

Abstract
Background
Diagnosis of Barrett's esophagus (BE) is typically done through morphologic analysis of esophageal tissue biopsy. Such samples contain several cell types. Laser capture microdissection (LCM) allows the isolation of specific cells from heterogeneous cell populations. The purpose of this study was to determine the degree of overlap of the two sample types and to define a set of genes that may serve as biochemical markers for BE.
Methods
We obtained biopsies from regions of the glandular tissue of BE and normal esophagus from 9 subjects with BE. Samples from 5 subjects were examined as whole tissue (BE [whole]; E [whole]), and in 4 subjects the glandular epithelium of BE was isolated using LCM (BE [LCM]) and compared to the averaged values (E [LCM]) for both basal cell (B [LCM]) and squamous cell (S [LCM]) epithelium.
Results
Gene expression revealed 1797 probesets between BE [whole] and E [whole] (fold change > 2.0; p<0.001). Most (74%) were also differentially expressed between BE [LCM] and E [LCM], showing that there was high concordance between the two sampling methods. LCM provided a great deal of additional information (2113 genes) about the alterations in gene expression that may represent the BE phenotype.
Conclusions
There are differences in gene expression profiles depending on whether specimens are whole tissue biopsies or LCM dissected. Whole tissue biopsies should prove satisfactory for diagnostic purposes. Because the data from LCM samples delineated many more Barrett's specific genes, this procedure may provide more information regarding pathogenesis than whole tissue material.
Introduction
Barrett's esophagus (BE) is the precursor lesion for esophageal adenocarcinoma. Persons with BE are at a 40 fold increased risk of esophageal adenocarcinoma compared to the general population. The pathogenesis of BE as well as the predictors and markers of progression from BE to esophageal adenocarcinoma remain unclear.
Several studies examined global gene expression profiles of BE with and without cancer using cDNA microarray to distinguish the different stages of BE progression(1-3). These studies used RNA pooled specimens from several patients with similar morphological phenotype. Other studies examined RNA extracted and pooled from BE patients and reported clusters of genes that are specific to BE in comparison to normal esophageal, gastric, and small intestine tissue(4). We previously demonstrated the feasibility of conducting studies on endoscopically obtained tissues and described the biopsy protocol required to obtain sufficient RNA without pooling for microarray analysis(5). The very strong tissue effect where all phenotypically similar tissue obtained from different subjects clustered with each other indicated low intersubject heterogeneity for grossly similar tissues.
In this study, we evaluated the degree to which gene expression detected by gene arrays from BE [whole] obtained from mucosal biopsies is representative of gene expression from glandular tissue. Laser capture microdissection (LCM) can be used to specifically dissect glandular tissue to allow the isolation of specific cells from heterogeneous cell populations. However, the feasibility of using LCM to obtain adequate quantity and quality of RNA from individuals has not been established for this tissue type. We conducted this study to compare global gene expression profiles obtained from two sample types (whole tissue and LCM), and to confirm our previous finding that tissue biopsies can be used to identify a pattern of gene expression diagnostic of BE. These findings are important in guiding future work in using expression profiles to understand the pathogenesis of BE and to identify markers of BE as well as progression from BE to esophageal adenocarcinoma.
Methods
Patients and Procedures
Nine patients with known long segment BE and no dysplasia underwent an upper endoscopy. A single experienced endoscopist (HES) used a sterile disposable Juniper forceps to obtain mucosal biopsies from BE and the grossly normal esophagus (2-3 cm above the proximal margin of BE). Several (2-3) paired mucosal biopsies were taken from each site. One of the paired biopsies was placed in a separate vial containing 10% formalin for subsequent histological examination. A single gastrointestinal pathologist blinded to endoscopic findings examined all mucosal biopsies and confirmed the presence of intestinal metaplasia and the absence of dysplasia. The other paired biopsy was snap frozen in liquid nitrogen, embedded in Optimal Cutting Temperature (OCT) embedding medium (Tissue-Tek) for subsequent RNA extraction.
