

Abstract
Pak4 is a member of the B group of p21-activated (Pak) kinases, originally identified as an effector protein for Cdc42. Although Pak4 is expressed at low levels in most adult tissues, it is highly overexpressed in tumor cell lines. Here, we show that Pak4 is also overexpressed in primary tumors, including colon, esophageal, and mammary tumors. Overexpression of Pak4 also leads to tumor formation in athymic mice, whereas deletion of Pak4 inhibits tumorigenesis. Although a constitutively active Pak4 mutant was previously shown to promote oncogenic transformation in cultured cells, our results are the first to show that Pak4 also promotes tumorigenesis in experimental animals. Furthermore, these results show for the first time that not only constitutively active Pak4, but also wild-type Pak4, is transforming, when experimental animals are used. These results are highly significant because wild-type Pak4, rather than activated Pak4, is overexpressed in tumor cells. Our results suggest that overexpression or activation of Pak4 is a key step in oncogenic transformation, due to its ability to promote cell survival and subsequent uncontrolled proliferation. The finding that Pak4 is up-regulated in so many types of cancers indicates that Pak4 may play a vital role in a wide range of different types of cancer. This makes it an attractive candidate for drug therapy for different types of cancer.
Introduction
Normal development requires precisely regulated levels of cell survival, apoptosis, proliferation, and differentiation. Increased levels of cell survival, uncontrolled proliferation, or failure to differentiate are often associated with oncogenesis. Understanding the signaling pathways that control these cellular processes is essential for understanding the molecular basis of transformation. Protein kinases play key roles in the intracellular signaling pathways that regulate cell growth control. One group of protein kinases that has important roles in a number of different intracellular signaling pathways is the p21-activated kinase (Pak) family of serine/threonine kinases. The Paks were first identified as effector proteins for Cdc42 and Rac, members of the Rho GTPase family. More recently, they have also been found to have Rho GTPase–independent activators. The Paks fall into two categories, group A and group B, based on their sequences and functions. The group A family includes mammalian Pak1, Pak2, and Pak3 (1-3), whereas group B includes Pak4, Pak5, and Pak6 (4). All of the Paks have an amino-terminal regulatory domain and a carboxyl-terminal kinase domain, with a GTPase-binding domain within the regulatory domain. The group A and B Paks, however, differ significantly from each other in both sequence and function (4).
Among the group B Paks, Pak4 is highly expressed during development and is ubiquitously expressed at low levels in all adult tissues. In contrast, Pak5 and Pak6 are largely expressed in the brain. Pak4 was originally identified as a protein that promotes filopodia formation in response to activated Cdc42 and it is an important link between Cdc42 and filopodia formation (5). Pak4 also leads to the dissolution of stress fibers and subsequent loss of focal adhesions, possibly due to inhibition of Rho activity (6). Although Pak4 is expressed at low levels in most adult tissues, it is highly overexpressed in almost every tumor cell line that has been tested (7). This is in sharp contrast to Pak6, which is highly expressed in a few adult tissues but is not overexpressed in most tumor cell lines (7). This suggests an important role for Pak4 in cell growth, survival, and proliferation, all of which are important for tumorigenesis. In fact, we and others have found that like activated Cdc42 (8-10), a constitutively active Pak4 mutant promotes anchorage-independent growth when overexpressed in immortalized fibroblasts (6). Anchorage-independent growth is an important hallmark of oncogenic transformation (6). Although normal adherent cells stop growing or die when they are not attached to a surface, cancer cells can survive and proliferate even when detached, leading to anchorage-independent growth and often to metastasis. The transforming ability of activated Pak4 is quite dramatic. In fact, the constitutively active Pak4 mutant is as efficient as oncogenic Ras, a very strong oncogene, in promoting anchorage-independent growth in cultured cells (6). Consistent with this effect, dominant negative Pak4 partially inhibits the formation of anchorage-independent foci in response to oncogenic Dbl in fibroblasts (6), and in some cells it also inhibits transformation by oncogenic Ras (7).
The mechanism by which the activated Pak4 mutant transforms cultured cells is unknown, but one possibility is that its role in cell survival contributes to its role in transformation. When overexpressed, Pak4, as well as several other Paks, is associated with protection from apoptosis (11, 12). Conversely, cells lacking Pak4 have an increased susceptibility toward apoptosis (13). Pak4 promotes cell survival by different mechanisms, depending on the stimulus. In response to serum withdrawal, Pak4 protects cells via a kinase-dependent mechanism that is associated with its ability to phosphorylate the proapoptotic protein Bad (12), similar to Pak1, which also protects cells from apoptosis (14, 15). In response to stimuli that activate death domain–containing receptors, however, such as tumor necrosis factor or Fas ligand, Pak4 functions via a kinase-independent mechanism. In this case, Pak4 inhibits the formation of the DISC complex that forms on the cytoplasmic side of the receptor, thereby inhibiting caspase-8 recruitment and activation (11). Studies using cells that lack Pak4 have also shown that Pak4 has a role in activating cell survival pathways, which lead to nuclear factor-κB and extracellular signal-regulated kinase (ERK) activation (13).
It is interesting that although the activated Pak4 mutant is transforming in cultured NIH3T3 cells, wild-type Pak4 does not transform cells in culture (6). Furthermore, although anchorage-independent growth in cultured cells is an important predictor of oncogenesis, there are many other criteria that define a cell as transformed. One very important requirement for defining cells as malignant is the ability to form tumors in experimental animals. Formation of tumors in athymic (nude) mice is often used as a test to determine whether cells are malignant and whether an oncogene is transforming. Here, we show that overexpression of Pak4 leads to tumor formation in athymic mice. We were also able to use our Pak4 knockout cells to show that Pak4 has an essential role in transformation downstream to activated Cdc42 and also plays a role in mediating transformation by oncogenic Ras. Interestingly, when using athymic mice as a model, even wild-type Pak4 is highly transforming. This is the first work to indicate a role for wild-type Pak4 in actually causing tumors to form when it is overexpressed. As part of this study, we also show that Pak4 is overexpressed in primary tumors, including colon, esophageal, and mammary tumors, rather than only in tumor cell lines. Taken together, our results suggest that Pak4 is a causative agent in tumor formation and not simply a side effect of transformation. Because Pak4 is up-regulated in several types of cancers, our results suggest that it could serve as a general mediator of oncogenic transformation, playing a vital role in a range of different types of cancer.
