

Abstract
Bid is a ubiquitous pro-apoptotic member of the Bcl-2 family that has been involved in a variety of pathways of cell death. Unique among pro-apoptotic proteins, Bid is activated after cleavage by the apical caspases of the extrinsic pathway; subsequently it moves to mitochondria, where it promotes the release of apoptogenic proteins in concert with other Bcl-2 family proteins like Bak. Diverse factors appear to modulate the pro-apoptotic action of Bid, from its avid binding to mitochondrial lipids (in particular, cardiolipin) to multiple phosphorylations at sites that can modulate its caspase cleavage. This work addresses the question of how the lipid interactions of Bid that are evident in vitro actually impact on its pro-apoptotic action within cells. Using site-directed mutagenesis, we identified mutations that reduced mouse Bid lipid binding in vitro. Mutation of the conserved residue Lys157 specifically decreased the binding to negatively charged lipids related to cardiolipin and additionally affected the rate of caspase cleavage. However, this lipid-binding mutant had no discernable effect on Bid pro-apoptotic function in vivo. The results are interpreted in relation to an underlying interaction of Bid with lysophosphatidylcholine, which is not disrupted in any mutant retaining pro-apoptotic function both in vitro and in vivo.
Keywords: Apoptosis, Bid, Mitochondria, Phospholipids, Caspase 8
1. Introduction
The Bcl-2 family of proteins regulates the intrinsic apoptotic pathway by controlling permeabilization of the outer mitochondrial membrane (MOMP). This process causes the release of apoptogenic factors, such as cytochrome c and SMAC/Diablo, into the cytosol, thus activating effector caspases. Bcl-2 proteins can be grouped into three subfamilies based upon their function and the number of shared regions of homology (Bcl-2 homology, or BH, domains) [1]. Bax and Bak are multidomain pro-apoptotic effectors, which are directly responsible for MOMP [2]. The multidomain pro-survival proteins, including Bcl-2 and Bcl-XL, antagonize Bax and Bak. A third class of Bcl-2 proteins are the BH3-only proteins [3]. These regulate apoptosis by either inactivating the anti-apoptotic multidomain proteins [4], or by activating the pro-apoptotic ones [5]. Different BH3-only proteins function by responding to distinct apoptotic stimuli. Bad is phosphorylated by kinases downstream of growth factor receptors, resulting in its sequestration by 14-3-3 proteins in the cytosol [6]. Conversely, PUMA and Noxa are transcriptionally regulated following DNA damage induced p53 activation [7,8]. Bim is regulated both at transcriptional and post-translational levels [9,10]. Growth factor mediated phosphorylation of FOXO transcription factors suppresses Bim expression. At the same time, Erk mediated phosphorylation of Bim itself leads to its targeting to proteosomal mediated degradation.
Another BH3-only protein, Bid, is unusual in that it is largely activated by the apical caspase of the extrinsic, or death receptor-mediated, pathway [11,12]. Thus, ligand binding to receptors of the TNFR family leads to activation of caspase 8, which cleaves Bid at aspartate 59 (mouse sequence). The resulting truncated form, tBid, associates with the OMM, activating Bax and Bak. Several studies have indicated that Bid is subject to a number of other post-translational modifications and can be pro-apoptotic without caspase cleavage [13-15]. Bid can be phosphorylated on residues around the caspase cleavage site, which has been demonstrated to inhibit cleavage [13,16]. Other sites in Bid can be phosphorylated in response to DNA damage, suggesting that it may play additional roles besides OMM permeabilization in receptor-mediated cell death [17].
An important aspect of Bid function is its binding to mitochondrial lipids, in particular cardiolipin and related lysolipid metabolites [18-26]. Cardiolipin has been reported to modulate the concerted pro-apoptotic action of caspase-truncated Bid (tBid) with Bax or Bak in studies with isolated mitochondria [23,27]. Some studies have shown that cardiolipin is required for tBid targeting to mitochondria [18], whereas other studies have shown that manipulation of cardiolipin content in cells had little effect on Bid and tBid functions [28,29]. In most instances, lipids have been manipulated within the membrane itself, which might have indirect effects on MOMP. Hence, it remains unclear to what extent lipid binding is instrumental for the mitochondrial targeting and pro-apoptotic action of Bid under different stimuli of cell death.
To address this problem, we have undertaken site-directed mutagenesis of Bid aiming to dissect the lipid-binding properties from the pro-apoptotic action of the protein. We have found that one conserved amino acid in the C-terminal part of Bid may be crucial for the link between lipid binding and the pro-apoptotic action that can be measured in vitro. However, mutation of the lipid-binding site in Bid had no effect on its ability to induce cytochrome c release and apoptosis within cells, which we can refer to be in vivo. Our results are discussed in terms of the relationship between in vitro and in vivo properties of pro-apoptotic Bcl-2 proteins.
2. Materials and methods
2.1. Generation of recombinant wild type and mutant Bid proteins
The cDNA encoding full-length mouse Bid was cloned into pET-12b vector with a tag of six histidine at the N-terminus. The Bid recombinant proteins (wild type and mutants proteins) were expressed in BL21(DE3)pLysS Escherichia coli and purified by affinity chromatography (HiTrap chelating column, Healthcare). The protein fractions were concentrated, aliquoted and stored at −80 °C. Mutations that changed the positively charged residues in helices 6 (K157) and 8 (R187) to negatively charged ones (i.e. E) were performed by a Quick Change Site-Directed Mutagenesis Kit II (Stratagene). Mutation K157E was generated using the mutagenic oligonucleotide 5′-GACCATGCTGTTGGCCGAAAAAGTGGCCAGTCACGC-3′ containing an A to G base change that was used to replace lysine 157 to glutamic acid. Mutation R187E was generated using the mutagenic oligonucleotide 5′-CCAGAACCTATTCTCCTATGTGGAGAACTTGGTTAGAAACGAG-3′ containing an AG to GA base change that was used to replace arginine 187 to glutamic acid. All mutations were confirmed by DNA sequencing.
2.2. Generation of YFP-tagged wild type and mutant Bid proteins
Bid cDNA representing full length, p15 or p11 Bid was cloned into pEYFPN1 vector (Invitrogen). Mutagenesis reactions were performed using a QuickChange II XL Site-Directed Mutagenesis Kit (Stratagene) with the oligonucleotides described above. All mutations were confirmed by DNA sequencing.
2.3. Cell culture and transfections
FSK7 cells were cultured in Dulbecco’s modified Eagle’s medium/F-12 medium supplemented with 5 ng/ml epidermal growth factor, 5 μg/ml insulin, 1% penicillin/streptomycin and 2% foetal calf serum (FCS) in 5% CO2 at 37 °C. Cells were transfected using LipofectAMINE plus (Invitrogen) as instructed. Bid knockout and Bax/Bak double knockout Mouse Embryonic Fibroblast (MEF) cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% FCS, 1% penicillin/streptomycin, 1% non-essential amino acids and 5 μM β-mercaptoethanol in 5% CO2 at 37 °C. Cells were transfected using either LipofectAMINE plus (Invitrogen) or Transpass D2 (NEB) as instructed.
