

Abstract
The navel orangeworm, Amyelois transitella (Walker), is an agricultural insect pest that can be controlled by disrupting male-female communication with sex pheromones, a technique known as mating disruption. Insect pheromone-binding proteins (PBPs) provide fast transport of hydrophobic pheromones through aqueous sensillar lymph and promote sensitive delivery of pheromones to receptors. Here we present the three-dimensional structure of a PBP from Amyelois transitella (AtraPBP1) in solution at pH 4.5 determined by nuclear magnetic resonance (NMR) spectroscopy. Pulsed-field gradient NMR diffusion experiments, multi-angle light scattering, and 15N NMR relaxation analysis indicate that AtraPBP1 forms a stable monomer in solution at pH 4.5 in contrast to forming mostly dimers at pH 7. The NMR structure of AtraPBP1 at pH 4.5 contains seven α-helices (α1: L8-L23, α2: D27-F36, α3: R46-V62, α4: A73-M78; α5: D84-S100; α6: R107-L125; α7: M131-E141) that adopt an overall main chain fold similar to that of PBPs found in Antheraea polyphemus and Bombyx mori. The AtraPBP1 structure is stabilized by three disulfide bonds formed by C19/C54, C50/C108 and C97/C117, and salt bridges formed by H69/E60, H70/E57, H80/E132, H95/E141 and H123/D40. All five His residues are cationic at pH 4.5, whereas H80 and H95 become neutral at pH 7.0. The C-terminal helix (α7) contains hydrophobic residues (M131, V133, V134, V135, V138, L139 and A140) that contact conserved residues (W37, L59, A73, F76, A77, I94, V111, V115) suggested to interact with bound pheromone. Our NMR studies reveal that acid-induced formation of the C-terminal helix at pH 4.5 is triggered by a histidine protonation switch that promotes rapid release of bound pheromone under acidic conditions.
Keywords: AtraPBP1, NMR, pheromone-binding protein, Amyelois transitella, pheromone, navel orangeworm moth, multi-angle light scattering, histidine protonation switch, disulfide bridge
The navel orangeworm, Amyelois transitella (Walker) (Lepidoptera: Pyralidae), is the most serious insect pest of almonds and pistachios in California, and a major pest of walnuts, figs and a number of other crops. This agricultural pest is primarily controlled with pyrethroids and insects growth regulators, but alternative methods of control, including sex pheromone-based mating disruption, are sorely needed. A potential way of controlling insect pests is to disrupt detection of sex pheromones. The sex pheromone system of this species has been previously identified (1, 2), but some constituents are unstable thus requiring the development of stable alternatives (parapheromones) for practical applications. We aim at employing olfactory proteins to screen potential attractants (parapheromones), an approach termed “reverse chemical ecology” (3). Previously, we have identified olfactory proteins from the navel orangeworm, including a male antennae-specific pheromone-binding protein, AtraPBP1 (4). There is growing evidence in the literature suggesting that pheromone-binding proteins (PBPs) contribute to the sensitivity and possibly the selectivity of the insect's olfactory system (5).
A molecular mechanism for moth PBPs has been proposed based on the PBP from silkworm moth, BmorPBP1, for which a pH-dependent conformational change was shown to be involved in pheromone binding and release (6-8). Indeed, previous structural studies showed the C-terminal part of PBPs, which is unstructured in pheromone-PBP complex (9) and forms an α-helix at low pH that competes with pheromone for the binding pocket (10-12), thus enabling the delivery of the pheromone in acidic environment similar to that formed by the negatively charged dendrite surfaces of the olfactory receptor neurons (13). Functional studies also showed that BmorPBP1, when co-expressed with pheromone receptor BmorOR1 in the empty neuron system of Drosophila, enhanced the response to the pheromone, indicating that OBPs contribute to the inordinate sensitivity of the insect's olfactory system (5).
In this study, we aimed at getting a better understanding of the structural features of AtraPBP1 to explore its use as a molecular target in a reverse chemical ecology-based screening of parapheromones. Preliminary studies have suggested that AtraPBP1 undergoes a pH-dependent conformational change (4). Here, we present the NMR solution structure of AtraPBP1 at pH 4.5 determined by NMR spectroscopy. First, we determined if delipidation of AtraPBP1 had any effect on protein structure as suggested by (14). Delipidated and non-delipidated samples of AtraPBP1 exhibit NMR spectra (and hence structures) that are indistinguishable (see below). The overall main chain structure of AtraPBP1 is quite similar to that of ApolPBP1 in Antheraea polyphemus (11) and BmorPBP1 in Bombyx mori (10, 15). An important structural feature of AtraPBP1 is two pH-dependent salt bridges involving H80/E132 and H95/E141 (termed histidine protonation switch) that stabilize formation of the C-terminal helix at pH 4.5. The C-terminal helix in AtraPBP1 interacts intimately with hydrophobic core residues that are homologous to residues in BmorPBP1 shown previously to interact with bound pheromone (9, 12). We propose that pH-dependent formation of the C-terminal helix in AtraPBP1 at pH 4.5 disrupts the binding site of hydrophobic sex pheromones and thus may promote rapid release of pheromones to odorant receptors under acidic conditions.
Experimental Procedures
Protein Expression and Purification
Uniformly 15N-labeled and 13C,15N-labeled AtraPBP1 was expressed in Eschericia coli and purified by ion-exchange and gel-filtration chromatography as described previously (4, 16). Typically, 5 mg of purified protein was obtained from a 1-liter culture. Highly purified protein fractions were concentrated by Centricon-10, desalted on four 5-ml HiTrap desalting columns (GE Healthcare Bio-Sciences, Piscataway, NJ) in tandem with water as mobile phase, and analyzed by LC-ESI/MS. The purest fractions were used for NMR studies or combined and delipidated following a previous protocol (15), with small modifications. Hydroxyalkoxypropyl-dextran type VI resin (Sigma, St. Louis, MO) (1 g) was suspended in HPLC grade methanol (20 ml), transferred to a glass column (i.d., 8.5 mm) with a stopper, washed with methanol (60 ml), and then equilibrated with 50 mM citric acid buffer, pH 4.5, after washing with 60 ml of this buffer. The content of the column was transferred to a 15 ml Falcon tube. Pure AtraPBP1 fractions (ca. 2 mg per delipidation batch) were dissolved in 50 mM citric acid, pH 4.5, mixed with the equilibrated resin, and incubated for 1 h at room temperature in a High Speed Rotating Extractor (RT50, Taitec, Tokyo, Japan) at 50 r/min. Then, the mixture was transferred to the glass column. AtraPBP1 was eluted with citric acid buffer, and analyzed by LC-ESI/MS. The purest fractions were desalted on four 5 ml HiTrap columns (GE Healthcare, Bio-Sciences), analyzed by LC-ESI/MS, and the highest purity fractions (>99%) were used.
