

Abstract
We have previously shown that inhibiting protein-tyrosine kinase increased whereas inhibiting protein-tyrosine phosphatase (PTP) decreased renal outer medullary potassium channel 1 (ROMK1) channel activity (1). We have now used confocal microscopy, the patch clamp technique, and biotin labeling to further examine the role of tyrosine phosphorylation in regulating ROMK1 trafficking. Human embryonic kidney 293 cells were cotransfected with c-Src and green fluorescent protein-ROMK1, which has the same biophysical properties as those of ROMK1. Patch clamp studies have shown that phenylarsine oxide (PAO), an inhibitor of PTP, decreased the activity of ROMK1. Moreover, addition of PAO reduced the cell surface localization of green fluorescent protein-ROMK1 detected by confocal microscopy and diminished the surface ROMK1 density by 65% measured by biotin labeling. Also, PAO treatment significantly increased the phosphorylation of ROMK1. The notion that the effect of PAO is mediated by stimulating tyrosine phosphorylation-induced endocytosis of ROMK1 has also been supported by findings that mutating the tyrosine residue 337 of ROMK1 to alanine abolished the effect of PAO. Finally, the inhibitory effect of PAO on ROMK1 was completely blocked in the cells co-transfected with dominant negative dynamin (dynaminK44A). This indicates that the tyrosine phosphorylation-induced endocytosis of ROMK1 is dynamin-dependent. We conclude that inhibiting PTP increases ROMK1 phosphorylation and results in a dynamin-dependent internalization of the channel.
ROMK11 is located in the apical membrane of the cortical collecting duct (CCD) and is generally believed to be a key component of the native small conductance K+ (SK) channel (2-6). The SK channels are the major contributors to the apical K+ conductance and are responsible for K+ secretion (4, 7). One important factor for regulating K+ secretion is the dietary K+ intake; a high K+ intake increases whereas a low K+ intake decreases K+ secretion (7). The low K+ intake-induced decrease in K+ secretion is at least partially achieved by reducing the number of SK channels in the apical membrane of the CCD (8).
Our preceding experiments strongly indicated that the low K+ intake-induced decrease in SK channel number was mediated by protein-tyrosine kinase (PTK). This conclusion is supported by the observation that inhibition of PTK increased the number of the SK channels in the apical membrane of the CCD from rats on a K+-deficient diet (8). In contrast, inhibition of PTP decreased the number of SK channels in the CCD from rats on a high K+ diet (9). Because the effect of inhibiting PTP on channel activity was blocked by 20% sucrose, we speculated that inhibiting PTP increases the endocytosis of the SK channels whereas inhibiting PTK augments the exocytosis of the SK channels into the cell membrane. This notion is supported by observations that inhibiting PTP with PAO reduced whereas inhibiting PTK with herbimycin A increased the membrane location of ROMK1 in oocytes injected with GFP-ROMK1 and c-Src (1).
Although our previous results strongly suggested that inhibiting PTP increases the endocytosis of ROMK1, additional experiments are required to prove the hypothesis, because ROMK1 trafficking may not be the same in oocytes as in mammalian cells. In addition, it is difficult to distinguish the ROMK1 channels located in the cell membrane from those in the submembrane of oocytes. In the present study we have provided additional evidence to support the hypothesis that inhibiting PTP increases the tyrosine phosphorylation of ROMK1 channels and stimulates the endocytosis of ROMK1.
EXPERIMENTAL PROCEDURES
Construction of GFP-ROMK and c-Src
We used primers 5′ TGGGCCTAAAAGAATTCAGCTGCTGTGCAGACAAC (sense, nucleotides 81–115) and 5′ TTGTAGGTGGAAGGATCCCTGCTACATCTGGGTGTCG (antisense, nucleotides 1310–1346) for amplifying ROMK1 or mutant ROMK1, which was subcloned into pcDNA3.1. GFP-ROMK1 or GFP-R1Y337A was constructed by cloning the PCR products into the EcoRI and BamHI sites of plasmid-enhanced GFP-C1 expression vector (CLONTECH, Palo Alto, CA). The coding sequence of c-Src was cut from pGEM vector with HindIII and EcoRI and ligated into pCDNA3.1 expression vector (Invitrogen). All vector sequences were confirmed by automated DNA sequencing at the William Keck Biotechnology Laboratory at Yale University. dynaminK44A was a gift from Dr. Michael-Caplan at Yale University.
Transfection of HEK293 Cells and Confocal Microscopy
Transient transfection of HEK293 cells (American Type Culture Collection, Manassas, VA) was carried out using 3 μl of LT1 reagent (PanVera, Madison, WI) per 35-mm cell culture dish according to the manufacturer’s instructions. The cells were transfected with 1 μg of ROMK1+ 1 μg of c-Src, 1 μg of R1Y337A + 1 μg of c-Src, or 1 μg of ROMK1 + 1 μg of c-Src + 3 μg of dynaminK44A. The success rate of transient transfection was over 70%, and the experiments were carried out 48 h after transfection. After identifying the cells for the study, we recorded cell images of middle sections under control conditions and after PAO treatment using a Bio-Rad MRC1000 confocal microscope. GFP fluorescence was excited at 488 nm with an argon laser beam and viewed with an inverted Olympus microscope equipped with a ×60 oil lens. The cell image taken under control conditions served as a control picture. All images were acquired, processed, and printed with identical parameters before and after PAO treatment.