2.4. Immunocytochemistry
Cells grown on coverslips or collected by cytospin onto polysine slides were rinsed in PBS and fixed in 4% PFA in PBS for 20 min. Cells were immunostained with either mouse anti-cytochrome c (BD Biosciences, Cat # 556432) or mouse anti-mtHsp70 (Affinity Bioreagents) for 1 h, rinsed in PBS, then incubated with RXR-conjugated donkey anti-mouse IgG (Jackson Immunoresearch) for 1 h. All antibodies were diluted in 0.2% Triton X-100, 0.05% Tween-20, 0.1% horse serum, and 0.05% sodium azide in PBS. Nuclei were stained with Hoechst. Images were collected on an Olympus IX70 microscope, equipped with a Deltavision imaging system. Images were processed by constrained iterative deconvolution on softWoRx™ software (Applied Precision).
2.5. Anoikis experiments
FSK7 cells were transfected with YFP-tagged Bid proteins. The following day cells were left attached or detached onto polyHEMA (Sigma). In apoptosis experiments cells were detached for 5 h then collected by cytospin onto polysine slides at 400×g for 5 min. Cells were fixed in 4% paraformaldehyde (PFA) for 20 min and cell nuclei stained using Hoescht. To detect Bid on mitochondrial membranes cells were detached onto polyHEMA for 1 h then lysed in hypotonic buffer (10 mM Tris–HCl pH 7.6, 10 mM NaCl and 1.5 mM MgCl2) for 10 min followed by douce homogenization. Membranes were isolated by ultracentrifugation in a TLA110 rotor at 55,000 rpm for 30 min at 4 °C. The supernatant containing cytosolic proteins was collected. CHAPS soluble mitochondrial membranes were isolated by resuspending the membrane pellet in 1% CHAPS and further centrifugation at 55,000 rpm for 30 min at 4 °C. Detached cell fractions were analysed by western blot.
2.6. In vitro cleavage with caspase 8
Recombinant Bid proteins (wild type and mutants) were cleaved by recombinant human caspase 8 (Calbiochem) in a buffer containing 20 mM K-Hepes (pH 7.4), 0.25 M sucrose, 2 mM DTT and 1 mM EDTA (containing protease inhibitors). The mixture was incubated for 2 h at 30 °C as described [13].
2.7. In vivo cleavage with FasL
FSK7 cells were transfected with YFP-tagged Bid proteins and treated overnight (16 h) with 0.25 μg/ml FasL (Biolegend) and 25 μg/ml cycloheximide or DMSO as control. Cells were lysed and immunoblotted for Bid.
2.8. Lipids
Lipids and lysolipids were obtained from Sigma or Avanti lipids; MCL was prepared in-house using the procedure reported in [20] (modified from that described in [30]).
2.9. Intrinsic fluorescence measurements
The intrinsic fluorescence of Bid proteins (at a concentration of 0.5 or 1 μM) were measured at room temperature in filtered assay buffer (20 mM K-Hepes pH 7.4, 0.12 M mannitol, 80 mM KCl, 1 mM EDTA) using excitation wavelength at 270 nm and recording emission between 290 and 410 nm. Lysolipids, dissolved in ethanol, were added in a stepwise manner and fluorescence changes were measured after 2 min equilibration.
2.10. ADIFAB fluorescence quenching
The fluorescently labelled intestinal fatty acid protein ADIFAB (Molecular Probes, Invitrogen) [31] has been used dissolved in 10 mM Tris, pH 8.0, 0.15 M NaCl and 1 mM EGTA, at 0.2 μM alone or in the presence of Bid recombinant proteins. Quenching measurements were carried out at room temperature (22–24 °C) using a Perkin-Elmer LS-500 instrument. The emission spectra were recorded between 410 and 610 nm using excitation wavelength at 390 nm in the absence of ligands and with increasing concentrations of lipid ligands. Results were analysed by using Stern–Volmer plots.
2.11. NATIVE PAGE (evaluating of Bid interaction with lipids)
Fresh solutions of Bid was diluted to 20 μM in filtered assay buffer containing 1 mM DTT in the absence or presence of LPC-C16 1 mM (from concentrated ethanolic solutions). Samples were incubated for 60 min at room temperature and the reaction terminated by addition of sample buffer (20% glicerol, 0.1 M Tris–HCl, pH 6.8, 2 β-mercaptoethanol, 0.01% bromophenol blue). Results were visualized by Coomassie staining and analysed in comparison with controls containing appropriate levels of non-interacting detergents, e.g. 0.2% Tween 20. The high molecular weight species present in the native gel were identified as Bid aggregates by means of western blot analysis using a mouse-specific goat anti-mouse Bid (R&D Systems, Cat # AF860).
2.12. Cell-free assays
Mouse liver mitochondria were prepared essentially as described previously [19,32]. After being soaked with ice-cold PBS, mouse livers were cleaned of connective tissue, cut with scissors and suspended. The cleaned tissues were homogenized with a Teflon pestle homogenizer in isolation medium (10 mM K-Hepes, 0.25 M mannitol, 1 mM EGTA, 0.2% bovine serum albumin [BSA], pH 7.4), generally containing a cocktail of protease inhibitors (0.1% [vol/vol] of P3840 Sigma) and then centrifuged at 600×g for 5 min at 4 °C. The supernatants were filtered to remove fats and centrifuged at 10,000×g for 15 min at 4 °C. The pellet containing crude mitochondria was resuspended in isolation medium without BSA and protease inhibitors and subsequently re-centrifuged at 10,000×g for 15 min at 4 °C. Finally, the resulting pellet was resuspended in a minimal volume of 20 mM K-Hepes pH 7.4, 0.12 M mannitol, 80 mM KCl, 1 mM EDTA (assay buffer) and utilised for the experiments. The protein content was determined with Bio-Rad Bradford mini-assay in the presence of the non-ionic detergent Triton X-100. The detergent was added to a concentrated solution of mitochondria to solubilize completely the membrane proteins; the sample was then diluted for Bradford analysis (final Triton X-100 concentration<0.002%). Freshly isolated mitochondria were resuspended at 1 mg/ml in assay buffer and incubated at room temperature (22 °C) for 15 min with recombinant mouse Bid proteins 200 nM. Ethanol-dissolved lysolipids (Sigma) were added to the incubation mixture; subsequently, mitochondria were separated by centrifugation and the release of cytochrome c was measured in the supernatant by immunoblotting as described earlier [19].
2.13. Western blotting
Protein samples were separated with 12–15% SDS polyacrylamide gels and transferred to PVDF membranes (Amersham — GE Healthcare). The monoclonal antibody used for the cell free assay analysis was the purified mouse anti-cytochrome c from BD Biosciences (Cat # 556433). To monitor Bid proteins we have utilised a mouse-specific goat anti-mouse Bid (R&D Systems) or goat anti-GFP antibody (Rockland). Mitochondrial fractions were identified using mouse anti-mtHsp70 (Affinity Bioreagents). Membranes were visualized using enhanced chemiluminescence reagents (ECL-Plus, Amersham — GE Healthcare).
2.14. Statistical analysis
To determine statistical differences, Student’s t test was used to assess statistical significance in cytochrome c release between control and mutant recombinant (usually with n=3 densitometric determinations). p<0.05 was considered significant.
