

Abstract
Increases in arginase activity have been reported in a variety of disease conditions characterized by vascular dysfunction. Arginase competes with NO synthase for their common substrate arginine, suggesting a cause and effect relationship. We tested this concept by experiments with streptozotocin diabetic rats and high glucose (HG)-treated bovine coronary endothelial cells (BCECs). Our studies showed that diabetes-induced impairment of vasorelaxation to acetylcholine was correlated with increases in reactive oxygen species and arginase activity and arginase I expression in aorta and liver. Treatment of diabetic rats with simvastatin (5 mg/kg per day, subcutaneously) or l-citrulline (50 mg/kg per day, orally) blunted these effects. Acute treatment of diabetic coronary arteries with arginase inhibitors also reversed the impaired vasodilation to acetylcholine. Treatment of BCECs with HG (25 mmol/L, 24 hours) also increased arginase activity. This effect was blocked by treatment with simvastatin (0.1 μmol/L), the Rho kinase inhibitor Y-27632 (10 μmol/L), or l-citrulline (1 mmol/L). Superoxide and active RhoA levels also were elevated in HG-treated BCECs. Furthermore, HG significantly diminished NO production in BCECs. Transfection of BCECs with arginase I small interfering RNA prevented the rise in arginase activity in HG-treated cells and normalized NO production, suggesting a role for arginase I in reduced NO production with HG. These results indicate that increased arginase activity in diabetes contributes to vascular endothelial dysfunction by decreasing l-arginine availability to NO synthase.
Keywords: arginine, coronary arteries, diabetes, endothelial nitric oxide synthase, oxidative stress, vascular endothelial function, vasodilation
Vascular dysfunction is a major cause of morbidity and mortality in diabetic patients.1 The pathological process is characterized by impaired endothelial cell production of the vasodilator and antiplatelet adhesion factor NO and/or decreased NO bioavailability. NO is a major regulator of vascular tone and integrity. In endothelial cells, NO is produced by activity of endothelial NO synthase (eNOS) on its substrate l-arginine.
Reduced availability of l-arginine to eNOS has been implicated in vascular dysfunction in diabetes and a variety of other disease conditions. Arginase, which metabolizes l-arginine to urea and ornithine, competes directly with NOS for l-arginine. Hence increases in arginase activity can decrease tissue and cellular arginine levels, reducing its availability to eNOS.2 This may lead to decreased NO production and increased production of superoxide by eNOS.3 Enhanced arginase activity has been implicated in a number of conditions characterized by vascular dysfunction, including diabetic erectile dysfunction, pulmonary hypertension, ischemia/reperfusion, atherosclerosis, and aging-associated endothelial dysfunction.4–9 During diabetes, impaired vascular function is closely associated with oxidative stress and vascular inflammation,10,11 both of which have been associated with increases in arginase activity and expression.4,12
Two types of mammalian arginase exist, arginase I and II. Each is encoded by a different gene.13,14 Arginase I, located in the cytoplasm, is expressed most abundantly in the liver as part of the urea cycle, whereas arginase II is a mitochondrial enzyme and is expressed primarily in kidney. Both arginase I and II have been found in different types of endothelial cells.15,16 The liver of diabetic rats has been found to show an increase in specific arginase activity as compared with nondiabetic rats.17,18 The increase in arginase activity may be explained by increased tissue manganese content.17,18 However, transcriptional upregulation of enzyme expression can also occur.19 It is well known that insulin represses expression of genes for urea synthesis pathways and that insulin signaling is impaired in both type 1 and type 2 diabetes. Thus, diabetes-induced increases in arginase activity could explain the decreased l-arginine levels reported in plasma from diabetic animals and patients,20,21 and in vascular tissue of streptozotocin (STZ) diabetic rats.20
Acute administration of supplemental l-arginine is reported to prevent or reverse endothelial dysfunction and restore endothelial-dependent vasodilation in diabetes, hypertension, and heart failure.22,23 However, a number of studies in animals and humans have found no benefit or worsening of adverse outcomes with prolonged administration of supplemental l-arginine.24,25 These negative outcomes may be related to the ability of l-arginine to activate and induce expression of arginase.26 We have shown previously that oral administration of l-arginine (200 mg/kg per day) to rabbits continuously for 3 days causes decreased NO production in response to acetylcholine (ACh), which was associated with increased arginase activity in both liver and aorta. In contrast, continuous treatment with l-citrulline for 3 days was beneficial in supporting NO production.27 l-Citrulline, the precursor of l-arginine and a byproduct in the formation of NO by NOS, is recycled back to l-arginine in many tissues and contributes to sustained l-arginine supply for NO production.28 l-Citrulline is also an allosteric inhibitor of arginase.29 Therefore, its use may suppress arginase activity.
In a previous study, we showed that STZ-induced diabetes in rats causes impaired coronary endothelial cell-dependent vasorelaxation.30 This dysfunction was prevented by treatment with simvastatin (5 mg/kg per day, subcutaneously) but not by l-arginine treatment (50 mg/kg per day, orally). Statins are known to prevent activation of small GTPases such as RhoA.31 Elevation of arginase activity has been shown to involve activation of the RhoA pathway in endothelial cells.32,33 Based on this and the evidence above that both diabetes and l-arginine treatment can increase arginase activity, we hypothesized that diabetes-induced vascular dysfunction involves increased levels of arginase activity and that simvastatin prevents vascular dysfunction because it reduces arginase activity. We tested this hypothesis and investigated the protective effects of statin treatment and l-citrulline supplementation in studies using STZ diabetic rats and high glucose (HG)-treated endothelial cells.
Materials and Methods
Male Sprague–Dawley rats (240 to 265 g) were rendered diabetic with STZ (50 mg/kg, intravenously). Control rats received injections of vehicle alone. Rats with blood levels >350 mg/dL were considered to be diabetic. Bovine coronary endothelial cells (BCECs) were from Cell Applications Inc.
An expanded Materials and Methods section is available in the online supplement at http://circres.ahajournals.org.
