

Abstract
Herpesviruses encode numerous microRNAs (miRNAs), most of whose functions are unknown. The Kaposi's sarcoma-associated herpesvirus (KSHV) encodes 17 known miRNAs as part of its latency program, leading to speculation that these RNAs might function, in part, to regulate the latent state. Here we show that at least one of these miRNAs, miRK9*, targets a sequence in the 3′ UTR of the mRNA encoding the major lytic switch protein (RTA), which controls reactivation from latency. Ectopic expression of this miRNA impairs RTA synthesis, while specific antagonism of miRK9* in latently infected cells enhances the frequency of spontaneous lytic reactivation 2-3 fold. Mutation of the recognition sequence in the RTA 3′UTR abolishes RTA downregulation by miRK9*. We propose that miRNA targeting of RTA, while not the primary regulator of the lytic switch, functions like a safety mechanism on the trigger of lytic reactivation, preventing stochastic variations in basal RTA transcription from triggering inappropriate entry into the lytic cycle.
Keywords: microRNA, KSHV, latency, herpesvirus replication, Kaposi's sarcoma
Introduction
MicroRNAs are ∼22 nt single-stranded RNAs that act post-transcriptionally to regulate the expression of large numbers of genes in eukaryotic genomes. (Filipowicz et al., 2008). It is estimated that human cells encode more than four hundred miRNAs (Bartel, 2009; Landgraf et al., 2007), and genome – wide transcriptome (Grimson et al., 2007; Lim et al., 2005) and proteome (Baek et al., 2008; Selbach et al., 2008) profiling have led to estimates that each miRNA may directly target 100-200 transcripts. These numbers suggest that the scope and complexity of miRNA action on the transcriptome is likely to be substantial.
A key determinant of miRNA targeting is the so-called seed sequence, nucleotides 2-7 of the microRNA (Bartel, 2009). MicroRNA targets usually display extensive complementarity to this seed region, most commonly in their 3′UTRs. However, the brevity of the seed means that it alone likely cannot account for all of the observed specificity of miRNA action – after all, any given 6 nt sequence occurs by chance every few kilobases in the mammalian genome. It seems likely that additional information outside the seed contributes to target recognition (Grimson et al., 2007), but our understanding of such contributions remains primitive.
Like their hosts, many human and animal viruses also encode miRNAs, (Cullen, 2009; Sullivan and Ganem, 2005); such miRNAs are particularly prominent in herpesviruses (Grundhoff et al., 2006; Pfeffer et al., 2005); Cai et al., 2005; Dunn et al., 2005; Grey et al., 2005; Grundhoff et al., 2006; Pfeffer et al., 2005; Samols et al., 2005; Umbach et al., 2008). Herpesviruses are large DNA viruses that can support two distinct genetic programs: latency, in which the viral genome persists in the cell but with very restricted gene expression, and lytic growth, in which all viral genes are expressed in a temporally regulated cascade, leading to death of the cell and progeny virus production.
The Kaposi's sarcoma-associated herpesvirus (KSHV) encodes 17 miRNAs, which are derived by processing from 12 pre-miRNAs, all of which are expressed during viral latency (Cai et al., 2005; Grundhoff et al., 2006; Samols et al., 2005). Because of their association with latency, it has been speculated that they may control or modulate the latent-lytic switch. In keeping with this idea, recent studies in HSV (Umbach et al 2008) and HCMV (Murphy et al., 2008) have identified viral miRNAs that regulate immediate-early regulatory proteins. However, because of the absence of tractable cell culture–based models of latency in these viruses, it has not yet been possible to examine the functional consequences of these interactions for the latent-lytic decision. Here we describe the identification of a KSHV miRNA that directly affects latency regulation by modulating the expression of the master regulator of the latent-lytic switch, the RTA protein (Replication and Transcription Activator).