3. Results
3.1. Mutations of the charged residues at the helices 4 and 6 of Bid: K157E and R187E
Using sequence analysis [33] we identified conserved residues within Bid that might play a role in the lipid-binding activity of the protein (Fig. 1A) [18,34]. By analysing Bid with the Q-sitefinder (http://www.bioinformatics.leeds.ac.uk/qsitefinder/), we identified hydrophobic pockets within the protein that can be potential binding sites for lipid molecules. Then, we verified the presence in these regions of charged residues that could be involved in stabilizing the interaction with the charged head groups of lipid molecules. In the C-terminal region (helix 6), there are two conserved lysine residues, K157 and K158, which have been studied previously for their importance in tBid function, in particular the interaction with mitochondrial membranes [34,35]. These residues are located in close proximity of a hydrophobic pocket. Liu et al. [25] generated mutants of various positive residues of human tBid, including these two lysines, which were substituted with hydrophobic residues. None of these mutated forms of tBid generated any apparent phenotype after trasfection into Hela cells and appeared to be equally targeted to mitochondria. No further in vitro report on the binding of Bid mutants to membrane lipids has been published to date.
Fig. 1.

Structural properties of Bid underline the mutations undertaken here. (A) Sequence alignment of Bid from mouse, human, bovine and Xenopus. (*) fully conserved amino acid; (:) conservative substitution. The mutated residues are represented in bold; lines under the sequences indicate α-helices. (B) NMR structure of mouse Bid (Protein Data Bank number 1DDB) (35). The structure shows the position of the two positive residues (K157 and R187) that have been mutated in this work. (C) Coomassie stained SDS-PAGE of expressed and purified wt, K157E and R187E Bid.
We have utilised recombinant mouse Bid (Fig. 1B). The first mutation introduced was the substitution of K157 with a negatively charged glutamic acid, to verify whether binding to the polar head of lipids requires a positive residue in this position. Lysine 157 faces an internal cavity pointing towards BH3 helix (Fig. 1B). The adjacent K158, equally conserved in many species (Fig. 1A), is more exposed to the surface in the NMR-deduced structure [36], and was excluded from our mutational analysis. The C-terminal region of Bid also contains an interesting positive residue, R187, which is conserved in many species except Xenopus laevis (Fig. 1A). R187 is located in another region of the protein recognised as a second potential binding pocket for hydrophobic substances, near K148 in the tertiary structure (a region that possesses a global positive charge, Fig. 1B [37]). This second region could be involved (together with other amino acids) in the recognition and binding of the negatively charged head groups that are present in phospholipid molecules. We mutated R187 with the negatively charged E that is present in the sequence of Xenopus Bid, considering that this naturally occurring substitution may be unable to compromise basic biological properties of Bid. Wild type and mutant Bid were expressed in E. coli and purified (Fig. 1C) as described in the Materials and Methods.
We next investigated whether the selected mutations modify Bid interaction with negatively charged lipids. Bid binding to lipids was determined by following changes in the intrinsic fluorescence of the protein, as previously described [20]. Overall, intrinsic fluorescence spectra of the K157E mutant and wild type protein were similar (Fig. 2A), even if a small red shift in the emission maximum of the mutant may reflect a partial modification the protein environment around the aromatic residues contributing to the intrinsic fluorescence of Bid. In contrast, the R187E mutant showed significantly lower intrinsic fluorescence than the wt protein (Fig. 2A), indicating that R187 may modulate the conformation or solvent exposure of the single tryptophan present in mouse Bid (W48), which is predominantly responsible for its intrinsic fluorescence. In all cases, these changes in the intrinsic fluorescence had little influence on the functional properties of Bid proteins, whereas another mutation, F171N, did cause a strong decrease in fluorescence emission indicating a loss of native conformation coupled to no lipid binding and pro-apoptotic action (data not shown).
Fig. 2.

Binding analysis of Bid to lysolipids in solution. Binding analysis of LPG and LPC-C16 were monitored through wild type and mutant Bid intrinsic fluorescence changes after the addition of LPG or LPC-C16. (A) Intrinsic fluorescence of recombinant wt (solid spectrum), K157E (dashed spectrum) and R187E (dot–dot–dashed spectrum) Bid in the absence of lipid. (B) Intrinsic fluorescence of recombinant wt Bid in the absence (solid spectrum) or presence of 2 μM LPG (dashed spectrum). (C) Intrinsic fluorescence of recombinant K157E Bid in the absence (solid spectrum) or presence of 2 μM LPG (dashed spectrum). (D) Intrinsic fluorescence of recombinant R187E Bid in the absence (solid spectrum) or presence of 2 μM LPG (dashed spectrum). (E) Histogram showing the intrinsic fluorescence variation (F–F0) at 348 nm where F0 is the intensity of intrinsic fluorescence in the absence of ligands, F is the intensity of intrinsic fluorescence in the presence of LPG. (F) Histogram showing the intrinsic fluorescence variation (F–F0) at 348 nm of recombinant wt, K157E and R187E Bid where F0 is the intensity of intrinsic fluorescence in the absence of ligands, F is the intensity of intrinsic fluorescence in the presence of 1 μM LPC-C16.
Because of the water insolubility of cardiolipin, we measured Bid binding to a related lipid, oleyl-lysophosphatidylglycerol (LPG-C18:1), that has been utilised to mimic monolysocardiolipin binding in previous studies [20] at concentrations below its critical micellar concentration (CMC). Wild type Bid bound LPG-C18:1, shown by the increase in fluorescence with added lipid (Fig. 2B, E). Both mutants had decreased LPG-C18:1 binding. Bid K157E showed the largest reduction in LPG-induced fluorescence changes and hence binding of this lipid (Fig. 2C, E), whereas Bid R187E exhibited attenuated changes in intrinsic fluorescence upon addition of LPG-C18:1, suggesting some reduced binding to LPG (Fig. 2D, E).
Dual lipid specificity has been reported for full length Bid [38], since the protein has been found to bind and transport also lysolipids derived from PC, such as palmitoyl-phosphatydylcholine (LPC-C16). Therefore, we investigated the interaction of Bid mutants with LPC-C16 under the same conditions (Fig. 2F). Compared with LPG-C18:1, LPC-C16 induced a small change in the intrinsic fluorescence of the wt protein, as well as the R187E mutant. However, LPC-C16 significantly enhanced the intrinsic fluorescence of the K157E mutant with respect to the wt as well as the R187E mutant protein. In order to verify the non-specific effect of the addition of lysolipid molecules to the Bid protein in solution, we also measured the effect of short chain lipids like LPC-C12, which are more water-soluble and have surfactant properties similar to zwitterionic detergents [21]. These compounds did not significantly influence the fluorescence emission of Bid, even if they did bind to either the wt or mutant proteins (see below, cf. [39]).
3.2. Binding to lysolipids induces Bid aggregates
To evaluate Bid binding to lipids, we used another approach based upon the principle that tight binding to lipids could induce changes in the electrophoretic mobility of the protein (gel shifts) under non-denaturing conditions, as previously reported for some lipid-binding proteins [20]. Interaction of Bid with LPC-C16 and MCL was evaluated by NATIVE PAGE after incubating 20 μM wild type or mutant Bid with 1 mM lysolipids, ensuing stable micellar structures mimicking membrane lipid surfaces. The electrophoretic migration of Bid wild type was significantly retarded by prior incubation with LPC-C16 producing diffuse aggregates that apparently migrated as oligomeric forms. Similar oligomers/aggregates were observed in the presence of monolysocardiolipin (MCL) (not shown).
