 14% yield among 44 unrelated BrS patients, who are included in the current compendium.
Here, through this international multicenter study, we provide an expanded compendium of 293 unique (200 novel) BrS1-associated SCN5A mutations derived from over 2,100 unrelated patients referred for BrS genetic testing. The overall yield was 21% and ranged from 11% to 28% among the 9 centers. The differences in yield between the centers may be due to technical differences in mutational analysis methods used among laboratories, but it more likely reflects phenotypic differences among cohorts. For example, the cohorts with the lowest yield may have a preponderance of sporadic cases compared with familial cases. In 2003, Schulze-Bahr et al8 reported that although none of their 27 sporadic BrS cases hosted an SCN5A mutation, 38% of their index cases with clearly familial BrS were positive. The number of sporadic versus familial BrS patients for each center’s cohort is currently unknown. Alternatively, some of the lower yield centers may have accepted a greater proportion of weaker clinical cases for BrS genetic testing because no particular litmus test was demanded before acceptance of a sample.
Eight of the 9 centers represent research-based genetic testing laboratories, where most often the cohorts for such laboratories are composed of phenotypically robust cases of BrS. However, 1 center (center 5 in Table 1) represents a clinical laboratory offering the commercially available, fee-based genetic test for both BrS1 and LQTS. In this setting, the level of clinical suspicion and the usage of the genetic test by the referring physician are unknown for each patient sample submitted. Among their first 195 BrS referral cases submitted for clinical genetic testing, 22.6% were identified with a possible pathogenic SCN5A mutation, which is in line with the higher point estimate of a 20% prevalence of SCN5A mutations among clinically strong cases of BrS patients,7 and consistent with the noncommercial centers in this compendium. In contrast, this laboratory has reported a yield of 36% for the first 2,500 consecutive unrelated LQTS referral cases tested compared with a yield of 75% among clinically strong cases of LQTS.27 These observations suggest that in clinical practice, prescribing cardiologists are submitting higher-probability BrS cases for BrS1 genetic testing compared with LQTS.
In this compendium, 3% of the genotype-positive patients hosted multiple putative pathogenic SCN5A mutations, absent in control subjects. Akin to genotype–phenotype observations in LQTS,28 patients hosting multiple SCN5A mutations were younger at diagnosis (29.7 ± 16 years) than those having a single mutation. Nearly half of the 13 cases (all male) with multiple mutations were younger than 25 years of age, with the youngest presenting at 2 years of age. Whether carriers of multiple mutations had a more severely expressed phenotype, such as having more multiple syncopal events, aborted cardiac arrest, or stronger family history of SCD, than single-mutation carriers is unknown.
Mutations in this compendium overwhelmingly represent “private” mutations, meaning they were seen only once. Nearly 80% of the 293 unique mutations were identified in a single unrelated case. Fewer than 20 mutations were seen in more than 3 unrelated BrS patients. However, nearly 10% of the 438 unrelated SCN5A mutation-positive patients hosted 1 of 4 mutations: E1784K (14 patients), F861WfsX90 (11 patients), D356N (8 patients), and G1408R (7 patients). In this compendium, more than one-third of the genotype-positive patients had radical or non-missense mutations (i.e., frameshift, nonsense, and splicing errors) that represent extremely high-probability case mutations (only 1 such variant was found in 1 of the 1,300 healthy volunteers) and would predictably cause a significant loss of sodium channel function through a mechanism of haploinsufficiency. Recently, Meregalli et al29 reported that BrS patients with truncation mutations, caused by radical mutations, had a more severe phenotype characterized as a higher propensity for syncope and prevalence of SCD among young first-degree relatives, than those BrS patients hosting missense mutations functionally characterized with ≤90% peak sodium current reduction.