Results
Screening for KSHV miRNAs that target RTA
The default pathway of gene expression in KSHV is latency; however, latently infected cells can be induced to undergo lytic replication by a variety of chemical or genetic stimuli. Induction is controlled by expression of the virus-encoded transcription factor RTA, the master regulator of the latent-lytic switch. Ectopic expression of RTA triggers reactivation from latency, and mutational inactivation of RTA ablates reactivation (Lukac et al., 1999; Lukac et al., 1998; Sun et al., 1998; Xu et al., 2005). Because it resides at a critical node in KSHV's regulatory network, we reasoned that it would be a likely target of miRNA regulation. To determine if any of the KSHV-encoded microRNAs regulate RTA expression, we cloned the 3′UTR of RTA downstream of the Renilla luciferase gene in a reporter plasmid psiCHECK-2. Following cotransfection of the plasmid with each of the individual synthetic KSHV miRNA precursors (or a control miRNA) into 293 cells, the Renilla luciferase activity in the cells was measured, and normalized to the amount of a control Firefly luciferase gene bearing its own 3′UTR expressed from the same plasmid. The normalized luciferase value observed in the presence of a control miRNA was set to 1.0, and the levels of luciferase generated in the presence of the KSHV miRNAs were expressed relative to this value. Although there was some scatter in this assay, only one viral miRNA, miRK9*, consistently reduced luciferase expression from the LUC/RTA chimera (Figure 1A); this reduction was highly statistically significant (p<0.005, by T-Test). The level of this inhibition, ca. 30%, is typical of that observed by known miRNA-target interactions in this assay [see (Ziegelbauer et al., 2009) and references therein]. (The smaller inhibitions seen with miRs K8, K9 and K11 in Fig 1A were not reproducible in other replicates of this experiment).
Figure 1. miR-K9* targets the RTA 3′ UTR and inhibits RTA function.
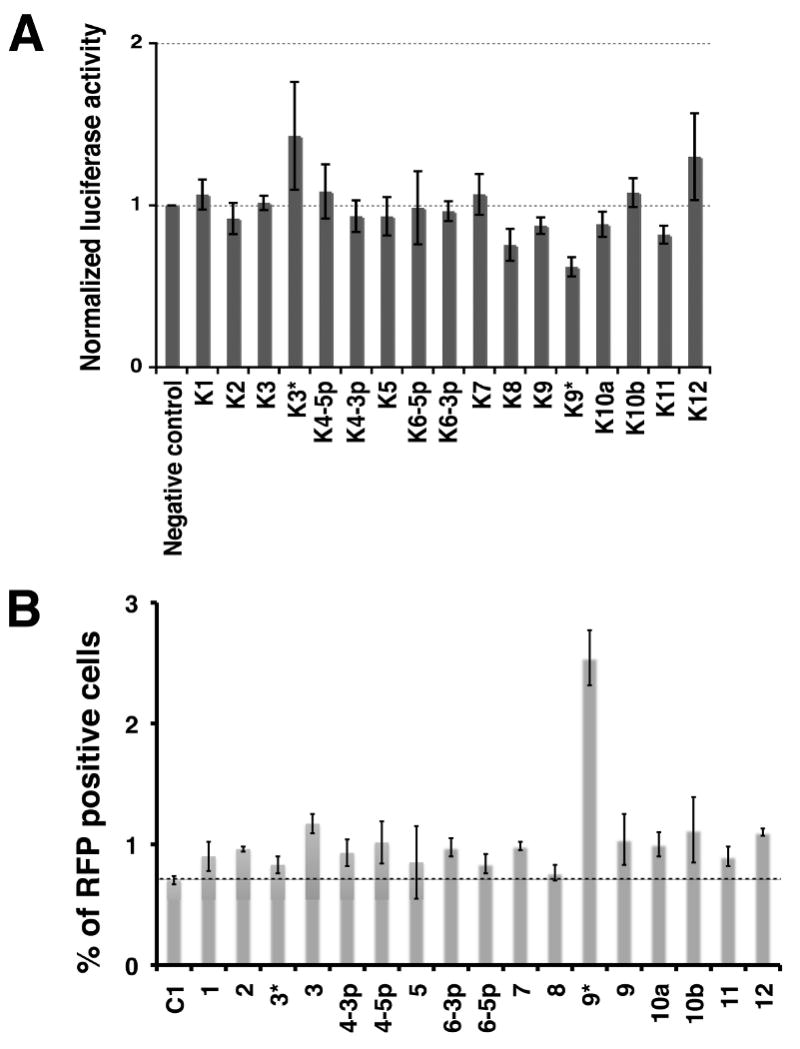
A. HEK293 cells were cotransfected with a psiCHECK2 vector in which the 3′UTR of RTA was cloned downstream of the Renilla luciferase gene, and individual KSHV-encoded microRNAs (indicated along the X-axis). The Renilla/Firefly ratio was normalized to a vector that did not contain the 3′UTR and to a negative control microRNA. Bars depict the mean values of the normalized luciferase activities from triplicate independent transfections. The difference between the negative control and miRK9* samples is highly significant (p= 0.003 by t-test).