Biotinylation, Immunoprecipitation, and Western Blot Analysis
The change in cell surface ROMK1 upon treatment of the cells with PAO was quantitated by labeling the cells with sulfo-NHS-SS-biotin (Pierce) according to the instructions provided by the manufacturer. HEK cells from each 35-mm dish were trypsinized with trypsin-EDTA (Fisher) and lysed with 200 μl of cold RIPA lysis buffer (1× PBS, 1% Igepal CA-630 (Sigma), 0.1% SDS) containing 0.5% deoxycholate, 1 mm sodium molybdate, 1 mm sodium fluoride, 1 μm phenylmethylsulfonyl fluoride and 100 μl of protease inhibitor mixture (Sigma) per ml of lysis buffer. The mixture was clarified at 14,000 rpm for 10 min at 4 °C, and the resultant supernatant was collected. GFP-ROMK1 fusion protein was immunoprecipitated by incubating the resultant supernatant with 1 μg of GFP monoclonal antibody (CLONTECH, Palo Alto, CA) for 2 h at 4 °C. Protein A/G (20 μl)-agarose (Santa Cruz Biotechnology) was added to the tube containing the mixture, and the tube was rotated overnight at 4 °C. The protein A/G-agarose immune complex was collected by centrifugation at 14,000 rpm for 2 min and washed twice with 1× PBS containing protease/phosphatase inhibitors. After centrifugation, 1× SDS (50 μl) sample buffer (2% SDS, 60 mm Tris-HCl (pH 6.8), 20% glycerol, 10% 2 mercaptoethanol, 0.05% bromphenol blue) was added to the resultant pellet followed by boiling the mixture for 5 min. Protein concentrations were determined using the Bio-Rad protein assay kit. The proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride membrane (Bio-Rad). The biotin-labeled GFP-ROMK1 proteins were detected by using NeutrAvidin (horseradish peroxidase-conjugated) (Pierce). ROMK1 or ROMK1 mutant protein was detected by using a polyclonal ROMK1 antibody (1:200; Alomone Laboratories, Ltd., Jerusalem, Israel) followed by anti-rabbit horseradish peroxidase (1:15,000; Amersham Biosciences, Inc.). The tyrosine-phosphorylated proteins were detected using PY20, an antibody that reacts with tyrosine-phosphorylated proteins (Upstate Biotechnology, Lake Placid, NY).
We also studied the endocytosis of ROMK1 following the protocol described by Collazo et al. (10). HEK cells were labeled with sulfo-NHS-SS-biotin (Pierce) followed by treatment of cells with 1 μm PAO for 15 min at 37 °C to stimulate endocytosis. The cells were washed two times with PBS, and the surface biotin was cleaved with the cell-impermeant reducing agent, TCEP (Tris [2-carboxyethyl] phosphine hydrochloride) (50 mm). The internalized proteins labeled by biotin were protected from TCEP cleavage. After washing two times with PBS, the cells were lysed with RIPA lysis buffer containing protease inhibitors. The lysate was treated as detailed above.
Patch Clamp Technique
An Axon200A patch clamp amplifier was used to record channel current. The current was low pass-filtered at 1 KHz by an eight-pole Bessel filter (902LPF; Frequency Devices, Haverhill, MA) and digitized by an Axon interface (Digitada1200). Data were acquired by an IBM-compatible Pentium computer (Gateway 2000) at a rate of 4 KHz and analyzed using the pClamp software system 6.04 (Axon Instruments, Burlingame, CA). Channel activity was defined as NPo that was calculated from data samples of 60-s duration in the steady state as shown in Equation 1,
| (Eq. 1) |
where ti is the fractional open time spent at each of the observed current levels.
Experimental Solution and Statistics
The bath solution for the patch clamp study was composed of the following (in mm): 140 NaCl, 5 KCl, 1.8 MgCl2, 1.8 CaCl2, and 10 HEPES (pH 7.4). The pipette solution was composed of the following (in mm): 140 mm KCl, 1.8 MgCl2, and 10 Hepes (pH 7.4). Phenylarsine oxide was purchased from Sigma and added directly to the bath to reach the final concentration. We present data as mean ± S.E. The student’s t test was used to determine the significance.
RESULTS
We first examined the properties of the GFP-ROMK fusion protein using Western blot and the patch clamp technique. Fig. 1 is a typical Western blot showing that ROMK antibody detects a 71-kDa protein harvested from the immunoprecipitation of the lysate of cells transfected with GFP-ROMK1 with GFP antibody. Moreover, we had previously carried out the patch clamp study in oocytes injected with cRNA encoding GFP-ROMK1 and detected an inward-rectifying K+ channel with an inward conductance of 40 picosiemens (1). This finding is also confirmed in the present investigation in which we have detected an inward-rectifying K+ channel with an inward slope conductance of 40 picosiemens in HEK293 cells transfected with GFP-ROMK1 (data not shown). The K+ channel has a high open probability and similar channel kinetics to that of ROMK1.
Fig. 1. A Western blot showing that both GFP antibody (left panel) and ROMK antibody (right panel) detect a 71-kDa protein harvested by immunoprecipitation (IP) of the lysate of HEK293 cells transfected with GFP-ROMK1.
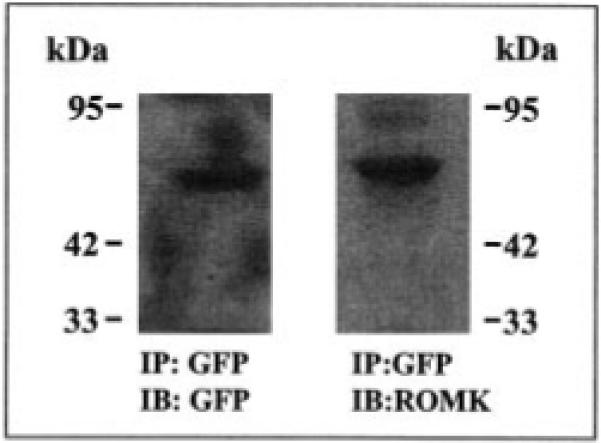
IB, immunoblotting.
After confirming that GFP-ROMK1 has the same biophysical properties as those of ROMK1, we examined the effect of PAO on the activity of ROMK1 in HEK293 cells transfected with GFP-ROMK1 and c-Src. Fig. 2 is a typical recording showing that addition of 1 μm PAO inhibits the activity of ROMK1 by 95 ± 9%, and NPo falls from 0.93 to 0.04 (n = 4). This observation is consistent with our previous finding that inhibiting PTP decreased the activity of the native ROMK1-like channels in the CCD (9). Moreover, we have demonstrated previously that the inhibitory effect of PAO was absent in the CCDs treated with hypertonic solution, indicating that the effect of PAO on channel activity was mediated by stimulation of endocytosis (9). This hypothesis was further investigated using confocal microscopy. Fig. 3 depicts typical confocal images obtained from HEK293 cells transfected with GFP-ROMK1 + vector (A–D) or ROMK1 + c-Src (E–H). Application of PAO had no significant effect on the location of ROMK1 in the cells transfected with vector (Fig. 3, A–D). In contrast, addition of PAO altered the location of ROMK1 in the cells transfected with c-Src. From inspection of Fig. 3, E–H, it is apparent that the membrane location of ROMK1 diminished whereas the density of ROMK1 in the intracellular compartment increased significantly within 15 min following PAO treatment. This strongly indicates that inhibiting PTP stimulates the endocytosis of ROMK1 in cells transfected with c-Src and GFP-ROMK1.