The SCN5A-encoded cardiac voltage-gated sodium channel (Nav1.5), which is responsible for the initial fast upstroke of the cardiac action potential and accordingly plays a vital role in the excitability of myocardial cells and the proper conduction of the electrical pulsation of the heart, consists of 4 homologous domains (DI-DIV) that are connected by intracellular linkers. Each domain contains 6 transmembrane-spanning segments (S1-S6). Although case mutations were scattered throughout Nav1.5, there was clustering of putative BrS1-associated mutations whereby nearly three-fourths localized to the transmembrane and pore-forming domains compared with <20% of the rare variants found among the control subjects. Further, nearly 10% of patients with clinically suspected BrS had a mutation localizing to the channel’s pore/selectivity filter (segments S5 and S6 and the interconnecting P-loops) compared with <0.5% of the control subjects. Because of the extreme rarity of pore-localizing missense variants among the healthy control subjects, such missense mutations found in cases are high probability BrS1-causative mutations. However, whether BrS1 patients with pore mutations behaved more poorly than those with mutations localizing to other domains was unable to be gleaned in this study.
In contrast, only 25 patients hosted a single missense mutation residing in either the DI-DII or DII-DIII linker domains. The observed preponderance (over 50% of which localize to these 2 IDLs) of the 42 unique rare missense variants identified in the healthy control subjects suggest that some of the 21 IDL localizing, possible BrS1-associated missense mutations that were identified in 25 cases may in fact represent false positives.11,13 Despite being absent in over 1,300 control subjects, either cosegregation or functional studies on these DI-DII and DII-DIII linker-localizing mutations should be considered before upgrading their status from a rare variant of uncertain significance to a probable BrS1-causative missense mutation. For example, 1 of the 21 missense mutations, G615E, has been previously characterized as having no significant changes in current density or kinetics compared with wild type, casting some doubt on its level of causality.25
Whereas loss-of-function mutations in SCN5A have been shown to serve as a pathogenic basis for BrS, gain-of-function mutations in SCN5A provide the pathogenic substrate for LQT3, a clinically and mechanistically distinct arrhythmia syndrome from BrS. Interestingly, 10 mutations identified in this BrS compendium have been implicated previously in LQTS, including the most commonly observed mutation, E1784K. In fact, E1784K represented the most commonly observed SCN5A mutation (4/26, ~15%) among a cohort of 541 unrelated LQTS patients.28 Although the clinical phenotype for the patients hosting 1 of these 10 mutations could have been assigned incorrectly, it is far more likely that these represent overlap syndrome/mixed phenotype syndrome–associated SCN5A mutations.
For example, E1784K represents the quintessential example of a cardiac sodium channel mutation with the capacity to provide for a mixed clinical phenotype of LQT3, BrS, and conduction disorders.30 Makita et al30 reported recently a high prevalence of LQT3/BrS/conduction phenotype overlap among 41 E1784K carriers from 15 kindreds of diverse ethnic background. Of the 41 cases, 93% displayed a prolonged QTc, 22% with a diagnostic indicator (ST-segment elevation or positive provocation test) of BrS, and 39% had sinus node dysfunction. Functional characterization of mutant E1784K sodium channels displayed unique biophysical and pharmacological properties consistent with other mutations that yield a mixed phenotype, including a negative shift of steady-state sodium channel activation and enhanced tonic block in response to sodium channel blockers, leading to an additional BrS/sinus node dysfunction phenotype in conjunction with a prolonged QTc.30 This particular functional effect may influence the pharmacological management of patients with E1784K-SCN5A disease.30
Similarly, Bezzina et al31 in 1999, described an LQT3/BrS overlap phenotype in a large 1795insD-SCN5A mutation–positive 8-generation kindred characterized with a high incidence of sudden nocturnal death, QT-interval prolongation, and Brugada ECG. Whether or not the 9 other mutations, identified here in BrS patients and elsewhere in patients purported to have LQTS, have a similar functional characteristic that provides a substrate for producing a mixed/overlapping clinical phenotype remains to be seen.