B. HFF cells latently infected with rKSHV.219 were transfected with individual inhibitors to individual KSHV-encoded microRNAs (indicated along the X-axis). 48 hours later, spontaneous lytic reactivation was measured as a % of RFP-positive cells from a population of healthy, GFP-expressing (infected) cells. Mean values of the normalized % RFP-positive cells were from five independent transfections for miRK9* and negative control inhibitors and from two independent transfections for the other microRNA inhibitors. The P-value between the negative control and miRK9* samples is 0.0013 as determined by T-test.
Inhibiting microRNA K9* in infected cells stimulates spontaneous lytic reactivation
To explore whether miRK9* might target RTA in the context of an authentic latent viral infection, we set up a second screen in which 2′O-methyl modified oligonucleotides antisense to each of the 17 viral miRNAs were transfected into cells bearing a latent KSHV genome. The virus genome employed in this experiment (rKSHV.219 (Vieira and O'Hearn, 2004)) is genetically marked, bearing a GFP gene driven by a cellular promoter that can be expressed in latently infected cells, and an RFP gene that is under the control of an RTA-responsive lytic promoter and is turned on only in lytically infected cells expressing RTA. Accordingly, if a miRNA targets RTA, transfection with its cognate inhibitor should upregulate RTA expression and trigger an increase in RFP-positive cells. To avoid complications from exogenous (nonphysiologic) chemical inducers, we first looked at spontaneous lytic reactivation. We tested two cell lines latently infected with rKSHV.219 – these were derived by infection of human foreskin fibroblasts (HFF) and SLK endothelial cells, respectively. Following transfection with each miRNA antagonist, cells were examined by flow cytometry for GFP and RFP expression. Figure 1B shows the results for HFF. As can be seen, only the antagonist of miRK9* appreciably upregulated RFP expression in this context, nearly tripling the frequency of spontaneous lytic reactivation (p<0.005). Similar results were obtained in SLK cells (data not shown). In a separate experiment with SLK cells, titration of the miRK9* inhibitor showed there was up to a four fold increase in RFP expression within about 72 hours after transfection. This experiment was carried out with a negative control inhibitor as well as additional control inhibitors to miRK4-3p and miRK12 that showed a negligible effect on lytic reactivation (Supplementary Figure S1). These results strongly indicate that miRK9*, when expressed at physiologic levels in latency, downregulates RTA.
Although one might expect the effects of miRNAs on RTA expression to be overwhelmed by strong transcriptional induction of the target gene, we nonetheless asked if it was possible to detect effects of miRK9* under conditions of induced lytic reactivation, again using HFF/rKSHV.219 cells. Sodium butyrate, an HDAc inhibitor, is a potent chemical inducer of lytic reactivation. Cells were induced with 10mM sodium butyrate, and examined at several time points post infection for RTA expression by immunoblotting with antisera specific to this protein. As shown in Fig 2, transfection of the inhibitor to miRK9* resulted in clearly elevated levels of RTA accumulation at both 18 and 26 hours post infection; this elevation was much reduced at 47 hours, indicating that, as expected, the effect of miRK9* can be overcome by transcriptional induction of its target mRNA, which is quite strong by this time (Lukac et al 1999). Consistent with this, butyrate-induced cells that had been pre-treated with the miRK9* antagonist display a ∼35% increase in RFP expression compared to similarly induced cells pre-treated with the negative control inhibitor (Supplementary Fig S2). By contrast, transfection of a mix of inhibitors to miRK9, miRK5, miRK10a and miRK10b (which regulate BCLAF-1; Ziegelbauer et al 2009) caused the expected modest antagonism of RFP expression in the butyrate-induced cells (Supplementary Fig S2).
Fig 2. Inhibition of miRK9* during lytic growth raises levels of RTA.

HFF/rKSHV.219 cells were transfected with the indicated antagomir, then induced with butyrate. Extracts prepared at the indicated times post-induction were immunoblotted for RTA (and for β-actin as a loading control).
miR-K9* inhibits RTA protein expression in a transient transfection assay
In order to directly measure the effect of KSHV-encoded microRNAs on RTA expression, transient transfections of the individual microRNAs together with a plasmid expressing RTA were done in 293 cells. The RTA-expressing plasmid contained genomic KSHV DNA from the start of the RTA coding region to the polyadenylation site of the RTA transcript, expressing a spliced RTA message that mimics that observed in infected cells. Following transfections, lysates from the cells were analyzed by Western blotting. We found that of all the microRNAs, miRK9* most strongly inhibited the expression of RTA (Figure 3); quantification of this data revealed that miRK9* produced a 45% reduction in RTA accumulation. (We also noted a significant inhibition of RTA accumulation in the presence of miRK12, the basis of which is presently unclear. This miRNA did not score in the luciferase suppression assay of Fig 1A, nor did its inhibitor score in the lytic induction assay (Fig 1B). Clearly, further work will be necessary to determine if miR-K12 is truly a regulator of RTA).