Laser Capture Microdissection and RNA Isolation
LCM was conducted to isolate the glandular cells of BE (BE [LCM]) and to isolate basal (B [LCM]) separate from squamous (S [LCM]) epithelial cells in normal esophageal tissue. These layers are normal combined in a tissue sample obtained via endoscopic biopsies. Tissue samples were encased in the embedding medium, Cryomatrix (Thermo Fisher Scientific, Waltham, MA) and frozen on dry ice in a plastic cryomold. The frozen tissue samples were cut with the Cryotome (Thermo Fisher Scientific, Waltham, MA), stained with Histogene Staining Solution (Molecular Devices Corporation, Sunnyvale, CA) in RNA-free surroundings. CapSure Macro caps (Molecular Devices Corporation, Sunnyvale, CA) were placed on the tissue sections. Microdissection was conducted using the PixCell IIe Laser Capture Microdissection system (Molecular Devices Corporation, Sunnyvale, CA), which directs the low-power infrared laser through the cap to melt the film onto the cells of interest, which were then lifted from the tissues section
RNA was extracted from whole tissue specimens as well as LCM treated samples at the Microarray Core Facility of the Texas Medical Center Digestive Disease Center at Baylor College of Medicine using the Qiagen RNeasy Extraction kit (Qiagen, Inc, Valencia, CA, USA). The RNA integrity, quality, and concentration were measured using the Agilent 2100 Bioanalyzer and Lab-on-a-Chip technology (Agilent Technologies, Palo Alto, CA).
Microarray Hybridization
Gene expression analysis of whole tissue specimens was performed using the Human Affymetrix HG_U133A GeneChip® (Affymetrix Inc, Santa Clara, CA, USA) while gene expression analysis of LCM obtained specimens was performed using the Human Affymetrix HG_ Hgu133plus2 ® (Affymetrix Inc, Santa Clara, CA, USA).
After the reverse transcription, the cDNA concentration was measured, followed by in vitro transcription. In a quality control step, the cRNA was checked for size range and quantity prior to fragmentation and hybridization. Subsequently, first strand cDNA was prepared from 5 μg of total RNA from each sample and labeled with Biotinylated dUTP as described by Affymetrix GeneChip Expression Analysis Technical Manual(6). After double-stranded cDNA was produced, the GeneChip®s were washed and stained with streptavidin and phycoerythrin prior to scanning at the GeneChip® Scanner (Affymetrix Inc, Santa Clara, CA, USA).
Data Transformation Methods
The raw signal intensity data in the CEL files were normalized using the CG-RMA method implemented in the Bioconductor (http://www.bioconductor.org/) package of R(7). Each set (whole tissue or LCM) was normalized separately and a combined expression set was constructed using an indexing method in R. The indexing method creates new equivalent probeset lists between arrays that use the same Affy ID for their probesets. The combined expression set (22,277 probesets) was normalized again by CG-RMA to account for differences in probeset architecture of the two chips, and the data was analyzed using the limma package (8) in Bioconductor.
We compared BE [whole] vs. E [whole], and BE [LCM] vs. E [LCM] generating contrast matrices in the limma model(8). We considered the mean expression values for S [LCM] and B [LCM] to represent E [LCM]. The raw p-values were adjusted by the Benjamnin-Hochberg method for 5% false discovery rate control. Gene lists for Figure 1 were generated in Agilent Genespring 7.1 using the combined expression set filtered for fold change of >2.0 and p-value <0.001.
Figure 1.