Given that SCN5A remains the most common BrS genotype despite accounting for only 20% of BrS, genetic heterogeneity of the disease is evident and the role of genetic background in the pathophysiology of BrS is important.32 Recently, mutations in the glycerol-3-phosphate dehydrogenase 1–like protein encoded by GPD1L have been shown to affect trafficking of the sodium channel to the plasma membrane, thus reducing overall sodium current, giving rise to the BrS phenotype.33 Recently, mutations involving the L-type calcium channel alpha and beta subunits encoded by the CACNA1C and CACNB2b genes, respectively, were implicated in nearly 11% of BrS cases.34 Other minor causes of BrS include mutations in the sodium channel beta 1 subunit encoded by SCN1B and in a putative beta subunit of the transient outward potassium channel (Ito) encoded by KCNE3.35,36 The most recent gene associated with BrS is the SCN3B-encoded beta-3 subunit of the cardiac sodium channel.37
A number of these minor BrS-susceptibility genes function in part as sodium channel interacting proteins (ChIPs). The cardiac sodium channel is now understood to be a part of a macromolecular complex, with numerous ChIPs regulating its expression, localization, and function. As exemplified by GPD1L, SCN1B, and SCN3B, other genes, which encode other sodium ChIPs whose disruption would portend a loss of function sodium channel phenotype, would warrant examination as candidate BrS disease or disease-modifying genes. These 6 minor BrS-susceptibility genes (GPD1L, CACNA1C, CACNB2B, SCN1B, SCN3B, and KCNE3) have not been examined among the remaining 1,673 SCN5A-negative unrelated cases represented in this compendium, but it is predicted that these minor genes will explain <10% of this cohort. Thus, the majority of BrS still remains genetically elusive.
Study limitations
For the purpose of this compendium of identified mutations, only minimal demographic information from each center’s cohort was made available because the focus was on the prevalence, spectrum, and localization of SCN5A mutations among suspected cases of BrS rather than an attempt to establish any particular genotype–phenotype correlates. Nevertheless, there is significant clinical value due to the data in aggregate. For example, the rare missense mutations seen numerous times among these cases referred for BrS genetic testing, and still not among the control subjects, indicate high-probability BrS-associated mutations.
Furthermore, despite the lack of clinical information, the physical distribution of mutations/variants among cases and control subjects is very telling, whereby the power in numbers can come from the series of mutations/variants clustering in a region, even if the individual mutations/variants have only been observed rarely. For example, even without cosegregation data or in vitro function analyses, the next rare, nonmissense mutation as well as the next transmembrane-localizing missense mutation represent high-probability pathogenic substrates. Conversely, those rare single amino acid substitutions localizing to the domain I-II and II-III linkers should be buttressed with either clinical cosegregation data or in vitro functional data before being upgraded from this list of possible deleterious mutations to highly probable deleterious mutations.
Additionally, this analysis focused on only identifying coding and splicing region single-nucleotide mutations and small insertion/deletion mutations by molecular techniques that often do not detect larger rearrangements, insertions, and deletions. SCN5A promoter region mutations, deep intronic mutations, epigenetic methylation mutations, and large genomic rearrangements, all of which could predictably produce a loss-of-function phenotype, would have escaped detection by the mutational analyses performed herein. However, such alterations are quite uncommon when compared with changes in the known coding sequences of the genes.
Despite these limitations, this compendium of nearly 300 distinct BrS-associated mutations provides key observations that may assist in the further interrogation of the cardiac sodium channel biology and serve as a foundation for the development of algorithms to assist in distinguishing pathogenic mutations from similarly rare but otherwise innocuous ones.
Conclusion
Since the sentinel discovery of BrS as a cardiac channelopathy in 1998, our genomic understanding of this potentially lethal disorder has matured from a phase of discovery to one of translational medicine. This international consortium of BrS genetic testing centers has tripled the catalog of possible BrS1-associated mutations with the addition of 200 new mutations to the public domain and has provided a template to draw upon for further genetic testing interpretation and biological inquiry.