Figure 3. miR-K9* inhibits the expression of RTA protein in a transient transfection assay.
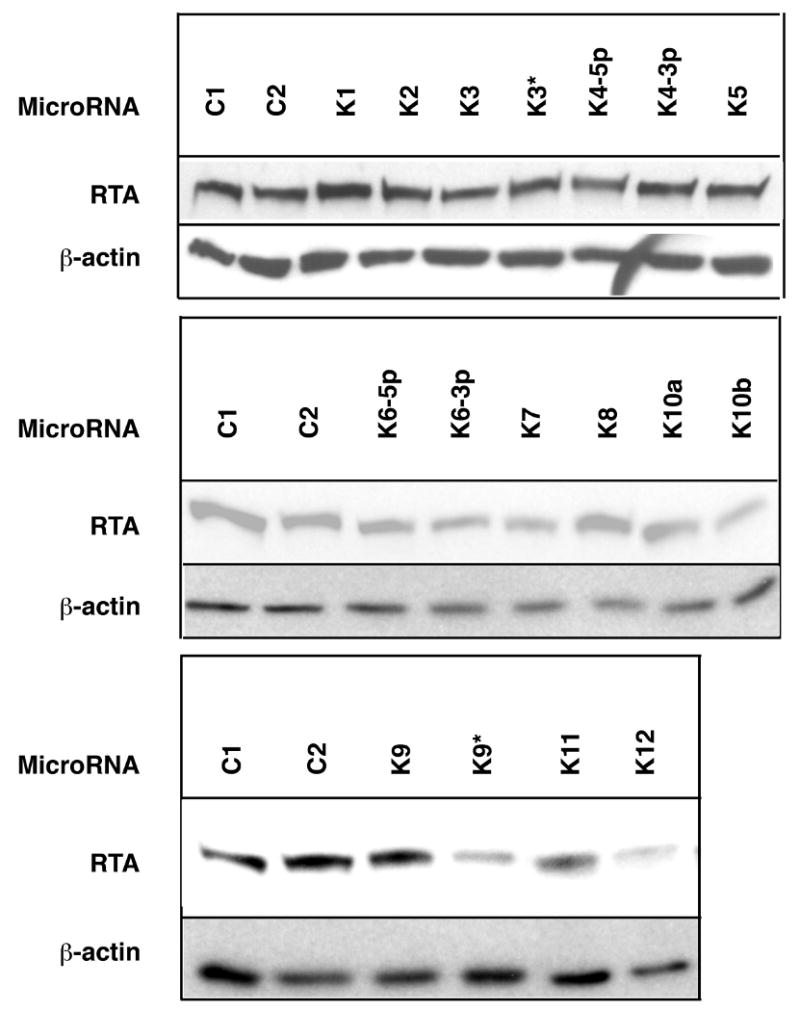
A genomic RTA expression vector bearing the full 3′UTR sequences was cotransfected with the indicated individual KSHV-encoded microRNAs; 48 hrs later, lysates were prepared and tested for RTA protein by immunoblotting. As a loading control, the same blots were probed with antibodies against β-actin.