Molecular mass analysis
Size exclusion chromatography (SEC) was performed on a Superdex 75 HR 10/30 column (GE Healthcare) at 4 °C equilibrated in buffers containing either 10 mM phosphate (pH 7.0) or 10 mM acetate (pH 4.5). A 0.1 ml aliquot of protein (500 μM) was loaded onto the column and eluted at a flow rate of 0.5 ml/min. Molecular masses were analyzed by analytical SEC performed in-line with a multi-angle light-scattering (MALS) miniDawn instrument with a 690-nm laser (Wyatt Technologies, Inc.) coupled to refractive index instrument (Optilab Rex, Wyatt Technologies, Inc.). The molar mass of chromatographed protein was calculated from the observed light scattering intensity and differential refractive index (17) using ASTRA software (Wyatt Technologies, Inc.) based on Zimm plot analysis using a refractive index increment, dn/dc = 0.185 L g-1 (18).
NMR Spectroscopy
Samples of AtraPBP1 for NMR analysis consisted of 15N-labeled or 13C/15N-labeled protein (0.5 mM) in 0.3 ml of a 95% H2O/5% 2H2O solution containing 10 mM sodium acetate (pH 4.5). All NMR experiments were performed at 25°C on Bruker Avance III 600 or 800 MHz spectrometers equipped with a four-channel interface and triple-resonance cryo-probe (TCI) with pulsed field gradients. The 15N-1H HSQC spectra were recorded on a sample of 15N-labeled AtraPBP1 (in 95% H2O, 5% 2H2O). The number of complex points and acquisition times were: 256, 180 ms (15N (F1)); and, 512, 64 ms (1H (F2)). All triple-resonance experiments were performed, processed and analyzed as described (19, 20) on a sample of 13C/15N-labeled AtraPBP1 (in 95% H2O, 5% 2H2O) with the following number of complex points and acquisition times: HNCO {15N (F1) 32, 23.7 ms; 13CO (F2) 64, 42.7 ms; 1H (F3) 512, 64 ms}; HNCACB {15N (F1) 32, 23.7 ms; 13C (F2) 48, 6.3 ms; 1H (F3) 512, 64 ms}; CBCACONNH {15N (F1) 32, 23.7 ms; 13C (F2) 48, 6.3 ms; 1H (F3) 512, 64 ms}; and, HBHACONNH {15N (F1) 32, 23.7 ms, 1Hab (F2) 64 21 ms, 1H (F3) 512, 64 ms}. 15N-edited and 13C-edited NOESY-HSQC and TOCSY-HSQC experiments were performed as described previously (21, 22). Stereospecific assignments of chiral methyl groups of valine and leucine were obtained by analyzing 1H-13C HSQC experiments performed on a sample that contained 10% 13C labeling of AtraPBP1 (23).
Triple resonance and NOESY spectra measured above were analyzed to determine resonance assignments and secondary structure of AtraPBP1. The chemical shift index (see (24) for detailed description of the chemical shift index), 3JNHα coupling constants, and Nuclear Overhauser Effect (NOE) connectivity patterns for each residue were analyzed and provided a measure of the overall secondary structure. Small 3JNHα coupling constants (<5 Hz), strong NOE connectivities (NN(i,i+1), αβ(i,i+3) and αN(i,i+3)), and positive chemical shift index are characteristic of residues in an α-helix. Conversely, large 3JNHα coupling constants (>8 Hz), strong αN(i,i+1) and weak NN(i,i+1) NOE connectivities, and negative chemical shift index are characteristic of residues in a β-strand. The results of the secondary structure analysis and topology of AtraPBP1 are summarized schematically in Fig. 1.
Figure 1.

Amino acid sequence alignment of AtraPBP1 with other moth PBPs. Residues forming histidine protonation switch are highlighted in bold and red. Hydrophobic residues interacting with α7 and implicated in pheromone binding are colored blue. Secondary structural elements at pH 4.5 indicated schematically (helices in red and light-red; strands in yellow) were derived from the analysis of NMR data (3JHNHα, chemical shift index, and sequential NOE patterns).
Two-dimensional 15N-1H long-range HMQC (LR-HMQC) experiments were performed to correlate the histidine ring nitrogen-15 resonances (Nδ1 and Nε2) with carbon attached ring protons, Hδ2 and Hε1 (Fig. 7). A dephasing delay of 45 ms was chosen to select the desired two-bond J-couplings (2JNH = 11.35 Hz) in the histidine ring and suppress unwanted signals from one-bond 1JNH amide couplings (25). The LR-HMQC spectra for uniformily 15N-labeled AtraPBP1 were collected at 298 K with 1H and 15N carrier frequencies at 4.70 and 210 ppm, respectively. Spectra were collected on samples having pH values 4.5, 5.0, 5.5, 6.0, 6.5 and 7.0. The 15N dimension had a sweep width of 110 ppm with 128 complex points, and the 1H dimension had a sweep width of 12 ppm with 2048 complex points. Decoupling of 15N was accomplished with the GARP sequence (26) using a 1.39 kHz field.
Figure 7.

2D 15N-1H LR-HMQC spectrum of AtraPBP1 at pH 4.5 (black) and pH 7.0 (red). The 15Nε2 resonances of H80 and H95 are marked and exhibit large pH-dependent chemical shift changes. Inset shows a plot of 15Nε2 resonance frequency for H80 (black squares) and H95 (open circles) vs. pH.
15N NMR Relaxation Measurements
15N R1, R2, and {1H}-15N NOE experiments were performed on AtraPBP1 at 25°C using standard pulse sequences described previously (27). Longitudinal magnetization decay was recorded using seven different delay times: 0.01, 0.05, 0.15, 0.2, 0.3, 0.4, and 0.8 s. Transverse magnetization decay was recorded with eight different delays: 0.0, 0.016, 0.032, 0.048, 0.064, 0.08, 0.096, and 0.112 s. To check sample stability, transverse magnetization decay at 0.032 s was verified unchanged before and after each set of measurements. A recycle delay of 1.5 s was employed in measurements of both 15N R1 and R2 experiments. Steady-state {1H}-15N NOE values were obtained by recording two sets of spectra in the presence and absence of a 3 s proton saturation period. The NOE experiments were repeated three times to calculate average and standard deviation of the NOE values. The overall rotational correlation time for backbone amide motion was determined using the protocol described previously (28).
