

Abstract
Purpose:
The miR-34 family is directly transactivated by tumor suppressor p53 which is frequently mutated in human epithelial ovarian cancer (EOC). We hypothesized that miR-34 expression would be decreased in EOC and that reconstituted miR-34 expression might reduce cell proliferation and invasion of EOC cells.
Experimental designs:
miR-34 expression was determined by quantitative RT-PCR and in situ hybridization in a panel of 83 human EOC samples. Functional characterization of miR-34 was accomplished by reconstitution of miR-34 expression in EOC cells with synthetic pre-miR molecules followed by determining changes in proliferation, apoptosis and invasion.
Results:
miR-34a expression is decreased in 100%, and miR-34b*/c in 72%, of EOC with p53 mutation, while miR-34a is also downregulated in 93% of tumors with wild-type p53. Furthermore, expression of miR-34b*/c is significantly reduced in stage 4 tumors compared to stage 3 (p=0.0171 and p=0.0029, respectively). Additionally, we observed promoter methylation and copy number variations at mir-34. In situ hybridization demonstrated that miR-34a expression is inversely correlated with MET immunohistochemical staining, consistent with translational inhibition by miR-34a. Finally, miR-34 reconstitution experiments in p53 mutant EOC cancer cells resulted in reduced proliferation, motility and invasion, the latter of which was dependent on MET expression.
Conclusions:
Our work suggests that miR-34 family plays an important role in EOC pathogenesis and reduced expression of miR-34b*/c may be particularly important for progression to the most advanced stages. Part of miR-34 effects on motility and invasion may be explained by regulation of MET, which is frequently overexpressed in EOC.
Keywords: microRNA, MET, ovarian cancer, translational research, tumor suppressor
Introduction
Ovarian cancer is the most deadly malignancy and will lead to almost 15,000 deaths in the USA in 2009 (1). While survival has increased slightly over the past 25 years, 5-year survival remains below 50%. A major factor for low survival is our poor understanding of the initiating events that lead to ovarian cancer and how the disease progresses. Due to asymptomatic development and few screening options, almost 70% of women present at late stages of carcinogenesis. At an advanced stage, treatment options are severely limited, with palliative treatment most often administered in the form of debulking surgery and paclitaxel and platinum based therapeutics. However, work over the past decade using human cancer samples and mouse models have revealed new insights into the molecular basis of ovarian cancer, particularly its most common form epithelial ovarian cancer (EOC). For example, it is well established that over 50% of high grade serous type EOC contain p53 mutations and alterations in the RB pathway (reviewed in 2, 3). Consistently, conditional inactivation of p53 and Rb in the mouse ovarian surface epithelium (OSE) leads to development of poorly differentiated serous ovarian adenocarcinomas (4), while K-Ras, Pten and Wnt/beta-catenin are implicated in carcinogenesis of the endometrioid EOC subtype (5, 6).
In recent years, the involvement of small non-coding RNAs called microRNAs (miRNAs) in cancers of many types has become unambiguous, including in ovarian cancer (7, 8). Although the precise roles they play during carcinogenesis are still being dissected, it is clear that miRNAs can act as tumor suppressors and oncogenes by regulating processes such as proliferation and the cell cycle, apoptosis, invasion and metastasis (reviewed in 9). miRNAs have been found to be dysregulated in cancer by DNA copy number changes and epigenetic alterations, altered processing by the miRNA biogenesis machinery and through altered transcriptional activation. In particular, the transcription factor and tumor suppressor p53 has been independently shown by several laboratories to directly transactivate genes of the miR-34 family, which is comprised of three members (10–15). Gene encoding miR-34a is located on human chromosome 1p36, while miR-34b and miR-34c are co-transcribed from one transcription unit on chromosome 11q23. The miR-34 family downregulates numerous important regulatory proteins and thereby presumably mediates tumor suppression (reviewed in 16).
Previously, we have demonstrated that conditional inactivation of p53 results in miR-34 downregulation in mouse OSE (10). To evaluate the potential roles of miR-34 family in human EOC, we have determined their expression level in a panel of 83 cancer tissues and found that miR-34 expression is frequently decreased in EOC, is associated with metastatic clinical stage and increased expression of receptor protein tyrosine kinase MET. Furthermore, reconstitution of miR-34 expression in EOC cells leads to reduced proliferation and invasion, as well as decreased MET levels.