The increased aggregate formation of K157E mutant in the presence of LPC-C16 (Fig. 3) confirmed the evidence previously obtained with intrinsic fluorescence analysis that the mutant has a stronger interaction with this lipid. Conversely, R187E has partially lost its LPC-C16 binding capacity as deduced by the decrease in its aggregate formation (Fig. 3). We have performed the same experiments with LPG too; however, this lipid was not able to induce similar aggregates (data not shown) despite its binding to Bid proteins (see Figs. 2 and 4). It is interesting to note that the mobility of the mutant Bid proteins slightly increased with respect to that of the wild type protein in NATIVE PAGE due to the change in global electric charge (positive residues substituted with a negative one, E).
Fig. 3.

Binding to lysolipids induces Bid aggregates. (A) NATIVE PAGE profile of wild type and mutant Bid proteins in the absence and in the presence of 1 mM LPC-C16 and with 0.2% Tween 20 as a negative control. (B) Densitometric analysis of Bid proteins mobility in the presence of LPC-C16. Note the large shift in mobility for the K157E mutant.
Fig. 4.

Mutations affecting lipid interaction also change caspase cleavage. (A) Stern–Volmer plots of the quenching of ADIFAB fluorescence in the presence of MCL (left panel), LPC-C16 (right panel) or LPG-C18:1 (bottom panel). Filled circles, no Bid (i.e. binding to ADIFAB alone); dark grey triangles, with wt Bid; light grey diamonds, with Bid K157E; empty squares, with Bid R187E. F0/F is the ratio between the fluorescence in the absence of lipids and the fluorescence in the presence of ligands and its increase as a function of lipid ligand concentration is reduced in the presence of Bid competing with ADIFAB for the same ligand (20). (B) Cleavage of recombinant wt Bid, Bid K157E and Bid R187E by recombinant caspase 8 after 10 min incubation. Bid K157E is cleaved more after 10 min than either wt or Bid R187E. (C) The graph shows the time course of Bid cleavage by caspase 8 as in B.
3.3. Altering the lipid-binding site results in altered caspase 8 sensitivity of Bid in vitro
Next we studied lipid interactions of Bid using the indirect method based on the reporter ADIFAB, which allows the evaluation of the binding to MCL [20]. In this approach, the Stern–Volmer plots of the changes in ADIFAB fluorescence fit a linear slope that is inversely proportional to the dissociation constant for a lipid ligand in the presence of a competing binding protein like Bid [20]. The Stern–Volmer plot of our results obtained following the fluorescence quenching of ADIFAB in the presence and absence of Bid indicated that the mutant K157E had a reduced affinity for MCL (Kd value changed from 1.3 μM in wt Bid to 1.8 μM), while R187E had approximately the same Kd as the wt (1.4 μM, Fig. 4A). A similar affinity profile has been obtained in the presence of LPG-C18 (Fig. 4A), indicating a higher Kd for the mutant K157E (0.2 μM) than for the R187E mutant and wt protein (0.1 μM and 0.08 μM, respectively). On the contrary, Bid binding to LPC analogues appeared to behave in the opposite way, since the mutant K157E showed the same (or even slightly increased) affinity for the binding to LPC-C16 with respect to the wild type protein (Kd values around 4 μM), while the mutant R187E showed a reduced affinity (Kd = 5.5 μM) (Fig. 4A and data not shown). These results confirmed the findings obtained by following the intrinsic fluorescence of the protein (Fig. 2), in that the mutation K157E had an opposite effect on the affinity of Bid for CL derivatives and LPC derivatives, while the comparable substitution R187E had negligible effects on lipid binding.
Next we evaluated whether the specific alteration in lipid affinity of the K157E mutant was related to changes in its pro-apoptotic function. Because this function is strongly enhanced by caspase cleavage of the full length protein [12], we first compared how Bid mutants were cleaved in vitro by purified caspase 8. Interestingly, we found that the initial rate of cleavage was faster for the K157E mutant than for either the wt or the R187E mutant protein (Fig. 4B). This difference progressively faded along the time course of proteolytic cleavage (Fig. 4C), suggesting that the K157E mutation could facilitate the accessibility of the protease for its major cleavage site, rather than changing the overall efficiency of cleavage.
3.4. Bid mutations and cell free assays of cytochrome c release
We asked if the properties of the K157E mutant of Bid extended to its capacity to release cytochrome c from isolated mitochondria. We analysed cytochrome c release using a cell free assay utilising isolated liver mitochondria [12,20,38,40]. After addition of full-length (FL) proteins to isolated mitochondria, both wt Bid and R187E Bid were able to induce some release of cytochrome c (Fig. 5A). In contrast, K157E Bid did not release cytochrome c, similar to the negative control and the Bid protein mutated in BH3-domain (G94E/D95E in Fig. 5A, cf. [38]).
Fig. 5.

Bid mutants differentially release cytochrome c from mitochondria in vitro. Western blot analysis of cytochrome c release from intact mouse liver mitochondria was undertaken after incubation with 200 nM recombinant Bid proteins (A) in the absence of exogenous lysolipids and (B) in the presence of 2 μM LPC-C16. The results shown are representative of >3 independent experiments. Histograms reported the densitometric analysis of a set of blots as shown. (C) Western blot analysis of cytochrome c release from intact mouse liver mitochondria after incubation with 20 nM recombinant caspase 8-cleaved Bid (tBid) proteins in the absence or in the presence of 2 μM LPC-C16. The results shown are representative of >3 independent experiments. Histograms reported the densitometric analysis of a set of blots as shown. (+) positive control (500 μM LPC-C16); (−) negative control (without Bid).
To verify whether these results were related to a different association of Bid proteins to mitochondria, we analysed the distribution of Bid after incubation with mitochondrial membranes (thus containing the natural complement of cardiolipin). We found no major difference in the association of wt and mutant proteins to mitochondria and their membranes, in agreement with recent data [29].
The pro-apoptotic activity of FL Bid is limited in comparison with that of tBid, but it is strongly enhanced by addition of selected lipids, in particular LPC [21]. We thus repeated the cell free assays in the presence of LPC-C16 and found that this lipid increased the cytochrome c releasing capacity of all Bid proteins we have analysed (Fig. 5B), with the exception of the BH3-domain mutant (G94E/D95E). This mutant displayed less efficient LPC binding and at the same time a complete loss of pro-apoptotic activity (not shown), confirming a previous work with an equivalent BH3-defective mutant [38]. Nevertheless, it is worth noting that while the wt and R187E FL proteins showed approximately a 2-fold increase in their capacity of releasing cytochrome c, the K157E mutant displayed a comparatively much stronger enhancement in the presence of LPC compared with its relative inactivity in the absence of LPC (Fig. 5A, B). Hence, the LPC-induced ‘activation’ of Bid [21] is stronger for the K157E mutant than for the other mutants tested, as well as the wt protein.