Structure Calculation
Backbone and side chain NMR resonances were assigned as described previously (20). Analysis of NOESY data determined nearly 2000 interproton distance relationships throughout the protein (19). The NMR-derived distances and dihedral angles then served as constraints (see Table 1) for calculating the three-dimensional structure of the protein using distance geometry and restrained molecular dynamics. Structure calculations were performed using the YASAP protocol within X-PLOR (29, 30), as described previously (31). A total of 1856 interproton distance constraints were obtained as described (20) by analysis of 13C-edited and 15N-edited NOESY-HSQC spectra (100 ms mixing time) of 13C,15N-labeled AtraPBP1. In addition to the NOE-derived distance constraints, the following additional constraints were included in the structure calculation: 196 dihedral angle constraints (φ and ψ) and 122 distance constraints for 61 hydrogen bonds verified by identifying slowly exchanging amide protons in hydrogen-deuterium exchange experiments (32). Fifty independent structures were calculated and the 15 structures of lowest energy were selected. The average total and experimental distance energies are 3361 ±359 and 187 kcal mol-1. The average root mean square (RMS) deviations from an idealized geometry for bonds and angles are 0.0081 Å and 1.98°. None of the distance and angle constraints were violated by more than 0.40 Å and 4°, respectively.
Table 1.
Structural statistics for the ensemble of 15 calculated structures of AtraPBP1.
| NOE restraints (total) | 1856 |
| Intra (|i − j| = 0) | 650 |
| Medium (1 ≤ |i − j| ≤ 4) | 422 |
| Long (|i − j| > 4) | 784 |
| Dihedral angle restraints (φ and ψ) | 196 |
| Hydrogen bond restraints in α-helical regions | 122 |
| RMSD from ideal geometry | |
| Bond length (Å) | 0.0081 ± 0.00012 |
| Bond angles (°) | 1.98 ± 0.00095 |
| Ramachandran plot | |
| Most favored region | 87% |
| Allowed regions | 12% |
| Disallowed regions | 1% |
| RMSD of atom position from average structure | |
| β-sheet and α-helical regions (main chain atoms) | 0.68 ± 0.09 Å |
| β-sheet and α-helical regions (non-hydrogen atoms) | 1.27 ± 0.08 Å |
Results and Discussion
Delipidation has no Effect on the Structure of AtraPBP1
Before beginning detailed NMR structural studies on AtraPBP1, we first examined whether protein delipidation has any effect on structure as suggested by (14). Two-dimensional 15N-1H HSQC NMR spectra of both delipidated (red) and non-delipidated (black) samples of AtraPBP1 are overlayed in Fig. 2. The assigned peaks in the spectra represent main chain and side-chain amide groups that serve as fingerprints of overall conformation. The NMR spectra are indistinguishable for both the delipidated and non-delipidated forms and indicate that delipidation of AtraPBP1 has no detectable effect on protein structure. The NMR structural studies below were performed on non-delipidated samples.
Figure 2.

Two-dimensional 15N-1H HSQC NMR Spectra of delipidated (red) and non-delipidated (black) samples of 15N-labeled AtraPBP1 at pH 4.5. Peaks corresponding to the NH2-groups of the side chain amides of Gln and Asn residues are connected by dotted lines. Sequence-specific assignments are indicated.
pH-dependent Dimerization of AtraPBP1
Previous studies have suggested a pH-dependent dimerization of BmorPBP1 (33). We performed NMR relaxation studies and SEC-MALS analysis to examine the oligomerization state of AtraPBP1 as a function of pH. A summary of 15N NMR relaxation and heteronuclear NOE data at pH 4.5 are presented in Fig. 3. The average 15N R1 and R2 values from residues in structured regions are 1.28 (± 0.05) s-1 and 13.3 (± 0.5) s-1, respectively. Elevated R1 values and decreased {1H}-15N NOEs (<0.65) are apparent for the first 8 residues from the N-terminus, consistent with significant backbone flexibility in this region. Assuming isotropic tumbling of AtraPBP1, the overall rotational correlation time was obtained from R1/R2 ratios of all residues within 1 standard deviation of the average value (34). Thus, the average rotational correlation time was calculated to be 8.7 ±0.5 ns at 298 K, indicating the protein is monomeric in solution at pH 4.5. A Zimm plot analysis of the SEC in-line MALS data in Fig. 4 determines the molar mass of AtraPBP1 in solution to be 16 ±2 kDa at pH 4.5 and 29 ±3 kDa at pH 7, indicating that AtraPBP1 becomes dimeric in solution at pH 7.0 under NMR conditions. NMR spectra of AtraPBP1 change quite dramatically upon increasing the pH from 4.5 to 7.0, consistent with a pH-dependent conformational change between two distinct conformational states of moth PBPs as described previously (4). The pH-dependent spectral changes can be titrated as two sets of NMR peaks (rather than a single averaged peak), indicating the two pH-dependent conformational states are in slow exchange on the NMR chemical shift time scale.
Figure 3.

15N NMR relaxation data for AtraPBP1 at pH 4.5. Steady-state {1H}-15N NOEs (A), spin-lattice relaxation rate constants (B) and spin-spin relaxation rate constants (C) are plotted as a function of residue number. All data were determined at 800 MHz 1H frequency and 298 K.
Figure 4.
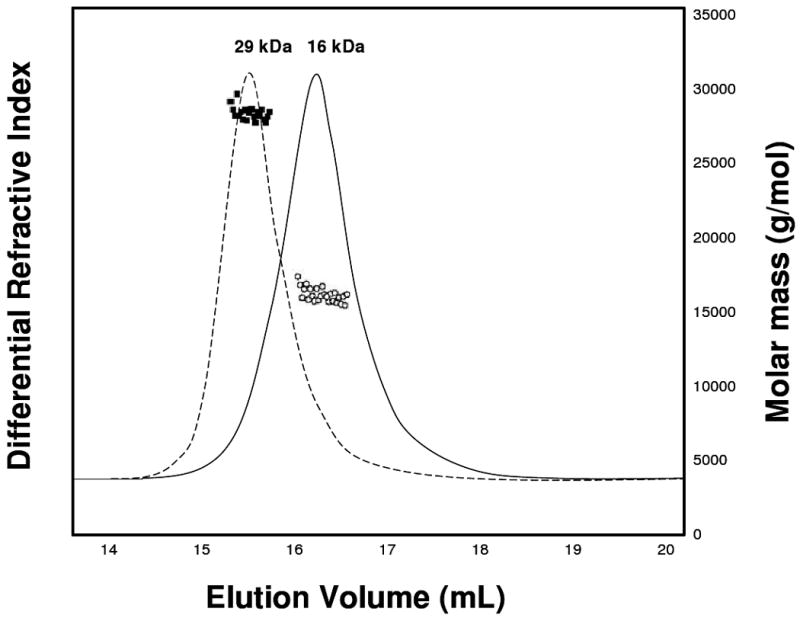
SEC-MALS analysis of AtraPBP1 at pH 4.5 (solid) and pH 7.0 (dashed). The molar masses of AtraPBP1 in solution at pH 4.5 (circles) and pH 7.0 (squares) were calculated from a Zimm plot analysis of the observed light scattering intensity using a refractive index increment, dn/dc = 0.185 L g-1 (17, 18). Protein concentrations are at 500 μM.
