

Abstract
Base-line urinary potassium secretion in the distal nephron is mediated by small conductance rat outer medullary K (ROMK)-like channels. We used the patch clamp technique applied to split-open cortical collecting ducts (CCDs) isolated from rats fed a normal potassium (NK) or low potassium (LK) diet to test the hypothesis that AngII directly inhibits ROMK channel activity. We found that AngII inhibited ROMK channel activity in LK but not NK rats in a dose-dependent manner. The AngII-induced reduction in channel activity was mediated by AT1 receptor (AT1R) binding, because pretreatment of CCDs with losartan but not PD123319 AT1 and AT2 receptor antagonists, respectively, blocked the response. Pretreatment of CCDs with U73122 and calphostin C, inhibitors of phospholipase C (PLC) and protein kinase C (PKC), respectively, abolished the AngII-induced decrease in ROMK channel activity, confirming a role of the PLC-PKC pathway in this response. Studies by others suggest that AngII stimulates an Src family protein-tyrosine kinase (PTK) via PKC-NADPH oxidase. PTK has been shown to regulate the ROMK channel. Inhibition of NADPH oxidase with diphenyliodonium abolished the inhibitory effect of AngII or the PKC activator phorbol 12-myristate 13-acetate on ROMK channels. Suppression of PTK by herbimycin A significantly attenuated the inhibitory effect of AngII on ROMK channel activity. We conclude that AngII inhibits ROMK channel activity through PKC-, NADPH oxidase-, and PTK-dependent pathways under conditions of dietary potassium restriction.
It is well established that urinary potassium excretion falls in the face of dietary potassium restriction (1, 2). This renal response is achieved, at least in part, by a reduction in the potassium secretory capacity of the distal nephron (3, 4). Two types of apical K channels have been identified in the distal nephron and specifically the cortical collecting duct (CCD),4 the ROMK (Kir1.1)-like small conductance K (ROMK) channel (5), and the high conductance maxi-K channel (6). The prevalence of the ROMK channel in the CCD and its high open probability at the resting membrane potential (7) has led to the belief that this channel mediates potassium secretion under base-line conditions. In contrast, the maxi-K channel, activated by membrane stretch, depolarization, and increases in intracellular Ca2+concentration (8) has been proposed to mediate flow-stimulated potassium secretion (9, 10). Recent evidence indicates that protein-tyrosine kinase (PTK) (11) and protein kinase C (PKC) (12) mediate the reduction in the number of conducting ROMK channels resident on the apical membrane of the CCD in response to dietary potassium restriction.
The intratubular renin-angiotensin system plays a major role in the control of salt and water transport within the kidney. Dietary potassium restriction stimulates the release of renal renin and AngII (13, 14). AngII exerts its physiological effects by binding to AT1 and AT2 receptors. AT1 receptor (AT1R) binding activates a number of signaling molecules, including PKC and PTK (cSrc) (15), whereas the precise nature of the signaling pathways activated by the AT2 receptor are still poorly understood (16). Although the effect of AngII on the ROMK channel is unknown, luminal perfusion of distal tubules with AngII stimulates sodium absorption yet suppresses potassium secretion (17), suggesting that AngII may inhibit apical K channels in the CCD.
The purpose of the present study was to directly test whether AngII regulates ROMK channel activity in the CCD. Using an electrophysiological approach (patch clamp technique), we have shown that AngII significantly inhibits ROMK channel activity in tubules isolated from rats fed a low potassium (LK) diet and that this effect is mediated by the AT1R. Furthermore, our studies identify a novel pathway involving NADPH oxidase by which AngII regulates the ROMK channel and thus urinary potassium excretion during dietary potassium restriction.
EXPERIMENTAL PROCEDURES
Preparation of CCDs
Pathogen-free Sprague-Dawley rats of either sex (5–6 weeks old; Taconic Farms, Inc., Germantown, NY) were fed either a normal potassium (NK; standard rat chow, Laboratory Diet, 1.1% w/w potassium content) or a low potassium (LK; Harland, 0.001–0.003% w/w potassium content) diet for 4–8 days. Animals were allowed free access to tap water. In one series of experiments, either losartan (10 mg/kg/day) or vehicle (ethanol in 4:1,000 final dilutions) was added to the drinking water. Animal use protocols were reviewed and approved by the Institutional Animal Care and Use Committee of New York Medical College and Mount Sinai School of Medicine.
Rats were killed by cervical dislocation. Kidneys were immediately removed, and several thin coronal slices were cut with a razor blade and placed in ice-cold Ringer’s solution for micro-dissection of CCDs, as previously described (18). The Ringer’s solution contained (in mM) 140 NaCl, 5 KCl, 1.8 CaCl2, 1.8 MgCl2, and 10 HEPES (pH 7.4). To immobilize tubules for patch clamp or immunolabeling, CCDs were affixed to 5 × 5 mm coverglasses coated with poly-D-lysine.
Patch Clamp Recording
The basic patch clamp methods have been described previously (18). In brief, each isolated CCD affixed to a coverglass was transferred to a chamber (1000 μl total volume) mounted on the stage of a Nikon inverted microscope. The chamber was filled with Ringer’s solution. The CCD was cut open with a sharpened micropipette to expose the apical membrane. Patch clamp pipettes were pulled using a vertical pipette puller (model PP-83; Narishige Scientific Instrument Laboratory, Tokyo, Japan) in two stages with borosilicate glass capillaries (Dagan Corp., Minneapolis, MN). The pipettes were fire-polished and had resistances of 2–5 MΩ when filled with a solution containing (in mM) 140 KCl, 1.8 MgCl2, and 5 HEPES (pH 7.4). Experiments were performed at room temperature. Currents originating in each cell-attached patch were recorded using an Axon200B patch clamp amplifier (Axon instruments Inc.). The currents were low pass-filtered at 1 kHz. Data were digitized using an Axon interface (Digidata 1200) and stored on the hard drive of an equipped computer.