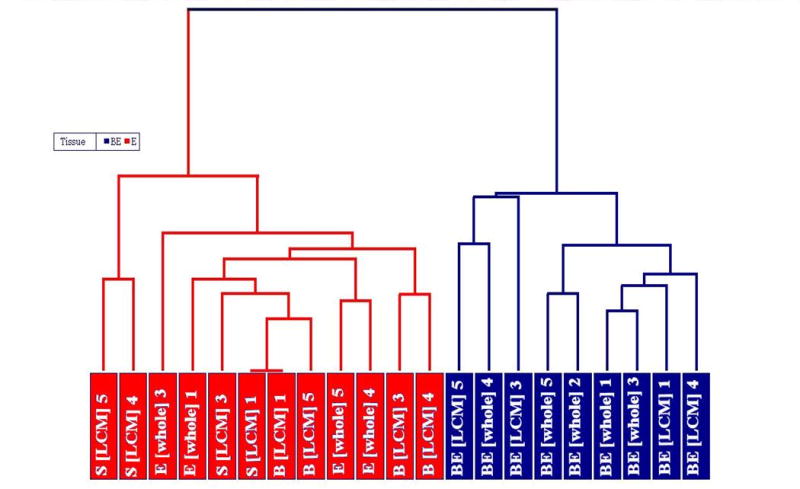
Dendrogram showing the correlation in gene expression in tissues subjected to LCM (n=4) (BE [LCM], S [LCM] and B [LCM]) and whole tissues (n=5) (BE [whole] and E [whole]). This graph was generated by hierarchal clustering analysis of GC-RMA batch normalized data of common probesets for LCM and whole tissue samples. Red: Normal esophagus E, S, B, Blue: Barrett's esophagus (BE), Number: patient ID.
We performed hierarchical cluster analysis with Parteks Genomics Suite 6.3. Briefly, GC-RMA normalized expression data of common probesets generated in Bioconductor was imported into Partek Genomics Suite. A mixed model ANOVA was used to adjust for the clustering within individual subjects(9).
Results
Nine patients with BE participated in the study. We obtained 5 BE [whole] tissue samples from five patients, but only four E [whole] tissue samples. The amount of RNA isolated from whole specimens ranged from 12.7 to 28 μg. LCM dissected samples from 3 sites BE [LCM], S [LCM] and E [LCM] in each of four patients were included in the final analysis. The amount of RNA isolated from LCM dissected samples ranged between 0.01 and 0.1 μg.
Hierarchical cluster analysis was performed using Partek Genomics Suite 6.3 (9) on all LCM and whole tissue samples using the common probesets and taking the method of sampling into account. The cluster analysis reveals that gene expression in similar tissues obtained from different patients clustered closely together based on similarity in their patterns of gene expression indicating low intersubject heterogeneity for grossly similar tissues (Figure 1).
The differential gene expression profile between BE [whole] and E [whole] tissue specimens was compared to that obtained in LCM obtained specimens (BE [LCM] vs. E [LCM]. Each set was normalized separately and a combined expression set was subsequently constructed using the indexing method described above. From the original number of 54,681 (Hgu133plus2) and 22,283 (Hgu133A) gene probes present on the arrays, 22,277 genes were shared by both array platforms and these were subjected to further analysis.
A Venn diagram indicating the number of genes (805) upregulated in BE compared to E that were shared (i.e., overlapped) between whole and LCM dissected tissues is shown in Figure 2, while the number of genes (526) upregulated in E compared to BE that were shared between whole and LCM dissected tissues is shown in Figure 3.
Figure 2.

72% of the genes that were more highly expressed in Barrett's whole biopsy tissue compared to normal esophagus were also more highly expressed in Barrett's LCM samples compared to the averaged values of S [LCM] and B [LCM].
Figure 3.
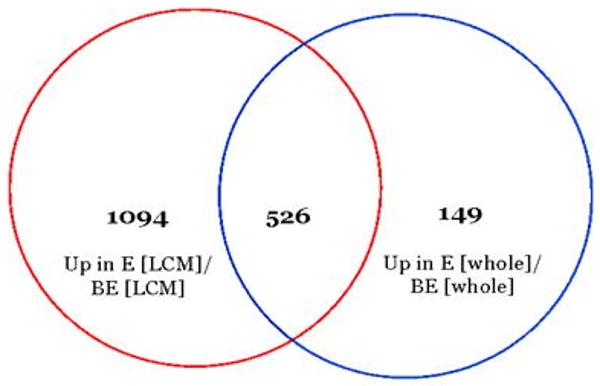
78% of the genes that were more highly expressed in E [whole] compared to BE [whole] were also more highly expressed in B [LCM] and S [LCM] samples compared to BE [LCM] samples.