We next analysed the pro-apoptotic activity of the caspase-cleaved forms of Bid mutants, i.e. p15 tBid (cf. Fig. 4B), in cell free assays with isolated mitochondria. The results indicated that the p15 form of the K157E mutant was slightly more active than the wild type and the R187E mutant, but showed a similar activity as wt in the presence of LPC (Fig. 5C). Hence, Bid K157E mutation had an effect on the in vitro activities of Bid only in its full length, native form. Once cleaved by caspase 8, this and other Bid mutants were essentially as effective as the native protein in both lipid binding and release of cytochrome c from mitochondria. Hence, only a detailed study of how Bid mutants affect cell properties in vivo could clarify the significance of the changes we have observed in vitro.
3.5. Expression of Bid mutants in Bax and Bak DKO indicates similar subcellular distribution
One way in which lipid binding has been proposed to regulate Bid function is to control its subcellular targeting. Binding to CL and possibly other lipids has been reported to enhance the association of Bid with intracellular membranes, especially mitochondria [20]. Because protein interactions of Bid with Bak or Bax may also play a role in its association with mitochondria, we initially studied the localisation of the in vitro characterized Bid mutants in mouse embryonic fibroblasts that lacked both Bax and Bak (DKO MEFs). In order to follow localisation, we expressed wt Bid, R187E Bid and K157E Bid as YFP fusions (Fig. 6A). DKO for Bax and Bak MEFs expressing FL-Bid YFP fusions were fixed and co-stained for mitochondrial Hsp70 (mtHsp70). All three FL-Bid YFP fusions were distributed throughout the cytoplasm with no discernable co-localisation with mtHsp70 (Fig. 6B). We then expressed the p15 Bid truncations of wt Bid, R187E Bid and K157E Bid in DKO cells (Fig. 6A, C). All three p15 Bid YFP fusions co-localised perfectly with mitochondria. It was conceivable that the exposed BH3-domain of p15 Bid was driving its localisation through an interaction with some anti-apoptotic Bcl-2 proteins, such as Blc-2 or Bcl-XL, on the OMM. We have previously demonstrated that a further truncated form of Bid, p11, lacking BH3-domain and therefore unable to interact with multidomain Bcl-2 proteins, still localises to the OMM [15]. We hypothesised that as the lipid-binding region lies within this p11 fragment, its localisation may be dependent upon these interactions. However, wild type and Bid mutants expressed as p11 YFP fusions all localised perfectly to the OMM (Fig. 6C), in agreement with previous results with comparable mutations [25].
Fig. 6.

Bid mutants localise with mitochondria in vivo. C-terminal YFP-tagged wt Bid, R187E Bid and K157E Bid were transiently expressed in Bax/Bak DKO MEFs as FL, p15 and p11 truncated forms. (A) Western blot analysis of whole cell lysates showing expression of FL-, p15- and p11-Bid YFP fusion proteins. Lysates were immunoblotted (IB) with anti-Bid. p15-Bid YFP runs as a doublet due to internal initiation at methionine 97. (B) DKO MEFs expressing full-length (FL) wt Bid YFP, K157E Bid YFP or R187E Bid YFP, fixed and immunostained with anti-mtHsp70. Nuclei were stained with Hoechst. Bid YFP fusion proteins all show a cytosolic localisation in the absence of apoptotic stimuli. (C) DKO MEFs expressing p15 truncated (tBid, initiated at G60) forms of wt Bid YFP, K157E Bid YFP or R187E Bid YFP, fixed and immunostained with anti-mtHsp70. p15-Bid YFP fusion proteins all show a mitochondrial localisation. (D) DKO MEFs expressing p11 truncated (initiated at M97) forms of wt Bid YFP, K157E Bid YFP or R187E Bid YFP, fixed and immunostained with anti-mtHsp70. p11-Bid YFP fusion proteins all show a mitochondrial localisation.
3.6. Mutation of the lipid-binding sites does not alter Bid’s pro-apoptotic function
We next asked if there was any difference in the ability of these Bid mutants to induce cytochrome c release and apoptosis in cells. We expressed wt Bid, R187E Bid and K157E Bid as FL, p15 and p11 YFP fusions in Bid knock out (Bid−/−) MEFs. 24 h after transfection, cells were fixed and apoptosis was quantified by analysing nuclear morphology (Fig. 7A). Low levels of apoptosis were observed in Bid−/− MEFs expressing all three FL proteins. Similarly, all three p11 truncations showed the same low level of apoptosis. Conversely, all three p15 Bid proteins induced significant levels of apoptosis. Furthermore, p15 wt Bid, R187E Bid and K157E Bid all induced apoptosis (measured by nuclear fragmentation) to the comparable extents, with no significant difference for the lipid-binding mutants (p>0.05, one-way ANOVA with Bonferroni’s Multiple Comparison Test). We then examined cytochrome c release in Bid−/− cells expressing the p11 and p15 mutants. Cells transiently transfected with the Bid YFP fusions were fixed and immunostained with anti-cytochrome c. As the p15 Bid constructs induced a high level of apoptosis, the culture medium was supplemented with 100 μM zVAD-fmk post-transfection. All three p15 Bid proteins had a punctuate distribution in Bid−/− MEFs (Fig. 7B) and also induced release of cytochrome c. In contrast, all three p11 Bid YFP fusions did not release cytochrome c, even though they localised to mitochondria (Fig. 7C).
Fig. 7.

Expression of Bid mutants induces apoptosis in Bid−/− cells. C-terminal YFP-tagged wt Bid, Bid K157E and Bid R187E were transiently expressed in Bid−/− MEFs as FL, p15 and p11 truncated forms. (A) Bid−/− MEFs transiently expressing the indicated Bid YFP fusion proteins were fixed and stained 24 h post transfection as in the Materials and methods. Apoptosis was quantified by nuclear morphology. Error bars indicate standard error of the mean. (B) Representative images of cytochrome c immunostaining (red) in Bid−/− MEF transiently expressing p15 constructs of mouse Bid as in Fig. 6C. (C) Representative images of cytochrome c immunostaining (red) in Bid−/− MEF after transfection of p11 constructs of mouse Bid as in Fig. 6D. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
In vitro assays that indicated a role for cardiolipin in the pro-apoptotic activity of Bid used recombinant Bax, whereas liver mitochondria and Bid−/− MEFs also have Bak present. We therefore transiently co-transfected DKO MEFs with p15 YFP fusions of wt Bid, R187E Bid or K157E Bid along with mRFP-Bax. Full length and p11 wt Bid were also expressed with mRFP-Bax. We then assessed apoptosis by nuclear morphology (Fig. 8A). This co-expression with wt Bid proteins resulted in similar levels of apoptosis to that seen in Bid−/− cells expressing the Bid mutants alone (cf. Fig. 7A). Similarly, all three p15 Bid proteins induced high levels of apoptosis when co-expressed with mRFP-Bax. There was no significant difference between the p15 Bid proteins (p>0.05, one-way ANOVA with Bonferroni’s Multiple Comparison Test).
Fig. 8.