Immunohistochemical Staining and Confocal Microscopy
Isolated CCDs affixed to a coverglass, as described above, were fixed with 2.5% paraformaldehyde prepared in 1× phosphate-buffered saline (PBS) plus 0.05% picric acid (pH 7.4) for 2–3 min and then rinsed with base buffer (1×PBS containing 1% bovine serum albumin and 0.1% glycine). Thereafter, tubules were permeabilized with permeabilization solution (0.1% Triton X-100 dissolved in the base buffer) for 1 h at room temperature and then incubated with rabbit polyclonal IgG anti-AT1R antibody (1:200 dilution in permeabilization solution) overnight at 4 °C. Tubules were rinsed three times with the base buffer and then incubated for 1 h with a goat anti-rabbit IgG secondary antibody conjugated to Alexa-488 at a 1:1000 dilution in 1× PBS containing 1% bovine serum albumin at room temperature. After rinsing with three volumes of 1× PBS, the tubules were co-labeled with the principal cell marker, Dolichos biflorus agglutinin (DBA, conjugated to rhodamine; 1:200 dilution in 1× PBS) by incubation for 30 min at room temperature. Following a final rinse, a single drop of antifade reagent was deposited on each tubule, and the coverglass was sealed onto the slide with clear nail polish. Fluorescence imaging was performed with an inverted Zeiss laser scanning microscope (LSM; 510-META) using a ×40 oil immersion objective. All images were saved in a tag information file format (TIFF).
Relative Quantitation of p22phox and p47phox mRNA in Single CCDs
CCDs were microdissected in 1× PBS containing 10 mM vanadyl ribonucleoside complexes for no longer than 1 h after the death of the animal. Approximately 10 mm total length of CCDs was pooled for each sample. RNA was extracted from the microdissected CCDs and cDNA synthesized using oligo(dT) primers as described previously (19).
Both p22phox and p47phox transcript expression were analyzed by semiquantitative real time PCR using the TaqMan™ technique and an ABI Prism 7900HT sequence detection system (Applied Biosystems, Foster City, CA). Both primers and probes for p22phox and p47phox were synthesized by Applied Biosystems (Table 1) based on the sequences reported for GenBank™ accession numbers U18729 and AY029167, respectively. The 5′ end of each probe was labeled with 6-FAM dye, and the 3′ end was labeled with carboxytetramethylrhodamine. GAPDH was chosen as the internal positive reference control; TaqMan™ rodent GAPDH primers and probes were obtained from Applied Biosystems.
TABLE 1.
Primers and probes used for real-time PCR
| p22phox | |
| Forward | 5′-ACCTGACCGCTGTGGTGAA-3′ |
| Reverse | 5′-GTGGAGGACAGCCCGGA-3′ |
| Probe | 5′-6FAM-CTGTTCGGGCCCCTCACCAGAAATTACT-TAMRA-3′ |
| P47phox | |
| Forward | 5′-ACGCTCACCGAGTACTTCAACA-3′ |
| Reverse | 5′-TCATCGGGCCGCACTTT-3′ |
| Probe | 5′-6FAM-CCCGCTGCCCACACCTCTTGAACT-TAMRA-3′ |
Real-time PCR was performed as follows. To 2 μl of cDNA sample was added 8 μl of a mixture containing 0.05 μl of Platinum TaqDNA® polymerase, 0.1 unit AmpErase uracil N-glycosylase (UNG), dNTPs with dUTP, 0.2 μl of passive reference ROX dye 0.2 μl (20 pM) of forward and reverse primers, and TaqMan™ probe, finally adding nuclease-free water to a total volume of 10 μl. Each plate was then covered with optical adhesive film, and after the initial steps of 50 °C for 2 min and 95 °C for 10 min, 40 cycles of 95 °C for 15 s (melt) and 60 °C for 1 min (anneal/extend) were performed.
Chemicals
Herbimycin A (HA), U73122, and phorbol 12-myristate 13-acetate (PMA) were purchased from Biomol Corp. (Plymouth Meeting, PA). Diphenyliodonium (IDP) was obtained from Calbiochem (La Jolla, CA). Losartan was a generous gift from Dr. Charles T. Stier (New York Medical College). AngII, PD123319, calphostin C, and other chemicals, including vanadyl ribonucleoside complexes, were purchased from Sigma. The anti-AT1R antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Alexa-488-conjugated goat anti-rabbit IgG secondary antibody was purchased from Invitrogen and rhodamine-conjugated DBA from Vector Laboratories (Burlingame, CA). The antifade reagent (Prolong Gold) was obtained from Molecular Probes (Eugene, Oregon). Platinum TaqDNA® polymerase and passive reference ROX dye were purchased from Invitrogen and AmpErase UNG® and dNTPs with dUTP from Applied Biosystems (Foster City, CA).
Data Analysis and Statistics
Data are all expressed as mean ± S.E.; n equals the number of cell-attached patches or animals for real-time PCR. Patch clamp data were analyzed by using the pCLAMP software system version 6.04 (Axon Instrument, Burlingame, CA). Channel activity was defined as NPo, the product of open probability (Po) and channel number (N), which was calculated from data samples of 60-s duration in the steady state as follows: NPo = Σ(t1 + 2t2 +···iti), where ti was the fractional open time spent at each of the observed current levels. Statistical analysis was performed using SigmaPlot (Systat Software Inc., Chicago, IL). A paired or unpaired Student’s t test was performed to determine statistical significance, as appropriate.
Real-time PCR data were analyzed using SDS version 2.1 software for calculation of the threshold values (CT) of p22phox and p47phox. Using the 2−ΔΔCT method (20), the data are represented as the fold change in mRNA expression, normalized to GAPDH, relative to the control. The standard deviation of differences was calculated from the standard deviations (S.D.) of the p22phox, p47phox, and GAPDH values using the formula s = √S12 + S22. Statistical significance was taken as p < 0.05.