Results
Coronary Artery Relaxation
Our previous study showed that 4 weeks of STZ-induced diabetes in rats causes impaired coronary endothelial cell-dependent vasorelaxation that was prevented by treatment with simvastatin (5 mg/kg per day, subcutaneously) but not by l-arginine treatment (50 mg/kg per day, orally).30 In the present study, we extended the period of diabetes and treatment with simvastatin to 8 weeks and also determined the effects of l-citrulline (50 mg/kg per day, orally) instead of l-arginine treatment on coronary endothelial-dependent vasorelaxation. ACh produced a concentration-dependent vasorelaxation in coronary arteries from all groups, with a maximal relaxation (Emax) of 73±11% and an EC50 value of 94.0±1.4 nmol/L in arteries from control rats (Figure 1). However, coronary arteries from 8-week diabetic rats exhibited a markedly reduced Emax to ACh (32±3%) and a slight rightward shift in the concentration–response curve with an EC50 value of 110±1.5 nmol/L. Simvastatin treatment of diabetic rats significantly improved the Emax in relaxation to ACh to 60±3.6%, with an EC50 of 106±4 nmol/L. l-Citrulline supplementation for 8 weeks was equally effective because simvastatin and significantly improved the Emax in relaxation to ACh to 61±3.3% and reduced the EC50 to 64±1.8 nmol/L (Figure 1). The vasorelaxant response to ACh was mostly a result of NO production because treatment with NG-nitro-l-arginine methyl ester (l-NAME) (3 mmol/L) reduced the Emax in all groups to ≈22% (supplemental Table I).
Figure 1.
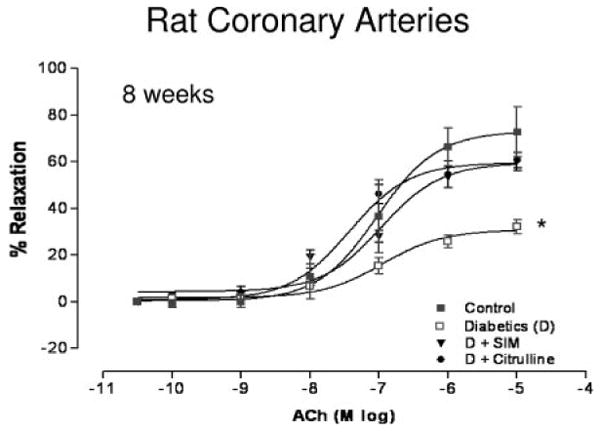
Concentration–response curves for the effect of ACh on coronary arteries from control, diabetic (D), simvastatin-treated diabetic (D+SIM), and l-citrulline–supplemented diabetic (D+Citrulline) rats at 8 weeks (n=8/group). Simvastatin treatment and l-citrulline supplementation improved the Emax to ACh compared with diabetic-untreated vessels. Values are expressed as means±SEM. *P<0.05 vs control, simvastatin-treated, and l-citrulline–supplemented diabetic rats.
Effect of Diabetes on Tissue Arginase Activity
To examine the role of arginase in this diabetes-induced vascular dysfunction, we determined the effect of diabetes and simvastatin, l-arginine, or l-citrulline treatment on tissue arginase activity. After 4 or 8 weeks of diabetes, both vascular and hepatic arginase activity were substantially increased as compared with the controls (Figure 2). These diabetes-induced increases in arginase activity were completely blocked by simvastatin treatment. Supplemental l-arginine treatment for 4 weeks did not prevent the diabetes-induced increases in tissue arginase activity (Figure 2A). However, supplemental l-citrulline treatment for 8 weeks completely inhibited the increases in both vascular and hepatic arginase activity (Figure 2B).
Figure 2.

Arginase activity in aortas and livers from control, diabetic, diabetic simvastatin–treated (Diab/Simv), diabetic l-arginine–supplemented (Diab/L-Arg), and diabetic l-citrulline–supplemented (Diab/L-Cit) rats at 4 (A) or 8 (B) weeks (n=8/group). Arginase activity is significantly increased in diabetic and l-arginine–supplemented diabetic tissues. Simvastatin and l-citrulline significantly decreased arginase activity in both aortas and livers from diabetic rats. Values are expressed as means±SEM. *P<0.05 vs control, diabetic simvastatin–treated, and diabetic l-arginine–supplemented rats.
Effect of Diabetes on Arginase Protein Levels
To determine whether increases in arginase activity in the diabetic rats are associated with any change in levels of arginase I and II protein levels, we analyzed aortae and livers of 8-week diabetic rats by Western blotting. This study showed significant increases in arginase I protein in both aorta (Figure 3A) and liver (Figure 3B) of diabetic rats. Treatment of diabetic rats with simvastatin prevented the rise in arginase I expression in both tissues. l-Citrulline treatment also blocked the increase in vascular arginase I but was slightly less effective in the liver. Arginase II protein was barely detectable in aortas of control and diabetic rats (Figure I in the online data supplement), suggesting that arginase I is the predominant isoform. Arginase II levels in liver were not examined.
Figure 3.
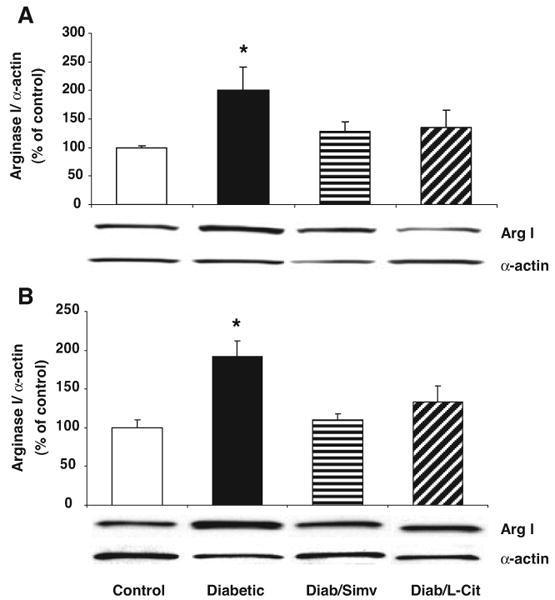
Western blot analysis of arginase I expression in aortas (A) and livers (B) of control, diabetic, diabetic simvastatin–treated (Diab/Simv), diabetic l-arginine–supplemented (Diab/L-Arg), and diabetic l-citrulline–supplemented (Diab/L-Cit) rats at 8 weeks (n=5/group). Results are quantified by densitometry. Simvastatin and l-citrulline decreased arginase I expression in both aortas and livers from diabetic rats. Values are expressed as means±SEM. *P<0.05 vs control and diabetic simvastatin–treated rats.
Effects of Arginase Inhibition on Coronary Relaxation
To further determine the role of arginase activity in diabetes-induced vascular dysfunction, we tested the efficacy of inhibiting arginase activity in preserving NO-mediated vasodilation in coronary vessels isolated from 8-week diabetic rats. Incubation of diabetic coronary vessels with the arginase inhibitor difluoromethyl ornithine (DFMO) (50 μmol/L, 1 hour) resulted in an increase in Emax to ACh from 30±4% before treatment to 74±7% after treatment (Figure 4). This enhanced response was not different from responses to ACh in control (nondiabetic) vessels with or without DFMO exposure or in vessels from diabetic/simvastatin-treated rats exposed to DFMO. Similar responses were observed in diabetic coronary arteries pretreated with another arginase inhibitor, l-norvaline (50 μmol/L) (supplemental Figure II).