Acknowledgments
Support for data analysis for this project was provided by the Mayo Clinic Windland Smith Rice Comprehensive Sudden Cardiac Death Program (Dr. Ackerman), grant HL47678 from the National Institutes of Health (Dr. Antzelevitch), New York State and Florida Free and Accepted Masons, the GIS Institut des Maladies Rares, the AFM (ANR-06-MRAR-022, PG, Dr. Schott), The Health Sciences Research Grants (H18, Research on Human Genome, 002) and the Research Grant for the Cardiovascular Diseases (21C-8) from the Ministry of Health, Labor, and Welfare of Japan (Dr. Shimizu), The Fondation Leducq Trans-Atlantic Network of Excellence Grant (05 CVD 01, Preventing Sudden Death, Dr. Schott), ANR grant ANR/-05-MRAR-028-01 (Dr. Schott), grant from the Fondation pour la recherche Medicale (Dr. Schott), FIS-ISCiii (Dr. Brugada), CNIC (Dr. Brugada), Ramon Brugada Sr. Foundation (Dr. Brugada), Leducq Foundation, grant 05 CVD, Alliance against Sudden Cardiac death (Drs. Wilde, Schott, and Schulze-Bahr), and Deutsche For-schungsgemeinschaft (Dr. Schulze-Bahr). All mutational analyses performed in this study were conducted at individual centers. Dr. Ackerman is a consultant for PGxHealth. Intellectual property derived from Dr. Ackerman’s research program resulted in license agreements in 2004 between Mayo Clinic Health Solutions (formerly Mayo Medical Ventures) and PGxHealth (formerly Genaissance Pharmaceuticals).
ABBREVIATIONS
- BrS
Brugada syndrome
- BrS1
Brugada syndrome type 1
- ChIPs
sodium channel interacting proteins
- ECG
electrocardiographic
- IDL
interdomain linker
- INa
available sodium current
- LQT3
long-QT syndrome type 3
- LQTS
long-QT syndrome
- MAF
minor allele frequency
- PCR
polymerase chain reaction
- SCD
sudden cardiac death
References
- 1.Chen PS, Priori SG. The Brugada syndrome. J Am Coll Cardiol. 2008;51:1176–1180. doi: 10.1016/j.jacc.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 2.Meregalli PG, Wilde AAM, Tan HL. Pathophysiological mechanisms of Brugada syndrome: depolarization disorder, repolarization disorder, or more? Cardiovasc Res. 2005;67:367–378. doi: 10.1016/j.cardiores.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 3.Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: report of the second consensus conference. Heart Rhythm. 2005;2:429–440. doi: 10.1016/j.hrthm.2005.01.005. Erratum: Heart Rhythm 2005;2:905. [DOI] [PubMed] [Google Scholar]
- 4.Tester DJ, Ackerman MJ. Cardiomyopathic and channelopathic causes of sudden unexplained death in infants and children. Annu Rev Med. 2009;60:69–84. doi: 10.1146/annurev.med.60.052907.103838. [DOI] [PubMed] [Google Scholar]
- 5.Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20:1391–1396. doi: 10.1016/0735-1097(92)90253-j. [DOI] [PubMed] [Google Scholar]
- 6.Priori SG, Napolitano C, Gasparini M, et al. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: a prospective evaluation of 52 families. Circulation. 2000;102:2509–2515. doi: 10.1161/01.cir.102.20.2509. [DOI] [PubMed] [Google Scholar]
- 7.Priori SG, Napolitano C, Gasparini M, et al. Natural history of Brugada syndrome: insights for risk stratification and management. Circulation. 2002;105:1342–1347. doi: 10.1161/hc1102.105288. [DOI] [PubMed] [Google Scholar]