Methods
Clinical samples
Informed consent was obtained from patients undergoing surgery for ovarian cancer at Fox Chase Cancer Center, Philadelphia, PA and M.D. Anderson Cancer Center, Houston, TX. Sample collection was performed after approval by an Institutional Review Board and a portion of tumor tissue not required for diagnostic purposes was snap frozen in liquid nitrogen and stored at −80°C. Surgical evaluation was used to determine clinical stage and presence of metastases, while histopathological analysis by gynecologic pathologists was performed to assess cancer type and subtype. Only tumors found to contain over 70% tumor cells were used in the study and tissue sample and clinical data was available for 83 patients (Table 1). Additional formalin-fixed paraffin embedded specimens were obtained from New York-Presbyterian Hospital/Weill Cornell Medical Center, New York, NY.
Table 1.
Characteristics of 83 patients with epithelial ovarian cancer
| Characteristic | Value | |
|---|---|---|
| Age (years) | Mean | 60.8 |
| Range | 28–87 | |
| Race (%) | White | 64 (77.1) |
| African American | 5 (6) | |
| Other non-white | 6 (7.2) | |
| Unknown | 2 (2.4) | |
| Tumor stage (%) | I | 2 (2.4) |
| II | 4 (4.8) | |
| III | 46 (55.4) | |
| IV | 18 (21.7) | |
| TX | 13 (15.7) | |
| Histology | Serous | 62 (74.7) |
| Mucinous | 2 (2.4) | |
| Endometrioid | 3 (3.6) | |
| Clear cell | 1 (1.2) | |
| Adenocarcinoma, NOS/undifferentiated | 4 (4.8) | |
| Mixed | 11 (13.3) |
miR-34 nomenclature
miRNA nomenclature has recently been revised such that the miR-34b sequence has been renamed miR-34b* (i.e. the passenger strand) (17). Nevertheless, “star” or passenger strands of the miRNA duplex have previously been shown to be biologically important (e.g. miR-199a* and miR-10* (18, 19). Although both miR-34b strands are likely to be functional, the miR-34b strand is not predicted to bind the 3′ UTR of MET and our studies have therefore focused on the miR-34b* strand. It is worth noting that our qRT-PCR data demonstrate that both strands are present at equal quantities and are highly correlated (Supplementary Figure 1); they therefore might be renamed miR-34b-3p and miR-34b-5p, respectively, consistent with mouse miR-34b nomenclature.
p53 mutation screening
The IARC protocol (http://www-p53.iarc.fr) was followed for p53 mutation screening. DNA was isolated by Qiagen DNeasy mini kit (Qiagen, Valencia, CA) and exons 4–11, including splice junctions, were amplified by PCR and sequenced with both forward and reverse primers (Supplementary Table 1). In the case sequencing data was unclear, T-vector cloning was performed, and three clones sequenced by T7 and SP6 primers.
Quantitative RT-PCR
Total RNA was isolated using mirVana miRNA Isolation Kit (Ambion, Austin, TX) according to the manufacturer’s protocol and RNA concentration and purity determined by NanoDrop analysis. Stem-loop qRT-PCR for mature miR-34 and miR-199a* miRNAs was performed as previously described (20). For MET qRT-PCR, cDNA was prepared from 100 ng total RNA using SuperScript III (Invitrogen, Carlsbad, CA) and amplified with TaqMan primer/probes. All PCR reactions were performed in triplicate on an AB 7500 Real Time PCR system (Applied Biosystems Inc, Foster City, CA) and miRNA and mRNA expression normalized to RNU6B and GAPDH, respectively, using the 2−ΔΔCt method (21).
MET immunohistochemistry
Paraffin sections of formalin-fixed tissue were stained according to modified avidin-biotin-peroxidase technique (22). The antibody used for detection of MET was CVD13 from Zymed Laboratories (dilution 1:200).