NMR Spectroscopy of AtraPBP1
The 1H-15N-HSQC NMR spectrum of 15N-labeled AtraPBP1 at pH 4.5 (Fig. 2) exhibited close to the expected number of backbone amide resonances (135 out of 142). The large chemical shift dispersion and uniform peak intensities indicate that the protein is structurally homogeneous and stably folded. More than 95% of the NMR resonances in the 15N-1H HSQC spectrum of AtraPBP1 at pH 4.5 were assigned (16) and the assignments have been deposited to the BioMagResBank (BMRB) repository (accession no. 15561). NMR assignments could not be obtained for the first four residues from the N-terminus, because of weak NMR intensities perhaps due to chemical exchange broadening caused by the unstructured N-terminus. Three-dimensional protein structures of AtraPBP1 were derived from the NMR assignments and calculated on the basis of NOE data, slowly exchanging amide NH groups, chemical shift analysis, and 3JNHα spin-spin coupling constants (see Methods). The analysis of chemical shift index (CSI) (35), 3JNHα (36) and hydrogen-deuterium exchange rates of NH groups (37) determined the secondary structure shown in Fig. 1. The final three-dimensional structures of AtraPBP1 derived from the NMR data are illustrated in Figs. 5-6 (atomic coordinates have been deposited in the RCSB Protein Databank2). Table 1 summarizes the structural statistics calculated for 15 lowest energy conformers.
Figure 5.
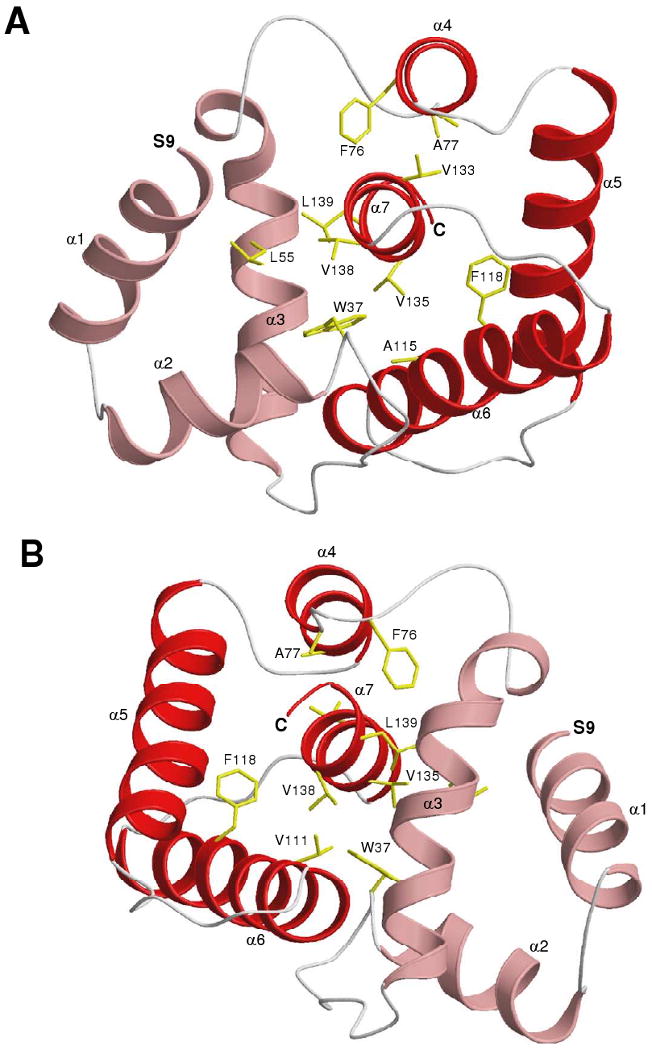
NMR-derived structures of AtraPBP1 in solution at pH 4.5 determined by NMR. (A) Ribbon representation of the energy-minimized average main chain structure. (B) ∼180° rotation of A. The N-terminal residues (1-8) are unstructured and not shown. α-helices are highlighted red and light-red, and hydrophobic side chains are yellow.
Figure 6.
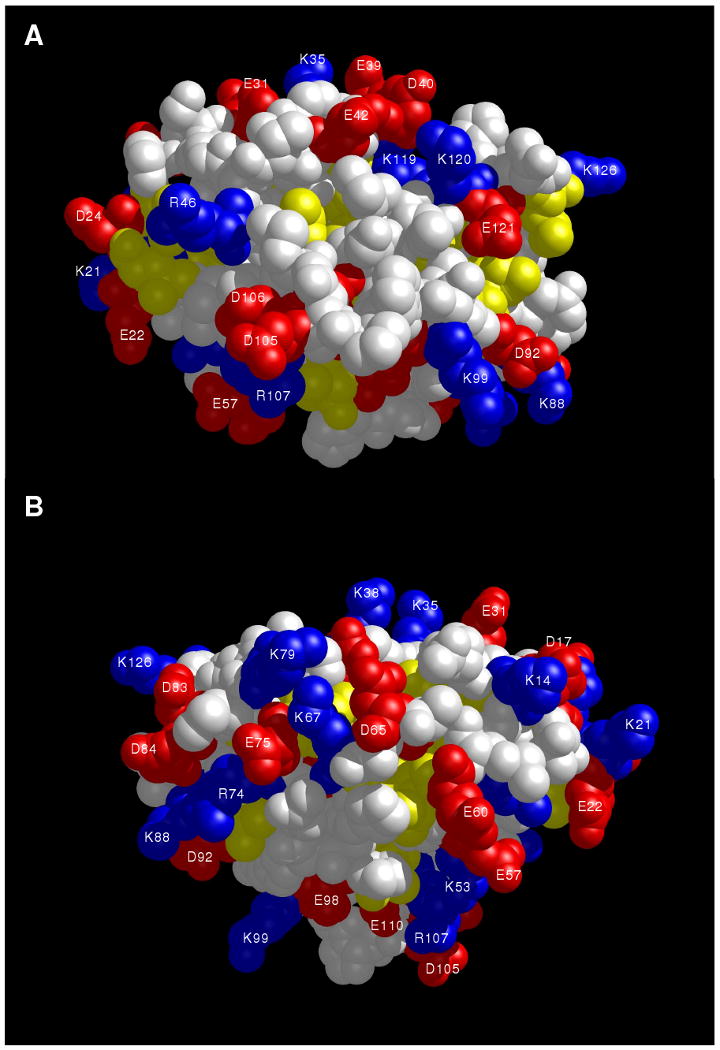
Space-filling representation of AtraPBP1 with same view as shown in Fig. 5. Acidic residues (Glu and Asp), basic residues (Lys and Arg), and hydrophobic residues (Leu, Ile, Phe, Trp, Val) are colored red, blue and yellow, respectively.