- 8.Schulze-Bahr E, Eckardt L, Breithardt G, et al. Sodium channel gene (SCN5A) mutations in 44 index patients with Brugada syndrome: different incidences in familial and sporadic disease. Hum Mutat. 2003;21:651–652. doi: 10.1002/humu.9144. Erratum: Hum Mutat 2005;26:61. [DOI] [PubMed] [Google Scholar]
- 9.Remme CA, Wilde AAM. SCN5A overlap syndromes: no end to disease complexity? Europace. 2008;10:1253–255. doi: 10.1093/europace/eun267. Comment. [DOI] [PubMed] [Google Scholar]
- 10.Zimmer T, Surber R. SCN5A channelopathies—an update on mutations and mechanisms. Prog Biophys Mol Biol. 2008;98(2–3):120–136. doi: 10.1016/j.pbiomolbio.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 11.Ackerman MJ, Splawski I, Makielski JC, et al. Spectrum and prevalence of cardiac sodium channel variants among black, white, Asian, and Hispanic individuals: implications for arrhythmogenic susceptibility and Brugada/long QT syndrome genetic testing. Heart Rhythm. 2004;1:600–607. doi: 10.1016/j.hrthm.2004.07.013. see Comment. [DOI] [PubMed] [Google Scholar]
- 12.Spiegelman JI, Mindrinos MN, Oefner PJ. High-accuracy DNA sequence variation screening by DHPLC. BioTechniques. 2000;29:1084–1092. doi: 10.2144/00295rr04. [DOI] [PubMed] [Google Scholar]
- 13.Kapa S, Tester DJ, Salisbury BA, et al. Genetic testing for long QT syndrome—distinguishing pathogenic mutations from benign variants. Circulation. 2009;120(18):1752–1760. doi: 10.1161/CIRCULATIONAHA.109.863076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.den Dunnen JT, Antonarakis SE. Nomenclature for the description of human sequence variations. Hum Genet. 2001;109:121–124. doi: 10.1007/s004390100505. [DOI] [PubMed] [Google Scholar]
- 15.Murray A, Donger C, Fenske C, et al. Splicing mutations in KCNQ1: a mutation hot spot at codon 344 that produces in frame transcripts. Circulation. 1999;100:1077–1084. doi: 10.1161/01.cir.100.10.1077. [DOI] [PubMed] [Google Scholar]
- 16.Zhuang Y, Weiner AM. A compensatory base change in U1 snRNA suppresses a 5′ splice site mutation. Cell. 1986;46:827–835. doi: 10.1016/0092-8674(86)90064-4. [DOI] [PubMed] [Google Scholar]
- 17.Rogan PK, Svojanovsky S, Leeder JS. Information theory-based analysis of CYP2C19, CYP2D6 and CYP3A5 splicing mutations. Pharmacogenetics. 2003;13:207–218. doi: 10.1097/00008571-200304000-00005. [DOI] [PubMed] [Google Scholar]
- 18.Splawski I, Shen J, Timothy K, Vincent GM, Lehmann MH, Keating MT. Genomic structure of three long QT syndrome genes: KVLQT1, HERG, and KCNE1. Genomics. 1998;51:86–97. doi: 10.1006/geno.1998.5361. [DOI] [PubMed] [Google Scholar]
- 19.Wang Q, Li Z, Shen J, Keating MT. Genomic organization of the human SCN5A gene encoding the cardiac sodium channel. Genomics. 1996;34:9–16. doi: 10.1006/geno.1996.0236. [DOI] [PubMed] [Google Scholar]
- 20.Neyroud N, Richard P, Vignier N, et al. Genomic organization of the KCNQ1 K+ channel gene and identification of C-terminal mutations in the long-QT syndrome. Circ Res. 1999;84:290–297. doi: 10.1161/01.res.84.3.290. [DOI] [PubMed] [Google Scholar]
- 21.Wehrens XHT, Rossenbacker T, Jongbloed RJ, et al. A novel mutation L619F in the cardiac Na+ channel SCN5A associated with long-QT syndrome (LQT3): a role for the I-II linker in inactivation gating. Hum Mutat. 2003;21:552. doi: 10.1002/humu.9136. [DOI] [PubMed] [Google Scholar]
- 22.Ruan Y, Liu N, Bloise R, Napolitano C, Priori SG. Gating properties of SCN5A mutations and the response to mexiletine in long-QT syndrome type 3 patients. Circulation. 2007;116:1137–1144. doi: 10.1161/CIRCULATIONAHA.107.707877. [DOI] [PubMed] [Google Scholar]