Results
Pak4 Is Overexpressed in Tumors
Previous studies showed that Pak4 is overexpressed in cancer cell lines (7). Cell lines, however, have gone through numerous passages and can be far removed from the actual primary tumor. We therefore cannot rule out the possibility that Pak4 overexpression is somehow a result of the culturing of these tumor cell lines. We were therefore interested in determining whether Pak4 is also overexpressed in primary tumors. To test this, we carried out Western blot analysis of primary tumor tissues from four sources. The first was primary human esophageal squamous cell carcinoma (16); the second was mouse colon tumor tissue; the third was rat mammary tumor tissue (17); and the fourth was human breast tumor tissue. For every condition, we also examined the corresponding normal tissue as a control. Although there was variability in the amount of overexpression, we found that in all of these types of cancer, there was a higher level of Pak4 protein in the tumor tissue compared with the normal control tissue (see Fig. 1). These results indicate that Pak4 is in fact overexpressed in primary tumors in addition to nearly every tumor or cell line that has been tested. Thus, Pak4 seems to be broadly expressed in different types of tumors rather than limited to a specific type of cancer.
FIGURE 1.
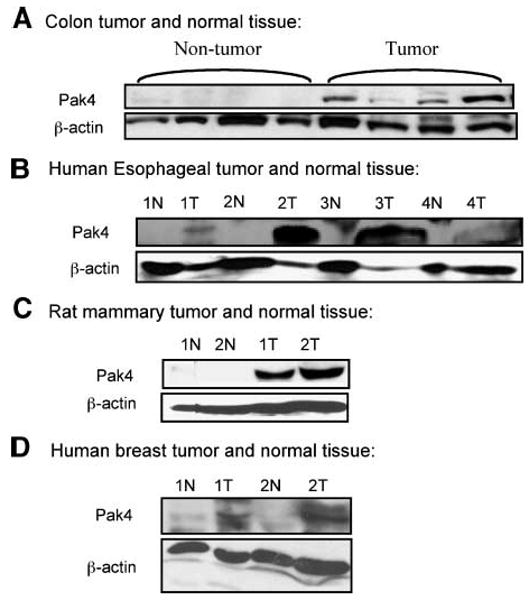
Pak4 is overexpressed in primary tumors. Western blot analysis illustrates Pak4 expression in tumor and normal tissues. The following tumor tissues were analyzed: A. Mouse colon tumor isolated from azoxymethane-treated CF-1 female mice; nontumor tissue was adjacent to normal tissue. B. Human esophageal squamous cell carcinoma (T) and normal tissues (N). Paired normal tissue was pathologically normal and from the same patient. C. Rat mammary tissue was originally isolated from N-methylnitrosourea treated Sprague-Dawley female rats; normal tissue was isolated from nontreated rats. D. Human breast tumor and normal tissues. Paired normal tissue was pathologically normal and from the same patient. In all cases, 100 μg of protein extract were used; β-actin served as a loading control.
Overexpression of Pak4 Leads to Tumor Formation in Athymic Mice
We next wanted to determine whether Pak4 is sufficient to promote oncogenesis. We previously showed that a constitutively active mutant of Pak4 promotes anchorage-independent growth in cultured immortalized fibroblasts (6). We did not, however, observe anchorage-independent growth in fibroblasts overexpressing wild-type Pak4. This was somewhat surprising because wild-type Pak4 promotes cell survival (11, 12), which is associated with tumorigenesis. We therefore determined whether wild-type Pak4 promotes tumorigenesis in the athymic mouse system, in addition to the activated Pak4 mutant.
NIH3T3 cells that stably overexpress wild-type Pak4, activated Pak4, or empty vector (pLPC; ref. 6) were injected subcutaneously into the flanks of athymic mice. Western blot analysis of a sample of each of these stable cell lines is shown in Fig. 2A. The different cell lines were injected s.c. into the flanks of athymic mice to determine whether they led to tumor formation. After 16 days, we found that cells containing activated Pak4 formed large tumors in the mice, whereas the control cells [containing empty vector (pLPC)] and the cells overexpressing wild-type Pak4 did not lead to tumor formation (see Fig. 2B). By 44 days, however, the cells overexpressing wild-type Pak4 also formed large tumors. The control cells still did not form tumors at this time point (see Fig. 2C) or at any time point tested. Tumor volumes and tumor weights at different time points are illustrated in Fig. 2D and E. From these results, it is clear that both wild-type and activated Pak4 can promote tumor formation in athymic mice.
FIGURE 2.
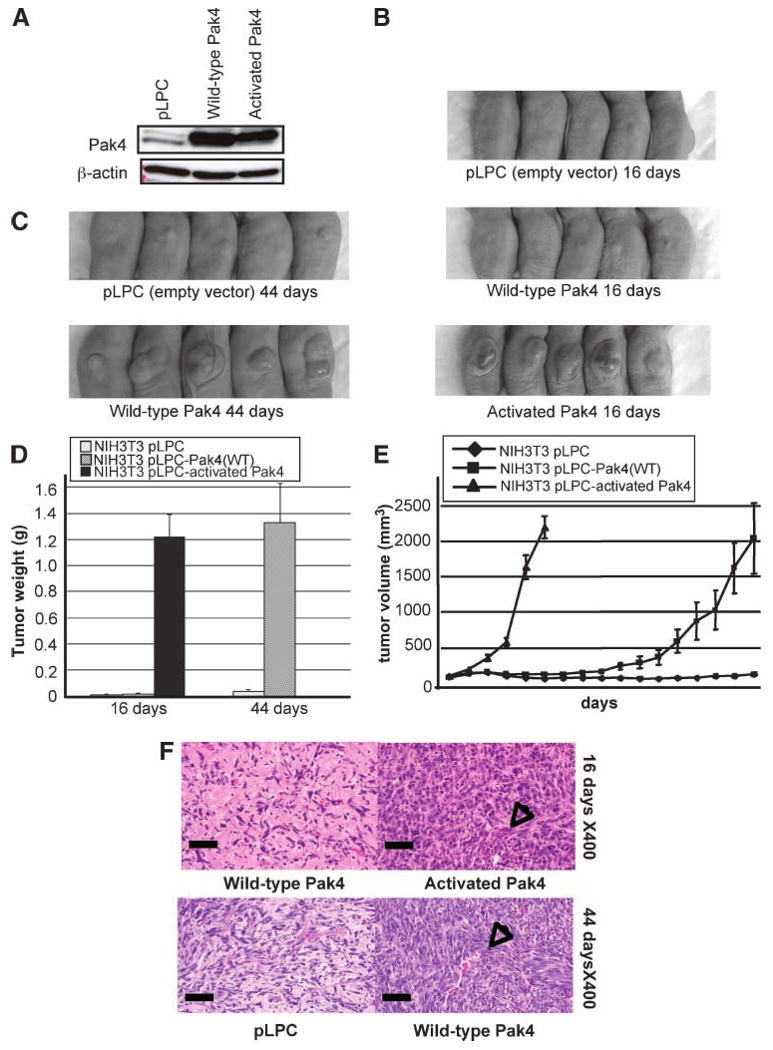
Ectopic overexpression of Pak4 leads to tumor formation. A. NIH3T3 cells were transfected with empty vector (pLPC), wild-type Pak4, or activated Pak4–containing pLPC-puro retroviral vectors. Cell extracts were prepared for Western blotting with the indicated antibodies. In each case, 20 μg of protein extract were used and β-actin served as a loading control. B. Cells containing activated Pak4 lead to tumor formation in athymic mice by 16 d postinjection. C. By 44 d, cells containing wild-type Pak4 also form tumors in athymic mice. D and E. Tumor weights and volumes from mice injected with NIH3T3 cells containing empty vector, wild-type Pak4, or activated Pak4 were assessed (activated Pak4-injected mice were sacrificed after day 16). F. Histology of tumor tissue isolated from mice injected with cells overexpressing wild-type or activated Pak4 (top), or empty vector or wild-type Pak4 (bottom), 16 or 44 d after injection. Arrows, areas with large amounts of spindle-shaped cells typical of sarcoma tissue. Scale bars, 50 μm.