Fig. 2. A channel recording shows the effect of 1 μm PAO on the activity of ROMK1 in HEK293 cells transfected with GFP-ROMK1 and c-Src.
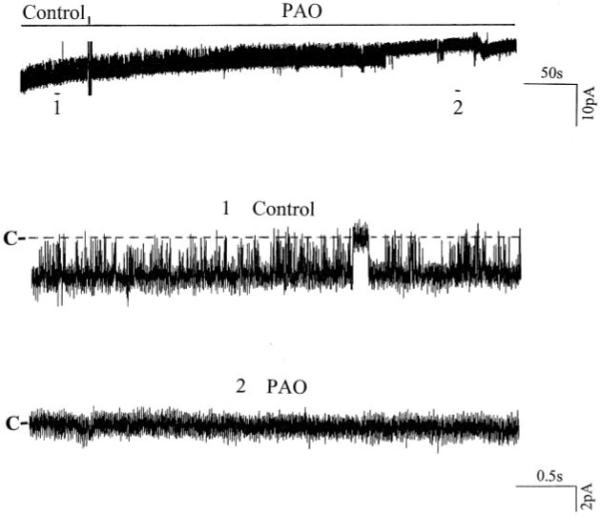
The top trace shows the time course of the experiment, and two parts of the data indicated by numbers are expanded to demonstrate the fast time resolution. The pipette holding potential was 0 mV, and the experiment was performed in a cell-attached patch. The channel closed level is indicated by C.
Fig. 3. A typical confocal image showing the effect of PAO on the ROMK1 location in HEK293 cells transfected with GFP-ROMK1 alone (A–D) or with GFP-ROMK1 + c-Src (E–H).
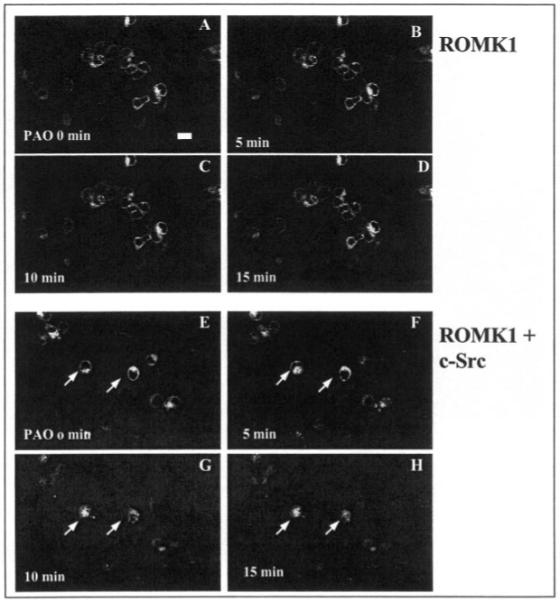
The magnification of the picture is ×600, and the length of the bar represents 10 μm. The cell image before addition of PAO is demonstrated in A whereas B, C, and D show the ROMK1 location following addition of 1 μm PAO at 5, 10, and 15 min, respectively. E shows the cell image under control conditions in cells transfected with GFP-ROMK1 and c-Src. F, G, and H demonstrate the changes in ROMK1 location following addition of PAO at 5, 10, and 15 min, respectively. Arrows indicate the cells in which a clear relocation of ROMK1 has been observed.
To further confirm that PAO enhanced the endocytosis of ROMK1, we used the biotin labeling technique to study the effect of PAO on ROMK1 membrane location. The cells were treated with PAO or vehicle at 37 °C for 15 min followed by labeling the surface proteins with biotin at 4 °C. The ROMK1 channels were harvested by immunoprecipitation of the cell lysate with GFP antibody and detected by the ROMK antibody whereas the biotin-labeled ROMK1 channels were identified with neutravidin (Fig. 4A). Clearly, treatment of cells with 1 μm PAO significantly reduced the number of the biotin-labeled ROMK1. In 11 experiments, the density of biotin-labeled ROMK1 channels decreased by 65 ± 3% in the cells treated with PAO in comparison to those without PAO. This strongly indicates that inhibiting PTP facilitates the internalization of ROMK1. This hypothesis is also supported by experiments in which the effect of PAO on ROMK1 membrane location was studied using sulfo-NHS-SS-biotin. After labeling the surface proteins with biotin, the cells were treated with PAO for 15 min at 37 °C to stimulate endocytosis and then incubated with TCEP, a reducing agent, to cleave the surface biotin (Fig. 4B). Thus, only internalized ROMK1 channels were labeled with biotin whereas the ROMK1 channels located in the cell membrane did not bear biotin. From inspection of Fig. 4B, it is apparent that inhibiting PTP significantly increased the internalization of ROMK1 channels. The amount of biotin-labeled ROMK1 in the PAO-treated group was 75 ± 9% (n = 4) of the control (no TCEP and PAO treatment) whereas the amount of the biotin-labeled ROMK1 in the absence of PAO was only 20 ± 4% (n = 4) of the control value.
Fig. 4. A Western blot shows the effect of PAO on the density of ROMK1 in the membrane of HEK293 cells transfected with GFP-ROMK1 and c-Src.