Identifying the target site of miRK9* in the 3′UTR of RTA
A decisive factor influencing targeting by a microRNA is the presence of a sequence (usually within the target 3′UTR) that is strongly complementary to the microRNA seed region (nucleotides 2-7) (Bartel, 2009). When we searched the 3′UTR of RTA mRNA for seed complementarity, we identified a 6-mer complementary to positions 2 through 7 of miRK9*, located near the 3′ extremity of the UTR. Using the luciferase/RTA chimeric reporter, we mutated the nucleotides in this element that were complementary to positions 2, 3 and 4 of miRK9* so as to reduce base pairing with the seed region of the microRNA (Figure 4A). In addition, we designed a version of miRK9* which contained mutations that compensated for the above changes in the 3′UTR so as to restore Watson-Crick base pairing (Figure 4A). Each wildtype (WT) or mutant chimera was cotransfected with either a control miRNA, miRK9* or the mutant miRK9* bearing the compensating mutations, and luciferase activity measured (Figure 4B). As expected, wildtype miRK9* inhibited luciferase activity from the WT RTA chimera by about 30% (as also observed in Figure 1A); however, mutating either the miRNA or the mRNA target in this region abolished the downregulation (Fig 4B). Gratifyingly, however, when the mutated UTR was cotransfected with the seed-complementary mutant miRK9*, the inhibition of luciferase expression was efficiently rescued (Figure 4B). The more robust inhibition in the case of the mutated UTR and microRNA is likely due to the lower base-pairing stability at positions 2, 3 and 4 of the microRNA duplex (A-U instead of C-G) which allows it to be incorporated more efficiently into the RISC complex (Schwarz et al., 2003). These observations corroborate that RTA mRNA is a direct target of miRK9*, and define the predominant molecular target of its action.
Figure 4. Mapping the target site for miR-K9* within the 3′UTR of RTA.
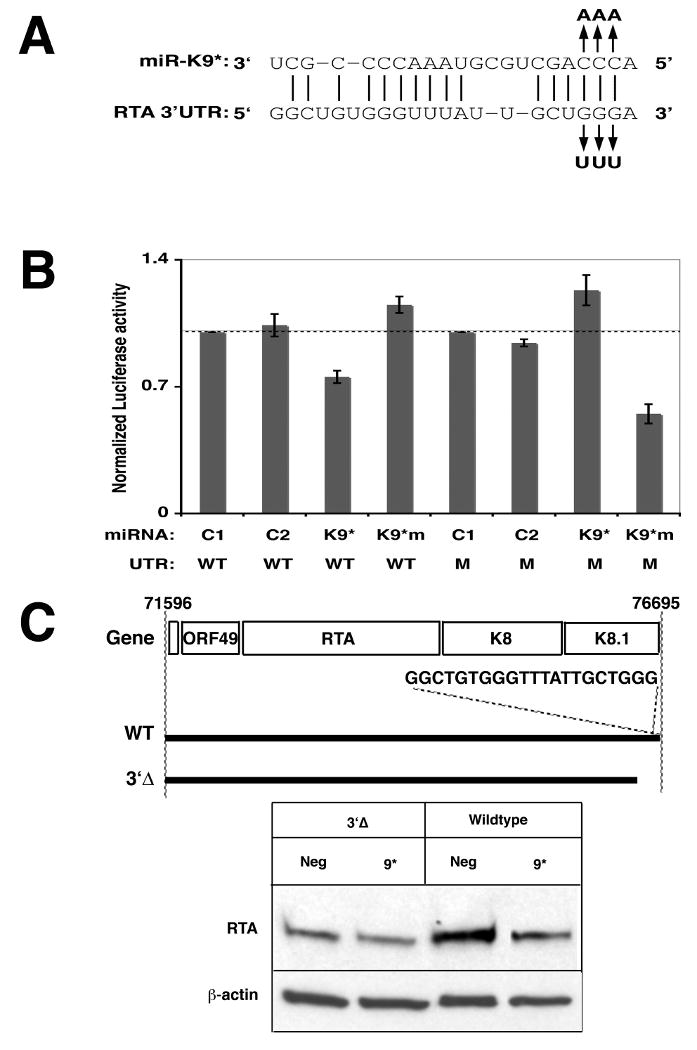
A. Predicted base pairing between miR-K9* and its putative site within the 3′UTR of RTA is shown. Watson-Crick base pairings are shown as vertical lines. Mutations made in the 3′UTR and miR-K9* are indicated above or below the wildtype sequences, respectively.
B. HEK293 cells were cotransfected with negative control 1 (C1), negative control 2 (C2), miR-K9* (K9*) or mutated miR-K9* (K9*m) microRNA together with the luciferase reporter plasmid containing the wildtype (W) or mutated (M) 3′UTR of RTA. The Renilla/Firefly ratio was normalized to a vector that did not contain the 3′UTR and to a negative control microRNA. Mean values of the normalized luciferase activities are from triplicate independent transfections. P-values taken from a t-test between C1 and K9* transfected with wildtype 3′UTR is 0.019 and between C1 and K9*m transfected with mutated 3′UTR is 0.014, reflecting statistically significant values.