Tumors Formed by Pak4 Have a Morphology Characteristic of Sarcomas
Next, we observed the morphologies of the tumors formed by wild-type or activated Pak4. Tumors and the corresponding normal tissues were sectioned, stained with H&E, and visualized under a microscope. Histologic analysis indicates that at day 16 postinjection, tumors from the mice injected with cells containing activated Pak4 had an appearance typical of sarcomas, arranged in interlacing fascicles of pleomorphic giant multinucleated cells, which are spindle-shaped cells with a stringy appearance (see Fig. 2F, top). Tissues taken from the injection sites of the mice injected with wild-type Pak4-expressing cells appeared normal at day 16, but by 44 days postinjection, wild-type Pak4 also promoted tumors with features typical of sarcomas (see Fig. 2F, bottom). These data support the finding that both wild-type and activated Pak4 promote tumors when overexpressed.
Pak4-Induced Tumors Have a Decreased Level of Apoptosis Compared with Control Tissue
The results described above are the first indication that wild-type Pak4 on its own is sufficient to induce tumors. Next, we were interested in determining the mechanism by which Pak4 promotes tumors. Previously, we have found that Pak4 protects cells from apoptosis (11, 12). One possibility is that the ability to protect cells from apoptosis and thus increase cell survival may be part of the mechanism by which Pak4 triggers tumorigenesis. We therefore examined apoptosis in the tumors formed by Pak4. This was done by examining the levels of activated caspase-3 as an end point of apoptosis (18, 19). Tumors formed by cells overexpressing wild-type Pak4 were isolated, as well as the corresponding regions from mice that had been injected with empty vector–containing cells. Lysates were prepared from the tissue samples and used to carry out Western blots using an antibody that recognizes cleaved (activated) caspase-3. The level of activated caspase-3 was significantly lower in the tumors compared with control samples (see Fig. 3A). Immunohistochemistry analysis of tissue sections also revealed a decrease in active caspase-3 and hence a decrease in apoptosis in the tumors overexpressing Pak4 (see Fig. 3B). In fact, tissue sections taken from mice injected with the wild-type Pak4-overexpressing cells showed a decrease in active caspase-3 levels as early as day 16 postinjection, although tumors had not even begun to form at this time point (see Fig. 3B). They continued to show a decrease in caspase-3 activity by day 44, at which point tumors were seen. Thus, inhibition of apoptosis seems to begin early, before Pak4 actually causes tumors to form.
FIGURE 3.
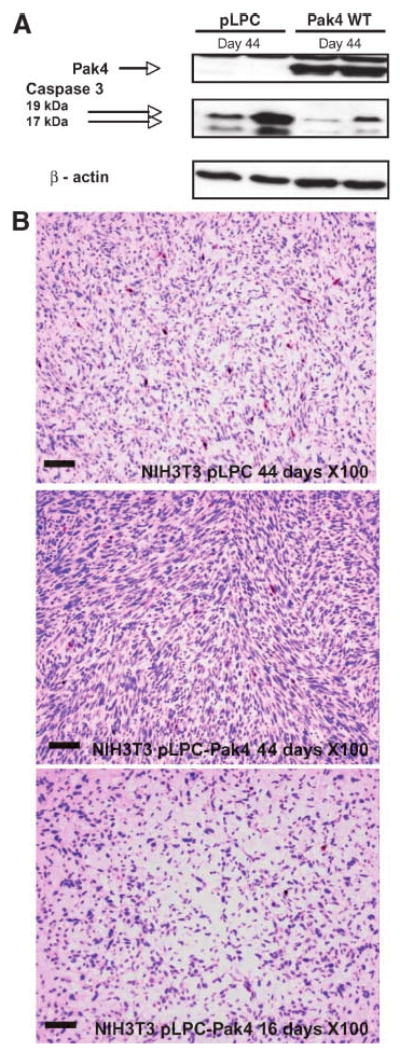
Activated caspase-3 levels are abrogated in tumors derived from Pak4-overexpressing cells. A. Activated caspase-3 (cleaved into 19 and 17 kDa fragments) was assessed by Western blot analysis of tumor or normal tissue that was isolated from mice injected with cells overexpressing empty vector (pLPC) or wild-type Pak4, 44 d after injection. The antibody recognizes only cleaved (activated) caspase-3. Pak4 expression and β-actin expression were also assessed. B. Activated caspase-3 (cleaved into 19 and 17 kDa fragments) was assessed by immunostaining of sections from tumors that were isolated from mice injected with cells overexpressing wild-type Pak4. Tissue sections from mice injected with cells containing empty vector (pLPC) were analyzed as a control. Tumor tissues were examined 16 and 44 d after injection. Areas containing active caspase-3 appear as red spots on the H&E-stained sections. Cells in individual sections were counted and at day 44; an average of 1.32% of the cells in control tissue stained positively for active caspases-3, whereas only 0.25% of the cells in the tumor tissue formed by wild-type Pak4 stained positively. Even at 16 d postinjection, as seen above, only 0.26% of the cells from the injection sites of mice injected with wild-type Pak4 stained positively for caspase-3, although tumors had not begun to form at that time point. In both cases, the difference between Pak4-containing cells and empty vector cells is statistically significant. Scale bars, 100 μm.