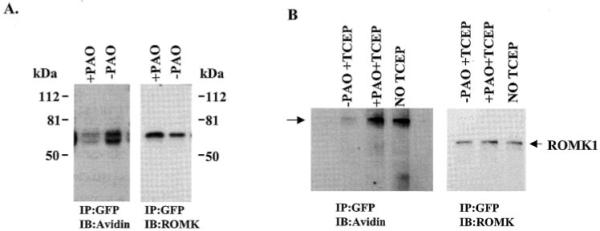
A, the cells were treated with 1 μm PAO for 15 min followed by biotinylation of the surface protein at 4 °C. IP, immunoprecipitation; IB, immunoblotting. B, the surface proteins of the cells were labeled with sulfo-NHS-SS-biotin followed by a 15-min treatment of cells with PAO at 37 °C to stimulate endocytosis. After PAO treatment, the surface biotin was removed by incubating the cells with TCEP. The ROMK1 channels were harvested by immunoprecipitation of the cell lysate with GFP antibody. The biotin-labeled protein was identified by neutravidin horseradish peroxidase (left panel), and the total ROMK1 was detected by ROMK1 antibody (right panel). As shown in the left panel, avidin recognizes two bands. However, only the top band is detected by ROMK1 antibody (right panel). The data was normalized according to the total ROMK1 level.
Inhibiting PTP is expected to enhance the tyrosine phosphorylation of ROMK1, which has been shown to be a substrate of c-Src.2 Therefore, we examined the effect of PAO on the tyrosine phosphorylation of ROMK1 in cells transfected with GFP-ROMK1 and c-Src. After immunoprecipitation of ROMK1 with the GFP antibody, we detected the tyrosine phosphorylated ROMK1 with PY20, an antibody that reacts with tyrosine-phosphorylated proteins. Fig. 5 is a typical recording from 10 such experiments in which the effect of inhibiting PTP on the tyrosine phosphorylation of ROMK1 was studied. Although the same amount of ROMK1 was present in the cells treated or not treated with PAO, inhibiting PTP significantly increased the fraction of the tyrosine-phosphorylated ROMK1 by 110 ± 8% in comparison to those without PAO treatment. Therefore, the data strongly suggest that PAO increases the tyrosine phosphorylation of ROMK1 and in turn stimulates the internalization of ROMK1.
Fig. 5. A Western blot illustrating the effect of PAO on the tyrosine phosphorylation level of ROMK1 in the presence and absence of PAO.
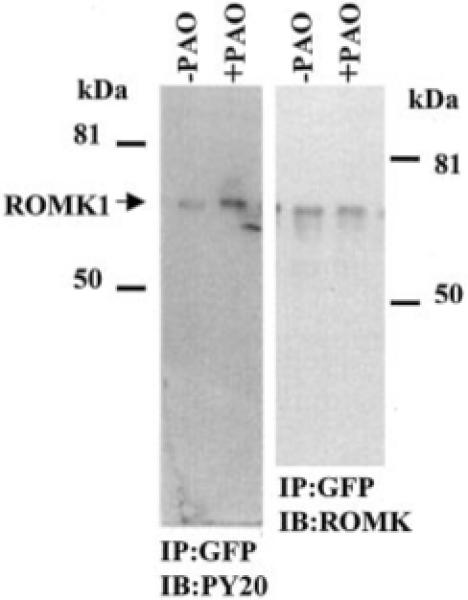
The cells were treated with PAO or vehicle for 15 min. The ROMK1 channels were harvested by immunoprecipitation (IP) of the cell lysate with GFP antibody. The phosphorylated ROMK1 was detected with PY20 (left panel), and the total ROMK1 is recognized by ROMK antibody (right panel). The protein phosphorylation level is normalized by comparison to the relative amount of the total ROMK1 protein. IB, immunoblotting.
Our previous experiments indicated that the tyrosine residue 337 in the C terminus was critical for mediating c-Src-induced phosphorylation.2 Moreover, the observation that mutation of the tyrosine residue to alanine abolished the inhibitory effect of PAO on K+ current in oocytes injected with R1Y337A and c-Src suggested that tyrosine residue 337 is essential for initiating PAO-induced endocytosis (1). To further test this hypothesis, we investigated the effect of PAO on the membrane location of ROMK1 mutant, R1Y337A. Fig. 6 is a typical confocal image from eight such experiments showing the effect of PAO on the distribution of R1Y337A in HEK293 cells cotransfected with c-Src and R1Y337A. In contrast to ROMK1, addition of PAO did not change the pattern of R1Y337A distribution, and the intensity of membrane fluorescence was not significantly altered in the presence of PAO. The possibility that tyrosine residue 337 is essential for facilitating the endocytosis of ROMK1 is also supported by experiments in which the effect of PAO on the membrane density of ROMK1 or R1Y337A was studied using the biotin labeling technique (Fig. 7). The cells transfected with c-Src + ROMK1 or c-Src + R1Y337A were incubated in the presence or in the absence of PAO at 37 °C for 15 min followed by biotin labeling at 4 °C. Similar to the results shown in Fig. 4, inhibiting PTP significantly diminished the amount of ROMK1 located in the cell membrane as shown by the fact that the intensity of the biotin-labeled ROMK1 decreased by 65 ± 3% in comparison to those in the absence of PAO, although the same amount of ROMK1 was present in cells treated or not treated with PAO. In contrast, PAO did not significantly reduce the number of R1Y337A in the cell membrane (95 ± 9% of the control value) in comparison to those in the absence of PAO in cells transfected with c-Src and R1Y337A.
Fig. 6. A confocal image showing the effect of PAO on the distribution of R1Y337A, a ROMK1 mutant in which tyrosine residue 337 was mutated to alanine.
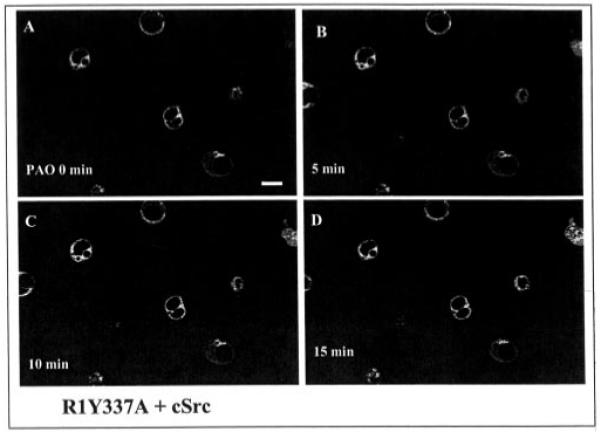
The magnification of the picture is ×600, and the length of the bar represents 10 μm. The cell image under control conditions (A) was taken right before addition of PAO. B, C, and D show the channel location following addition of PAO at 5 min (B), 10 min (C), and 15 min (D), respectively.