C. Top: Schematic physical map of the WT and 3′Δ deletion mutant of KSHV RTA region. Bottom: HEK293 cells were cotransfected with plasmids encoding WT or 3′Δ mutant transcript of RTA and with negative control or miR-K9* (K9*) microRNA. 48 hours later cell lysates were probed for RTA protein and for β-actin as a loading control.
To examine the importance of the target site in the authentic RTA mRNA (as opposed to the reporter constructs employed above), we cotransfected miRK9* with a genomic RTA clone containing either (i) the full-length 3′UTR, or (ii) one in which the terminal 278 nucleotides of the UTR (including the mapped miRK9* target sequence) was deleted (3′Δ, Figure 4C, top). We observed that while miRK9* inhibited RTA expression from the full-length transcript, it was unable to regulate RTA when the transcript lacked the mapped target site (Figure 4C, bottom). When the genomic RTA clone expressing the full-length transcript was cotransfected with the miRK9* or the seed-mutant version of miRK9*, decrease in RTA protein expression was observed only in the presence of the wildtype miRK9* (Supplemental Figure S3) again pointing to the importance of the seed sequence in the microRNA.
Discussion
Reactivation from latency is a critical step in the replication and spread of herpesviruses. In gammaherpesviruses like KSHV and EBV, latency is the default pathway following de novo infection of most cell types. Reactivation from latency is required for spread of virus from cell to cell in an individual, and from person to person in the population (Ganem, 2006). So a full accounting of the molecular mechanisms regulating the latent-lytic switch is important to a nuanced understanding of KSHV biology.
A possible role for microRNAs in such regulation has recently been suggested by several observations. First, studies in HSV (Umbach et al 2008) and HCMV (Murphy et al., 2008) have identified viral microRNAs that regulate immediate-early genes that are needed for efficient lytic replication. It has been postulated that these interactions may modulate the latent-lytic switch; however, neither virus system has tractable in vitro latency models that allow experimental validation of this suggestion, which has therefore remained conjectural. Second, in a recent study of cellular targets of KSHV microRNAs, we found that targeting of the host BCLAF-1 transcript by miR-K5 had clear effects on the latent-lytic switch – in that case, sensitizing cells to lytic reactivation. With these precedents in mind, we set out to screen for viral microRNAs that could regulate the latent-lytic switch to stabilize latency. Our initial screens focused on RTA, the dominant viral factor controlling this switch.
In earlier work, Murphy et al (2008) employed a sophisticated bioinformatic algorithm to predict potential viral targets of HCMV-encoded miRNAs. Application of this algorithm to KSHV led them to propose that the 3′UTR of KSHV RTA might be targeted by miRK6-3p. However, this prediction could not be experimentally validated in our study. Rather, we found that miRK9* consistently impaired RTA expression, whether from a chimeric reporter gene or the authentic transcript, and it did so in the context of an authentic viral infection as well as in transfected cells. Moreover, genetic studies allowed us to unambiguously assign the major target site mediating this interaction. These observations underscore the difficulties and limitations of bioinformatic predictions of miRNA targets. We expect that continuing efforts to experimentally identify and characterize miRNA target sequences will help provide the information necessary to productively refine such algorithms and increase their predictive power.
We note with interest a recent study of KSHV clinical isolates that examined their degree of sequence variability in the region encoding the microRNAs (Marshall et al., 2007). In general, the KSHV-encoded miRNAs were highly conserved, but the pre-microRNA for miRK9* showed a somewhat larger number of sequence polymorphisms than most of the other KSHV pre-microRNAs. Most of these changes were outside of the mature miRNA; many may simply be polymorphisms with no phenotypic consequence, but some could theoretically affect processing efficiency of the pre-microRNA, and thereby the levels of mature miRK9*. Only 1 of 22 sequenced KSHV isolates had a (single) lesion in the seed sequence of miRK9* (C→U at position 2 of the miRNA); interestingly, this polymorphism would still support a G-U basepair at this position with the RTA transcript. Thus, based on their positions in the miRNA, most of the described changes would be predicted to have little consequence on RTA targeting. [The seed mutated variant, which comes from a B cell (effusion lymphoma) cell line, might have more substantial effects, especially since this miRNA has numerous other changes in the mature and pre-miRNA K9* sequence – however, the significance of this cannot be rigorously discerned in the absence of the sequence of its cognate RTA gene].