Overall, there were 1797 differentially expressed probe sets in whole tissue BE [whole] vs. E [whole] (fold change > 2.0; p<0.001). Most of these genes (74%) were also differentially expressed in LCM collected tissue, BE [LCM] vs. E [LCM]. Many more genes were found to be differentially expressed in the LCM derived specimens (BE [LCM] vs. E [LCM]) than in the whole tissue biopsies; 3443 in total, of which 2113 were revealed only in the LCM specimens as they did not reach significance in the whole tissue samples.
Characterization of Differential Expressed Genes
We used the 805 genes significantly upregulated in BE vs. E in both whole tissue and LCM derived tissue (Figure 2) to identify potential BE biomarkers. We identified 52 genes with a >100 - fold difference in the average level of mRNA expression between BE [LCM] and E [LCM]. Next, we identified 41 genes where at least 10-fold increased expression was observed in all 9 patients (whole or LCM) (Figure 4). Table 1 displays these 41 genes with highest differential level of expression between BE and E. These genes (or their products) could serve as potential biomarkers for BE.
Figure 4.

Flow diagram showing the selection process for candidate genes over expressed in BE. From the 805 genes that were significantly upregulated in both BE vs. E and BE [LCM] vs. E [LCM], there were 52 which were at least 100 fold more highly expressed in BE [LCM] vs. E [LCM]. Among these 52 genes, there were 41 which showed at least 10 fold higher expression in all nine patients and were considered to be the most likely biomarkers for BE.
Table 1.
Candidate gene markers for BE based on universal elevation in 9 patients
| GENE NAME | MOLECULAR FUNCTION | BIOLOGICAL PROCESS | CELLULAR COMPONENT |
|---|---|---|---|
| Annexin A10 | Ca ion binding Ca-dependent phospholipid binding |
Mitochondrion | |
| Aldolase B, fructose-bisphosphate | Fructose-bisphosphate aldolase activity lyase activity |
fructose metabolic process glycolysis metabolic process |
Cytoplasm |
| Amiloride binding protein 1 (amine oxidase (copper-containing)) | Amine oxidase activity calcium ion binding copper ion binding drug binding heparin binding oxidoreductase activity |
Metabolic process | Perixome |
| Calmodulin-like 4 | Calcium ion binding | ||
| Cathepsin E | cathepsin E activity pepsin A activity peptidase activity |
antigen processing and presentation of exogenous peptide antigen via MHC class II digestion proteolysis |
Endosome |
| claudin 18 | identical protein binding structural molecule activity |
calcium-independent cell-cell adhesion | cell junction integral to membrane membrane tight junction |
| fatty acid binding protein 1, liver | chromatin binding lipid binding lipid transporter activity transporter activity |
cell-cell signaling fatty acid metabolic process organ morphogenesis transport |
cytoplasm |
| GATA binding protein 6 | RNA polymerase II transcription factor activity, enhancer binding chromatin binding metal ion binding protein binding sequence-specific DNA binding transcription activator activity zinc ion binding |
liver development muscle development positive regulation of transcription positive regulation of transcription from RNA polymerase II promoter regulation of transcription, DNA-dependent transcription |
nucleus |
| glycoprotein A33 (transmembrane) | Receptor activity | integral to plasma membrane plasma membrane |
|
| golgi phosphoprotein 2 | |||
| hephaestin | copper ion binding copper ion transmembrane transporter activity iron ion binding metal ion binding oxidoreductase activity |
copper ion transport ion transport iron ion transport |
integral to membrane membrane |
| hydroxysteroid (17-beta) dehydrogenase 2 | estradiol 17-beta-dehydrogenase activity oxidoreductase activity |
estrogen biosynthetic process metabolic process |
endoplasmic reticulum membrane integral to membrane |
| keratin 20 | structural constituent of cytoskeleton | Biological process | cytoplasm intermediate filament |
| lectin, galactoside-binding, soluble, 4 (galectin 4) | Sugar binding | Cell adhesion | cytosol plasma membrane |
| mucin 13, cell surface associated | integral to membrane | ||
| N-acetyltransferase 2 (arylamine N-acetyltransferase) | acetyltransferase activity arylamine N-acetyltransferase activity transferase activity |
Metabolic process | cytoplasm |
| nuclear receptor subfamily 1, group I, member 2 | metal ion binding protein binding sequence-specific DNA binding steroid hormone receptor activity transcription coactivator activity transcription factor activitytranscription repressor activity zinc ion binding |
negative regulation of transcription regulation of transcription, DNA-dependent signal transduction steroid metabolic process transcription xenobiotic metabolic process |
nucleus |
| olfactomedin 4 | latrotoxin receptor activity | membrane | |