Fig. 7. A Western blot shows the effect of PAO on the membrane fraction of ROMK1 or R1Y337A labeled by biotin.
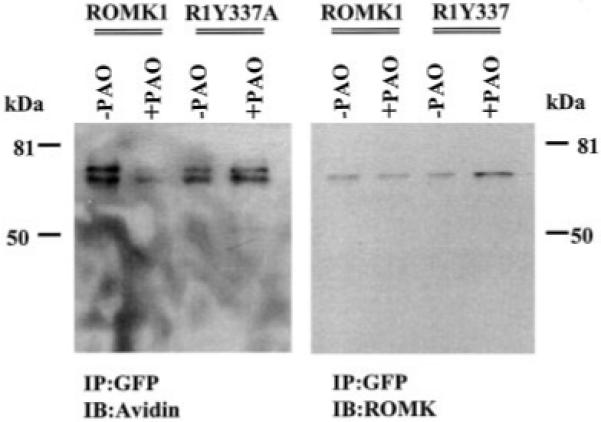
The cells transfected with ROMK1/R1Y337A + c-Src were treated with PAO for 15 min followed by biotin labeling at 4 °C. The ROMK1/R1Y337A proteins were harvested by immunoprecipitation (IP) of cell lysate with GFP antibody. The membrane fraction of the K+ channels was detected with neutravidin horseradish peroxidase (left panel), and the total ROMK1/R1Y337A proteins were identified with ROMK antibody (right panel). The data were normalized according to the total K+ channel protein level. IB, immunoblotting.
After establishing that inhibiting PTP increases the endocytosis of ROMK1 and that tyrosine residue 337 plays a key role in mediating the effect of PAO, we expanded the study to determine whether the effect of inhibiting PTP requires the involvement of dynamin. We first cotransfected the HEK293 cells with dominant negative dynamin (dynaminK44A), c-Src, and ROMK1 and found that the cells expressing ROMK1 also successfully expressed c-Src and dynaminK44A (Fig. 8). The effect of PAO on the ROMK1 distribution has also been studied using confocal microscopy and biotin labeling techniques. Fig. 9 is a typical confocal image demonstrating the effect of PAO on ROMK1 distribution in the cells transfected with GFP-ROMK1, c-Src, and dynaminK44A at a ratio of 1:1:3. It is apparent that PAO-induced internalization of ROMK1 is absent in the cells transfected with dynaminK44A (n = 7). The possibility that dynamin is required for the PAO-induced endocytosis of ROMK1 is also supported by the observation that PAO did not significantly decrease the number of ROMK1 in the membrane of cells transfected with dynaminK44A (Fig. 10). In 10 experiments we have observed that inhibiting PTP reduced the amount of the biotin-labeled ROMK1 by 10 ± 8% in comparison to those without PAO treatment in cells transfected with dynaminK44A. The difference is not significant. Therefore, this indicates that inhibiting PTP-induced endocytosis of ROMK1 requires the involvement of dynamin.
Fig. 8. A cell image of fluorescence microscopy showing the co-localization of GFP-ROMK1, c-Src, and dynaminK44A in HEK293 cells transfected with the three genes.
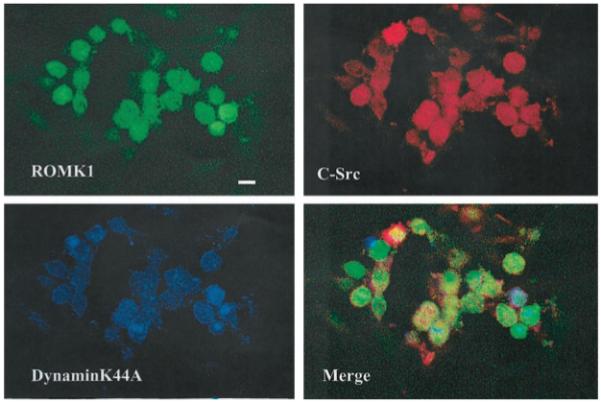
The magnification of the picture is ×600.
Fig. 9. A confocal image showing the effect of PAO on the distribution of ROMK1 channels in HEK293 cells transfected with dynaminK44A, GFP-ROMK1, and c-Src.
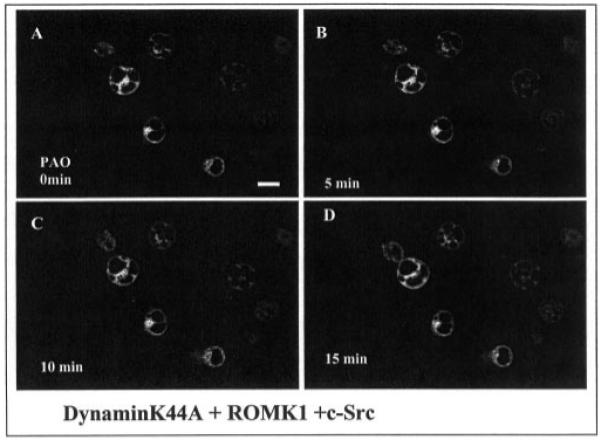
The magnification of the picture is ×600, and the length of the bar represents 10 μm. The cell image under control conditions (A) was taken right before addition of PAO. B, C, and D show the channel location following addition of PAO at 5 min (B), 10 min (C), and 15 min (D), respectively.
Fig. 10. A Western blot shows the effect of PAO on the membrane fraction of ROMK1 labeled with biotin.