Figure 4.

Effect of the arginase inhibitor DFMO on ACh concentration–response curves of diabetic and control coronary arteries. A, In vitro treatment of diabetic coronary arteries for 1 hour with DFMO caused a significant increase in Emax to ACh compared with diabetic-untreated vessels (n=8/group) (open symbols). This enhanced response with DFMO was not different from responses in control (nondiabetic) vessels with or without DFMO exposure or from vessels of diabetic/simvastatin-treated rats exposed to DFMO (n=5 to 6/group). Values are expressed as means±SEM. *P<0.05 vs diabetic untreated rats.
Effect of Diabetes and High Glucose on Oxidative Stress
Our previous study had demonstrated increases levels of oxidative stress in the 4 week diabetic rat heart as shown by elevated levels of lipid peroxidation (malonic dialdehyde formation) and nitrotyrosine formation, a marker for ONOO− production.30 Similar to those results, 8-week diabetic rats displayed elevated lipid peroxidation and nitrotyrosine formation in the heart by 37% and 39%, respectively (supplemental Figure III). Treatment with either simvastatin or l-citrulline prevented the rise of both oxidant markers.
To evaluate oxidative stress levels in the coronary arteries of the diabetic hearts and to identify potential sources of reactive oxygen species formation, we performed dihydroethidium (DHE) imaging of fresh frozen sections of the cardiac ventricular septum of 8-week rats. Under identical reaction conditions, the DHE signal was much more intense within and around the coronary arteries of the diabetic rats than the controls. This increase in DHE staining was blocked by treatment with either L-NAME (3 mmol/L) or apocynin (30 μmol/L), indicating that sources of superoxide production in diabetic vessels include both NOS and NADPH oxidase (Figure 5A). Specificity of the reaction for superoxide was demonstrated by complete blockade of the signal by SOD.
Figure 5.
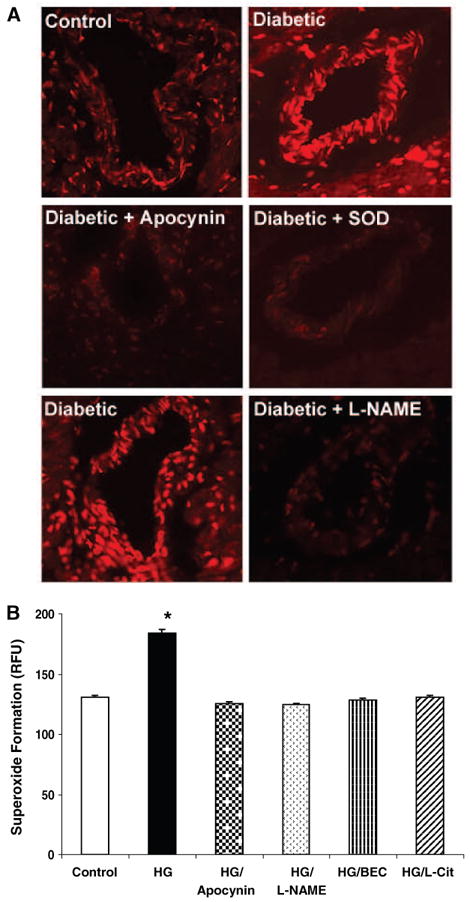
Superoxide production in rat coronary arteries from control and diabetic rats (A) and BCEC exposure to HG (B). A, Superoxide production was assessed by intensity of DHE staining of fresh frozen sections of ventricular septum from control and diabetic rats (8 weeks) (n=4/group). Effects of pretreatment of slides with L-NAME (3 mmol/L) or apocynin (30 μmol/L) were also assessed. Inhibition of the signal by superoxide dismutase (SOD) shows specificity of the reaction for superoxide anion. B, Superoxide production was measured using the luminance dye L-012 in BCECs exposed to HG (25 mmol/L) for 24 hours without and with concurrent treatment with L-NAME or apocynin (same concentrations as in A), BEC (100 μmol/L), or l-citrulline (1 mmol/L). Values are expressed as means±SEM. *P<0.05 vs diabetic untreated rats.
To further evaluate effects of the diabetic condition on coronary endothelial cells, we exposed BCECs to 25 mmol/L d-glucose (HG) (24 hours) and analyzed superoxide formation by chemiluminescence. The HG-treated cells had significant increases in superoxide levels as compared with control cells in 5 mmol/L d-glucose (Figure 5B). This HG effect was completely blocked by l-NAME (3 mmol/L) or apocynin (30 μmol/L), confirming that NOS and NADPH oxidase are prominent sources of superoxide formation in the HG-treated cells. Furthermore, treatment with the arginase inhibitor S-2-boronoethyl-l-cysteine (BEC) (100 μmol/L) or l-citrulline (1 mmol/L) was equally effective in preventing the HG effect of superoxide formation, suggesting that limiting arginase activity and increasing l-arginine availability reduces superoxide formation.
Effects of High Glucose on Arginase Activity and NO Formation
To further define the role of arginase in diabetes-induced vascular dysfunction, we next determined the effects of HG on arginase activity in BCECs. HG treatment for 24 hours resulted in a significant increase in arginase activity above the control levels (Figure 6). Because simvastatin treatment and l-citrulline supplementation prevented diabetes-induced elevation in vascular and hepatic arginase activity, we next determined simvastatin and l-citrulline effects on arginase activity in the HG-treated BCECs. Furthermore, because simvastatin reduces levels of active RhoA and RhoA activation has been associated with elevation of arginase activity,31,32,34,35 we also tested the effect of pretreatment with the Rho kinase inhibitor Y-27362. Exposure of cells to HG in the presence of either simvastatin (0.1 μmol/L) or Y-27362 (10 μmol/L) completely prevented the HG effect in increasing arginase activity. We also observed a prominent increase in levels of active RhoA in the HG-treated BCECs (inset of Figure 6). This effect was prevented by cotreatment with simvastatin. These data indicate involvement of activated RhoA in HG-induced activation of arginase. Concurrent exposure of BCECs to HG with l-citrulline (1 mmol/L) or apocynin (30 μmol/L) substantially reduced the rise in arginase activity, implying the involvement of NOS and NADPH oxidase. Levels of arginase I were not altered by the HG treatment (supplemental Figure IV).