RESULTS
We used the patch clamp technique to study the effect of AngII on the ROMK channel in principal cells of the CCD. AngII significantly inhibited ROMK channel activity (NPo) in LK-fed animals in a dose-dependent manner (Figs. 1 and 2). AngII in concentrations of 1, 10, and 100 nM reduced NPo by 45 ± 16% (n = 8, p < 0.05), 58 ± 20% (n = 6, p < 0.05) and 70 ± 15% (n = 10, p < 0.01), respectively. This inhibitory effect on channel activity was partially reversible when AngII was washed out from the bathing solution (Fig. 1). However, AngII was without a consistent effect in CCDs isolated from rats fed a NK diet; only 2 of 7 patches responded to 100 nM AngII with a reduction in ROMK channel activity.
FIGURE 1. Representative traces of the inhibitory effect of AngII (100 nM) on the apical ROMK channel in a CCD from LK-fed rats.
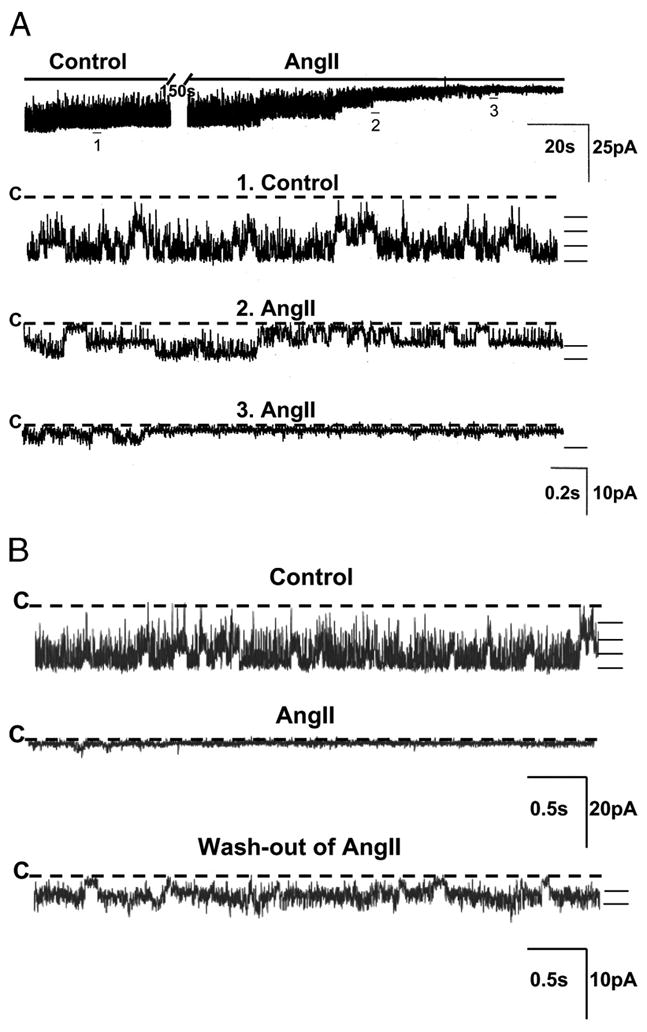
A, the top trace shows the time course of the experiment. Three parts of the trace are expanded (traces 1–3) to show detailed channel activity at faster time resolution. The channel closed state (“C”) is indicated by a dashed line. Current levels are indicated by the short bars on the right of each trace. The experiment was performed in a cell-attached patch at a holding potential of 0 mV. B, channel recording showing that the inhibitory effect of AngII is partially reversible after washout of AngII from bathing solution.
FIGURE 2. Dose-response curves for the AngII-induced inhibition of the ROMK channel in CCDs.

AngII led to a reduction in NPo of ROMK channels in CCDs from LK (open circles)- but not from NK (closed circles)-fed rats at concentrations ≥ 1 nM. *, p < 0.05; **, p < 0.01 versus 0.1 nM AngII. #, p < 0.05 versus 100 nM AngII in NK-fed rats only.
To identify which subtype of AngII receptor (AT1 or AT2) mediated the inhibitory effect of AngII on the ROMK channel, CCDs from LK rats were pretreated with losartan (20 μM), an AT1R antagonist, before exposure to AngII. Losartan completely abolished the inhibitory effect of AngII on the ROMK channel (n = 9, p = NS (not significant)) (Fig. 3, A and B). In CCDs pretreated with the AT2 receptor antagonist PD123319 (10 μM), AngII reduced ROMK channel activity by 75 ± 17% (n = 6, p < 0.05), a degree of inhibition identical to that observed in CCDs not treated with any receptor antagonist (Fig. 3B).
FIGURE 3. Effect of AT1 and AT2 receptor antagonists on the AngII-induced inhibition of ROMK channels in CCDs from LK-fed rats.