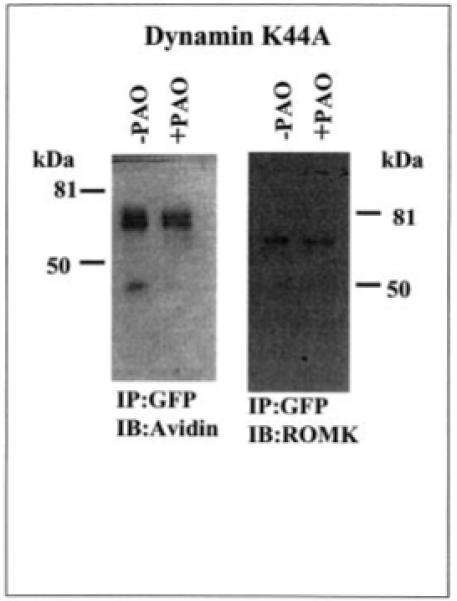
The cells transfected with ROMK1 + c-Src + dynaminK44A/vector were treated with PAO for 15 min followed by biotin labeling at 4 °C. The ROMK1 proteins were harvested by immunoprecipitation (IP) of cell lysate with GFP antibody. The membrane fraction of ROMK1 was detected with neutravidin (left panel), and the total ROMK1 proteins were identified with ROMK antibody (right panel). The data were normalized according to the total K+ channel protein level. IB, immunoblotting.
DISCUSSION
The main finding of the present investigation is that inhibiting PTP increases the internalization of ROMK1 and that the endocytosis of ROMK1 is a dynamin-dependent process. We used PAO as a tool to study the effect of tyrosine phosphorylation on ROMK1. Although it is possible that PAO may have effects other than inhibiting PTP, three lines of evidence indicated that the effect of PAO on channel activity results from inhibiting PTP-induced tyrosine phosphorylation. First, our present study shows that PAO treatment increased the tyrosine phosphorylation of ROMK1. Second, mutation of tyrosine residue 337 of ROMK1 to alanine abolished the effect of PAO on ROMK1 channel distribution. This finding is consistent with our previous observation that PAO did not affect channel activity in oocytes injected with R1Y337A (1). Third, our previous investigation has shown that inhibiting PTK can also abolish the effect of PAO on the activity of ROMK1 (1).
ROMK1 is an inwardly rectifying K+ channel that is located in the apical membrane of the CCD (3, 5, 6). The CCD is responsible for K+ secretion and the hormone-regulated sodium reabsorption (7, 12). The K+ secretion is a two-step process; K+ enters the cell via the basolateral Na+-K+-ATPase and then diffuses into the lumen across the apical K+ channels (7). Two types of K+ channels, an SK channel and a Ca2+-activated large conductance K+ channel (13-18), have been identified in the apical membrane. The SK channel is mainly responsible for K+ secretion, because the SK channel has a high density and a high channel open probability (4). Although it is still not completely understood whether ROMK1 alone is sufficient to form the native SK channel, it is well established that ROMK1 is a key component of the native SK channel in the CCD (4). Therefore, ROMK1 plays a key role in regulating K+ secretion in the kidney.
K+ secretion is regulated by hormones such as aldosterone and by a dietary K+ intake (7, 12). The stimulatory effect of aldosterone on K+ secretion may be mediated by increasing the driving force for K+ diffusion across the apical membrane rather than altering the K+ conductance (19). This view is supported by the observation that application of aldosterone alone did not increase the number of the functional SK channels in the CCD (20). The effect of the dietary K+ intake on K+ secretion is well established; a low K+ intake decreases whereas a high K+ intake increases K+ secretion in the CCD (7, 12).
The effect of the dietary K+ intake on K+ secretion is at least in part achieved by changing the number of apical SK channels. For instance, we and others (14, 20) have observed that the number of ROMK1 channels increased in the CCD harvested from rats on a high K+ diet. Moreover, the effect of a high K+ intake on ROMK1 channels was not mediated by increasing transcription or translation, because neither ROMK mRNA nor ROMK protein in the kidney harvested from rats on a high K+ diet was changed in comparison to those on a normal K+ diet (21). This indicates that post-translation factors are involved in regulating the effect of the dietary K+ intake on K+ secretion. However, the nature of the factors responsible for mediating the effect of the K+ intake on ROMK1 is not completely understood.
We have observed previously that a low K+ intake increases whereas a high K+ intake decreases the expression of c-Src and cYes PTK in the kidneys (8), suggesting the role of PTK in mediating the effect of dietary K+ intake on K+ secretion. This notion is further supported by observations that inhibiting PTK increased the number of functional ROMK1 channels in the CCD obtained from rats on a K+-deficient diet (8, 22). In contrast, inhibiting PTP decreased the number of ROMK1 channels in the tubule from rats on a high K+ diet (9). Moreover, a low K+ intake increases the PTK-sensitive recycling pool of ROMK1 channels (8). Therefore, tyrosine phosphorylation or dephosphorylation of ROMK1 is an important mechanism by which a dietary K+ intake changes the ROMK1 density in the cell membrane. It is highly possible that an increase in tyrosine phosphorylation of ROMK1 diminishes whereas a decrease in tyrosine phosphorylation of ROMK1 augments the number of the ROMK1 channels in the cell membrane. This hypothesis has been supported by the preceding findings that PAO inhibits whereas herbimycin A increases the K+ current in oocytes injected with ROMK1 and c-Src (1).
Four lines of evidence obtained from the previous and the present investigations strongly suggest that the inhibitory effect of PTK on ROMK1 channels is mediated by increasing the internalization of ROMK1 rather than by a direct inhibition induced by tyrosine phosphorylation. First, we have previously shown that addition of exogenous c-Src to the bath facing cytosolic membrane of inside-out patches did not block the channel activity (1). Second, previous investigations have found that the inhibitory effect of PAO on K+ current in oocytes injected with ROMK1 and c-Src was abolished by hypertonic solution or concanavalin A (1, 9); both maneuvers have been shown to block the endocytosis of the membrane proteins (23, 24). Third, we have previously used GFP-ROMK1 as a tool to demonstrate that PAO decreases whereas herbimycin A increases the fluorescence intensity of the membrane in oocytes injected with c-Src and GFP-ROMK1 (1). This observation has also been confirmed by the present finding that inhibiting PTP decreased the number of ROMK1 in the membrane of HEK cells using confocal microscopy and the biotin labeling technique. Finally, we have shown in the present study that the effect of PAO on the ROMK1 surface distribution was absent in the cells transfected with dominant negative dynamin.