Three-dimensional Structure of AtraPBP1 at pH 4.5
The NMR-derived structures of AtraPBP1 (Figs. 5-6) reveal an overall fold similar to that of BmorPBP1 (10, 12) and ApolPBP1 (11). The RMS deviation of the main chain atoms is 1.18 Ǻ in comparing AtraPBP1 to BmorPBP1 and 1.22 Ǻ in comparing AtraPBP1 to ApolPBP1. The entire main chain structure of AtraPBP1 has been defined except for the unstructured N-terminal region (residues 1-8). These unstructured residues are poorly defined due to a lack of long-range NOE contacts as well as chemical shift and {1H}-15N NOE data (Fig. 3) indicating an unstructured, random coil secondary structure. The main chain fold (residues 9-142) contains a total of seven α-helices and two β-strands: α1 (residues 9-23), α2 (residues 27-36), α3 (residues 46-62), α4 (residues 76-81), α5 (residues 84-100), α6 (107-126), α7 (residues 131-141), β1 (residues 68-70), β2 (residues 71-73) (Fig. 1A). The overall fold is stabilized by three disulfide bonds located between Cys19 and Cys54, Cys50 and Cys108, and Cys97 and Cys117. The disulfide bonds were determined by NOE patterns between the linked Cys residues as well as by the characteristic chemical shift of the beta-methylene carbon-13 resonance for each linked Cys residue (38). The overall structure of AtraPBP1 can be divided into two helical sub-domains: Three helices α1, α2, and α3 are grouped as an N-terminal sub-domain (shaded light red in Fig. 5) flanked by a four helix bundle sub-domain, comprised of α4, α5, α6 and α7 (dark red in Fig. 5). The α3 helix forms an interface between the two sub-domains with many side-chains in the protein hydrophobic core.
The C-terminal helix (α7) contains hydrophobic side-chains (M131, V133, V134, V135, V138, L139 and A140) that form intimate NOE contacts with conserved residues (W37, L59, A73, F76, A77, I94, V111, V115) implicated previously to interact with bound pheromone (9, 12). The C-terminal helix also forms close contacts with a β-turn structure (residues 68 – 73) that resembles a flap to cover and stabilize the C-terminal helix at pH 4.5. The flap contains two exposed His residues (H69 and H70) that form close contacts with E57 and E60. The C-terminal helix contains E132 and E141 located at each end that forms NOE contacts with H80 and H95, respectively.
A surface representation of AtraPBP1 is shown in Fig. 6. The protein surface contains almost exclusively charged residues, which explains the very high solubility of this protein at pH 4.5. H80, H95 and H123 on the surface are charged at pH 4.5, but these His residues are expected to become neutral and deprotonated at pH 7 (see below), which lowers the surface charge and could explain in part the pH-dependent dimerization (Fig. 4). Indeed, exposed hydrophobic residues (L59, M61, A64, V91, I114, V142), located near the exposed His residues are predicted to form an extended hydrophobic surface at pH 7 that could facilitate protein dimerization.
Protonation State of His Residues
The NMR chemical shifts of nitrogen-15 resonances of the histidine ring are characteristic of their protonation state and thus indicate whether a histidine side-chain is cationic or in a particular neutral tautomer (39, 40). AtraPBP1 contains five histidines (H69, H70, H80, H95 and H123) represented by five sets of resolved peaks in 15N-1H LR-HMQC spectra at pH 4.5 that can be assigned to individual proton/nitrogen-15 correlations in the histidine ring (Fig. 7 and Table 2). The 15N-1H LR-HMQC spectra of delipidated AtraPBP1 (see supplemental Fig. 1) look identical to spectra of non-delipidated AtraPBP1 (Fig. 7), indicating that protein delipidation has no effect on histidine protonation state. The histidine spectral assignments were carried out first by correlating the assigned backbone amide proton resonance of each His with the corresponding Hδ2 ring resonance in 15N-edited NOESY spectra. The Hδ2 resonance assignments were confirmed by verifying correlations between assigned β-methylene 13C resonances of each His with the corresponding Hδ2 ring resonance in CB(CGCD)HD spectra. The Hδ2 resonances of H80, H95, H69, H123, and H70 were assigned at 6.51, 7.18, 7.22, 7.29, and 7.34 ppm, respectively. The assignment of each Hδ2 resonance then allowed assignment of the corresponding 15Nδ1 and 15Nε2 resonances for each His residue in 15N-1H LR-HMQC (Fig. 7 and Table 2). The Nε2-Hδ2 correlations (two-bond J-coupling) have stronger peak intensities than the Nδ2-Hδ2 correlations (three-bond J-coupling), enabling separate assignment of 15Nδ1 and 15Nε2 resonances. The chemical shifts of both 15Nδ1 and 15Nε2 are less than 180 ppm for each His in AtraPBP1 at pH 4.5 (Fig. 7 and Table 2), indicating that all five His residues are in the fully protonated, cationic state (both Nδ1 and Nε2 are protonated) at pH 4.5.
Table 2.
NMR Chemical shifts of histidine imidazole side-chain resonances in AtraPBP1.
| Residue # | 1Hδ2 (ppm) | 1Hε1 (ppm) | 15Nδ1 (ppm) | 15Nε2 (ppm) | Tautomer |
|---|---|---|---|---|---|
| pH 4.5 | |||||
| H69 | 7.22 | 8.58 | 176 | 173 | cationic |
| H70 | 7.34 | 8.54 | 176 | 174 | cationic |
| H80 | 6.51 | 8.48 | 176 | 176 | cationic |
| H95 | 7.18 | 8.60 | 175 | 175 | cationic |
| H123 | 7.29 | 8.57 | 178 | 172 | cationic |
| pH 7.0 | |||||
| H69 | 7.24 | 8.44 | 181 | 174 | cationic |
| H70 | 7.27 | 7.95 | 180 | 173 | cationic |
| H80 | 6.93 | 7.57 | 249 | 165 | ε-NH |
| H95 | 6.98 | 7.11 | 236 | 170 | ε-NH |
| H123 | 7.04 | 8.18 | 195 | 178 | ε-NH |
ε-NH represents epsilon-N-H tautomer.