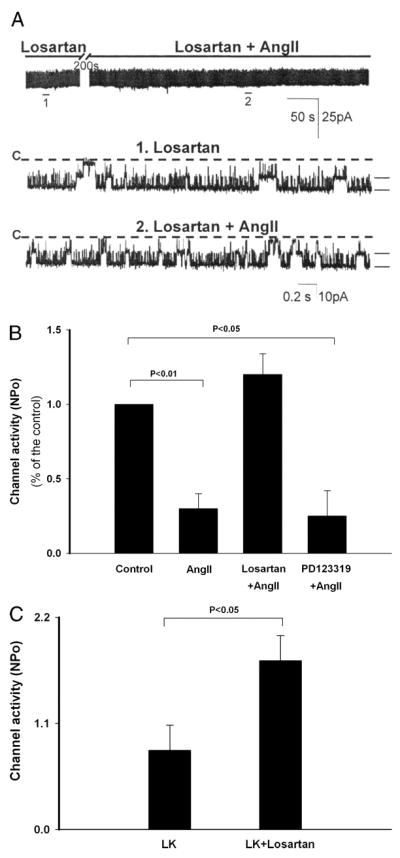
A, representative channel recording obtained in a principal cell-attached patch at a holding potential of 0 mV. The inhibitory effect of AngII was completely blocked by pretreatment of the CCD with losartan (20 μM), an AT1 receptor antagonist. The top trace demonstrates the time course of the experiment; the recording is expanded below (traces 1 and 2) to show fast time resolution. The channel-closed state (“C”) is indicated by a dashed line. Current levels are indicated by the short bars on the right of each trace. B, bar graph summarizing the effects of antagonists of the AT1 receptor (losartan) and AT2 receptor (PD123319) on the AngII-induced reduction of ROMK channel activity. C, bar graph summarizing the effect of oral losartan administration (10 mg/kg/day added to the drinking water) in LK-fed rats. ROMK channel activity (NPo) in losartan-treated rats significantly exceeded that measured in LK-fed animals provided vehicle alone in drinking water.
To confirm the presence of the AT1R and determine its localization in the CCD of LK rats, we performed immunofluorescence labeling of isolated nephron segments using antibodies directed against the AT1R, visualized with an Alexa-488-conjugated secondary antibody. Antibody binding was present along the apical membranes of cells that expressed DBA, a principal cell-specific marker. We observed abundant apical expression of immunodetectable AT1R in tubules isolated from LK-fed rats (Fig. 4). Immunolabeling of CCDs isolated from NK rats (n = 20 tubules from four animals) using a protocol identical to that in CCDs from LK rats showed inconsistent and weak apical expression (data not shown).
FIGURE 4. Apical membrane expression of AT1 receptors in rat CCD.

CCD from LK rat was co-labeled with an anti-AT1 receptor antibody and visualized with an Alexa-488-conjugated secondary antibody (green) and rhodamine-conjugated DBA (red), a marker of distal nephron principal cells. The CCD exhibits abundant apical membrane expression of AT1 receptors.
To test the in vivo relevance of the in vitro observation of AngII-induced inhibition of the ROMK channel activity, the effect of oral administration of losartan (10 mg/kg/day) to LK-fed rats for 6–8 days of treatment was examined. We found that ROMK channel activity (NPo) in CCDs from losartan-treated animals (1.75 ± 0.26, n = 16) significantly exceeded that measured in vehicle-treated animals (0.85 ± 0.26, n = 12, p < +0.05) (Fig. 3C). These results support the notion that AngII plays a physiologically relevant role in regulating channel activity in vivo.
An early event following AngII binding to the AT1R is activation of phospholipase C (PLC) leading to stimulation of PKC (15). Activation of PKC has previously been reported to inhibit the ROMK channel (21). In fact, we confirmed that PMA (10 μM), an agonist of PKC, inhibited ROMK channel activity by 51 ± 12% (n = 7, p < 0.01) (Fig. 5).
FIGURE 5.

The inhibitory effect of AngII on ROMK channel activity in CCDs is mediated by activation of PLC-PKC. In CCDs from LK-fed rats, PMA (10 μM), an activator of PKC, inhibited ROMK channel activity. Application of inhibitors of PLC (U73122; 10 μM) or PKC (calphostin C; 100 nM) alone had no effect on ROMK channel activity but prevented the AngII-induced reduction in NPo. In CCDs pretreated with U73122 and exposed to AngII, subsequent PMA application inhibited channel activity due to direct stimulation of PKC.
To test whether the AngII-induced inhibition of ROMK channel activity was mediated by activation of the PLC-PKC pathway, we first examined the effect of AngII on ROMK channels in CCDs pretreated with U73122 (10 μM), an inhibitor of PLCβ. Application of U73122 alone had no effect on ROMK channel activity. However, in U73122-pretreated tubules, AngII failed to inhibit channel activity (n = 9, p = NS) (Fig. 5). We next sought to confirm the role of PKC in the AngII response. The addition of PMA to CCDs pretreated, as above, with U73122 and AngII led to the inhibition of ROMK channel activity by 69 ± 16% (n = 6, p < 0.05) (Fig. 5). To further explore the role of PKC in the AngII response, CCDs were treated with calphostin C (100 nM), an inhibitor of PKC, which alone had no effect on ROMK channel activity (Fig. 5), as we have previously reported (22). AngII failed to reduce the NPo in calphostin C-treated CCDs (n = 9, p = NS) (Fig. 5). These data strongly suggest a role of PLC and PKC in the AngII-induced inhibition of ROMK channel activity.
We have previously reported that the reduction in number of apical ROMK channels in LK animals is due to an increase in PTK (cSrc) activity, which enhances endocytosis of the channel (23). We confirmed that inhibition of cSrc with HA (1 μM) increases NPo of the channel (Fig. 6) (24). cSrc is stimulated by AngII in vascular smooth muscle cells (25). To examine whether the AngII-mediated effects on the ROMK channel involves PTK signaling, CCDs were pretreated with HA to inhibit cSrc, and the effect of AngII on channel activity was examined. In HA-treated CCDs, AngII significantly reduced ROMK channel activity (n = 5, p < 0.05 versus HA-treated or untreated control CCDs). The AngII-induced PTK-independent reduction of ROMK channel activity presumably reflects other PKC-associated effects on the channel, such as that regulated by phosphatidylinositol 4,5-bisphosphate (PIP2) (26). Of note was our observation that application of PMA to HA-treated CCDs failed to inhibit the ROMK channel (data not shown), suggesting that PTK is required for activation of PKC (27, 28).
FIGURE 6. The inhibitory effect of AngII on ROMK channel activity is mediated, in part, by activation of PTK.