Dynamin has been shown to be involved in clathrin-mediated vesicular trafficking processes (25) and endocytosis of a variety of proteins such as GLUT 4 (26, 27) and glutamate receptor (28). In the kidney, dynamin is required for initiating endocytosis of epithelial sodium channels (29) and Na+/H+ exchanger (10). It has been shown that dynamin is a substrate of PTK, and phosphorylation of dynamin is essential for inducing the internalization of β2-adrenergic receptor (30, 31). We need further experiments to determine whether dynamin phosphorylation is required for inducing the endocytosis of ROMK1. However, the observation that expression of dynaminK44A abolished the effect of PAO on the internalization of ROMK1 channels strongly indicates that endocytosis of ROMK1 requires dynamin.
In the present study we have demonstrated that the tyrosine phosphorylation of ROMK1 is an important step for initiating the endocytosis of ROMK1, because mutation of tyrosine residue 337 of ROMK1 completely abolished the PAO-induced stimulation of ROMK1 endocytosis. We have previously demonstrated that ROMK1 is a substrate of c-Src, and tyrosine residue 337 is the main site of PTK-induced tyrosine phosphorylation (11). However, it is possible that phosphorylation of tyrosine residue is not the only requirement for initiating the endocytosis of ROMK1. This conclusion is based on our observation that PAO did not inhibit the activity of ROMK2 channels in oocytes.3 Moreover, inhibiting PTP did not inhibit the ROMK-like K+ channels in the thick ascending limb (11) where ROMK2 is located. This indicates that the N terminus of ROMK1 may also be required for starting the endocytosis. We need further experiments to explore the role of the N terminus in inducing the endocytosis of ROMK1.
The physiological significance of the present investigation is to define a mechanism by which a low K+ intake decreases the apical K+ conductance and K+ secretion. We have demonstrated previously that the amount of tyrosine-phosphorylated ROMK channels was significantly larger in the kidney from rats on a K+-deficient diet than those on a high K+ diet (11). This finding may be used to explain the observation that the positive response of ROMK1-like K+ channels in the CCD to herbimycin A was over 80% in the CCD from rats on a K+-deficient diet. In contrast, the response of the K+ channel to PTK inhibitor was absent in the CCD from rats on a high K+ diet (8). Fig. 11 is a model of a CCD principal cell illustrating the mechanism by which low K+ intake decreases the apical SK channels. A low K+ intake increases the expression and activity of PTK and in turn enhances the tyrosine phosphorylation of ROMK1. As a consequence of tyrosine phosphorylation of ROMK1, the K+ channel is internalized, and the apical K+ conductance falls.
Fig. 11. A model of CCD principal cell illustrating the mechanism by which low K+ intake decreases the number of ROMK1 channels in the cell membrane.
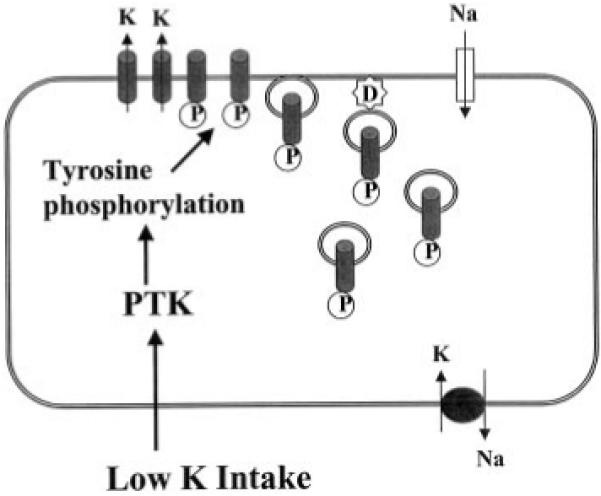
D, dynamin, P, phosphorylation.
We conclude that inhibiting PTP increases the tyrosine phosphorylation of ROMK1 and enhances the dynamin-dependent endocytosis and that tyrosine residue 337 of ROMK1 is essential for the effect of inhibiting PTP.
Acknowledgment
We thank Melody Steinberg for helping in the preparation of the manuscript.
Footnotes
The work was supported in part by National Institutes of Health Grants DK 47402 and DK 54983.
- ROMK
- renal outer medullary potassium channel
- CCD
- cortical collecting duct
- SK
- small conductance K
- PTK
- protein-tyrosine kinase
- PTP
- protein-tyrosine phosphatase
- PAO
- phenylarsine oxide
- GFP
- green fluorescent protein
- HEK
- human embryonic kidney
- PBS
- phosphate-buffered saline
Lin, D. H., Sterling, H., Lerea, K. M., Welling, P., Jin, L., Giebisch, G., and Wang, W. H. (2001) J. Am. Soc. Nephrol. 12, 28.
Unpublished observation.