| prominin 1 | response to stimulus visual perception |
brush border extracellular space integral to plasma membrane membrane microvillus |
|
| serine peptidase inhibitor, Kazal type 1 | protein binding serine-type endopeptidase inhibitor activity |
||
| serine peptidase inhibitor, Kazal type 4 | serine-type endopeptidase inhibitor activity | ||
| solute carrier family 3 (cystine, dibasic and neutral amino acid transporters, activator of cystine, dibasic and neutral amino acid transport), member 1 | L-cystine transmembrane transporter activity basic amino acid transmembrane transporter activity catalytic activity cation binding |
L-cystine transport amino acid metabolic process basic amino acid transport carbohydrate metabolic process transport |
integral to plasma membrane membrane membrane fraction mitochondrial inner membrane |
| sulfotransferase family, cytosolic, 1C, member 1 | |||
| tetraspanin 1 | cell adhesion cell motility cell proliferation |
integral to membrane membrane |
|
| transmembrane channel-like 5 | integral to membrane | ||
| trefoil factor 1 (breast cancer, estrogen-inducible sequence expressed in) | growth factor activity protein binding |
carbohydrate metabolic process cell differentiation defense response digestion negative regulation of cell proliferation |
|
| Trefoil factor 3 | defense response digestion |
extracellular region secretory granule |
|
| trinucleotide repeat containing 9 | DNA binding | regulation of transcription, DNA-dependent | chromatin nucleus |
| UDP glucuronosyltransferase 2 family, polypeptide B15 | glucuronosyltransferase activity | metabolic process steroid metabolic process xenobiotic metabolic process |
endoplasmic reticulum integral to membrane membrane microsome |
| leucine rich repeat containing 31 | protein binding | ||
| ornithine carbamoyltransferase | amino acid binding ornithine carbamoyltransferase activity transferase activity |
amino acid biosynthetic process arginine biosynthetic process urea cycle |
mitochondrial inner membrane mitochondrial matrix mitochondrion ornithine carbamoyltransferase complex |
| phosphatidylinositol-4-phosphate 5-kinase, type I, beta | 1-phosphatidylinositol-4-phosphate 5-kinase activity kinase activity protein binding transferase activity |
phosphatidylinositol metabolic process phosphorylation |
cellular_component membrane |
| MAWD binding protein | catalytic activity IEA isomerase activity |
biological_process biosynthetic process |
cellular_component |
| villin 1 | actin binding calcium ion binding protein binding |
actin filament bundle formation actin filament severing barbed-end actin filament capping cytoskeleton organization and biogenesis protein complex assembly |
F-actin capping protein complex cytoplasm cytoskeleton |
| chromosome 1 open reading frame 121 | |||
| alcohol dehydrogenase 1A (class I) | alcohol dehydrogenase activity, zinc-dependent metal ion binding oxidoreductase activity zinc ion binding |
Alcohol metabolic process | cytoplasm |
| Keratin 8 | protein binding structural molecule activity |
cytoskeleton organization and biogenesis | intermediate filament |
| BCL2-like 14 (apoptosis facilitator) | protein binding | regulation of apoptosis | cytoplasm cytosol intracellular organelle membrane |
| melanophilin | Rab GTPase binding actin binding metal ion binding microtubule plus-end binding myosin V binding protein binding zinc ion binding |
melanocyte differentiation melanosome localization pigmentation protein targeting |
actin cytoskeleton cytoplasm |
| dopa decarboxylase (aromatic L-amino acid decarboxylase) | aromatic-L-amino-acid decarboxylase activity carboxy-lyase activity lyase activity pyridoxal phosphate binding |
amino acid and derivative metabolic process carboxylic acid metabolic process catecholamine biosynthetic process |
|
| IQ motif containing GTPase activating protein 2 | GTPase inhibitor activity Ras GTPase activator activity actin binding calmodulin binding |
regulation of small GTPase mediated signal transduction signal transduction |
actin cytoskeleton intracellular |
Discussion
By assessing the number of genes that were found to be differentially expressed between BE and normal esophagus, it is clear that the LCM samples (3443 differentially expressed genes compared to controls) provides a more in depth picture of the BE phenotype than is obtained from the whole tissue biopsy samples (1797 differentially expressed genes). For studies of molecular pathways that may be important to the development of BE from squamous epithelium, LCM samples would be the methodology of choice as it leads to better BE phenotype identification at the molecular level. For the identification of markers of BE, our studies show that the LCM derived samples not only had a larger number of differentially expressed genes, but showed more genes that had a high magnitude of differential expression.