Increased Proliferation in Pak4-Induced Tumors
Mice were injected with NIH3T3 cells containing empty vector (pLPC), wild-type Pak4, activated Pak4, or oncogenic RasV12, as a positive control. Tissue sections were taken from the injection sites of all of the mice 16 or 44 days after injection. Tumor sections and control tissues were stained with H&E and with antibody against Ki67, which recognizes proliferating cells. The results, as shown in Fig. 4A, indicate that wild-type and activated Pak4 lead to an increased number of proliferating cells in the tissue sections. The increased number of proliferating cells was also seen as early as 16 days in the tissue injected with cells containing wild-type Pak4, before the tumors started to form. Increased proliferation is often associated with increased phosphorylation of the ERK mitogen-activated protein kinase. To determine whether ERK phosphorylation is increased in the tumors, we carried out Western blot analysis of tumor and control lysate using anti–phospho-ERK antibody and anti–total ERK antibody. Interestingly, we saw a slight increase in the level of phospho-ERK, relative to the amount of total ERK. This was especially noticeable for ERK2 (p44; see Fig. 4B). As a negative control, we also examined phosphorylation of p38, another mitogen-activated protein kinase, and did not see an increase in its phosphorylation in the tumors (see Fig. 4B). Thus, increased proliferation and increased ERK activation may be another contributing factor by which Pak4 leads to transformation, and may occur early, during the lag time before Pak4 actually produces tumors.
FIGURE 4.
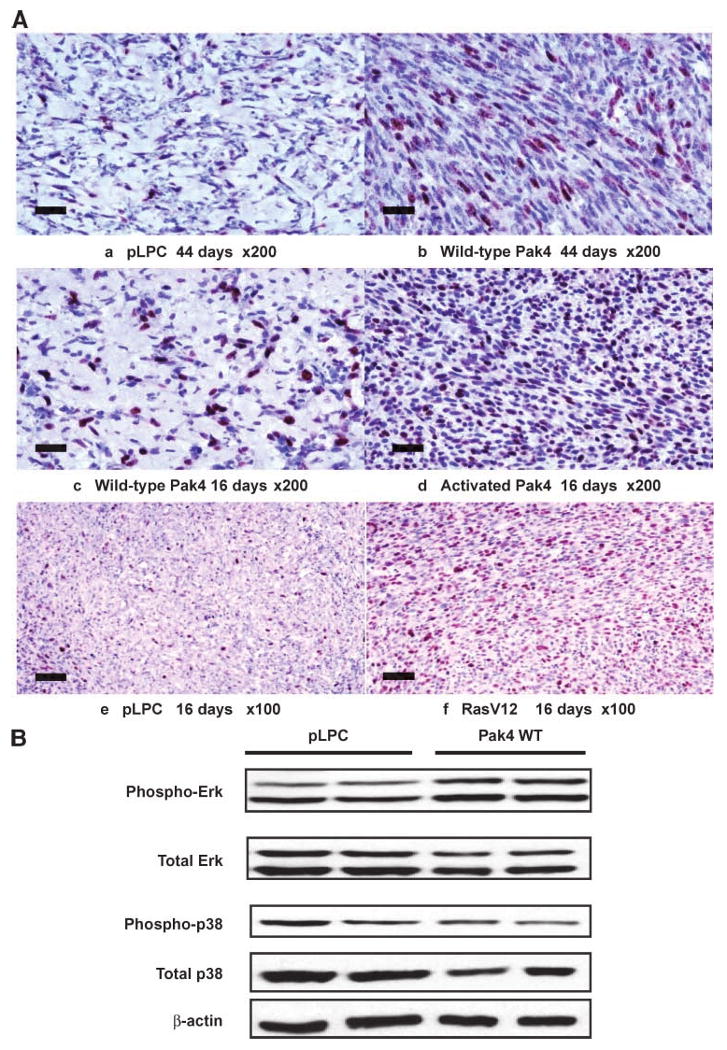
Increased proliferation is in response to Pak4 expression. Mice were injected with NIH3T3 cells containing empty vector (pLPC), wild-type Pak4, activated Pak4, or oncogenic RasV12, as a positive control. A. Tissue sections were taken from the injection sites of all of the mice 16 or 44 d after injection. Sections were stained with H&E and with antibody against Ki67, which recognizes proliferating cells (dark purple). The results indicate that wild-type and activated Pak4 lead to an increased number of proliferating cells in the tissue sections. Cells in individual sections were counted and at day 44; an average of 20.4% of the cells from control tissue stained positively for Ki67, whereas 36.4% of cells from tumors derived from wild-type Pak4 stained positively. At 16 d, 31.7% of cells from the injection sites of mice injected with wild-type Pak4 stained positively for Ki67. In both cases, the difference between Pak4-containing cells and empty vector cells was statistically significant. This was seen even at 16 d, at which point wild-type Pak4 had not yet caused tumors to form. Scale bars, 50 μm. B. Phospho-ERK and total ERK levels were assessed by Western blot analysis of tumor or normal tissue that was isolated from mice injected with cells overexpressing empty vector (pLPC) or wild-type Pak4, 44 d after injection. Top band, ERK2; bottom band, ERK1. As a negative control, phosphorylation of p38 was also examined by Western blot. The results indicated that wild-type Pak4 can lead to a slight increase in phosphorylation of the ERK mitogen-activated protein kinase pathway, which is often associated with increased proliferation.
Tumor Formation in Response to Oncogenic Ras Is Attenuated in Pak4 Null Cells
The above studies indicate that Pak4 is sufficient to form tumors in nude mice. An equally important question is whether Pak4 is also necessary for tumor formation. To address this question, we used fibroblasts that had been isolated from Pak4 null embryos or wild-type controls (20) and immortalized by serial passaging. The Pak4 null and wild-type fibroblasts (which grow at similar rates) were infected with a retrovirus containing oncogenic RasV12, a strong oncogene, or with empty vector (pLPC). Western blot analysis of the wild-type (Pak+/+) and Pak4 knockout (Pak4−/−) cells infected with either empty vector or RasV12 is shown in Fig. 5A. When injected into nude mice, RasV12; Pak4+/+ cells (wild-type cells infected with oncogenic Ras) formed large tumors by 18 d. At this time point, RasV12; Pak4−/− cells (Pak4 null cells infected with oncogenic Ras) formed significantly smaller tumors (see Fig. 5B, top). Because of the size of the tumors, mice injected with RasV12; Pak4+/+ cells were sacrificed at the 18-d time point. However, mice injected with RasV12; Pak4−/− cells were maintained for 6 more days, by which time larger tumors began to form even in these mice (see Fig. 5B, bottom). In some cases, these Pak4−/− tumors were especially bloody in appearance (see Fig. 5B, bottom). Figure 5C and D shows graphs indicating the weights and volumes of tumors at different time points. The results indicate that although the RasV12; Pak4−/− cells formed tumors, they grew quite a bit more slowly and they never obtained the same weight as the RasV12; Pak4+/+ cells.