REFERENCES
- 1.Moral Z, Deng K, Wei Y, Sterling H, Deng H, Ali S, Gu RM, Huang XY, Hebert SC, Giebisch G, Wang WH. J. Biol. Chem. 2001;276:7156–7163. doi: 10.1074/jbc.M008671200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boim MA, Ho K, Schuck ME, Bienkowski MJ, Block JH, Slightom JL, Yang Y, Brenner BM, Hebert SC. Am. J. Physiol. 1995;268:F1132–F1140. doi: 10.1152/ajprenal.1995.268.6.F1132. [DOI] [PubMed] [Google Scholar]
- 3.Ho K, Nichols CG, Lederer WJ, Lytton J, Vassilev PM, Kanazirska MV, Hebert SC. Nature. 1993;362:31–38. doi: 10.1038/362031a0. [DOI] [PubMed] [Google Scholar]
- 4.Wang WH, Hebert SC, Giebisch G. Annu. Rev. Physiol. 1997;59:413–436. doi: 10.1146/annurev.physiol.59.1.413. [DOI] [PubMed] [Google Scholar]
- 5.Xu JZ, Hall AE, Peterson LN, Bienkowski MJ, Eessalu TE, Hebert SC. Am. J. Physiol. Renal Physiol. 1997;273:F739–F748. doi: 10.1152/ajprenal.1997.273.5.F739. [DOI] [PubMed] [Google Scholar]
- 6.Mennitt PA, Wade JB, Ecelbarger CA, Palmer LG, Frindt G. J. Am. Soc. Nephrol. 1997;8:1823–1830. doi: 10.1681/ASN.V8121823. [DOI] [PubMed] [Google Scholar]
- 7.Giebisch G. Am. J. Physiol. Renal Physiol. 1998;274:F817–F833. doi: 10.1152/ajprenal.1998.274.5.F817. [DOI] [PubMed] [Google Scholar]
- 8.Wei Y, Bloom P, Lin DH, Gu RM, Wang WH. Am. J. Physiol. Renal Physiol. 2001;281:F206–F212. doi: 10.1152/ajprenal.2001.281.2.F206. [DOI] [PubMed] [Google Scholar]
- 9.Wei Y, Bloom P, Gu RM, Wang WH. J. Biol. Chem. 2000;275:20502–20507. doi: 10.1074/jbc.M000783200. [DOI] [PubMed] [Google Scholar]
- 10.Collazo R, Fan L, Hu MC, Zhao H, Wiederkehr MR, Moe OW. J. Biol. Chem. 2000;275:31601–31608. doi: 10.1074/jbc.M000600200. [DOI] [PubMed] [Google Scholar]
- 11.Gu RM, Wei Y, Falck JR, Krishna UM, Wang WH. Am. J. Physiol. Cell. Physiol. 2001;281:C1188–C1195. doi: 10.1152/ajpcell.2001.281.4.C1188. [DOI] [PubMed] [Google Scholar]
- 12.Palmer LG. Am. J. Physiol. Renal Physiol. 1999;277:F821–F825. doi: 10.1152/ajprenal.1999.277.6.F821. [DOI] [PubMed] [Google Scholar]
- 13.Frindt G, Palmer LG. Am. J. Physiol. Renal Physiol. 1989;256:F143–F151. [Google Scholar]
- 14.Wang W, Schwab A, Giebisch G. Am. J. Physiol. Renal Physiol. 1990;259:F494–F502. doi: 10.1152/ajprenal.1990.259.3.F494. [DOI] [PubMed] [Google Scholar]
- 15.Satlin LM, Palmer LG. Am. J. Physiol. Renal Physiol. 1997;272:F397–F404. doi: 10.1152/ajprenal.1997.272.3.F397. [DOI] [PubMed] [Google Scholar]
- 16.Schlatter E, Lohrmann E, Greger R. Pflugers Arch. 1992;420:39–45. doi: 10.1007/BF00378639. [DOI] [PubMed] [Google Scholar]
- 17.Hunter M, Lopes AG, Boulpaep EL, Giebisch G. Proc. Natl. Acad. Sci. U. S. A. 1984;81:4237–4239. doi: 10.1073/pnas.81.13.4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frindt G, Palmer LG. Am. J. Physiol. Renal Physiol. 1987;252:F458–F467. doi: 10.1152/ajprenal.1987.252.3.F458. [DOI] [PubMed] [Google Scholar]
- 19.Schafer JA, Troutman SL, Schlatter E. Am. J. Physiol. Renal Physiol. 1990;258:F199–F210. doi: 10.1152/ajprenal.1990.258.1.F199. [DOI] [PubMed] [Google Scholar]
- 20.Palmer LG, Antonian L, Frindt G. J. Gen. Physiol. 1994;105:693–710. doi: 10.1085/jgp.104.4.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frindt G, Zhou H, Sackin H, Palmer LG. Am. J. Physiol. Renal Physiol. 1998;274:F525–F531. doi: 10.1152/ajprenal.1998.274.3.F525. [DOI] [PubMed] [Google Scholar]
- 22.Wang WH, Lerea KM, Chan M, Giebisch G. Am. J. Physiol. Renal Physiol. 2000;278:F165–F171. doi: 10.1152/ajprenal.2000.278.1.F165. [DOI] [PubMed] [Google Scholar]
- 23.Luttrell LM, Daaka Y, Della-Rocca GJ, Lefkowitz RJ. J. Biol. Chem. 1997;272:31648–31656. doi: 10.1074/jbc.272.50.31648. [DOI] [PubMed] [Google Scholar]
- 24.Beaumont V, Hepworth MB, Luty JS, Kelly E, Henderson G. J. Biol. Chem. 1998;273:33174–33183. doi: 10.1074/jbc.273.50.33174. [DOI] [PubMed] [Google Scholar]
- 25.Marks B, Stowell MH, Vallis Y, Mills IG, Gibson A, Hopkins CR, McMahon HT. Nature. 2001;410:231–235. doi: 10.1038/35065645. [DOI] [PubMed] [Google Scholar]
- 26.Kao AW, Ceresa BP, Santeler SR, Pessin JE. J. Biol. Chem. 1998;273:25450–25457. doi: 10.1074/jbc.273.39.25450. [DOI] [PubMed] [Google Scholar]
- 27.Al-Hasani H, Hinck CS, Cushman SW. J. Biol. Chem. 1998;273:17504–17510. doi: 10.1074/jbc.273.28.17504. [DOI] [PubMed] [Google Scholar]
- 28.Carroll RC, Beattie EC, Xia H, Luescher C, Altschuler Y, Nicoll RA, Malenka RC, von Zastrow M. Proc. Natl. Acad. Sci. 1999;96:14112–14117. doi: 10.1073/pnas.96.24.14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shimkets RA, Lifton RP, Canessa CM. J. Biol. Chem. 1997;272:25537–25541. doi: 10.1074/jbc.272.41.25537. [DOI] [PubMed] [Google Scholar]
- 30.Foster-Barber A, Bishop M. Proc. Natl. Acad. Sci. 1998;95:4673–4677. doi: 10.1073/pnas.95.8.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller WE, Maudsley S, Ahn S, Khan KD, Luttrell LM, Lefkowitz RJ. J. Biol. Chem. 2000;275:11312–11319. doi: 10.1074/jbc.275.15.11312. [DOI] [PubMed] [Google Scholar]