The pKa of each His side-chain was determined by measuring the resonance frequencies of 15Nδ1 and 15Nε2 as a function of pH (Fig. 7). The resonance frequency of 15Nδ1 for each His residue changed very little as a function of pH from 4.5 to 7.0, indicating that Nδ1 remains protonated in this pH range. By contrast, the resonance frequency of 15Nε2 in H80 and H95 both increased from ∼180 ppm to ∼250 ppm as the pH was raised from 4.5 to 7.0, indicating that Nε2 in these residues becomes deprotonated at pH 7.0. Identical results were observed using delipidated AtraPBP1 (see supplemental Fig. 1), showing that protein delipidation has no effect on the pKa of histidines. The 15Nε2 resonance frequencies for H80 and H95 are plotted as a function of pH and reveal that Nε2 in both H80 and H95 becomes deprotonated with a pKa of ∼6 (Fig. 7, inset). The resonance frequency of 15Nε2 in H69 and H70 increased by a much smaller amount (< 2%) as the pH was raised from 4.5 to 7.0, suggesting that H69 and H70 both remain cationic (fully protonated) at pH 7. The resonance frequency of 15Nε2 in H123 increased from 178 ppm to 195 ppm at pH 7, which is half way between the chemical shifts expected for a protonated vs. unprotonated nitrogen (39). This intermediate 15N chemical shift suggests that Nε2 in H123 exists as an equilibrium mixture of protonated and depronated states or perhaps forms a strong hydrogen bond as the pH is raised from 4.5 to 7.0. The deprotonation of H80 and H95 (pKa ∼ 6.0) correlates well with the overall pH-dependent conformational change described previously (pKa ∼ 5.7, (4)).
Implications for pH-sensitive Pheromone Detection
The goal of this study was to investigate the structural mechanism of pH-dependent pheromone binding to AtraPBP1 (4). We present here the NMR solution structure of AtraPBP1 at pH 4.5 (Figs. 5-6) and propose a histidine protonation switch to explain pH-dependent pheromone binding (Fig. 8). At pH 4.5, AtraPBP1 adopts a helical fold similar to that of ApolPBP1 (11) and BmorPBP1 (10, 15). The C-terminal helix (α7) in AtraPBP1 interacts intimately with conserved residues implicated previously in pheromone binding (9, 12). Hydrophobic side-chains of residues in α7 form NOE contacts with the side-chains of conserved residues (W37, L59, A73, F76, A77, I94, V111, V115) suggested to interact with bound pheromone. We propose an insertion of the C-terminal helix (α7) inside the protein hydrophobic core at pH 4.5 (Figs. 5 and 8) that structurally resembles hydrophobic pheromones and thus serves to block the binding of pheromone under acidic conditions.
Figure 8.
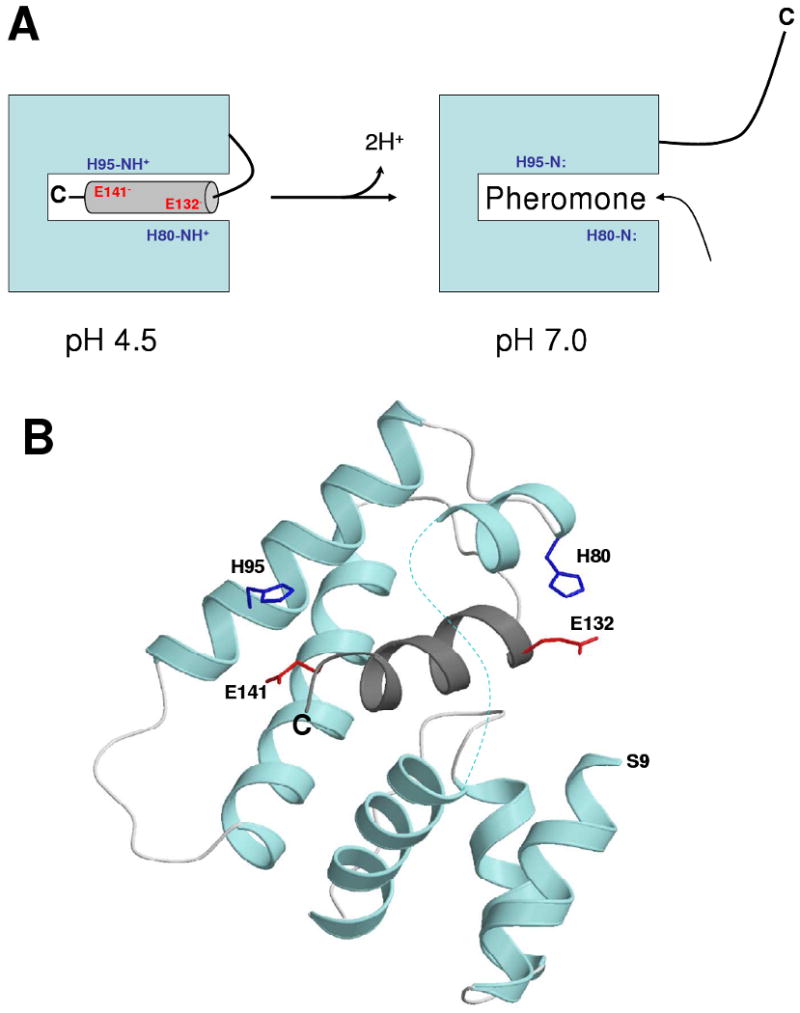
Schematic model showing pH-dependent pheromone binding regulated by a histidine protonation switch (A) and atomic structure highlighting histidine salt bridges (B). Acidic-induced salt bridges (H80/E132 and H95/E141) stabilize insertion of the C-terminal helix (α7) at pH 4.5. At pH 7, H80 and H95 become unprotonated (neutral) and promote extrusion of the C-terminal helix. A hydrophobic sex pheromone structurally resembles the C-terminal helix and binds inside the hydrophobic cavity at pH 7.
Our NMR analysis demonstrates two acid-induced salt bridges (H80/E132 and H95/E141) that promote the formation of the C-terminal helix (α7) at pH 4.5, referred to as a histidine protonation switch (Fig. 8). We propose that this switch provides important stabilizing forces for the insertion of the C-terminal helix at pH 4.5. The two ends of the C-terminal helix form direct salt links (H80/E132 and H95/E141) that position the C-terminal helix inside the pheromone binding site. A remote salt link (H123/D40) also helps orient the C-terminal helix inside the hydrophobic core. The salt bridges, H69/E60 and H70/E57, stabilize the β-turn “flap” that shields the C-terminal helix on the opposite side. Mutations that substitute uncharged residues in the histidine protonation switch (H95A, D132N and E141A) dramatically affect the pH-dependent binding of bombykol pheromone to BmorPBP1 (41), demonstrating the functional importance of the switch. In this study, we show that all five histidines are cationic at pH 4.5 (Fig. 7 and Table 2). The deprotonation of H80 and H95 at pH 7.0 disables salt bridges at the two ends of the C-terminal helix (Fig. 8), promoting the extrusion of the helix outward from the hydrophobic cavity to enable binding of hydrophobic sex pheromones at neutral pH.