FIGURE 5.
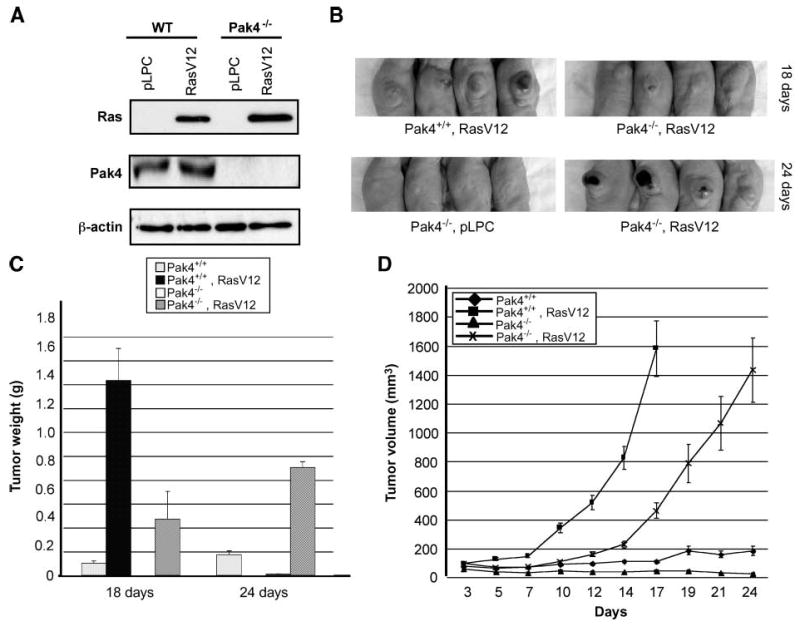
Tumor formation is attenuated in Pak4 null cells when transfected with Ras. A. Immortalized wild-type and Pak4−/− fibroblasts were infected with either empty vector (pLPC) or RasV12-containing pLPC retroviral vectors. Two days after infection, cells were selected with puromycin for 2 wk, after which cell extracts were prepared for Western blotting with the indicated antibodies. In each case, 20 μg of protein extract were used. β-Actin served as a loading control. B. Top, mice were injected with Pak4+/+ cells (wild-type) or Pak4−/− cells that were infected with oncogenic Ras. Tumors were examined 18 d after injection (for both cell types, cells infected with empty vector did not form tumors; not shown). Bottom, mice were injected with Pak4−/− cells containing empty vector (pLPC; left), or with RasV12 (right). Tumors were examined 24 d after injection (the Pak4+/+, RasV12 condition is not shown at 24 d because the tumors became too large and the mice had to be sacrificed). C and D. Tumor weight and tumor volume change in mice injected with Pak4+/+ and Pak4−/− cells with or without RasV12 were assessed (tumor weight in mice injected RasV12; Pak4+/+ cells were only analyzed at day 18 because the mice had to be sacrificed due to the large size of the tumors).
Tumor Formation in Response to Activated Cdc42 Is Almost Completely Abolished in the Absence of Pak4
Pak4 was originally identified as an effector for Cdc42 (5). We therefore reasoned that Pak4 may function downstream to Cdc42 during tumorigenesis. Wild-type or Pak4 knockout immortalized fibroblasts were infected with activated Cdc42 (Cdc42V12) or empty vector. Western blot analysis of the cells is shown in Fig. 6A. Cdc42V12-transfected cells resulted in large tumors when injected into the athymic mice. Strikingly, however, no tumors formed in mice that were injected with the Pak4 knockout cells, even those infected with Cdc42V12 (see Fig. 6B and C). Our results indicate that Pak4 has a direct role downstream to Cdc42 in oncogenic transformation.
FIGURE 6.
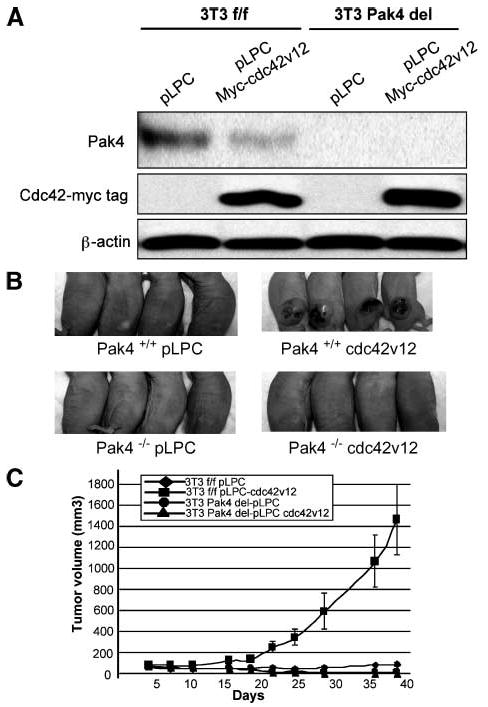
Tumor formation is completely abrogated in Pak4 null cells when transfected with Cdc42V12. A. Immortalized wild-type and Pak4 conditional knockout fibroblasts were infected with either empty vector (pLPC) or Cdc42V12-containing pLPC retroviral vectors. Two days after infection, cells were selected with puromycin for 2 wk, after which cell extracts were prepared for Western blotting with the indicated antibodies. In each case, 20 μg of protein extract were used. β-Actin served as a loading control. B. Top, mice were injected with Pak4+/+ cells (wild-type) that were infected with empty vector (pLPC; left) or with Cdc42V12 (right). Bottom, mice were injected with Pak4−/− cells containing empty vector (pLPC; left) or with Cdc42V12 (right). Tumors were examined 38 d after injection. For both cell types, cells infected with empty vector did not form tumors. Wild-type cells infected with Cdc42V12 formed tumors; however, tumor formation were completely abrogated in Pak4 null cells when transfected with Cdc42V12. C. Tumor volumes in mice injected with Pak4+/+ and Pak4 conditional knockout cells with or without Cdc42V12 were assessed at the indicated time points.
Discussion
Pak4 was originally identified as a protein that plays a role in regulating cytoskeletal organization and cell shape (5). Later, it was also found to have a role in regulating cell growth and survival (6, 11-13). Although Pak4 is expressed at high levels in embryos, it is expressed at low levels in most normal adult tissues. It is overexpressed, however, in a large number of different tumor cell lines (7), suggesting an important role for Pak4 in cancer. Here, we examined Pak4 expression levels in three different types of primary tumors (colon, mammary, and esophagus), and found that, in every case, tumor tissues contained more Pak4 than the corresponding control tissues. This suggests that overexpression of Pak4 is not simply a by-product resulting from the culture of the tumor cells because it is found directly in the primary tumors.