Figure 6.
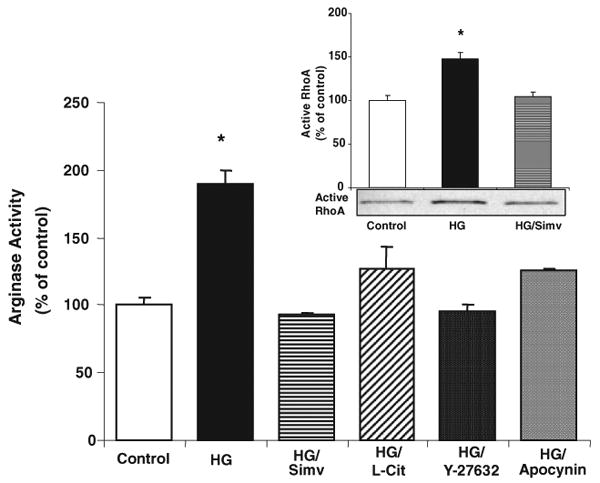
Effect of HG on arginase activity and active RhoA levels in BCECs. HG (25 mmol/L d-glucose) increased arginase activity in BCECs after exposure for 24 hours without modification of arginase I protein expression (n=6/group). Concurrent treatment with simvastatin (Simv) (0.1 μmol/L) or the Rho kinase inhibitor Y-27632 (1 μmol/L) prevented HG-induced increased arginase activity. Cotreatment with l-citrulline (L-Cit) (1 mmol/L) or apocynin (30 μmol/L) significantly inhibited the rise in activity cause by HG. HG exposure for 24 hours also raised active RhoA levels in BCECs, an effect prevented by simvastatin cotreatment (inset). Values are expressed as means±SEM. *P<0.05 vs all other groups.
To directly determine the role of arginase I in the HG effect, we used small interfering (si)RNA techniques to downregulate arginase I in BCECs and determined the effect on arginase activity and NO production following exposure to HG. BCECs were transfected with arginase I siRNA or scrambled (SC) siRNA as a control and exposed to HG for 24 hours. Western blotting confirmed that arginase I protein expression was significantly decreased (≈60%) in arginase I siRNA-transfected BCECs (Figure 7, inset) but was unaltered by SC siRNA. HG treatment increased arginase activity similarly in both nontransfected cells and in control cells transfected with SC siRNA (Figure 7A). Transfection with arginase I siRNA completely blocked the HG-induced increase in arginase activity and reduced arginase activity in the control cultures by nearly 50%, indicating that arginase I is a major source of the HG-induced increases in arginase activity. The residual activity may be attributable to the remaining arginase I protein or to the activity of arginase II.
Figure 7.
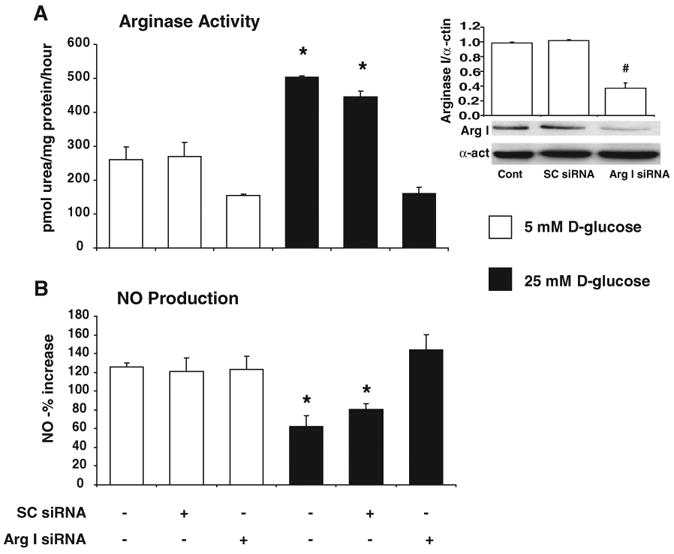
Effect of arginase I siRNA transfection on BCECs exposed to normal glucose (NG) (5 mmol/L) or HG (25 mmol/L) for 24 hours (n=4/group). Arginase I protein expression was significantly decreased in arginase I siRNA-transfected BCECs (inset). #P<0.05 vs control and SC siRNA]. A, HG-induced increases in arginase activity were inhibited in BCECs transfected with arginase I siRNA but were not altered in cells transfected with SC siRNA. B, HG-induced decreases in NO production were prevented in BCECs transfected with arginase I siRNA but not in cells transfected with scrambled siRNA (SC siRNA). Values are expressed as means±SEM. *P<0.05 vs normal glucose controls.
To demonstrate the role of arginase I activity in HG-mediated endothelial cell dysfunction, we determined the effects of arginase I knockdown on NO formation in the HG-treated BCECs. This study showed that HG exposure decreased NO production by 50% as compared with the control cells. This inhibitory effect of HG was completely blocked in cells transfected with arginase I siRNA (Figure 7B), implying the critical involvement of increased arginase I activity in the HG-mediated decreases in NO formation.
Discussion
We previously observed significant impairment of endothelium-dependent coronary vasodilation after 4 weeks of STZ-induced diabetes, which was substantially reduced by simvastatin treatment. However, no protection was observed when diabetic rats were given supplemental l-arginine.30 The results of our present studies show that the diabetes-induced impairment of coronary artery vasodilation is associated with increased arginase activity in both vascular tissue and liver. These increases in enzyme activity were correlated with significant increases in tissue levels of arginase I protein. Treatment with simvastatin or l-citrulline diminished arginase I expression, normalized arginase activity, and restored endothelial-dependent vasorelaxation responses. l-Arginine treatment was without effect on arginase activity. Treatment with the arginase inhibitors DFMO and l-norvaline significantly improved endothelium-dependent vasorelaxation in the diabetic vessels, supporting a causal role for arginase activity in coronary dysfunction. Analyses of superoxide formation in coronary arteries and BCECs showed that NOS and NADPH oxidase are prominent sources of diabetes and HG-induced reactive oxygen species formation. Studies using isolated coronary endothelial cells showed that exposure to HG conditions in vitro also caused an increase in arginase activity, which was blocked by simvastatin and l-citrulline. The Rho kinase inhibitor Y-27632 also normalized arginase activity, implying the involvement of RhoA in the HG effect. Knocking down arginase I expression with siRNA transfection blocked the action of HG in increasing arginase activity and decreasing NO production, providing further support for the causal role of increased arginase I activity in diabetes/HG-induced impairment of vascular endothelial function.