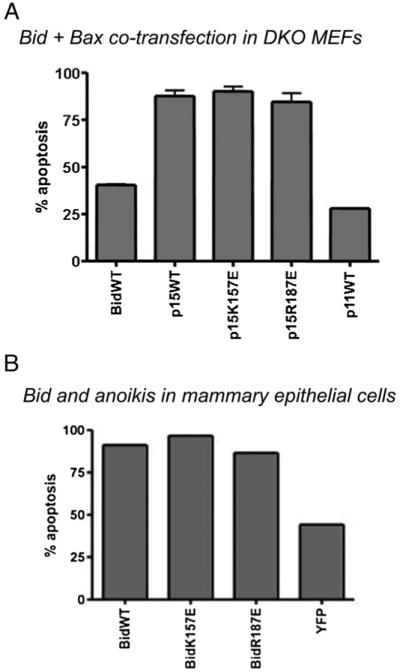
Bid mutants induce Bax dependent apoptosis in DKO MEFs and mammary epithelial cells. (A) Apoptosis in DKO MEFs. C-terminal YFP-tagged FL wt Bid, p15 wt Bid, p15 Bid K157E, p15 Bid R187E or p11 wt Bid were transiently expressed in DKO MEFs along with mRFP-Bax. 24 h post transfection, cells were fixed and nuclei were stained with Hoechst. Apoptosis was quantified by nuclear morphology. Error bars indicate standard error of the mean. (B) Bid and anoikis in epithelial cells. C-terminal YFP-tagged FL wt tBid, FL Bid K157E, FL Bid R187E or YFP alone was expressed in FSK-7 mouse mammary epithelial cells. 18 h post transfection, cells were detached from ECM and replated onto a non-adhesive substrate for 5 h, before collection by cytospinning onto glass slides, fixing and staining with Hoechst. Apoptosis was quantified by nuclear morphology.
Finally, we examined the pro-apoptotic role of the lipid-binding mutants as FL proteins. We have previously shown that during epithelial cell anoikis, FL Bid translocates to the OMM in the absence of caspase-8 cleavage and induces cell death [15]. We expressed FL wt and the two mutant Bid in mammary epithelial cells (MEC) and detached these from ECM as described previously [15]. MEC expressing YFP alone showed around 40% apoptosis after 5 h, whereas over-expression of wt Bid YFP sensitised the cells to anoikis and increased cell death to around 85%, as previously shown [15]. There was no difference between wt Bid and the full length form of R187E Bid YFP or K157E Bid YFP in the amount of sensitisation to anoikis.
Together, these results suggest that the ability of Bid to bind to mitochondrial membrane lipids, in particular CL, may not be essential for its pro-apoptotic function in vivo.
4. Discussion
In this study we report the first point mutation of Bid that specifically alters its interaction with the polar heads of phospholipids. Mutation of the conserved lysine residue at position 157 in the mouse sequence (Fig. 1A) decreases the binding of Bid to the glycerolphosphate head of (lyso)lipids, since it affects binding to LPG and MCL but not LPC (Figs. 2 and 4A). These results confirm previous indications for a dual lipid specificity of full length Bid [38], which can now be dissected at the molecular level by changing the positive charged residue conserved of position 157. Other conserved residues with positive charge like R187 do not show an equivalent alteration of lipid binding and essentially behave as the wild type protein in all in vitro tests (Figs. 2-5).
Both our in vitro and cellular studies indicate that the mutation K157E, which affects binding to negatively charged lipids in vitro (Figs. 2 and 4A), does not change the association of the protein to mitochondria, either as the full length or cleaved protein (Figs. 6 and S1). In addition, the mutation K157E has no evident effect on the pro-apoptotic capacity in vivo, as shown by both the extent of apoptosis observed and the ability to be cleaved by caspase 8 following FasL treatment (Figs. 7, 8 and S2). We have also analysed the distribution of Bid after incubation with mitochondrial membranes (i.e. cardiolipin-containing membranes); again, no significant change in the membrane association of wt and mutant Bid was observed (Fig. S3).
Two alternative interpretations can account for the difference that the same mutation produces in vivo and in vitro. The first interpretation would be that there is no direct relationship between the in vitro binding to membrane lipids and the pro-apoptotic function of Bid within cells, for instance because the binding affinities for CL-related lysolipids in solution and for CL itself in the mitochondria may not be the same. However, recent work has established that tBid binding to cardiolipin-containing membranes is the first crucial event in the coordinated action with other Bcl-2 proteins that leads to membrane permeabilization [41]. Consequently, in vitro studies of Bid–lipid interactions have relevance to the biological pro-apoptotic action of the protein.
The alternative explanation is that the interaction with LPC is much more important for the pro-apoptotic function of Bid than that with negatively charged lipids related to cardiolipin. The latter interpretation is also supported by the results of protein aggregation obtained here with NATIVE PAGE (Fig. 3). Conformational changes and self-aggregation of Bcl-2 proteins is now established to be fundamental for MOMP [41].
The modulating role of cardiolipin has been considered in a recent paper by Gonzalvez et al. [29], which showed that cardiolipin is required for the processing of Bid by caspase 8 on mitochondria but not for the association of Bid to mitochondria and its pro-apoptotic action in vitro. These observations indirectly support our suggestion for a crucial role of LPC in modulating the subcellular distribution and action of Bid proteins. Indeed, BH3-confined mutations that abolish the pro-apoptotic action of Bid also disrupt its binding to LPC. For instance, the addition of LPC-C16 to the G94E-D95E double mutant does not cause any significant changes in Bid fluorescence emission (Fig. 2 and data not shown). Previous observations with similar BH3-confined mutants [38] and native Bid [21] did indicate an exquisite interaction of Bid with LPC that directly affects its pro-apoptotic function and may direct the full length protein towards LPC-enriched membranes, especially mitochondria. It should be considered that two different binding sites are present in the Bid protein but only one site is probably sufficient for the pro-apoptotic activity. A double mutant that inactivates both sites can be useful to clarify this and we are currently screening such a mutant, also for establishing further the essential role of LPC interaction proposed here.
Caspase cleavage removes the specificity of LPC interaction, as we have demonstrated here with different mutant proteins (Fig. 5, cf. [20]). Of note, when the interaction of Bid with lipids has been modulated by altering the levels of cardiolipin in mitochondria and cells, results have been negative or unclear [28,29]. By contrast, induced increase in mitochondrial LPC has been shown to clearly enhance the membrane association and pro-apoptotic action of Bid [21,26].
The K157E mutation affected the rate of cleavage of FL Bid by caspase 8 in vitro. This novel data suggests that interaction of Bid with negatively charged lipids like LPG may affect the rate of caspase cleavage within cells, especially during the initial activation of caspases. Consequently, binding to negatively charged lipids related to CL may add another layer of regulation to the critical activation step associated with protease cleavage of the protein, which depends upon the action of diverse kinases in different cellular contexts [16,20]. This would fit the recent evidence that cardiolipin may modulate Bid cleavage by caspase 8 around or in mitochondria [29]. Our results also seem to draw a parallel with the recent report that Bax cleavage by general proteases is modulated by interaction with cholesterol, another membrane lipid [42].