- 23.Kambouris NG, Nuss HB, Johns DC, Tomaselli GF, Marban E, Balser JR. Phenotypic characterization of a novel long-QT syndrome mutation (R1623Q) in the cardiac sodium channel. Circulation. 1998;97:640–644. doi: 10.1161/01.cir.97.7.640. [DOI] [PubMed] [Google Scholar]
- 24.Wei J, Wang DW, Alings M, et al. Congenital long-QT syndrome caused by a novel mutation in a conserved acidic domain of the cardiac Na+ channel. Circulation. 1999;99:3165–3171. doi: 10.1161/01.cir.99.24.3165. [DOI] [PubMed] [Google Scholar]
- 25.Yang P, Kanki H, Drolet B, et al. Allelic variants in long-QT disease genes in patients with drug-associated torsades de pointes. Circulation. 2002;105:1943–1948. doi: 10.1161/01.cir.0000014448.19052.4c. [DOI] [PubMed] [Google Scholar]
- 26.Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–296. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 27.Kapplinger JD, Tester DJ, Salisbury BA, et al. Spectrum and prevalence of mutations from the first 2500 consecutive unrelated patients referred for the FAMILION© long QT syndrome genetic test. Heart Rhythm. 2009;6:1297–1303. doi: 10.1016/j.hrthm.2009.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 29.Meregalli PG, Tan HL, Probst V, et al. Type of SCN5A mutation determines clinical severity and degree of conduction slowing in loss-of-function sodium channelopathies. Heart Rhythm. 2009;6:341–348. doi: 10.1016/j.hrthm.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 30.Makita N, Behr E, Shimizu W, et al. The E1784K mutation in SCN5A is associated with mixed clinical phenotype of type 3 long QT syndrome. J Clin Invest. 2008;118:2219–2229. doi: 10.1172/JCI34057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bezzina C, Veldkamp MW, van den Berg MP, et al. A single Na+ channel mutation causing both long-QT and brugada syndromes. Circ Res. 1999;85:1206–1213. doi: 10.1161/01.res.85.12.1206. [DOI] [PubMed] [Google Scholar]
- 32.Probst V, Wilde AAM, Barc J, et al. SCN5A Mutations and the role of genetic background in the pathophysiology of brugada syndrome. Circ Cardiovasc Genet. 2009 doi: 10.1161/CIRCGENETICS.109.853374. accepted manuscript, in press. [DOI] [PubMed] [Google Scholar]
- 33.London B, Michalec M, Mehdi H, et al. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation. 2007;116:2260–2268. doi: 10.1161/CIRCULATIONAHA.107.703330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Antzelevitch C. Genetic basis of Brugada syndrome. Heart Rhythm. 2007;4:756–757. doi: 10.1016/j.hrthm.2007.03.015. Comment. Erratum: Heart Rhythm 2007;4:990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watanabe H, Koopman TT, Scouarnec SL, et al. Sodium channel β1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest. 2008;118:2260–2268. doi: 10.1172/JCI33891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delpon E, Cordeiro JM, Nunez L, et al. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythmia Electrophysiol. 2008;1:209–218. doi: 10.1161/CIRCEP.107.748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu S, Barajas-Martinez H, Burashnikove E, et al. A mutation in the β3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ Cardiovasc Genet. 2009;2:270–278. doi: 10.1161/CIRCGENETICS.108.829192. [DOI] [PMC free article] [PubMed] [Google Scholar]