Given that arginase is a major participant in l-arginine catabolism and its activity is increased by diabetes, it is not surprising that l-arginine supplementation failed to improve the impaired endothelium-dependent vasodilation in our previous study. Although l-arginine supplementation has been reported to be beneficial in preventing vascular dysfunction in some settings,36,37 other studies have shown either no benefit or worsening of cardiovascular disease with chronic l-arginine treatment.24,25 We previously have observed that continuous oral supplementation of l-arginine to rabbits over 3 days caused increased arginase activity in liver and aortas, which was associated with decreases in NO production in response to ACh.27 In contrast, supplemental oral l-citrulline was beneficial in supporting NO production. Our present results suggest that these adverse effects of l-arginine supplementation involve stimulation of arginase activity.
Multiple factors are likely to contribute to elevated arginase activity during diabetes. Increased arginase activity is associated with inflammatory cytokines and reactive oxygen species, both of which are increased during diabetes.38,39 In particular, oxidative stress seems to be the major means by which activity of arginase is increased. H2O2 and peroxynitrite have been shown to activate arginase in endothelial cells,40,41 and these actions are prevented by antioxidants or FeTTPs, a decomposition catalyst for peroxynitrite. There is strong evidence that oxidative stress activates RhoA.42,43 Active RhoA, in turn, has been shown to activate arginase.32,33 The lineage of events appears to be oxidative stress→activation of RhoA→activation and enhanced expression of arginase. Further study is needed to elucidate other molecular mediators in this pathway.
Our vasorelaxation studies using coronary vessels from rats diabetic for 8 weeks showed that severe impairment in endothelium-dependent vasodilation was correlated with significantly enhanced arginase activity and elevated arginase I protein levels in both liver and vascular tissue. The simvastatin treatment significantly improved the vasodilation with a beneficial effect similar to that seen in the four week diabetic rats in our previous study30 and also prevented the rise in arginase expression and activity. Similar to the actions of simvastatin in blocking the effects of diabetes on vascular arginase activity, simvastatin treatment of coronary endothelial cells reduced the elevation in arginase activity in response to HG exposure. Statins block isoprenylation and activation of small GTPases34 and their vasoprotective effects are known to involve the enhancement of eNOS expression and activity and suppression of NADPH oxidase assembly and activity. Increased arginase activity also has been associated with activation of the small GTPase RhoA,32,33 which is known to occur during diabetes.44,45 Thus, statin blockade of Rho and Rac GTPases46 could also improve NOS function by blocking diabetes effects in increasing arginase activity and thereby increasing l-arginine availability and NO production. Our data showing that HG raised active RhoA levels in coronary endothelial cells and that the Rho kinase inhibitor Y-27632 and simvastatin were equally effective in preventing the rise in arginase activity in the HG-treated cells imply that RhoA activation has a critical role in HG-induced activation of arginase.
Interestingly, l-citrulline supplementation enhanced endothelial-dependent coronary vasodilation in 8-week diabetic rats to a level similar to that seen in the simvastatin-treated diabetic rats. l-Citrulline, the precursor of l-arginine, is a byproduct in the formation of NO and is recycled back to l-arginine in many tissues, contributing to sustained l-arginine supply for NOS activity.28 We believe that supplemental l-citrulline provides sufficient l-arginine to endothelial NOS for robust NO production and prevents NOS uncoupling and superoxide production. Because l-citrulline is also an allosteric inhibitor of arginase,29 its use may suppress arginase activity. In fact, l-citrulline prevented the increase in arginase activity in hepatic and vascular tissue from 8-week diabetic rats and in HG-treated coronary endothelial cells. Moreover, arginase I protein levels tended to decrease in both vascular and hepatic tissues of the l-citrulline-treated diabetic rats. Improvement of endothelium-dependent vasodilation in diabetic coronary vessels after treatment with the arginase inhibitors further demonstrates a role for arginase in impaired coronary vasorelaxation in diabetic rats. Given that acute treatment of the diabetic coronaries with 2 different arginase inhibitors had a vasorelaxant-enhancing effect comparable with chronic l-citrulline and simvastatin treatment of the animals and that arginase inhibitor treatment did not further improve the vasorelaxant responses in vessels from the simvastatin treated rats, our data strongly suggest that the protective actions of l-citrulline and simvastatin are mediated by blockade of diabetes-induced arginase activation.
We speculate that the decreases in arginase activity in the simvastatin and l-citrulline–treated diabetic rats and cultured endothelial cells are attributable to decreased oxidative stress. In support of this idea, simvastatin or l-citrulline treatment of the diabetic rats prevented the elevation of vascular and cardiac lipid peroxidation and protein tyrosine nitration, a measure of ONOO− formation from superoxide combining with NO. We believe that the resultant increase in NO availability is a cardinal effect of both treatments. Enhanced NO production could reduce superoxide production by NADPH oxidase because of NO-mediated S-nitrosylation of the enzyme, which inhibits its activity.47 Furthermore, the decrease in arginase activity would increase the availability of arginine to NOS, reducing superoxide formation attributable to NOS uncoupling. This reasoning is supported by the results of our DHE imaging and chemiluminescence analyses of superoxide formation in diabetic coronary arteries and HG-treated coronary endothelial cells. The data showing that diabetes- and HG-induced increases in superoxide formation are prevented by treatment with L-NAME or apocynin indicate that NOS and NADPH oxidase are both sources of superoxide formation in the diabetic vessels.
Our studies using arginase I siRNA in coronary endothelial cells provide further support for the competitive role of arginase I in decreasing l-arginine availability to NOS. The treatment with arginase I siRNA markedly reduced arginase I protein expression, completely blocked the actions of HG in increasing arginase activity, and restored NO formation to normal levels. These data indicate that arginase I is critically involved in endothelial NOS dysfunction under HG conditions. Although we were unable to determine the effects of diabetes on arginase I protein levels in the coronary vessels, the role of this enzyme in diabetes vascular complications is supported by our data showing that diabetes-induced increases in arginase activity in the aorta are correlated with increases in arginase I protein levels.
It is possible that arginase II may also contribute to the increase in arginase activity in diabetes. Increases in arginase II mRNA have been reported in human aortic endothelial cells exposed to oxidized low-density lipoprotein.48 However, our data indicate that arginase II protein expression is low in rat aorta and BCECs. Others have shown that arginase I is the predominant form in rat aortic endothelial cells and the form responsible for reciprocal regulation of NOS in aorta of aging rats.2,9 Our observations that HG-induced increase in arginase activity is totally blocked in the arginase I siRNA-transfected coronary endothelial cells imply that arginase I is very likely to be involved in the diabetes-induced endothelial cell dysfunction.