The finding that Pak4 is overexpressed in tumors is compelling, and these results led us to determine whether overexpression of Pak4 is also sufficient to promote tumorigenesis. Interestingly, the expression of a constitutively active mutant of Pak4 promotes anchorage-independent growth in cultured cells, but wild-type Pak4 has no effect (6). Anchorage-independent growth in cultured cells is an important predictor of oncogenesis because normal adherent cells need to attach to a surface to survive, whereas cancer cells lose this anchorage dependence. However, there are numerous other tests to determine whether a cell is transformed and whether a gene promotes tumorigenesis in the cells that express it, including decreased density dependence, decreased serum requirement, loss of cell cycle control, loss of differentiation, and resistance to apoptosis. One very important requirement for defining cells as malignant is that they form tumors in experimental animals. Formation of tumors in athymic mice is often used as a test to determine whether cells are malignant and whether an oncogene is transforming. To test whether Pak4 promotes tumors in athymic mice, we used NIH3T3 cells that overexpress Pak4. NIH3T3 cells normally only have a low level of Pak4 but the stable cell lines we generated express Pak4 to a level comparable with what is seen in tumor cell lines. Interestingly, we found that overexpression of Pak4 is sufficient to cause tumor formation in athymic mice. These results are quite exciting because they show for the first time that Pak4 promotes tumorigenesis in animals and not only in cultured cells. The results are especially intriguing because we found that in mice, even wild-type Pak4 is tumorigenic when overexpressed. Our results provide support for the idea that wild-type Pak4 is not only overexpressed in cancer cells but also sufficient to cause tumorigenesis. It should be noted that these experiments were all carried out using NIH3T3 cells. NIH3T3 cells are frequently used as a model to study tumorigenesis. However, because most tumors are of epithelial origin rather than fibroblasts, in future studies it will be important to determine whether Pak4 also plays an important role in transformation using epithelial cells as a model.
An important question is, “What is the mechanism by which Pak4 promotes tumors?” Previously, we found that Pak4 inhibits apoptosis and thus promotes cell survival (11, 12). Interestingly, we have found that there is a decreased level of apoptosis in Pak4-induced tumors compared with control tissues, as assessed by examining caspase-3 levels. Conversely, we see an increase in proliferation in the cells of the Pak4-induced tumors. We propose, therefore, that inhibition of apoptosis and subsequent increased cell survival and cell growth play a key role in Pak4-induced tumorigenesis. Previously, we found that Pak4 inhibits apoptosis by two different mechanisms. The first is a kinase-independent mechanism, mediated by inhibition of initiator caspases (11); the second is a kinase-dependent mechanism, mediated by phosphorylation of Bad (12). Interestingly, we have found that Pak4 (K350M), a kinase dead mutant, can promote tumorigenesis as efficiently as wild-type Pak4 (data not shown). This supports the idea that Pak4 can operate via the first mechanism, the kinase-independent mechanism, to promote cell survival and subsequent tumorigenesis. However, although the Pak4 (K350M) mutant and wild-type Pak4 were both equally transforming, the activated Pak4 mutant, Pak4 (S445N), had an even more dramatic effect on tumor formation. Thus, although kinase activity may not be required for Pak4-induced transformation, it may contribute to cell survival and tumorigenesis. In future experiments, it will be interesting to examine activated caspase-8 levels in the tumors as well as caspase-3, because Pak4 was shown to inhibit initiator caspases (such as caspase-8), as well as initiator caspases (like caspase-3), via a kinase-independent mechanism (11). It is interesting that there is a lag time before wild-type Pak4-expressing cells form tumors; yet, once tumors begin to form, they do so quite rapidly and grow to a large size. We have found that the decrease in apoptosis and increase in proliferation is seen early, even before tumors form in response to wild-type Pak4. These results suggest that increased proliferation and survival are events that occurs early, during the lag time before tumorigenesis begins in response to wild-type Pak4, and that this eventually primes the cells to proliferate without control and become tumor cells. The sequence of events that must occur before wild-type Pak4 causes tumors to form may require the complex environment of an animal model system. This may explain why transformation in response to wild-type Pak4 is not seen in a simpler cell culture system.
In addition to inhibiting apoptosis, we previously showed that Pak4 specifically activates prosurvival pathways, which lead to activation of pathways such as the nuclear factor-κB pathway and the ERK mitogen-activated protein kinase pathway. Cells lacking Pak4 thus have decreases in nuclear factor-κB and ERK activities (13). Consistent with this, we see an increase in phosphorylated ERK in the Pak4-induced tumors and this is associated with an increase in proliferation. We propose that cells overexpressing Pak4 have a decrease in apoptosis, an increase in cell survival pathways, and consequently an increase in signaling pathways such as the ERK pathway that promote cell proliferation.
Another issue addressed in this study is whether Pak4 is necessary for tumorigenesis. We found that although RasV12 led to tumor formation, even in the absence of Pak4, the tumors grew more slowly and grew to a smaller size. It is interesting that some of the tumors derived from the RasV12; Pak4-/- cells were especially bloody in appearance (see Fig. 5). This raises the intriguing possibility that the Pak4−/− tumors are undergoing apoptosis or necrosis and are thus stopped from growing any larger, presumably because of the lack of Pak4. This would be consistent with a critical role for Pak4 in cell survival and inhibition of apoptosis. In contrast to oncogenic Ras, the absence of Pak4 led to nearly complete abrogation of tumors in response to Cdc42V12. This is consistent with the role for Pak4 as a Cdc42 effector protein (5) and suggests the presence of a Pak4-dependent signaling pathway leading to Cdc42 and its activators to transformation.
It is interesting that Pak4 is highly expressed during embryogenesis but expressed at low levels in most adult tissues (20). This is consistent with a role for this protein in cancer. Although Pak4 most likely plays an important role in the rapid cell growth that occurs during embryogenesis, in adults, proliferation levels decrease and high levels of Pak4 may no longer be needed. Instead, when improperly overexpressed or activated in adult tissues, Pak4 may promote increased cell survival, uncontrolled growth, and tumorigenesis. Because Pak4 is almost undetectable in most normal tissues but up-regulated in several tumors, it may be an attractive candidate for drug therapy for a number of different types of cancer.