HA (1 μM), an inhibitor of PTK, increased ROMK channel activity in CCDs from LK-fed rats. In CCDs pretreated with HA, AngII led to a partial reduction in channel activity. In this figure, mean NPo is presented relative to that measured in untreated control CCDs.
cSrc has been reported to be a downstream signal of reactive oxygen species (ROS, including , H2O2) (29). and related products are increased in CCDs during dietary potassium restriction (30). Decreasing by tempol, a scavenger of anions, reduces expression of cSrc in CCDs, which increases ROMK channel activity and potassium excretion in LK rats (30). The immunodetection of components of NADPH oxidase in the CCD suggests that NADPH oxidase may be a source of intracellular ROS in this nephron segment (31). AngII stimulates production by activating NADPH oxidase via AT1R in kidneys (32). If AngII activates this signaling pathway in the CCD, blockade of NADPH oxidase should abolish the effect of AngII on the ROMK channel. We used IDP (10 μM), a specific inhibitor of NADPH oxidase, to explore the role of this pathway in the AngII-induced reduction of ROMK channel activity. The inhibitory effect of AngII on the channel was absent in CCDs pretreated with IDP (n = 7, p = NS) (Fig. 7). To confirm the role of NADPH oxidase in the AngII-mediated effect, we pre-treated CCDs with apocynin (100 μM), which prevents the transfer of the soluble cytosolic subunits p47phox and p67phox of NADPH oxidase to the cell membrane and blocks the function of the enzyme (33). We found that apocynin also blocked the effect of AngII on the ROMK channel (Fig. 7).
FIGURE 7. The inhibitory effect of AngII on ROMK channel activity is mediated, in part, by NADPH oxidase.

A, pretreatment of CCDs from LK rats with one of two NADPH oxidase inhibitors, IDP (10 μM) or apocynin (100 nM), blocked the effect of AngII on the ROMK channel. Neither one of these inhibitors had a significant effect on base-line channel activity. CCDs pretreated with IDP failed to respond to PMA with an inhibition of ROMK channel activity, suggesting that AngII inhibits the channel via the activation of the PKC-NADPH oxidase pathway. B, relative expression of mRNA encoding p22phox and p47phox in CCDs from NK- and LK-fed rats. Dietary potassium restriction for 4 – 6 days increased expression of p47phox (but not p22phox) when compared with NK-fed rats.
To confirm that NADPH oxidase plays a role in the adaptation to a LK diet, we compared mRNA levels of both p22phox and p47phox in single CCDs isolated from LK (n = 3)- and NK (n = 3)-fed rats. We found that the p47phox mRNA transcript abundance in LK rats was 2.4-fold greater than that from NK rats (p < 0.05) (Fig. 7).
PKC stimulates NADPH oxidase in neutrophils (34). To test whether PKC stimulates this oxidase in the CCD and regulates ROMK channel activity, tubules were pretreated with IDP and then exposed to PMA. The observation that PMA failed to inhibit the channel activity in IDP-treated CCDs (Fig. 7) provides support for the role of both PKC and NADPH oxidase in the AngII-mediated reduction in channel activity in LK rats.
DISCUSSION
The results of the present in vitro and in vivo studies demonstrate that AngII plays an important role in the regulation of the ROMK channel in CCDs isolated from potassium-restricted animals. In this situation, AngII inhibits channel activity via a signaling cascade that is initiated by AT1R binding and involves activation of PLC-PKC, PTK, and NADPH oxidase (Fig. 8). We propose that this represents a novel pathway in regulating potassium secretion in the kidney.
FIGURE 8.

Proposed signaling pathway by which AngII inhibits the activity of ROMK channels in the CCD from LK-fed rats.
Dietary potassium intake is an important regulator of the ROMK channel, although its mechanism has not been fully clarified. Renal potassium secretion responds promptly to changes in dietary potassium intake (35). The reduction in potassium secretion in the distal nephron induced by dietary potassium restriction is achieved by a reduction in the number of ROMK channels resident in the apical membrane, a response mediated, at least in part, by PTK- and PKC-dependent pathways (11, 22, 26, 36). Potassium depletion increases expression of Src family PTK, including cSrc, which phosphorylates ROMK channels, thereby enhancing their endocytosis and reducing potassium secretion (11, 36). Dietary potassium restriction is also associated with an increase in the expression of PKCε (11). Stimulation of PKC by PMA inhibits ROMK channel activity (21), presumably also by mediating the internalization of the channel (12) as well as by reducing the membrane content of PIP2, a membrane phospholipid required to maintain the open state of the channel (26). The present study provides evidence to suggest that PTK and PKC co-operate in the AngII-induced reduction in ROMK channel activity.
All components of the renin-angiotensin system are present within kidney. AngII is locally produced in the proximal tubule (37), and concentrations within the low nanomolar range have been reported in tubular fluid sampled from this site (38, 39). AngII concentrations in distal tubular fluid remain unknown. However, the detection of renin and angiotensin-converting enzyme within the distal nephron (40, 41) and urinary AngII concentrations as high as 300 fmol/ml (14) suggest that the AngII concentration in the distal tubular fluid may influence distal tubule transport. Wang and Giebisch (17) found that AngII concentrations of as low as 10 fmol/ml (10−11 M) stimulated sodium transport and suppressed potassium secretion in late distal tubules/CCDs microperfused in vivo.
AngII exerts its physiologic effects by binding to AT1 and AT2 receptors. AT2 receptors are rarely detected in the adult but are expressed in high abundance in fetal tissues (42). Immunodetectable AT1R has been observed in the brush border and basolateral membrane of proximal tubule, thick ascending limb, and distal nephron segments, including the collecting duct (43). Indeed, our immunofluorescence studies (Fig. 4) demonstrate abundant apical expression of this receptor in CCDs from LK-fed rats. This luminal localization of AT1R provides compelling support for an important paracrine/autocrine function of luminal AngII. Relevant to the focus of the current study is the observation that dietary potassium restriction increases AT1R mRNA in rat renal cortex and proximal tubule (44). In addition, mRNA encoding AT1a (the main subtype of the AT1R in rodents) is more highly expressed in collecting ducts than in proximal tubules or thick ascending limbs (45).