In situ hybridization
Detection of miR-34 in a panel of serous adenocarcinomas was performed by the protocol adapted from Nelson et al. (23). In order to prevent the loss of miRNAs, we additionally applied 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) fixation as described in (24). In brief, 4 μm-thick sections of formalin-fixed paraffin-embedded material were deparaffinized, rehydrated, and fixed with EDC. After 1 hour prehybridization, a DIG-labeled LNA probe (Exiqon, Woburn, MA) was hybridized to Proteinase K-treated sections at 56°C for 16 hours. Slides were then incubated with anti-DIG-AP antibody (Roche, Indianapolis, IN), and microRNA expression was detected by NBT/BCIP method. Methyl green was used for nucleic counter staining.
Statistical analysis
Statistical tests used were two-sided Student’s t tests, with Welch’s correction for unequal variance where appropriate, using InStat 3.05 and Prism 4.03 software (Graphpad, Inc., La Jolla, CA).
Other methods, including Quantitative PCR, Methylation-specific PCR analysis, Cell culture experiments and Western blotting are described in Supplementary Data.
Results
miR-34 expression is reduced in EOC and is correlated with metastatic stage
To determine miR-34 family expression in EOC, we isolated total RNA from 83 EOC samples and compared expression levels to that in six wild type OSE primary cell samples (Table 1). We observed significantly reduced expression for all three family members in EOC compared to wild type (Figure 1A), with miR-34a most significantly reduced by 21.2-fold (p< 0.0001), while miR-34b* and miR-34c were reduced 2.3-fold (p=0.0172) and 3.4-fold (p=0.0002), respectively. It has been recently reported that Drosha and Dicer expression is deregulated in EOC (8, 25). To test whether alteration in miR-34 expression can be explained by Drosha/Dicer-mediated global changes in miRNA processing, we determined miR-199a* expression, which has been shown to be deregulated in EOC (7, 26). miR-199a* expression appears to be elevated, although this is not statistically significant (Supplementary Figure 2). No significant differences in expression of miR-34 family members were detected among different histological types of EOC, with an exception of significantly reduced miR-34a expression in endometrioid type as compared to serous adenocarcinoma (Supplementary Figure 3).
Figure 1. miR-34 is downregulated in EOC and associated with metastatic clinical stage.

A. qRT-PCR analysis reveals significantly reduced miR-34a, miR-34b* and miR-34c expression in a panel of EOC samples (open circles, n=83) relative to normal OSE samples (open triangles, n=6) (p<0.0001, p=0.0172 and p=0.0022, respectively). B. Ovarian cancer stage was determined by surgical evaluation during cryoreduction/debulking surgery. miR-34a, miR-34b* and miR-34c expression is shown for stage 3 (n=46, open triangles) and stage 4 (n= 18, open circles) tumors. miR-34b* and miR-34c expression is significantly decreased in stage 4 (distant metastasis) compared to stage 3 (localized to peritoneum) (p=0.0171 and p=0.0029, respectively). C. Adenocarcinomas (serous type, n=26) and wild type OSE samples (n=6) were analyzed for p53 mutational status by direct sequencing and correlated with miR-34 expression. While both p53 wild type (WT, circles) and p53 mutant (Mut, squares) EOC samples show significantly reduced miR-34a expression compared to wild type OSE (N, triangles), EOC samples with mutant p53 demonstrate most downregulated expression for miR-34a, miR-34b* and miR-34c (p=0.0012, p=0.0285 and p=0.0216, respectively). Error bars represent standard deviation.
Tumor staging is linked to survival, with stage 3 tumors having tumor cell dissemination in the peritoneum, while stage 4 tumors have distant metastasis, commonly to liver, and is indicative of poor prognosis. We compared gene expression in stage 3 and stage 4 tumors, and observed significantly reduced miR-34b* and miR-34c expression in stage 4 tumors (p=0.0171 and p=0.0033, respectively) (Figure 1B), suggesting that miR-34b* and miR-34c may be involved in metastatic progression. Interestingly however, change in miR-34a expression was not statistically significant (p=0.2574).