In conclusion, our whole animal and cellular studies have provided direct evidence for the role of arginase in coronary dysfunction in diabetes. Increased arginase activity and expression of arginase I appear to be associated with diabetes-induced increases in oxidative stress and activation of the RhoA pathway. The elevated arginase activity could initiate a feed-forward cycle of diminished NO levels and further oxidative stress. Arginase can be a novel therapeutic target in the treatment of diabetic endothelial dysfunction. l-Citrulline is also a promising adjunct therapy to treat diabetic cardiovascular complications because of its actions in limiting the activation of arginase and oxidative stress.
Supplementary Material
Supplemental Figure I. Western blot analysis of arginase II protein expression in aorta of Control, Diabetic Diab/Simv and, Diab/L-Cit rats at eight weeks.
Supplemental Figure II. Effect of the arginase inhibitor L-norvaline on ACh concentration-response curves of diabetic coronary arteries. In vitro treatment of diabetic coronary arteries (n =6 - 7 / group) for 2 hr with L-norvaline caused a significant increase in Emax to ACh compared with diabetic-untreated vessels. Values are expressed as means ± S.E.M.; *, p < 0.05 versus Diabetic-untreated.
Supplemental Figure III. Cardiac levels of lipid peroxides, as malondialdehyde (MDA) formation (A), and nitrotyrosine formation (B) in Control, Diabetic, simvastatin-treated diabetic (Diab/Simv) and, L-citrulline-supplemented diabetic (Diab/L-Cit) rats at 8 weeks (n = 8 / group). Values are expressed as means ± S.E.M.; *, p < 0.05 versus control, Diab/Simv and, Diab/L-Cit.
Supplemental Figure IV. Effect of high glucose (HG) on arginase I protein levels in BCEC. BCED were exposed to HG (25 mmol/L) or normal levels of glucose (control, 5 mmol/L) for 24 hours and arginase I levels were determined by Western blotting.
Supplemental Table I. Maximum vasorelaxant response after L-NAME treatment
Acknowledgments
We thank the many excellent contributions of Jennifer Iddings, Su Matragoon, and Dr Nai-Tsi Tsai to these studies and to the preparation of the manuscript.
Sources of Funding: This work was supported by NIH grants R01 HL 70215 and R01 EY 11766.
Footnotes
Disclosures: None.
References
- 1.Cosentino F, Luscher TF. Endothelial dysfunction in diabetes mellitus. J Cardiovasc Pharmacol. 1998;32(suppl 3):S54–S61. [PubMed] [Google Scholar]
- 2.Berkowitz DE, White R, Li D, Minhas KM, Cernetich A, Kim S, Burke S, Shoukas AA, Nyhan D, Champion HC, Hare JM. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation. 2003;108:2000–2006. doi: 10.1161/01.CIR.0000092948.04444.C7. [DOI] [PubMed] [Google Scholar]
- 3.Kaesemeyer WH, Ogonowski AA, Jin L, Caldwell RB, Caldwell RW. Endothelial nitric oxide synthase is a site of superoxide synthesis in endothelial cells treated with glyceryl trinitrate. Br J Pharmacol. 2000;131:1019–1023. doi: 10.1038/sj.bjp.0703665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bivalacqua TJ, Hellstrom WJ, Kadowitz PJ, Champion HC. Increased expression of arginase II in human diabetic corpus cavernosum: in diabetic-associated erectile dysfunction. Biochem Biophys Res Commun. 2001;283:923–927. doi: 10.1006/bbrc.2001.4874. [DOI] [PubMed] [Google Scholar]
- 5.Zhang C, Hein TW, Wang W, Miller MW, Fossum TW, McDonald MM, Humphrey JD, Kuo L. Upregulation of vascular arginase in hypertension decreases nitric oxide-mediated dilation of coronary arterioles. Hypertension. 2004;44:935–943. doi: 10.1161/01.HYP.0000146907.82869.f2. [DOI] [PubMed] [Google Scholar]
- 6.Hein TW, Zhang C, Wang W, Chang CI, Thengchaisri N, Kuo L. Ischemia-reperfusion selectively impairs nitric oxide-mediated dilation in coronary arterioles: counteracting role of arginase. FASEB J. 2003;17:2328–2330. doi: 10.1096/fj.03-0115fje. [DOI] [PubMed] [Google Scholar]
- 7.Morris CR, Morris SM, Jr, Hagar W, Van Warmerdam J, Claster S, Kepka-Lenhart D, Machado L, Kuypers FA, Vichinsky EP. Arginine therapy: a new treatment for pulmonary hypertension in sickle cell disease? Am J Respir Crit Care Med. 2003;168:63–69. doi: 10.1164/rccm.200208-967OC. [DOI] [PubMed] [Google Scholar]
- 8.Morris CR, Kato GJ, Poljakovic M, Wang X, Blackwelder WC, Sachdev V, Hazen SL, Vichinsky EP, Morris SM, Jr, Gladwin MT. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA. 2005;294:81–90. doi: 10.1001/jama.294.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.White AR, Ryoo S, Li D, Champion HC, Steppan J, Wang D, Nyhan D, Shoukas AA, Hare JM, Berkowitz DE. Knockdown of arginase I restores NO signaling in the vasculature of old rats. Hypertension. 2006;47:245–251. doi: 10.1161/01.HYP.0000198543.34502.d7. [DOI] [PubMed] [Google Scholar]
- 10.Csiszar A, Labinskyy N, Smith K, Rivera A, Orosz Z, Ungvari Z. Vasculoprotective effects of anti-tumor necrosis factor-alpha treatment in aging. Am J Pathol. 2007;170:388–398. doi: 10.2353/ajpath.2007.060708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wassmann S, Laufs U, Muller K, Konkol C, Ahlbory K, Baumer AT, Linz W, Bohm M, Nickenig G. Cellular antioxidant effects of atorvastatin in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2002;22:300–305. doi: 10.1161/hq0202.104081. [DOI] [PubMed] [Google Scholar]