Finally, it is noteworthy that the phenotype we describe here for miRK9* - the stabilization of latency by attenuation of RTA accumulation – is the opposite of that of miRK5, which acts to enhance susceptibility to lytic reactivation (Ziegelbauer et al 2009). As might be predicted from what is known about microRNA action generally (Baek et al., 2008; Grimson et al., 2007; Selbach et al., 2008), both phenotypes are subtle, consistent with the idea that miRNA regulation is about the fine-tuning of gene expression, acting as rheostats (Baek et al., 2008) that modulate the output of other components of the regulatory circuitry. We speculate that by promoting the ability to up- or down-regulate susceptibility to lytic reactivation, these latent miRNAs afford the virus the flexibility it needs to adjust reactivation frequency in response to different environmental circumstances.
We wish to underscore that microRNA-mediated regulation is not the primary determinant of the latent-lytic switch – that role clearly belongs to the transcriptional regulation of RTA expression. Ablation of miRK9* function results in a discernable increase in lytic reactivation (ca two to three fold), but this effect is small by comparison with inducers of RTA transcription (which can enhance reactivation 10-40 fold). How might a latent miRNA that targets RTA actually contribute to the regulation of latency? At first blush this seems paradoxical, since, according to conventional notions of KSHV regulation, the RTA target gene is not expressed in latency. But this is an oversimplification. Sensitive RT-PCR measurements indicate that very low basal levels of expression exist for many inducible transcripts in the absence of inducing stimuli. This basal level (sometimes called “transcriptional noise”) is subject to considerable stochastic variation (Blake et al., 2006; Raser and O'Shea, 2004; Weinberger et al 2005). We propose that latent miRK9* expression serves as a safety mechanism that prevents stochastic variations in the levels of basal RTA transcripts from triggering inappropriate lytic reactivation. The fact that we can observe modest increases in lytic reactivation in the presence of antagonists to miRK9* (Fig 1B) allows an estimate of the frequency of such inadvertent activation events. But it also reaffirms that microRNA-mediated modulation of lytic reactivation is not the central determinant of the latent-lytic switch, but a subsequent evolutionary modification designed for fine-tuning of its control.
Experimental Procedures
Cell culture, plasmid transfections, luciferase assays and immunoblotting were carried out as described in Supplementary Materials
Transfection of microRNA inhibitors
The KSHV-encoded microRNA inhibitors were from Ambion. HFF and SLK cell lines expressing the recombinant KSHV (rKSHV.214) were transfected with the miRNA inhibitors at about 77nM and 115nM respectively using Dharmafect 1 (Dharmacon). Transfections were done in a 12-well format and 3 μL of Dharmafect 1 was used for each well. The rest of the procedure was as described by the manufacturer. Flow cytometry analysis was done one, three or five days after transfection for the SLK cells and two days after transfection for the HFF cells. The mean values of % RFP-positive cells were derived from atleast three (for SLK cells) or two (for HFF cells) independent transfections. The inhibitors were transfected into BCBL-1 cells as done before (Ziegelbauer et al., 2009) except that sequential transfections about 48 hours apart were performed. Before the second transfection cells were split to 200,000 cells/mL. Immunoflourescence on the cells was done about 48 hours after the second transfection. Mean values of % ORF59-positive cells were from duplicate independent transfections and from at least 5 different fields within each well.
Immunofluorescence and Flow Cytometry
BCBL-1 cells were washed with PBS and 200 μL of cells (at a concentration of 200,000 cells/mL) were added to each well of a 6-well slide and allowed to settle down for 30 minutes. Cells were then fixed with 4% Paraformaldehyde for 30 minutes and then washed with PBS. The rest of the procedure was as done before (Lukac et al., 1998) using polyclonal antibody to Latency associated nuclear antigen (LANA, provided by A. Polson) and monoclonal antibody to ORF59 (Advanced Biotechnologies Inc.) or a polyclonal antibody to RTA (Lukac et. al, 1998). Secondary anti-mouse rhodamine and anti-rabbit fluorescein isothiocyanate (FITC) (Santacruz Biotechnology) were used at 1:300. For flow cytometry cells were fixed in 4% paraformaldehyde, washed and their green and red fluorescence was measured using a Becton Dickinson FACSCalibur.