AngII binding to the AT1R induces multiple signaling cascades, including those associated with phospholipids, protein-tyrosine phosphorylation, and activation of non-receptor tyrosine kinases, such as cSrc (15). An early event following AT1 receptor activation is PLC-dependent hydrolysis of PIP2 (15). This is associated with rapid production of inositol 1,4,5-trisphosphate and a more sustained release of diacylglycerol, which are involved in Ca2+ mobilization from internal stores and stimulation of PKC, respectively. Dietary potassium restriction significantly increases the expression of PKCε in renal cortex and outer medulla (22). Activation of PKC by PMA inhibits ROMK channel activity in CCD (Fig. 5) (21). In the present study, our observations that (i) U73122 and calphostin C abolished the AngII-induced inhibition of ROMK channel activity (Fig. 5) and (ii) PMA inhibited ROMK channel activity in U73122-pretreated CCDs (Fig. 5) confirm a role for PLC and PKC, respectively, in the response.
In addition to the effects of PKC on phosphorylation of the ROMK channel and reduction of membrane PIP2 content, as discussed above, PKC also activates NADPH oxidase leading to generation of the ROS family of intracellular second messengers (46). The kidney contains all components of NADPH oxidase (31). This multimolecular enzyme complex is composed of a membrane-associated 22-kDa α-subunit (p22phox) and a 91-kDa catalytic β-subunit (gp91phox/Nox2) with cytosolic components, including p47phox, p67phox, and p40phox and the small GTPase Rac (47). Upon activation (e.g. by PKC), the cytosolic components translocate to the membrane-associated complex composed of Nox2 (or its homologues)/p22phox to facilitate electron transfer from NADPH to FAD and produce (47). Nox4, a homologue of Nox2, is present in kidneys (48). Distal tubules, including the CCD, constitutively express p22phox, p47phox, and p67phox, suggesting that NAPDH oxidase may generate ROS at these sites (31).
Chronic (1 week) AngII infusion into rats stimulates oxidative stress via AT1R binding (32). This observation, along with the finding that LK diet increases in renal cortex from LK-fed rats and that ROS stimulates PTK activity (30), suggested to us that the reduction in ROMK channel activity in LK animals may be due to generation of ROS. Our finding that dietary potassium restriction increases p47phox mRNA abundance (Fig. 7B) and that inhibitors of NADPH oxidase block the AngII-induced reduction in ROMK channel activity (Fig. 7A) supports this notion. Furthermore, PKC activation failed to inhibit the channel in IDP-treated CCDs, suggesting that NADPH oxidase is a target of PKC and that AngII inhibits the ROMK channel via a PKC-NADPH oxidase pathway.
The finding that blockade of Src family PTK with HA did not completely abolish the inhibitory effect of AngII on ROMK channels (Fig. 6) implies that the cSrc may be only one of multiple effectors in the AngII response (Fig. 8). It is possible that an AngII-induced activation of PKC may also inhibit ROMK channels by a mechanism not directly associated with PTK. In this regard, it has been shown that a PKC-induced reduction in membrane PIP2 content may play a role in mediating the PKC-mediated inhibition of ROMK channels (26). Also it is possible that PTK is required for activation of PKC. In support of this argument is the finding that some PKCs (PKCδ and PKCε) are phosphorylated by cSrc (28, 49). The identity of the PKC isozymes involved in the regulation of the ROMK channel is unknown. The complexity of cellular PKC interaction in the regulation of the ROMK channel activity remains to be further explored.
Potassium secretion occurs in the late distal and connecting tubule and CCD by passive diffusion from epithelial cells into the urinary space down a favorable electrochemical gradient across apical potassium-secretory channels. Net potassium secretion in the distal nephron thus depends not only on the apical permeability to this ion, but also on the electrochemical gradient favoring its passive diffusion across the apical membrane. AngII directly stimulates epithelial sodium channel (41) and thus is expected to depolarize the apical membrane, thereby enhancing the electrochemical gradient and facilitating luminal potassium secretion. Our findings indicate that the ROMK channel is an important target of AngII regulation and provides an explanation for the clinical finding that losartan can increase urinary potassium excretion (50). However, the effect of AngII on net potassium secretion in the distal nephron and urinary potassium excretion remains to be directly measured in LK-fed animals.
Under general conditions, potassium secretion in the CCD is regulated to match the physiological needs of the body. Two of the most important physiological determinants of distal potassium secretion are the serum aldosterone concentration and the delivery of sodium to the distal nephron. Our identification of an effect of AngII on the ROMK channel in the CCD suggests a novel pathway to effectively fine-tune potassium secretion, especially under conditions of dietary potassium restriction, a state associated with low levels of circulating aldosterone and distal sodium delivery. In summary, the regulation of the ROMK channel by AngII provides yet another example of the complex and diverse pathways regulating renal potassium secretion.
Acknowledgments
The technical support of Dr. PokMan Chan is greatly appreciated. We acknowledge the support of the Mount Sinai School of Medicine Microscopy Shared Resource Facility, supported by NCI-National Institutes of Health Shared Resources Grant 5R24 CA095823-04, National Science Foundation Major Research Instrumentation Grant DBI-9724504, and National Institutes of Health Shared Instrumentation Grant 1 S10 RRO 9145-01.