- 12.Jiang M, Jia L, Jiang W, Hu X, Zhou H, Gao X, Lu Z, Zhang Z. Protein disregulation in red blood cell membranes of type 2 diabetic patients. Biochem Biophys Res Commun. 2003;309:196–200. doi: 10.1016/s0006-291x(03)01559-6. [DOI] [PubMed] [Google Scholar]
- 13.Cederbaum SD, Yu H, Grody WW, Kern RM, Yoo P, Iyer RK. Arginases I and II: do their functions overlap? Mol Genet Metab. 2004;81(suppl 1):S38–S44. doi: 10.1016/j.ymgme.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 14.Li H, Meininger CJ, Hawker JR, Jr, Haynes TE, Kepka-Lenhart D, Mistry SK, Morris SM, Jr, Wu G. Regulatory role of arginase I and II in nitric oxide, polyamine, and proline syntheses in endothelial cells. Am J Physiol Endocrinol Metab. 2001;280:E75–E82. doi: 10.1152/ajpendo.2001.280.1.E75. [DOI] [PubMed] [Google Scholar]
- 15.Zhang C, Hein TW, Wang W, Chang CI, Kuo L. Constitutive expression of arginase in microvascular endothelial cells counteracts nitric oxide-mediated vasodilatory function. FASEB J. 2001;15:1264–1266. doi: 10.1096/fj.00-0681fje. [DOI] [PubMed] [Google Scholar]
- 16.Chicoine LG, Paffett ML, Young TL, Nelin LD. Arginase inhibition increases nitric oxide production in bovine pulmonary arterial endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L60–L8. doi: 10.1152/ajplung.00194.2003. [DOI] [PubMed] [Google Scholar]
- 17.Bond JS, Failla ML, Unger DF. Elevated manganese concentration and arginase activity in livers of streptozotocin-induced diabetic rats. J Biol Chem. 1983;258:8004–8009. [PubMed] [Google Scholar]
- 18.Spolarics Z, Bond JS. Comparison of biochemical properties of liver arginase from streptozocin-induced diabetic and control mice. Arch Biochem Biophys. 1989;274:426–433. doi: 10.1016/0003-9861(89)90455-4. [DOI] [PubMed] [Google Scholar]
- 19.Li Z, Yarmush ML, Chan C. Insulin concentration during preconditioning mediates the regulation of urea synthesis during exposure to amino acid-supplemented plasma. Tissue Eng. 2004;10:1737–1746. doi: 10.1089/ten.2004.10.1737. [DOI] [PubMed] [Google Scholar]
- 20.Pieper GM, Dondlinger LA. Plasma and vascular tissue arginine are decreased in diabetes: acute arginine supplementation restores endothelium-dependent relaxation by augmenting cGMP production. J Pharmacol Exp Ther. 1997;283:684–691. [PubMed] [Google Scholar]
- 21.Hagenfeldt L, Dahlquist G, Persson B. Plasma amino acids in relation to metabolic control in insulin-dependent diabetic children. Acta Paediatr Scand. 1989;78:278–282. doi: 10.1111/j.1651-2227.1989.tb11070.x. [DOI] [PubMed] [Google Scholar]
- 22.Angulo J, Rodriguez-Manas L, Peiro C, Neira M, Marin J, Sanchez-Ferrer CF. Impairment of nitric oxide-mediated relaxations in anaesthetized autoperfused streptozotocin-induced diabetic rats. Naunyn Schmiedebergs Arch Pharmacol. 1998;358:529–537. doi: 10.1007/pl00005289. [DOI] [PubMed] [Google Scholar]
- 23.Pieper GM, Peltier BA. Amelioration by L-arginine of a dysfunctional arginine/nitric oxide pathway in diabetic endothelium. J Cardiovasc Pharmacol. 1995;25:397–403. doi: 10.1097/00005344-199503000-00008. [DOI] [PubMed] [Google Scholar]
- 24.Jeremy RW, McCarron H, Sullivan D. Effects of dietary L-arginine on atherosclerosis and endothelium-dependent vasodilatation in the hypercholesterolemic rabbit. Response according to treatment duration, anatomic site, and sex. Circulation. 1996;94:498–506. doi: 10.1161/01.cir.94.3.498. [DOI] [PubMed] [Google Scholar]
- 25.Schulman SP, Becker LC, Kass DA, Champion HC, Terrin ML, Forman S, Ernst KV, Kelemen MD, Townsend SN, Capriotti A, Hare JM, Gerstenblith G. L-arginine therapy in acute myocardial infarction: the Vascular Interaction With Age in Myocardial Infarction (VINTAGE MI) randomized clinical trial. JAMA. 2006;295:58–64. doi: 10.1001/jama.295.1.58. [DOI] [PubMed] [Google Scholar]
- 26.Morris SM., Jr Regulation of enzymes of urea and arginine synthesis. Annu Rev Nutr. 1992;12:81–101. doi: 10.1146/annurev.nu.12.070192.000501. [DOI] [PubMed] [Google Scholar]
- 27.Socha HM, Romero MJ, Caldwell RW. Oral citrulline administration enhances NO-dependent vasodilation. FASEB J. 2006;20:A1125. [Google Scholar]
- 28.Romero MJ, Platt DH, Caldwell RB, Caldwell RW. Therapeutic use of citrulline in cardiovascular disease. Cardiovasc Drug Rev. 2006;24:275–290. doi: 10.1111/j.1527-3466.2006.00275.x. [DOI] [PubMed] [Google Scholar]
- 29.Shearer JD, Richards JR, Mills CD, Caldwell MD. Differential regulation of macrophage arginine metabolism: a proposed role in wound healing. Am J Physiol. 1997;272:E181–E190. doi: 10.1152/ajpendo.1997.272.2.E181. [DOI] [PubMed] [Google Scholar]
- 30.Tawfik HE, El-Remessy AB, Matragoon S, Ma G, Caldwell RB, Caldwell RW. Simvastatin improves diabetes-induced coronary endothelial dysfunction. J Pharmacol Exp Ther. 2006;319:386–395. doi: 10.1124/jpet.106.106823. [DOI] [PubMed] [Google Scholar]
- 31.Liao JK, Laufs U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol. 2005;45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ming XF, Barandier C, Viswambharan H, Kwak BR, Mach F, Mazzolai L, Hayoz D, Ruffieux J, Rusconi S, Montani JP, Yang Z. Thrombin stimulates human endothelial arginase enzymatic activity via RhoA/ROCK pathway: implications for atherosclerotic endothelial dysfunction. Circulation. 2004;110:3708–3714. doi: 10.1161/01.CIR.0000142867.26182.32. [DOI] [PubMed] [Google Scholar]