Decreased miR-34 expression is associated with p53 mutation
Mutation of p53 is a common event in many human cancers but is particularly common in high grade serous EOC (3). We therefore took a subset of our serous EOC samples (n= 26) and sequenced p53 exons 4–11, where over 99% of p53 mutations are located (Supplementary Table 2). We have found that while miR-34a expression is reduced in both in samples with wild type or mutant p53, patients with mutant p53 demonstrate significantly lower expression of all miR-34 family members than patients with wild type p53 (p=0.0012, p=0.0285 and p=0.0216 for miR-34a, miR-34b* and miR-34c, respectively) (Figure 1C).
Regulation of miR-34 by promoter methylation and copy number alterations
Promoters of both mir-34a and mir-34b*/c are located in CpG islands and methylation has been reported to regulate miR-34a expression in several cancer cell lines and primary prostate tumors and melanomas, while miR-34b*/c expression in colorectal cancer is also epigenetically regulated (27, 28). Such methylation has not been reported in ovarian cancer, so we set out to determine the frequency of such methylation by methylation-specific PCR analysis. Methylation at the mir-34a and mir-34b*/c loci was observed in 27% (8/30) and 47% (14/30) EOC samples, respectively (Figure 2A). All samples (8/8) with mir-34a methylation demonstrate reduced miR-34a expression, while 57% (8/14) of samples with methylation at mir-34b*/c show reduced miR-34b*/c expression. Classification of samples based upon p53 mutation status revealed that 21% (3/14) and 50% (7/14) mutant p53 samples show promoter methylation at mir-34a and mir-34b*/c, respectively, while for samples with wild type p53, 38% (5/13) and 46% (6/13) show methylation, respectively.
Figure 2. mir-34 promoter methylation and copy number variations are common in EOC.
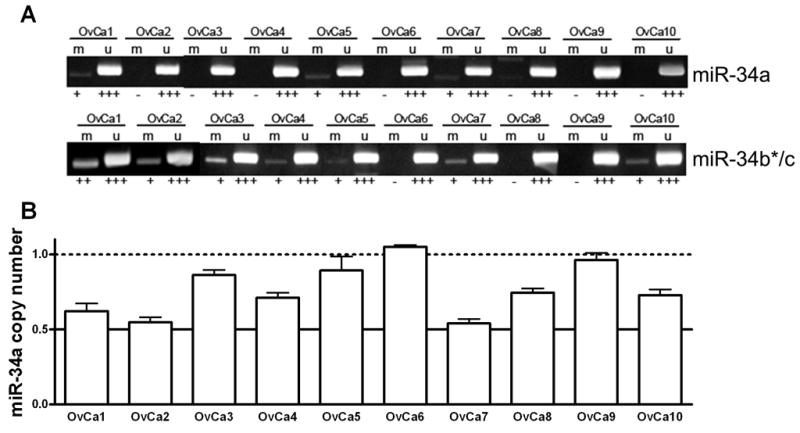
A, B. Representative examples of mir-34a and mir-34b*/c promoter methylation determined by methylation-specific PCR with primers specific for methylated (M) and unmethylated (U) DNA (A) and copy number changes at the mir-34a locus determined by qPCR (B). Error bars represent standard deviation.
We next raised the question of whether loss of heterozygosity (LOH) or copy number alterations could be responsible for reduced miR-34a expression and designed custom TaqMan primer and probes to amplify the mir-34a locus in a qPCR assay. Reduced copy number at the mir-34a locus was observed in 39% (13/33) of EOC samples (Fig. 2C), of which 92% (12/13) had reduced miR-34a expression. Three of these 12 samples (25%) with reduced mir-34a copy number and expression shown no p53 mutation or promoter methylation. Taken together, reduced miR-34a expression is associated with p53 mutation, mir-34a promoter methylation and/or copy number variation in 82% (27/33) of EOC samples.
miR-34 and MET expression in EOC paraffin sections
To explore miR-34 expression in EOC tissue, we performed miRNA in situ hybridization with paraffin-embedded tissue sections of human ovarian cancer. In the case of miR-34a probe, we observed positive signal in cytoplasm compared to control probe, whereas U6 snRNA was exclusively expressed in nucleus as expected (Figure 3A, 3B and 3C). We tested a total of 21 cases of serous EOC with LNA-miR-34a probe. Consistent with qRT-PCR data, 85.7% (18/21) of cases have weak or undetectable miR-34a expression.