Supplementary Material
Acknowledgments
We are grateful to Dr. Jeff de Ropp and Jerry Dallas for help with NMR experiments, Yunhong Li for assistance in sample preparation, Dr. Frits Abildgaard for providing NMR pulse-sequence programs, and Frank Delaglio for writing computer software for NMR data processing and analysis.
Abbreviations
- PBP
pheromone-binding protein
- ApolPBP1
Antheraea polyphemus pheromone-binding protein-1
- AtraPBP1
Amyelois transitella pheromone binding protein-1
- BmorPBP1
Bombyx mori pheromone-binding protein-1
- HMQC
heteronuclear multiple quantum coherence
- HSQC
heteronuclear single quantum coherence
- IPTG
isopropyl β-D-1-thiogalactoside
- ITC
isothermal titration calorimetry
- LR-HMQC
long-range heteronuclear multiple quantum coherence
- MALS
multi-angle light scattering
- NMR
nuclear magnetic resonance
- NOE
nuclear Overhauser effect
- NOESY
nuclear Overhauser effect spectroscopy
- RMSD
root-mean-squared deviation
- SDS PAGE
sodium dodecylsulfate polyacrylamide gel electrophoresis
- SEC
size exclusion chromatography
- TOCSY
total correlation spectroscopy
Footnotes
This work was supported by NIH grants EY012347 (J.B.A.) and RR11973 (UC Davis NMR), NSF grant 0234769 (WSL), USDA-AFRI grant 2009-05278 (WSL), the Almond Board of California, the California Pistachio Research Board, and UC Davis NMR facility.
Atomic coordinates have been deposited into the RCSB Protein Data Bank (accession no. 2kph.pdb).
Supporting Information Available: NMR spectra of delipidated AtraPBP1 at pH 4.5 and pH 7.0 are available free of charge online at http://pubs.acs.org.
References
- 1.Coffelt JA, Vick KW, Sonnet PE, Doolittle RE. Isolation, identification, and synthesis of a female sex pheromone of the navel orangeworm, Amyelois transitella (Lepidoptera: Pyralidae) J Chem Ecol. 1979;5:955–966. [Google Scholar]
- 2.Leal WS, Parra-Pedrazzoli AL, Kaissling KE, Morgan TI, Zalom FG, Pesak DJ, Dundulis EA, Burks CS, Higbee BS. Unusual pheromone chemistry in the navel orangeworm: novel sex attractants and a behavioral antagonist. Naturwissenschaften. 2005;92:139–146. doi: 10.1007/s00114-004-0598-5. [DOI] [PubMed] [Google Scholar]
- 3.Leal WS. Pheromone reception. Top Curr Chem. 2005;240:1–36. [Google Scholar]
- 4.Leal WS, Ishida Y, Pelletier J, Xu W, Rayo J, Xu X, Ames JB. Olfactory proteins mediating chemical communication in the navel orangeworm moth, Amyelois transitella. PloS ONE. 2009;4:e7235. doi: 10.1371/journal.pone.0007235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Syed Z, Ishida Y, Taylor K, Kimbrell DA, Leal WS. Pheromone reception in fruit flies expressing a moth's odorant receptor. Proc Natl Acad Sci U S A. 2006;103:16538–16543. doi: 10.1073/pnas.0607874103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wojtasek H, Leal WS. Conformational change in the pheromone-binding protein from Bombyx mori induced by pH and by interaction with membranes. J Biol Chem. 1999;274:30950–30956. doi: 10.1074/jbc.274.43.30950. [DOI] [PubMed] [Google Scholar]
- 7.Damberger FF, Nikonova L, Horst R, Peng G, Leal WS, Wuthrich K. NMR characterization of a pH-dependent equilibrium between two folded solution conformations of the pheromone-binding protein from Bombyx mori. Protein Sci. 2000;9:1038–1041. doi: 10.1110/ps.9.5.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leal WS, Chen AM, Ishida Y, Chiang VP, Erickson ML, Morgan TI, Tsuruda JM. Kinetics and molecular properties of pheromone binding and release. Proc Natl Acad Sci U S A. 2005;102:5386–5391. doi: 10.1073/pnas.0501447102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sandler BH, Nikonova L, Leal WS, Clardy J. Sexual attraction in the silkworm moth: structure of the pheromone-binding-protein-bombykol complex. Chem Biol. 2000;7:143–151. doi: 10.1016/s1074-5521(00)00078-8. [DOI] [PubMed] [Google Scholar]
- 10.Horst R, Damberger FF, Luginbuhl P, Guntert P, Peng G, Nikonova L, Leal WS, Wuthrich K. NMR structure reveals intramolecular regulation mechanism for pheromone binding and release. Proc Natl Acad Sci U S A. 2001;98:14374–14379. doi: 10.1073/pnas.251532998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Damberger FF, Ishida Y, Leal WS, Wuthrich K. Structural basis of ligand binding and release in insect pheromone-binding proteins: NMR structure of Antheraea polyphemus PBP1 at pH 4.5. J Mol Biol. 2007;373:811–819. doi: 10.1016/j.jmb.2007.07.078. [DOI] [PubMed] [Google Scholar]
- 12.Lautenschlager C, Leal WS, Clardy J. Bombyx mori pheromone-binding protein binding nonpheromone ligands: implications for pheromone recognition. Structure. 2007;15:1148–1154. doi: 10.1016/j.str.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keil TA. Surface coats of pore tubules and olfactory sensory dendrites of a silkmoth revealed by cationic markers. Tissue Cell. 1984;16:705–717. doi: 10.1016/0040-8166(84)90004-1. [DOI] [PubMed] [Google Scholar]
- 14.Katre UV, Mazumder S, Prusti RK, Mohanty S. Ligand binding turns moth pheromone-binding protein into a pH sensor: Effect on the Antheraea polyphemus PBP1 conformation. J Biol Chem. 2009 doi: 10.1074/jbc.M109.013383. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lautenschlager C, Leal WS, Clardy J. Coil-to-helix transition and ligand release of Bombyx mori pheromone-binding protein. Biochem Biophys Res Commun. 2005;335:1044–1050. doi: 10.1016/j.bbrc.2005.07.176. [DOI] [PubMed] [Google Scholar]
- 16.Xu X, Li Y, Rayo J, Ishida Y, Leal WS, Ames JB. 1H, 15N, and 13C Chemical shift assignments of the navel orange worm pheromone-binding protein-1 (Atra-PBP1) Biomol NMR Assign. 2008;2:105–106. doi: 10.1007/s12104-008-9096-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wyatt PJ. Combined differential light scattering with various liquid chromatography separation techniques. Biochem Soc Trans. 1991;19:485. doi: 10.1042/bst0190485. [DOI] [PubMed] [Google Scholar]