Footnotes
The abbreviations used are: CCD, cortical collecting duct; ROMK, rat outer medullary K (channel); NK, normal potassium; LK, low potassium; PLC, phospholipase C; PKC, protein kinase C; PTK, protein-tyrosine kinase; PMA, phorbol 12-myristate 13-acetate; HA, herbimycin A; AT1R, AT1 receptor; PBS, phosphate-buffered saline; IDP, diphenyliodonium; DBA, D. biflorus agglutinin; PIP2, phosphatidylinositol 4,5-bisphosphate; ROS, reactive oxygen species.
This study was supported by American Heart Association Scientist Development Grant 0530092N (to Y. W.).
References
- 1.Linas SL, Peterson LN, Anderson RJ, Aisenbrey GA, Simon FR, Berl T. Kidney Int. 1979;15:601–611. doi: 10.1038/ki.1979.79. [DOI] [PubMed] [Google Scholar]
- 2.Okusa MD, Unwin RJ, Velazquez H, Giebisch G, Wright FS. Am J Physiol. 1992;262:F488–F493. doi: 10.1152/ajprenal.1992.262.3.F488. [DOI] [PubMed] [Google Scholar]
- 3.Giebisch G. Am J Physiol. 1998;274:F817–F833. doi: 10.1152/ajprenal.1998.274.5.F817. [DOI] [PubMed] [Google Scholar]
- 4.Najjar F, Zhou H, Morimoto T, Bruns JB, Li HS, Liu W, Kleyman TR, Satlin LM. Am J Physiol. 2005;289:F922–F932. doi: 10.1152/ajprenal.00057.2005. [DOI] [PubMed] [Google Scholar]
- 5.Boim MA, Ho K, Shuck ME, Bienkowski MJ, Block JH, Slightom JL, Yang Y, Brenner BM, Hebert SC. Am J Physiol. 1995;268:F1132–F1140. doi: 10.1152/ajprenal.1995.268.6.F1132. [DOI] [PubMed] [Google Scholar]
- 6.Frindt G, Palmer LG. Am J Physiol. 1987;252:F458–F467. doi: 10.1152/ajprenal.1987.252.3.F458. [DOI] [PubMed] [Google Scholar]
- 7.Frindt G, Palmer LG. Am J Physiol. 1989;256:F143–F151. doi: 10.1152/ajprenal.1989.256.1.F143. [DOI] [PubMed] [Google Scholar]
- 8.Pacha J, Frindt G, Sackin H, Palmer LG. Am J Physiol. 1991;261:F696–F705. doi: 10.1152/ajprenal.1991.261.4.F696. [DOI] [PubMed] [Google Scholar]
- 9.Woda CB, Bragin A, Kleyman TR, Satlin LM. Am J Physiol. 2001;280:F786–F793. doi: 10.1152/ajprenal.2001.280.5.F786. [DOI] [PubMed] [Google Scholar]
- 10.Bailey MA, Cantone A, Yan Q, MacGregor GG, Leng Q, Amorim JB, Wang T, Hebert SC, Giebisch G, Malnic G. Kidney Int. 2006;70:51–59. doi: 10.1038/sj.ki.5000388. [DOI] [PubMed] [Google Scholar]
- 11.Wang W, Lerea KM, Chan M, Giebisch G. Am J Physiol. 2000;278:F165–F171. doi: 10.1152/ajprenal.2000.278.1.F165. [DOI] [PubMed] [Google Scholar]
- 12.Lin D, Sterling H, Lerea KM, Giebisch G, Wang WH. J Biol Chem. 2002;277:44278–44284. doi: 10.1074/jbc.M203702200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ray PE, Suga S, Liu XH, Huang X, Johnson RJ. Kidney Int. 2001;59:1850–1858. doi: 10.1046/j.1523-1755.2001.0590051850.x. [DOI] [PubMed] [Google Scholar]
- 14.Wang CT, Navar LG, Mitchell KD. J Hypertens. 2003;21:353–360. doi: 10.1097/00004872-200302000-00027. [DOI] [PubMed] [Google Scholar]
- 15.Touyz RM, Berry C. Braz J Med Biol Res. 2002;35:1001–1015. doi: 10.1590/s0100-879x2002000900001. [DOI] [PubMed] [Google Scholar]
- 16.Carey RM. Curr Opin Nephrol Hypertens. 2005;14:67–71. doi: 10.1097/00041552-200501000-00011. [DOI] [PubMed] [Google Scholar]
- 17.Wang T, Giebisch G. Am J Physiol. 1996;271:F143–F149. doi: 10.1152/ajprenal.1996.271.1.F143. [DOI] [PubMed] [Google Scholar]
- 18.Wei Y, Babilonia E, Sterling H, Jin Y, Wang WH. Am J Physiol. 2005;289:F1065–F1071. doi: 10.1152/ajprenal.00063.2005. [DOI] [PubMed] [Google Scholar]
- 19.Woda CB, Miyawaki N, Ramalakshmi S, Ramkumar M, Rojas R, Zavilowitz B, Kleyman TR, Satlin LM. Am J Physiol. 2003;285:F629–F639. doi: 10.1152/ajprenal.00191.2003. [DOI] [PubMed] [Google Scholar]
- 20.Livak KJ, Schmittgen TD. Methods (San Diego) 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 21.Wang WH, Giebisch G. Proc Natl Acad Sci U S A. 1991;88:9722–9725. doi: 10.1073/pnas.88.21.9722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sterling H, Lin DH, Chen YJ, Wei Y, Wang ZJ, Lai J, Wang WH. Am J Physiol. 2004;286:F1072–F1078. doi: 10.1152/ajprenal.00425.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moral Z, Dong K, Wei Y, Sterling H, Deng H, Ali S, Gu R, Huang XY, Hebert SC, Giebisch G, Wang WH. J Biol Chem. 2001;276:7156–7163. doi: 10.1074/jbc.M008671200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei Y, Wang WH. Am J Physiol. 2002;282:F680–F686. doi: 10.1152/ajprenal.00229.2001. [DOI] [PubMed] [Google Scholar]