Materials and Methods
Tumor Samples
Human esophageal squamous cell carcinomas were obtained from patients in Linzhou People's Hospital, Linzhou, Henan, China (16). Human breast tissues were obtained from the Cooperative Human Tissue Network of University of Pennsylvania medical center. Mouse colon tumor tissue was obtained from azoxymethane-treated CF-1 female mice 44 wk old (33 wk after the last injection of azoxymethane). The dosage of azoxymethane was 10 mg/kg, subcutaneously, and weekly injections were given starting at 5 wk of age. Rat mammary tumor tissue was isolated from N-methylnitrosourea–treated Sprague-Dawley female rats (17).
Plasmids
pLPC-Pak4 WT, pLPC-Pak4 (K350M), and pLPC-Pak4 (S445N) are described in refs. (6, 11). To construct pLPC-Ras, HindIII/XhoI fragments from pCAN-Myc2-Ras were inserted into the HindIII/XhoI sites of the pLPC vector. Cdc42V12 was inserted into the EcoRI/XhoI sites of the pLPC vector. pLPC is a retroviral vector with a puromycin resistance marker (6). The activated Pak4 mutant [Pak4(S445N)] has a single point mutation in which serine 445 is changed to asparagine (S445N). This mutation stabilizes the catalytic loop of the kinase domain and causes Pak4 to have a constitutively high level of kinase activity (6).
Cell Culture, Transfection, and Establishment of Stable Cell Lines
Pak4 null fibroblasts were isolated from Pak4 null embryos at embryonic day (E9.5), as described (20). Control fibroblasts were isolated from E9.5 wild-type littermates. All fibroblasts were cultured in DMEM (Invitrogen) containing 10% bovine calf serum. Eco Phoenix packaging cells were cultured in DMEM containing 10% fetal bovine serum. The medium was supplemented with 100 units of penicillin/mL, 100 μg of streptomycin/mL, and 1 mmol/L glutamine. Stable cell lines containing wild-type Pak4, activated Pak4, empty vector, or RasV12 were grown as described above in the presence of puromycin (2.0 μg/mL). Stable cell lines were generated by retroviral infection of the vectors, followed by selection with the selectable marker puromycin, as described (6). Briefly, Phoenix packaging cells were transfected with empty pLPC vector, pLPC-myc-Pak4, pLPC-myc-Pak4 (S445N), or pLPC-myc-RasV12by the calcium phosphate precipitation method. Supernatants containing the released viruses were collected from the packaging cells 2 d after transfection and filtered through a 0.45-μm pore size filter. The virus was then used to infect either NIH3T3 cells or Pak4 null or Pak4 wild-type fibroblasts. Cells were selected with puromycin (2 μg/mL). Expression of Pak4 or Ras was determined by Western blotting.
In vivo Tumorigenesis
Five-week-old Ncr nu/nu male mice were purchased from Taconic Farm. All animals were housed four to a plastic cage with filter top. The animal room was controlled at 20 ± 2°C, 50 ± 10% humidity, and a 12-h light/dark cycle. Fresh AIN-93G diet was replenished twice weekly. For all studies, the mice were allowed to acclimate at least 3 d after receipt of shipment. Before injection, the cells were washed with PBS, harvested by trypsinization, resuspended, and kept on ice until injection. Cells (1 × 106) in a 100 μL mixture containing Matrigel (BD Biosciences) and culture medium at a 1:1 ratio were injected subcutaneously to the both flanks of the mice. This is the same concentration that was reported to be used for assessing the tumorigenicity of oncogenic Ras in nude mice (21). Mice were monitored regularly until termination of the study. Tumor size (length and width) was measured by a caliper and calculated based on tumor volume = 0.5 × width × length (22). All animals were sacrificed by CO2 asphyxiation, and tumors were harvested and weighed. Half of the tissue was fixed in 10% formalin for histopathology analysis and the other half was frozen in liquid nitrogen for Western blot analysis.
Histologic Examination
Tumor tissues from athymic mice were fixed in 10% formalin and then dehydrated gradually in alcohol. The tissues were embedded in paraffin and were sectioned at a thickness of 5 μm. The sections were stained with H&E for histologic evaluation.
Immunohistochemistry
A standard avidin-biotin peroxidase complex method was used in this study, as described (23). In brief, after dewaxing and rehydrating, the slides were heated in a pressure cooker in sodium citrate buffer (0.01 mol/L, pH 6.0) for 3 min after reaching full pressure. Endogenous peroxidase was quenched using 3% hydrogen peroxide in methanol. Sections were then blocked for 1 h at room temperature in PBS containing 3% normal horse or goat serum, depending on the origin of the primary antibody. The sections were then immunostained with cleaved caspase-3 (1:200, Cell Signaling) or Ki-67 (1:50 DakoCytomation) antibodies overnight at room temperature. The antibodies were diluted in 10% goat serum. The sections were rinsed in PBS and incubated with a biotinylated secondary antibody and subsequently incubated in Vectorstain Elite ABC reagent for 30 min, using 3,3-diaminobenzidine (Vector Laboratories) as the chromogen. Sections were then counter-stained for 2to 3 min with hematoxylin (Sigma) and mounted with Permount.
Western Blot Analysis
Western blots were carried out as described (5). Horseradish peroxidase–conjugated secondary antibodies were from Sigma. Western blot protein bands were visualized by the enhanced chemiluminescence method (Amersham).
Antibodies
Polyclonal Pak4 antibody, cleaved caspase-3 antibody, phospho-ERK, total ERK, phospho-p38, p38, and myc tag antibodies were from Cell Signaling. Mouse monoclonal Ras antibody was from Calbiochem.
Generation of Conditional Pak4 Knockout Cells
Ad-Cre, an adenovirus that expresses Cre recombinase (24), was prepared in 293 cells as described (25). Immortalized mouse fibroblasts containing a floxed allele of Pak4 were used for these studies. These cells, referred to as Pak4Flox cells, have two LoxP sites flanking the Pak4 coding sequence (see Supplementary Fig. S1). The cells were immortalized by serial passaging. Pak4Flox cells were infected with Cre recombinase adenoviruses as described (24). The Cre recombinase causes the sequences between the LoxP sites to be deleted, resulting in a null allele of Pak4, referred to as Pak4Del. Cells were plated in a six-well plate and used when they were 70% confluent. Cells were incubated with AdCre in DMEM medium overnight at 37°C After infection, cells were maintained in DMEM medium containing 10% fetal bovine serum, 100 units of penicillin/mL, 100 μg of streptomycin/mL, and 1 mmol/L glutamine. Pak4 protein expression was then evaluated by Western blotting to confirm the deletion of Pak4. Pak4Del is referred to as Pak4−/−, and Pak4Flox is referred to as Pak4+/+ in Fig. 6.
Acknowledgments
We thank Dr. Mousumi Bose, for providing mouse colon and normal tissue; the Cooperative Human Tissue Network of University of Pennsylvania Medical Center for providing human breast cancer and normal tissue; and Dr. Lisa Epstein Wong for the Cdc42V12 construct.