- 18.Meyer M, Morganstern B. Characterization of gelatine and acid soluble collagen by size exclusion chromatography coupled with multi angle light scattering (SEC-MALS) Biomacromolecules. 2003;4:1727–1732. doi: 10.1021/bm0341531. [DOI] [PubMed] [Google Scholar]
- 19.Clore GM, Gronenborn AM. NMR structures of proteins and protein complexes beyond 20,000 M(r) Nat Struct Biol. 1997;4:849–853. [PubMed] [Google Scholar]
- 20.Tanaka T, Ames JB, Kainosho M, Stryer L, Ikura M. Differential isotype labeling strategy for determining the structure of myristoylated recoverin by NMR spectroscopy. J Biomol NMR. 1998;11:135–52. doi: 10.1023/a:1008212316986. [DOI] [PubMed] [Google Scholar]
- 21.Talluri S, Wagner G. An optimized 3D NOESY-HSQC. J Magn Reson B. 1996;112:200–205. doi: 10.1006/jmrb.1996.0132. [DOI] [PubMed] [Google Scholar]
- 22.Muhandiram DR, Farrow NA, Xu G, Smallcombe SH, Kay LE. A gradient NOESY-HSQC Experiment for Recording NOESY Spectra of Proteins Dissolved in H2O. J Magn Reson B. 1993;102:317–321. [Google Scholar]
- 23.Neri D, Szyperski T, Otting G, Senn H, Wuthrich K. Stereospecific nuclear magnetic resonance assignments of the methyl groups of valine and leucine in the DNA-binding domain of the 434 repressor by biosynthetically directed fractional 13C labeling. Biochemistry. 1989;28:7510–7516. doi: 10.1021/bi00445a003. [DOI] [PubMed] [Google Scholar]
- 24.Wishart DS, Sykes BD, Richards FM. The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry. 1992;31:1647–1651. doi: 10.1021/bi00121a010. [DOI] [PubMed] [Google Scholar]
- 25.Pelton JG, Torchia DA, Meadow ND, Roseman S. Tautomeric states of the active-site histidines of phosphorylated and unphosphorylated IIIGlc, a signal-transducing protein from Escherichia coli, using two-dimensional heteronuclear NMR techniques. Protein Sci. 1993;2:543–558. doi: 10.1002/pro.5560020406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shaka AJ, Barker P, Freeman R. GARP Decoupling Sequence. J Magn Reson. 1985;64:547–552. [Google Scholar]
- 27.Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Kay LE. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 28.Freedberg DI, Ishima R, Jacob J, Wang YX, Torchia DA. Rapid structural fluctuations of the free HIV protease flaps in solution: relationship to crystal structures and comparison with predictions of dynamics calculations. Protein Sci. 2002;11:221–232. doi: 10.1110/ps.33202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brünger AT. X-PLOR, Version 3.1: A System for X-Ray Crystallography and NMR. Yale University Press; New Haven, CT: 1992. [Google Scholar]
- 30.Badger J, Kumar RA, Yip P, Szalma S. New features and enhancements in the X-PLOR computer program. Proteins. 1999;35:25–33. [PubMed] [Google Scholar]
- 31.Bagby S, Harvey TS, Eagle SG, Inouye S, Ikura M. NMR-derived three-dimensional solution structure of protein S complexed with calcium. Structure. 1994;2:107–122. doi: 10.1016/s0969-2126(00)00013-7. [DOI] [PubMed] [Google Scholar]
- 32.Ames JB, Tanaka T, Stryer L, Ikura M. Secondary structure of myristoylated recoverin determined by three-dimensional heteronuclear NMR: implications for the calcium-myristoyl switch. Biochemistry. 1994;33:10743–53. doi: 10.1021/bi00201a023. [DOI] [PubMed] [Google Scholar]
- 33.Leal WS. Duality monomer-dimer of the pheromone-binding protein from Bombyx mori. Biochem Biophys Res Commun. 2000;268:521–529. doi: 10.1006/bbrc.2000.2158. [DOI] [PubMed] [Google Scholar]
- 34.Marion D, Driscoll PC, Kay LE, Wingfield PT, Bax A, Gronenborn AM, Clore GM. Overcoming the overlap problem in the assignment of 1H NMR spectra of larger proteins by use of three-dimensional heteronuclear 1H-15N Hartmann-Hahn-multiple quantum coherence and nuclear Overhauser-multiple quantum coherence spectroscopy: application to interleukin 1 beta. Biochemisty. 1989;28:6150–6156. doi: 10.1021/bi00441a004. [DOI] [PubMed] [Google Scholar]
- 35.Wishart DS, Sykes BD, Richards FM. Relationship between Nuclear Magnetic Resonance Chemical Shift and Protein Secondary Structure. J Mol Biol. 1991;222:311–333. doi: 10.1016/0022-2836(91)90214-q. [DOI] [PubMed] [Google Scholar]
- 36.Anglister J, Grzesiek S, Wang AC, Ren H, Klee CB, Bax A. 1H, 13C, 15N nuclear magnetic resonance backbone assignments and secondary structure of human calcineurin B. Biochemistry. 1994;33:3540–3547. doi: 10.1021/bi00178a010. [DOI] [PubMed] [Google Scholar]
- 37.Wuthrich K. NMR of Proteins and Nucleic Acids. John Wiley and Sons, Inc.; New York, NY: 1986. [Google Scholar]
- 38.Sharma D, Rajarathnam K. 13C NMR chemical shifts can predict disulfide bond formation. J Biomol NMR. 2000;18:165–171. doi: 10.1023/a:1008398416292. [DOI] [PubMed] [Google Scholar]
- 39.Farr S, Wong WYL, Gutheil WG, Bachovchin WW. Nitrogen-15 NMR chemical shifts for determining the protonation state of the histidine side chain. J Am Chem Soc. 1993;115:6813–6819. [Google Scholar]
- 40.Drohat AC, Xiao G, Tordova M, Jagadeesh J, Pankiewicz KW, Watanabe KA, Gilliland GL, Stivers JT. Heteronuclear NMR and crystallographic studies of wild-type and H187Q Escherichia coli uracil DNA glycosylase: electrophilic catalysis of uracil expulsion by a neutral histidine 187. Biochemistry. 1999;38:11876–11886. doi: 10.1021/bi9910880. [DOI] [PubMed] [Google Scholar]
- 41.Xu W, Leal WS. Molecular Switches for Pheromone Release from a Moth Pheromone-binding Protein. Biochem Biophys Res Commun. 2008;373:559–564. doi: 10.1016/j.bbrc.2008.05.087. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.