- 25.Ishida M, Marrero MB, Schieffer B, Ishida T, Bernstein KE, Berk BC. Circ Res. 1995;77:1053–1059. doi: 10.1161/01.res.77.6.1053. [DOI] [PubMed] [Google Scholar]
- 26.Zeng WZ, Li XJ, Hilgemann DW, Huang CL. J Biol Chem. 2003;278:16852–16856. doi: 10.1074/jbc.M300619200. [DOI] [PubMed] [Google Scholar]
- 27.Braiman L, Sheffi-Friedman L, Bak A, Tennenbaum T, Sampson SR. Diabetes. 1999;48:1922–1929. doi: 10.2337/diabetes.48.10.1922. [DOI] [PubMed] [Google Scholar]
- 28.Rosenzweig T, Aga-Mizrachi S, Bak A, Sampson SR. Cell Signal. 2004;16:1299–1308. doi: 10.1016/j.cellsig.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 29.Frank GD, Eguchi S, Yamakawa T, Tanaka S, Inagami T, Motley ED. Endocrinology. 2000;141:3120–3126. doi: 10.1210/endo.141.9.7630. [DOI] [PubMed] [Google Scholar]
- 30.Babilonia E, Wei Y, Sterling H, Kaminski P, Wolin M, Wang WH. J Biol Chem. 2005;280:10790–10796. doi: 10.1074/jbc.M414610200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chabrashvili T, Tojo A, Onozato ML, Kitiyakara C, Quinn MT, Fujita T, Welch WJ, Wilcox CS. Hypertension. 2002;39:269–274. doi: 10.1161/hy0202.103264. [DOI] [PubMed] [Google Scholar]
- 32.Chabrashvili T, Kitiyakara C, Blau J, Karber A, Aslam S, Welch WJ, Wilcox CS. Am J Physiol. 2003;285:R117–R124. doi: 10.1152/ajpregu.00476.2002. [DOI] [PubMed] [Google Scholar]
- 33.Engels F, Renirie BF, Hart BA, Labadie RP, Nijkamp FP. FEBS Lett. 1992;305:254–256. doi: 10.1016/0014-5793(92)80680-f. [DOI] [PubMed] [Google Scholar]
- 34.Cox JA, Jeng AY, Sharkey NA, Blumberg PM, Tauber AI. J Clin Investig. 1985;76:1932–1938. doi: 10.1172/JCI112190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang W. Annu Rev Physiol. 2004;66:547–569. doi: 10.1146/annurev.physiol.66.032102.112025. [DOI] [PubMed] [Google Scholar]
- 36.Wei Y, Bloom P, Lin D, Gu R, Wang WH. Am J Physiol. 2001;281:F206–F212. doi: 10.1152/ajprenal.2001.281.2.F206. [DOI] [PubMed] [Google Scholar]
- 37.Navar LG, Nishiyama A. Curr Opin Nephrol Hypertens. 2004;13:107–115. doi: 10.1097/00041552-200401000-00015. [DOI] [PubMed] [Google Scholar]
- 38.Seikaly MG, Arant BS, Jr, Seney FD., Jr J Clin Investig. 1990;86:1352–1357. doi: 10.1172/JCI114846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Braam B, Mitchell KD, Fox J, Navar LG. Am J Physiol. 1993;264:F891–F898. doi: 10.1152/ajprenal.1993.264.5.F891. [DOI] [PubMed] [Google Scholar]
- 40.Rohrwasser A, Morgan T, Dillon HF, Zhao L, Callaway CW, Hillas E, Zhang S, Cheng T, Inagami T, Ward K, Terreros DA, Lalouel JM. Hypertension. 1999;34:1265–1274. doi: 10.1161/01.hyp.34.6.1265. [DOI] [PubMed] [Google Scholar]
- 41.Komlosi P, Fuson AL, Fintha A, Peti-Peterdi J, Rosivall L, Warnock DG, Bell PD. Hypertension. 2003;42:195–199. doi: 10.1161/01.HYP.0000081221.36703.01. [DOI] [PubMed] [Google Scholar]
- 42.Grady EF, Sechi LA, Griffin CA, Schambelan M, Kalinyak JE. J Clin Investig. 1991;88:921–933. doi: 10.1172/JCI115395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harrison-Bernard LM, Navar LG, Ho MM, Vinson GP, el-Dahr SS. Am J Physiol. 1997;273:F170–F177. doi: 10.1152/ajprenal.1997.273.1.F170. [DOI] [PubMed] [Google Scholar]
- 44.Burns KD, Smith IB. Nephron. 1998;78:73–81. doi: 10.1159/000044885. [DOI] [PubMed] [Google Scholar]
- 45.Imanishi K, Nonoguchi H, Nakayama Y, Machida K, Ikebe M, Tomita K. Hypertens Res. 2003;26:405–411. doi: 10.1291/hypres.26.405. [DOI] [PubMed] [Google Scholar]
- 46.Sauer H, Wartenberg M, Hescheler J. Cell Physiol Biochem. 2001;11:173–186. doi: 10.1159/000047804. [DOI] [PubMed] [Google Scholar]
- 47.Babior BM. Blood. 1999;93:1464–1476. [PubMed] [Google Scholar]
- 48.Geiszt M, Kopp JB, Varnai P, Leto TL. Proc Natl Acad Sci U S A. 2000;97:8010–8014. doi: 10.1073/pnas.130135897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sampson SR, Cooper DR. Mol Genet Metab. 2006;89:32–47. doi: 10.1016/j.ymgme.2006.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmidt A, Gruber U, Bohmig G, Koller E, Mayer G. Nephrol Dial Transplant. 2001;16:1034–1037. doi: 10.1093/ndt/16.5.1034. [DOI] [PubMed] [Google Scholar]