Supplementary Material
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake WJ, Balazsi G, Kohanski MA, Isaacs FJ, Murphy KF, Kuang Y, Cantor CR, Walt DR, Collins JJ. Phenotypic consequences of promoter-mediated transcriptional noise. Molecular cell. 2006;24:853–865. doi: 10.1016/j.molcel.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Cai X, Lu S, Zhang Z, Gonzalez CM, Damania B, Cullen BR. Kaposi's sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:5570–5575. doi: 10.1073/pnas.0408192102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen BR. Viral and cellular messenger RNA targets of viral microRNAs. Nature. 2009;457:421–425. doi: 10.1038/nature07757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn W, Trang P, Zhong Q, Yang E, van Belle C, Liu F. Human cytomegalovirus expresses novel microRNAs during productive viral infection. Cellular microbiology. 2005;7:1684–1695. doi: 10.1111/j.1462-5822.2005.00598.x. [DOI] [PubMed] [Google Scholar]
- Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nature reviews. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- Ganem D. KSHV infection and the pathogenesis of Kaposi's sarcoma. Annual review of pathology. 2006;1:273–296. doi: 10.1146/annurev.pathol.1.110304.100133. [DOI] [PubMed] [Google Scholar]
- Grey F, Antoniewicz A, Allen E, Saugstad J, McShea A, Carrington JC, Nelson J. Identification and characterization of human cytomegalovirus-encoded microRNAs. Journal of virology. 2005;79:12095–12099. doi: 10.1128/JVI.79.18.12095-12099.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Molecular cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundhoff A, Sullivan CS, Ganem D. RNA. Vol. 12. New York, NY: 2006. A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpesviruses; pp. 733–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- Lukac DM, Kirshner JR, Ganem D. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. Journal of virology. 1999;73:9348–9361. doi: 10.1128/jvi.73.11.9348-9361.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukac DM, Renne R, Kirshner JR, Ganem D. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology. 1998;252:304–312. doi: 10.1006/viro.1998.9486. [DOI] [PubMed] [Google Scholar]
- Marshall V, Parks T, Bagni R, Wang CD, Samols MA, Hu J, Wyvil KM, Aleman K, Little RF, Yarchoan R, et al. Conservation of virally encoded microRNAs in Kaposi sarcoma--associated herpesvirus in primary effusion lymphoma cell lines and in patients with Kaposi sarcoma or multicentric Castleman disease. The Journal of infectious diseases. 2007;195:645–659. doi: 10.1086/511434. [DOI] [PubMed] [Google Scholar]
- Martin DF, Kuppermann BD, Wolitz RA, Palestine AG, Li H, Robinson CA. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. Roche Ganciclovir Study Group. The New England journal of medicine. 1999;340:1063–1070. doi: 10.1056/NEJM199904083401402. [DOI] [PubMed] [Google Scholar]
- Murphy E, Vanicek J, Robins H, Shenk T, Levine AJ. Suppression of immediate-early viral gene expression by herpesvirus-coded microRNAs: implications for latency. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:5453–5458. doi: 10.1073/pnas.0711910105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer S, Sewer A, Lagos-Quintana M, Sheridan R, Sander C, Grasser FA, van Dyk LF, Ho CK, Shuman S, Chien M, et al. Identification of microRNAs of the herpesvirus family. Nature methods. 2005;2:269–276. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- Raser JM, O'Shea EK. Control of stochasticity in eukaryotic gene expression. Science (New York, NY. 2004;304:1811–1814. doi: 10.1126/science.1098641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samols MA, Hu J, Skalsky RL, Renne R. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi's sarcoma-associated herpesvirus. Journal of virology. 2005;79:9301–9305. doi: 10.1128/JVI.79.14.9301-9305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- Sullivan CS, Ganem D. MicroRNAs and viral infection. Molecular cell. 2005;20:3–7. doi: 10.1016/j.molcel.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:10866–10871. doi: 10.1073/pnas.95.18.10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature. 2008;454:780–783. doi: 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira J, O'Hearn PM. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology. 2004;325:225–240. doi: 10.1016/j.virol.2004.03.049. [DOI] [PubMed] [Google Scholar]
- Weinberger LS, Burnett JC, Toettcher JE, Arkin AP, Schaffer DV. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell. 2005;122:169–182. doi: 10.1016/j.cell.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Xu Y, AuCoin DP, Huete AR, Cei SA, Hanson LJ, Pari GS. A Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. Journal of Virology. 2005;79:3479–3487. doi: 10.1128/JVI.79.6.3479-3487.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegelbauer JM, Sullivan CS, Ganem D. Tandem array-based expression screens identify host mRNA targets of virus-encoded microRNAs. Nature Genetics. 2009;41:130–134. doi: 10.1038/ng.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.