- 33.Horowitz S, Binion DG, Nelson VM, Kanaa Y, Javadi P, Lazarova Z, Andrekopoulos C, Kalyanaraman B, Otterson MF, Rafiee P. Increased arginase activity and endothelial dysfunction in human inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1323–G1336. doi: 10.1152/ajpgi.00499.2006. [DOI] [PubMed] [Google Scholar]
- 34.Eto M, Kozai T, Cosentino F, Joch H, Luscher TF. Statin prevents tissue factor expression in human endothelial cells: role of Rho/Rho-kinase and Akt pathways. Circulation. 2002;105:1756–1759. doi: 10.1161/01.cir.0000015465.73933.3b. [DOI] [PubMed] [Google Scholar]
- 35.Endres M. Statins and stroke. J Cereb Blood Flow Metab. 2005;25:1093–1110. doi: 10.1038/sj.jcbfm.9600116. [DOI] [PubMed] [Google Scholar]
- 36.Huynh NT, Tayek JA. Oral arginine reduces systemic blood pressure in type 2 diabetes: its potential role in nitric oxide generation. J Am Coll Nutr. 2002;21:422–427. doi: 10.1080/07315724.2002.10719245. [DOI] [PubMed] [Google Scholar]
- 37.Lerman A, Burnett JC, Jr, Higano ST, McKinley LJ, Holmes DR., Jr Long-term L-arginine supplementation improves small-vessel coronary endothelial function in humans. Circulation. 1998;97:2123–2128. doi: 10.1161/01.cir.97.21.2123. [DOI] [PubMed] [Google Scholar]
- 38.Hink U, Li H, Mollnau H, Oelze M, Matheis E, Hartmann M, Skatchkov M, Thaiss F, Stahl RA, Warnholtz A, Meinertz T, Griendling K, Harrison DG, Forstermann U, Munzel T. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res. 2001;88:e14–e22. doi: 10.1161/01.res.88.2.e14. [DOI] [PubMed] [Google Scholar]
- 39.Helmersson J, Vessby B, Larsson A, Basu S. Association of type 2 diabetes with cyclooxygenase-mediated inflammation and oxidative stress in an elderly population. Circulation. 2004;109:1729–1734. doi: 10.1161/01.CIR.0000124718.99562.91. [DOI] [PubMed] [Google Scholar]
- 40.Thengchaisri N, Hein TW, Wang W, Xu X, Li Z, Fossum TW, Kuo L. Upregulation of arginase by H2O2 impairs endothelium-dependent nitric oxide-mediated dilation of coronary arterioles. Arterioscler Thromb Vasc Biol. 2006;26:2035–2042. doi: 10.1161/01.ATV.0000233334.24805.62. [DOI] [PubMed] [Google Scholar]
- 41.El-Remessy AB, Tawfik HE, Matragoon S, Ali TK, Caldwell RB, Caldwell RW. Peroxynitrite mediates diabetes-induced endothelial dysfunction by reducing eNOS expression: possible role of Rho kinase (ROCK) activation. Circulation. 2006;114:II_330. Abstract. [Google Scholar]
- 42.Jin L, Ying Z, Hilgers RH, Yin J, Zhao X, Imig JD, Webb RC. Increased RhoA/Rho-kinase signaling mediates spontaneous tone in aorta from angiotensin II-induced hypertensive rats. J Pharmacol Exp Ther. 2006;318:288–295. doi: 10.1124/jpet.105.100735. [DOI] [PubMed] [Google Scholar]
- 43.Seko T, Ito M, Kureishi Y, Okamoto R, Moriki N, Onishi K, Isaka N, Hartshorne DJ, Nakano T. Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circ Res. 2003;92:411–418. doi: 10.1161/01.RES.0000059987.90200.44. [DOI] [PubMed] [Google Scholar]
- 44.Kovacs P, Stumvoll M, Bogardus C, Hanson RL, Baier LJ. A functional Tyr1306Cys variant in LARG is associated with increased insulin action in vivo. Diabetes. 2006;55:1497–1503. doi: 10.2337/db05-1331. [DOI] [PubMed] [Google Scholar]
- 45.Sandu OA, Ragolia L, Begum N. Diabetes in the Goto-Kakizaki rat is accompanied by impaired insulin-mediated myosin-bound phosphatase activation and vascular smooth muscle cell relaxation. Diabetes. 2000;49:2178–2189. doi: 10.2337/diabetes.49.12.2178. [DOI] [PubMed] [Google Scholar]
- 46.Brown JH, Del Re DP, Sussman MA. The Rac and Rho hall of fame: a decade of hypertrophic signaling hits. Circ Res. 2006;98:730–742. doi: 10.1161/01.RES.0000216039.75913.9e. [DOI] [PubMed] [Google Scholar]
- 47.Park JW. Attenuation of p47phox and p67phox membrane translocation as the inhibitory mechanism of S-nitrosothiol on the respiratory burst oxidase in human neutrophils. Biochem Biophys Res Commun. 1996;220:31–35. doi: 10.1006/bbrc.1996.0351. [DOI] [PubMed] [Google Scholar]
- 48.Ryoo S, Lemmon CA, Soucy KG, Gupta G, White AR, Nyhan D, Shoukas A, Romer LH, Berkowitz DE. Oxidized low-density lipoprotein-dependent endothelial arginase II activation contributes to impaired nitric oxide signaling. Circ Res. 2006;99:951–960. doi: 10.1161/01.RES.0000247034.24662.b4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure I. Western blot analysis of arginase II protein expression in aorta of Control, Diabetic Diab/Simv and, Diab/L-Cit rats at eight weeks.
Supplemental Figure II. Effect of the arginase inhibitor L-norvaline on ACh concentration-response curves of diabetic coronary arteries. In vitro treatment of diabetic coronary arteries (n =6 - 7 / group) for 2 hr with L-norvaline caused a significant increase in Emax to ACh compared with diabetic-untreated vessels. Values are expressed as means ± S.E.M.; *, p < 0.05 versus Diabetic-untreated.
Supplemental Figure III. Cardiac levels of lipid peroxides, as malondialdehyde (MDA) formation (A), and nitrotyrosine formation (B) in Control, Diabetic, simvastatin-treated diabetic (Diab/Simv) and, L-citrulline-supplemented diabetic (Diab/L-Cit) rats at 8 weeks (n = 8 / group). Values are expressed as means ± S.E.M.; *, p < 0.05 versus control, Diab/Simv and, Diab/L-Cit.
Supplemental Figure IV. Effect of high glucose (HG) on arginase I protein levels in BCEC. BCED were exposed to HG (25 mmol/L) or normal levels of glucose (control, 5 mmol/L) for 24 hours and arginase I levels were determined by Western blotting.
Supplemental Table I. Maximum vasorelaxant response after L-NAME treatment