Cluster analysis (Fig. 1) as well as the observation that genes most elevated in BE compared to normal esophageal tissues (Fig. 4) in all study subjects indicated that inter-patient variability was relatively low. In our studies, the major source of variation was cell or tissue type suggesting a distinct gene expression pattern for BE compared to any other cell type in the normal esophagus.
To obtain a list of genes that might serve as a BE specific set of markers, we selected genes whose expression differed by 100 fold or greater in the LCM derived samples and asked how many of the 9 patients in this study showed a significant (10-fold) change of these candidate markers. Of the 52 genes that formed a putative BE specific profile, 41 were indeed over expressed by at least 10 fold in all 9 patients under study regardless of method of sampling. This list of candidate marker/prognostic genes for further analysis in a larger study, and one or more of the 41 genes might ultimately be used as biomarkers for BE. These biomarkers could be used to distinguish BE without neoplasia (as in this case) for BE with neoplasia, or distinguish BE from contigious similar tissue such as the gastric cardia. However, replication of our findings is essential given the relatively small sample size and the possibilities of bias. The BE candidate gene list will serve as the basis for future studies of the proteins encoded by these mRNAs using immunohistochemistry (IHC).
The extent of overlap between LCM and whole biopsy samples was high, with 74% of the genes differentially expressed in whole tissue being confirmed as differentially expressed in LCM samples. Whole tissue specimens, which are much less technically demanding to obtain than LCM samples, will be more easily adapted to clinical settings both for RNA or IHC analysis.
Several of the 41 candidate markers of BE deserve additional comment. Trefoil factors, TFF1 and TFF3 were significantly upregulated in BE vs. E. Trefoil factors are a group of small protease resistant peptides secreted by gastrointestinal epithelial cells. TFF1 is expressed at a high level in gastric epithelial cells and several reports describe an important role in maintaining the integrity of gastric mucosa. TFF1-3 expression was associated with restitution of the intestinal mucosa in infants with necrotizing enterocholitis(10). Hoffmann (11)describes the many roles of TFFs in mucosal repair and notes that rapid repair reduces inflammation, which is associated with cancer progression. TFF1 knockout mice exhibit increased tumors of the intestinal tract implicating TFFs as tumor suppressors.(12) The fact that expression of TFFs are elevated in our samples of BE may suggest that tissue injury is associated with the presence of BE cells and that, in this premalignant cell type, TFF expression is a repair response to injury and/or inflammation. Consistent with the findings in knockout mice, the loss of TFF-1 was demonstrated late in the progression of BE to adenocarcinoma(13).
We also found GATA-6 to be significantly upregulated in BE vs. E. GATA-6 is likely to be responsible for the elevation of trefoil factor expression as consensus sites for GATA-6 have been identified in the promoter region of the TTF genes and cotranfection studies have shown that GATA-6 regulated expression of TFF1 and TFF2.(14) GATA-6 has been implicated in other tumor types as mRNA and protein were reported to be upregulated colon cancer, pediatric yolk sac tumors and teratomas(15;16).
Mucin 13 is one member of a large family of epithelial glycoproteins, many of which have been associated with cancers of the gastrointestinal tract(17). Mucin 13 elevation has been reported in colorectal cancers(18), gastric cancer, and intestinal metaplasia(19).