It has been widely proposed that the interaction of tBid with cardiolipin specifies both its subcellular targeting and its pro-apoptotic function. Lutter et al. [18], using a temperature sensitive mutant of phosphatidylglycerophosphate synthase, implicated a requirement for cardiolipin for tBid mitochondrial targeting and its ability to induce cytochrome c release. Kuwana et al. [23], using a reductionist approach, demonstrated that Bid cooperated with charged lipids, such as cardiolipin, for its interaction with the membrane and its ability to activate Bax. However, it may be that these lipids alter the response of pro-apoptotic Bcl-2 proteins to physico-chemical properties of the whole membrane (cf. [27]). Cardiolipin binding was proposed to target tBid to the contact sites between the inner and outer mitochondrial membranes [43], regulating Bid dependent reorganisation of mitochondrial cristae for cytochrome c release [24]. This reorganisation of mitochondrial membranes may be important for maximising cytochrome c release from the cristae, where up to 90% of the cytochrome c is found [44]. One interesting possibility is that Bid/cardiolipin interactions play a role in cristae remodelling and is not required for MOMP itself, a hypothesis supported by published data showing that tBid can induce cristae reorganisation in the absence of a functional BH3 domain [44].
A number of studies have indicated that cardiolipin is essential for Bid binding to liposomes. This may indeed be the case, but, as our new data indicates, Bid binding to CL-related lipids is not required for mitochondrial targeting in vivo. Bid may interact with proteins on the OMM that could facilitate its membrane insertion via lipid binding. Interactions with other proteins may therefore lead to OMM targeting, and proteins such as MTCH2 have been proposed for this [45]. The mitochondrial-targeting region of Bid has been shown to reside within helices 4–6, which contains K157 required for binding to CL-related lipids, but this may also contain binding sites for proteins. Both the p15 and p11 Bid constructs interact with proteins on the OMM (Valentijn and Gilmore, unpublished data), and it may well be that here are multiple mechanisms for targeting Bid. Indeed, it has also been proposed that tBid becomes myristoylated at its N-terminus following caspase 8 cleavage, providing another mechanism for membrane targeting [46].
Bid can clearly interact with the specific membrane environment of mitochondria. However, the precise role of mitochondrial lipids in Bid’s pro-apoptotic function remains open to debate. Emerging data suggests that Bid can be activated or influenced by a wider range of stimuli than previously thought [15,17]. There could be, therefore, an equally diverse number of ways by which it is targeted to its site of action.
Acknowledgements
Research in Verona was funded by the University of Verona (ex 60%), Italy. Work in Manchester was sponsored by BBSRC grant BB/C50846 in Degli Esposti laboratory and was supported by grants from The Breast Cancer Campaign in Andrew Gilmore laboratory.
Abbreviations
- ADIFAB
AcryloDated Intestinal Fatty Acid Binding Protein
- CL
cardiolipin
- FL Bid
full length Bid
- LPC
lysophosphatidylcholine
- LPC-C16
1-palmitoyl-LPC
- LPC-C12
1-dodecyl-LPC
- LPG
lysophosphatidylglycerol
- LPG-C18:1
1-oleyl-LPG
- MCL
monolysocardiolipin
- tBid
truncated (caspase-8-cleaved) Bid
- OMM
outer mitochondrial membrane
- MOMP
mitochondrial outer membrane permeabilization
References
- [1].Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- [2].Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bouillet P, Strasser A. BH3-only proteins — evolutionarily conserved proapoptotic Bcl-2 family members essential for initiating programmed cell death. J. Cell Sci. 2002;115:1567–1574. doi: 10.1242/jcs.115.8.1567. [DOI] [PubMed] [Google Scholar]
- [4].Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, Ierino H, Lee EF, Fairlie WD, Bouillet P, Strasser A, Kluck RM, Adams JM, Huang DC. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- [5].Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- [6].Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X (L) Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- [7].Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- [8].Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- [9].Gilley J, Coffer PJ, Ham J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J. Cell Biol. 2003;162:613–622. doi: 10.1083/jcb.200303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ley R, Ewings KE, Hadfield K, Howes E, Balmanno K, Cook SJ. Extracellular signal-regulated kinases 1/2 are serum-stimulated “BimEL Kinases” that bind to the BH3-only protein BimEL causing its phosphorylation and turnover. J. Biol. Chem. 2004;279:8837–8847. doi: 10.1074/jbc.M311578200. [DOI] [PubMed] [Google Scholar]
- [11].Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- [12].Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- [13].Degli Esposti M, Masdehors P, Boutin JA, Hickman JA, Dive C. Post-translational modification of Bid has differential effects on its susceptibility to cleavage by caspase 8 or caspase 3. J. Biol. Chem. 2003;278:15749–15757. doi: 10.1074/jbc.M209208200. [DOI] [PubMed] [Google Scholar]
- [14].Sarig R, Zaltsman Y, Marcellus RC, Flavell R, Mak TW, Gross A. BID-D59A is a potent inducer of apoptosis in primary embryonic fibroblasts. J. Biol. Chem. 2003;278:10707–10715. doi: 10.1074/jbc.M210296200. [DOI] [PubMed] [Google Scholar]
- [15].Valentijn AJ, Gilmore AP. Translocation of Full-length bid to mitochondria during anoikis. J. Biol. Chem. 2004;279:32848–32857. doi: 10.1074/jbc.M313375200. [DOI] [PubMed] [Google Scholar]
- [16].Desagher S, Osen-Sand A, Montessuit S, Magnenat E, Vilbois F, Hochmann A, Journot L, Antonsson B, Martinou JC. Phosphorylation of bid by casein kinases I and II regulates its cleavage by caspase 8. Mol. Cell. 2001;8:601–611. doi: 10.1016/s1097-2765(01)00335-5. [DOI] [PubMed] [Google Scholar]
- [17].Kamer I, Sarig R, Zaltsman Y, Niv H, Oberkovitz G, Regev L, Haimovich G, Lerenthal Y, Marcellus R, Gross A. Proapoptotic BID is an ATMeffector in the DNA-damage response. Cell. 2005;122:593–603. doi: 10.1016/j.cell.2005.06.014. [DOI] [PubMed] [Google Scholar]
- [18].Lutter M, Fang M, Luo X, Nishijima M, Xie X, Wang X. Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat. Cell Biol. Nat. Cell Biol. 2000;2:754–756. doi: 10.1038/35036395. [DOI] [PubMed] [Google Scholar]