Grant support: NIH R01 CA076342 and ACS RSG-04-178-01-DDC.
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Daniels RH, Bokoch GM. p21-Activated protein kinase: a crucial component of morphological signaling? Trends Biochem Sci. 1999;24:350–5. doi: 10.1016/s0968-0004(99)01442-5. [DOI] [PubMed] [Google Scholar]
- 2.Knaus UG, Bokoch GM. The p21Rac/Cdc42-activated kinases (PAKs) Int J Biochem Cell Biol. 1998;30:857–62. doi: 10.1016/s1357-2725(98)00059-4. [DOI] [PubMed] [Google Scholar]
- 3.Sells MA, Chernoff J. Emerging from the Pak: the p21-activated protein kinase family. Trends Cell Biol. 1997;7:162–7. doi: 10.1016/S0962-8924(97)01003-9. [DOI] [PubMed] [Google Scholar]
- 4.Jaffer ZM, Chernoff J. p21-Activated kinases: three more join the Pak. Int J Biochem Cell Biol. 2002;34:713–7. doi: 10.1016/s1357-2725(01)00158-3. [DOI] [PubMed] [Google Scholar]
- 5.Abo A, Qu J, Cammarano MS, et al. PAK4, a novel effector for Cdc42Hs, is implicated in the reorganization of the actin cytoskeleton and in the formation of filopodia. EMBO J. 1998;17:6527–40. doi: 10.1093/emboj/17.22.6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qu J, Cammarano MS, Shi Q, Ha KC, de Lanerolle P, Minden A. Activated PAK4 regulates cell adhesion and anchorage-independent growth. Mol Cell Biol. 2001;21:3523–33. doi: 10.1128/MCB.21.10.3523-3533.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Callow MG, Clairvoyant F, Zhu S, et al. Requirement for PAK4 in the anchorage-independent growth of human cancer cell lines. J Biol Chem. 2002;277:550–8. doi: 10.1074/jbc.M105732200. [DOI] [PubMed] [Google Scholar]
- 8.Lin R, Bagrodia S, Cerione R, Manor D. A novel Cdc42Hs mutant induces cellular transformation. Curr Biol. 1997;7:794–7. doi: 10.1016/s0960-9822(06)00338-1. [DOI] [PubMed] [Google Scholar]
- 9.Lin R, Cerione RA, Manor D. Specific contributions of the small GTPases Rho, Rac, and Cdc42 to Dbl transformation. J Biol Chem. 1999;274:23633–41. doi: 10.1074/jbc.274.33.23633. [DOI] [PubMed] [Google Scholar]
- 10.Qiu RG, Abo A, McCormick F, Symons M. Cdc42 regulates anchorage-independent growth and is necessary for ras transformation. Mol Cell Biol. 1997;17:3449–58. doi: 10.1128/mcb.17.6.3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gnesutta N, Minden A. Death receptor induced activation of initiator caspase-8 is antagonized by the serine/threonine kinase PAK4. Mol Cell Biol. 2003;23:7838–48. doi: 10.1128/MCB.23.21.7838-7848.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gnesutta N, Qu J, Minden A. The serine/threonine kinase PAK4 prevents caspase activation and protects cells from apoptosis. J Biol Chem. 2001;276:14414–9. doi: 10.1074/jbc.M011046200. [DOI] [PubMed] [Google Scholar]
- 13.Li X, Minden A. PAK4 functions in tumor necrosis factor (TNF) α-induced survival pathways by facilitating TRADD binding to the TNF receptor. J Biol Chem. 2005;280:41192–200. doi: 10.1074/jbc.M506884200. [DOI] [PubMed] [Google Scholar]
- 14.Schurmann A, Mooney AF, Sanders LC, et al. p21-Activated kinase 1 phosphorylates the death agonist bad and protects cells from apoptosis. Mol Cell Biol. 2000;20:453–61. doi: 10.1128/mcb.20.2.453-461.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang Y, Zhou H, Chen A, Pittman RN, Field J. The Akt proto-oncogene links Ras to Pak and cell survival signals. J Biol Chem. 2000;275:9106–9. doi: 10.1074/jbc.275.13.9106. [DOI] [PubMed] [Google Scholar]
- 16.Fang MZ, Jin Z, Wang Y, et al. Promoter hypermethylation and inactivation of O(6)-methylguanine-DNA methyltransferase in esophageal squamous cell carcinomas and its reactivation in cell lines. Int J Oncol. 2005;26:615–22. [PubMed] [Google Scholar]
- 17.Thompson HJ, Singh M. Rat models of premalignant breast disease. J Mammary Gland Biol Neoplasia. 2000;5:409–20. doi: 10.1023/a:1009582012493. [DOI] [PubMed] [Google Scholar]
- 18.Fadeel B, Orrenius S. Apoptosis: a basic biological phenomenon with wide-ranging implications in human disease. J Intern Med. 2005;258:479–517. doi: 10.1111/j.1365-2796.2005.01570.x. [DOI] [PubMed] [Google Scholar]
- 19.Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269–90. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- 20.Qu J, Li X, Novitch BG, et al. PAK4 kinase is essential for embryonic viability and for proper neuronal development. Mol Cell Biol. 2003;23:7122–33. doi: 10.1128/MCB.23.20.7122-7133.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kroger A, Dallugge A, Kirchhoff S, Hauser H. IRF-1 reverts the transformed phenotype of oncogenically transformed cells in vitro and in vivo. Oncogene. 2003;22:1045–56. doi: 10.1038/sj.onc.1206260. [DOI] [PubMed] [Google Scholar]
- 22.Cheung AM, Brown AS, Hastie LA, et al. Three-dimensional ultrasound biomicroscopy for xenograft growth analysis. Ultrasound Med Biol. 2005;31:865–70. doi: 10.1016/j.ultrasmedbio.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 23.Hao XP, Pretlow TG, Rao JS, Pretloww TP. β-Catenin expression is altered in human colonic aberrant crypt foci. Cancer Res. 2001;61:8085–8. [PubMed] [Google Scholar]
- 24.Troussard AA, Mawji NM, Ong C, Mui A, St-Arnaud R, II, Dedhar S. Conditional knockout of integrin-linked kinase demonstrates an essential role in protein kinase BAkt activation. J Biol Chem. 2003;278:22374–8. doi: 10.1074/jbc.M303083200. [DOI] [PubMed] [Google Scholar]
- 25.Negrete A, Chuan T, Lyddiatt A. Production of adenoviral vectors and its recovery. Process Biochem. 2007;42:1107–13. [Google Scholar]