Lastly, we found that keratin 20 was significantly upregulated in BE vs. E. Although the pattern of cytokeratin 20 staining allowed differentiation of BE from that of gastric IM (20;21) in some studies, others reported low test accuracy(22). Our data would suggest that keratin 20 deserves additional analysis as a Barrett's biomarker.
The differences in gene expression profiles depending on whether specimens are whole tissue biopsies or LCM can provide valuable information regarding the properties of Barrett's cells. A considerable number of differentially expressed genes were detected in LCM derived tissues that were not found in the data from whole tissue. We recommend that studies of prognostic markers should focus on genes that are commonly expressed in both whole tissue as well as LCM specimens. Further studies aiming at internal and external validation (e.g., employing RT-PCR, in situ hybridization and immunohistochemistry) of potential markers provided in the current study is warranted.
Acknowledgments
This work was supported in part by R21DK067366-02 (HES) and Public Health Service grant DK56338, which funds the Texas Medical Center Digestive Diseases Center
Reference List
- 1.Brabender J, Marjoram P, Salonga D, Metzger R, Schneider PM, Park JM, Schneider S, Holscher AH, Yin J, Meltzer SJ, Danenberg KD, Danenberg PV, et al. A multigene expression panel for the molecular diagnosis of Barrett's esophagus and Barrett's adenocarcinoma of the esophagus. Oncogene. 2004 Jun 10;23(27):4780–8. doi: 10.1038/sj.onc.1207663. [DOI] [PubMed] [Google Scholar]
- 2.Selaru FM, Zou T, Xu Y, Shustova V, Yin J, Mori Y, Sato F, Wang S, Olaru A, Shibata D, Greenwald BD, Krasna MJ, et al. Global gene expression profiling in Barrett's esophagus and esophageal cancer: a comparative analysis using cDNA microarrays. Oncogene. 2002 Jan 17;21(3):475–8. doi: 10.1038/sj.onc.1205111. [DOI] [PubMed] [Google Scholar]
- 3.Xu Y, Selaru FM, Yin J, Zou TT, Shustova V, Mori Y, Sato F, Liu TC, Olaru A, Wang S, Kimos MC, Perry K, et al. Artificial neural networks and gene filtering distinguish between global gene expression profiles of Barrett's esophagus and esophageal cancer. Cancer Res. 2002 Jun 15;62(12):3493–7. [PubMed] [Google Scholar]
- 4.Barrett MT, Yeung KY, Ruzzo WL, Hsu L, Blount PL, Sullivan R, Zarbl H, Delrow J, Rabinovitch PS, Reid BJ. Transcriptional analyses of Barrett's metaplasia and normal upper GI mucosae. Neoplasia. 2002 Mar;4(2):121–8. doi: 10.1038/sj.neo.7900221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El-Serag HB, Nurgalieva Z, Souza RF, Shaw C, Darlington G. Is genomic evaluation feasible in endoscopic studies of Barrett's esophagus? A pilot study. Gastrointest Endosc. 2006 Jul;64(1):17–26. doi: 10.1016/j.gie.2005.08.041. [DOI] [PubMed] [Google Scholar]
- 6.genechip. Expression Analysis Technical Manual. Affymetrix Inc.; 2004. [Google Scholar]
- 7.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003 Feb 15;31(4):e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 9.On-Line Help [computer program]. Version 6.3 beta, build 6.07.0905. 2008 [Google Scholar]
- 10.Vieten D, Corfield A, Carroll D, Ramani P, Spicer R. Impaired mucosal regeneration in neonatal necrotising enterocolitis. Pediatr Surg Int. 2005 Mar;21(3):153–60. doi: 10.1007/s00383-004-1312-6. [DOI] [PubMed] [Google Scholar]