- [19].Degli Esposti M, Erler ET, Hickman AJ, Dive C. Bid, a widely expressed pro-apoptotic protein of the Bcl-2 family, displays lipid transfer activity. Mol. Cell. Biol. 2001;21:7268–7276. doi: 10.1128/MCB.21.21.7268-7276.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Degli Esposti M, Cristea IM, Gaskell SJ, Nakao Y, Dive C. Proapoptotic Bid binds to monolysocardiolipin, a new molecular connection between mitochondrial membranes and cell death. Cell Death Differ. 2003;10:1300–1309. doi: 10.1038/sj.cdd.4401306. [DOI] [PubMed] [Google Scholar]
- [21].Degli Esposti M. Sequence and functional similarities between pro-apoptotic Bid and plant lipid transfer proteins. Biochim. Biophys. Acta. 2002;1553:331–340. doi: 10.1016/s0005-2728(02)00187-1. [DOI] [PubMed] [Google Scholar]
- [22].Epand RF, Martinou JC, Fornallaz-Mulhauser M, Hughes DW, Epand RM. tBid promotes leakage by altering membrane curvature. J. Biol. Chem. 2002;277:32632–32639. doi: 10.1074/jbc.M202396200. [DOI] [PubMed] [Google Scholar]
- [23].Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- [24].Kim TH, Zhao Y, Ding WX, Shin JN, He X, Seo YW, Chen J, Rabinowich H, Amoscato AA, Yin XM. Bid–cardiolipin interaction at mitochondrial contact site contributes to mitochondrial cristae reorganization and cytochrome c release. Mol. Biol. Cell. 2004;15:3061–3072. doi: 10.1091/mbc.E03-12-0864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu J, Durrant D, Yang HS, He Y, Whitby FG, Myszka DG, Lee RM. The interaction between tBid and cardiolipin or monolysocardiolipin. Biochem. Biophys. Res. Commun. 2005;330:865–870. doi: 10.1016/j.bbrc.2005.03.048. [DOI] [PubMed] [Google Scholar]
- [26].Tyurin VA, Tyurina YY, Osipov AN, Belikova NA, Basova LV, Kapralov AA, Bayir H, Kagan VE. Interactions of cardiolipin and lyso-cardiolipins with cytochrome c and tBid: conflict or assistance in apoptosis. Cell Death Diff. 2007;14:872–875. doi: 10.1038/sj.cdd.4402068. [DOI] [PubMed] [Google Scholar]
- [27].Terrones O, Antonsson B, Yamaguchi H, Wang HG, Liu J, Lee RM, Herrmann A, Basanez G. Lipidic pore formation by the concerted action of proapoptotic BAX and tBid. J. Biol. Chem. 2004;279:30081–30091. doi: 10.1074/jbc.M313420200. [DOI] [PubMed] [Google Scholar]
- [28].Choi SY, Gonzalvez F, Jenkins GM, Slomianny C, Chretien D, Arnoult D, Petit PX, Frohman MA. Cardiolipin deficiency releases cytochrome c from the inner mitochondrial membrane and accelerates stimuli-elicited apoptosis. Cell Death Differ. 2007;14:597–606. doi: 10.1038/sj.cdd.4402020. [DOI] [PubMed] [Google Scholar]
- [29].Gonzalvez F, Schug ZT, Houtkooper RH, MacKenzie ED, Brooks DG, Wanders RJA, Petit PX, Vaz FM, Gottlieb E. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J. Cell Biol. 2008;183:681–696. doi: 10.1083/jcb.200803129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Schlame M, Rustow B. Lysocardiolipin formation and reacylation in isolated rat liver mitochondria. Biochem. J. 1990;272:589–595. doi: 10.1042/bj2720589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Richieri GV, Ogata RT, Kleinfeld AM. A fluorescently labeled intestinal fatty acid protein. J. Biol. Chem. 1992;267:23495–23501. [PubMed] [Google Scholar]
- [32].Crimi M, Astegno A, Zoccatelli G, Degli Esposti M. Pro-apoptotic effect of maize lipid transfer protein on mammalian mitochondria. Arch. Biochem. Biophys. 2006;445:65–71. doi: 10.1016/j.abb.2005.10.024. [DOI] [PubMed] [Google Scholar]
- [33].Degli Esposti M. The roles of Bid. Apoptosis. 2002;7:433–440. doi: 10.1023/a:1020035124855. [DOI] [PubMed] [Google Scholar]
- [34].Oh KJ, Barbuto S, Meyer N, Kim RS, Collier RJ, Korsmeyer SJ. Conformational changes in BID, a pro-apoptotic BCL-2 family member, upon membrane binding. J. Biol. Chem. 2005;280:753–767. doi: 10.1074/jbc.M405428200. [DOI] [PubMed] [Google Scholar]
- [35].Hu X, Han Z, Wyche JH, Hendrickson EA. Helix 6 of tBid is necessary but not sufficient for mitochondrial binding activity. Apoptosis. 2003;8:277–289. doi: 10.1023/a:1023676906857. [DOI] [PubMed] [Google Scholar]
- [36].McDonnell JM, Fushman D, Milliman CL, Korsmeyer SJ, Cowburn D. Solution structure of the proapoptotic molecule BID: a structural basis for apoptotic agonists and antagonists. Cell. 1999;9:625–634. doi: 10.1016/s0092-8674(00)80573-5. [DOI] [PubMed] [Google Scholar]
- [37].Becattini B, Sareth S, Zhai D, Crowell KJ, Leone M, Reed JC, Pellecchia M. Targeting apoptosis via chemical design: inhibition of bid-induced cell death by small organic molecules. Chemistry & Biology. 2004;11:1107–1117. doi: 10.1016/j.chembiol.2004.05.022. [DOI] [PubMed] [Google Scholar]
- [38].Goonesinghe A, Mundy ES, Smith M, Khosravi-Far R, Martinou JC, Degli Esposti M. Pro-apoptotic Bid induces membrane perturbation by inserting selected lysolipids into the bilayer. Biochem. J. 2005;387:109–118. doi: 10.1042/BJ20041389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Darra E, Abdel-Azeim S, Manara A, Shoji K, Maréchal JD, Mariotto S, Cavalieri E, Perbellini L, Pizza C, Perahia D, Crimi M, Suzuki H. Insight into the apoptosis-inducing action of alpha-bisabolol towards malignant tumor cells: involvement of lipid rafts and Bid. Arch. Biochem. Biophys. 2008;476:113–123. doi: 10.1016/j.abb.2008.02.004. [DOI] [PubMed] [Google Scholar]
- [40].Gross A, Yin XM, Wang K, Wei MC, Jockel J, Milliman C, Erdjument-Bromage H, Tempst P, Korsmeyer SJ. Caspase cleaved BID targets mitochondria and is required for cytochrome c release while BCL-XL prevents this release but not tumor necrosis factor death. J. Biol. Chem. 1999;274:1156–1163. doi: 10.1074/jbc.274.2.1156. [DOI] [PubMed] [Google Scholar]
- [41].Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B, Andrews DW. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell. 2008;135:1074–1084. doi: 10.1016/j.cell.2008.11.010. [DOI] [PubMed] [Google Scholar]
- [42].Lucken-Ardjomande S, Montessuit S, Martinou JC. Bax activation and stress-induced apoptosis delayed by the accumulation of cholesterol in mitochondrial membranes. Cell Death Differ. 2008;15:484–493. doi: 10.1038/sj.cdd.4402280. [DOI] [PubMed] [Google Scholar]
- [43].Lutter M, Perkins GA, Wang X. The pro-apoptotic Bcl-2 family member tBid localizes to mitochondrial contact sites. BMC Cell Biol. 2001;2:22. doi: 10.1186/1471-2121-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, Korsmeyer SJ. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell. 2002;2:55–67. doi: 10.1016/s1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- [45].Grinberg M, Schwarz M, Zaltsman Y, Eini T, Niv H, Pietrokovski S, Gross A. Mitochondrial carrier homolog 2 is a target of tBID in cells signaled to die by tumor necrosis factor alpha. Mol. Cell Biol. 2005;25:4579–4590. doi: 10.1128/MCB.25.11.4579-4590.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zha J, Weiler S, Oh KJ, Wei MC, Korsmeyer SJ. Posttranslational N-myristoylation of Bid as a molecular switch for targeting mitochondria and apoptosis. Science. 2000;290:1761–1765. doi: 10.1126/science.290.5497.1761. [DOI] [PubMed] [Google Scholar]