- 11.Hoffmann W. Trefoil factors TFF (trefoil factor family) peptide-triggered signals promoting mucosal restitution. Cell Mol Life Sci. 2005 Dec;62(24):2932–8. doi: 10.1007/s00018-005-5481-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lefebvre O, Chenard MP, Masson R, Linares J, Dierich A, LeMeur M, Wendling C, Tomasetto C, Chambon P, Rio MC. Gastric mucosa abnormalities and tumorigenesis in mice lacking the pS2 trefoil protein. Science. 1996 Oct 11;274(5285):259–62. doi: 10.1126/science.274.5285.259. [DOI] [PubMed] [Google Scholar]
- 13.Fox CA, Sapinoso LM, Zhang H, Zhang W, McLeod HL, Petroni GR, Mullick T, Moskaluk CA, Frierson HF, Hampton GM, Powell SM. Altered expression of TFF-1 and CES-2 in Barrett's Esophagus and associated adenocarcinomas. Neoplasia. 2005 Apr;7(4):407–16. doi: 10.1593/neo.04715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Al-azzeh ED, Fegert P, Blin N, Gott P. Transcription factor GATA-6 activates expression of gastroprotective trefoil genes TFF1 and TFF2. Biochim Biophys Acta. 2000 Feb 29;1490(3):324–32. doi: 10.1016/s0167-4781(00)00013-0. [DOI] [PubMed] [Google Scholar]
- 15.Shureiqi I, Zuo X, Broaddus R, Wu Y, Guan B, Morris JS, Lippman SM. The transcription factor GATA-6 is overexpressed in vivo and contributes to silencing 15-LOX-1 in vitro in human colon cancer. FASEB J. 2007 Mar;21(3):743–53. doi: 10.1096/fj.06-6830com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siltanen S, Heikkila P, Bielinska M, Wilson DB, Heikinheimo M. Transcription factor GATA-6 is expressed in malignant endoderm of pediatric yolk sac tumors and in teratomas. Pediatr Res. 2003 Oct;54(4):542–6. doi: 10.1203/01.PDR.0000081295.56529.E9. [DOI] [PubMed] [Google Scholar]
- 17.Byrd JC, Bresalier RS. Mucins and mucin binding proteins in colorectal cancer. Cancer Metastasis Rev. 2004 Jan;23(12):77–99. doi: 10.1023/a:1025815113599. [DOI] [PubMed] [Google Scholar]
- 18.Walsh MD, Young JP, Leggett BA, Williams SH, Jass JR, McGuckin MA. The MUC13 cell surface mucin is highly expressed by human colorectal carcinomas. Hum Pathol. 2007 Jun;38(6):883–92. doi: 10.1016/j.humpath.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 19.Shimamura T, Ito H, Shibahara J, Watanabe A, Hippo Y, Taniguchi H, Chen Y, Kashima T, Ohtomo T, Tanioka F, Iwanari H, Kodama T, et al. Overexpression of MUC13 is associated with intestinal-type gastric cancer. Cancer Sci. 2005 May;96(5):265–73. doi: 10.1111/j.1349-7006.2005.00043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jovanovic I, Tzardi M, Mouzas IA, Micev M, Pesko P, Milosavljevic T, Zois M, Sganzos M, Delides G, Kanavaros P. Changing pattern of cytokeratin 7 and 20 expression from normal epithelium to intestinal metaplasia of the gastric mucosa and gastroesophageal junction. Histol Histopathol. 2002 Apr;17(2):445–54. doi: 10.14670/HH-17.445. [DOI] [PubMed] [Google Scholar]
- 21.Shen B, Ormsby AH, Shen C, Dumot JA, Shao YW, Bevins CL, Gramlich TL. Cytokeratin expression patterns in noncardia, intestinal metaplasia-associated gastric adenocarcinoma: implication for the evaluation of intestinal metaplasia and tumors at the esophagogastric junction. Cancer. 2002 Feb 1;94(3):820–31. doi: 10.1002/cncr.10215. [DOI] [PubMed] [Google Scholar]
- 22.Nurgalieva Z, Lowrey A, El-Serag HB. The use of cytokeratin stain to distinguish Barrett's esophagus from contiguous tissues: a systematic review. Dig Dis Sci. 2007 May;52(5):1345–54. doi: 10.1007/s10620-006-9399-3. [DOI] [PubMed] [Google Scholar]