Figure 3. miR-34a expression is inversely associated with MET expression in EOC.

Sections of formalin-fixed paraffin-embedded human EOC specimens were hybridized with DIG-labeled LNA control probe (A), U6 snRNA probe (B), miR-34a probe (C and E) or immunostained with MET antibody (D and F). EOC with strong miR-34a expression shows relatively low level of MET expression (C and D). On the contrary, EOC with weak miR-34a expression has strong staining for MET (E and F). Insets show high magnification of areas indicated with arrows. Similar structures of parallel sections (A and B, C and D, E and F) are indicated with arrowheads. Methyl green and hematoxylin were used for counterstaining of in situ hybridization and immunostaining, respectively. Scale bar = 100 μm.
One of the shared targets of miR-34 family is the receptor tyrosine kinase MET according to bioinformatic assessment and luciferase assays (15, 18). Furthermore, the majority of EOC express elevated levels of MET (29). Thus we decided to compare MET expression level in parallel sections of 17 cases with semi-quantitative immunohistochemistry analysis. EOC cases expressing moderate to strong miR-34a (Figure 3C) had a relatively weak expression of MET (Figure 3D). On the contrary, low miR-34a expressing EOC (Figure 3E) had strong expression of MET (Figure 3F). Based on semi-quantitative analysis, expression of miR-34a and MET had statistically significant inverse correlation (r=−0.5898, p=0.0162), confirming that miR-34a might play a role in regulating MET expression in EOC.
miR-34 reduces migration, invasion and proliferation in EOC cells
To determine the role of miR-34 in human ovarian cancer, we transfected synthetic miR-34 molecules either separately or in combination into SKOV-3, p53 null human ovarian adenocarcinoma cells (Figure 4 and Supplementary Figure 4). SKOV-3 cells express low endogenous levels of all three miR-34 family members (8, 10) and is therefore well suited to test functions of miR-34. We transfected SKOV-3 cells with 15 nM miR-34 individually or 5 nM combined and observed reduced amounts of MET protein and mRNA (Figure 4A and B). Even more significant reduction of Met levels was observed after transfection with 30 nM of miR-34 (Figure 4C).
Figure 4. miR-34 reconstitution decreases migration, invasion and proliferation in EOC cells.

A, B. Individual miR-34 family member (15 nM) or entire miR-34 family (5 nM each) were transfected and statistically significant reduction in mRNA observed for each treatment (miR-34a, p=0.028; miR-34b*, p=0.0017; miR-34c, p=0.0022; entire miR-34 family, p=0.0020), although reconstitution of entire miR-34 family does not further downregulate MET expression. C. MET siRNA and/or indicated miR-34 precursor molecule (30 nM) were transfected in SKOV-3 cell line. Transfected cell lysates were probed against MET, CDK4 and GAPDH in Western blot analysis. MET siRNA induced strong knockdown of MET protein with no effect on CDK4, while miR-34 downregulated CDK4 as well as MET. D, E. MET siRNA and/or miR-34 transfection induced significant reduction of cell migration (MET siRNA, p=0.0005; miR-34a, p=0.0003; miR-34c, p=0.0003; MET siRNA and miR-34a, p=0.0008; MET siRNA and miR-34c, p=0.0003) and invasion (MET siRNA, p=0.0013; miR-34a, p=0.0009; miR-34c, p=0.0011; MET siRNA and miR-34a, p=0.0009; MET siRNA and miR-34c, p=0.0009). F. Quantitative assessment of proliferation by BrdU incorporation 48 hours after transfection of SKOV-3 cells with either 15 nM synthetic miR-34a, miR-34b* or miR-34c Pre-miR individually, or 5 nM of each Pre-miR concurrently (miR-34a, p=0.0044; miR-34b*, p=0.0042; miR-34c, p=0.0106; miR-34 in combination, p=0.0021). Error bars represent standard deviation.
The miR-34 family has been shown to reduce cell invasion in gastric and hepatocellular carcinoma cells, at least partially through downregulation of MET (18, 30). To examine the role of miR-34 family in invasion and motility in ovarian cancer, we performed transwell motility and Matrigel invasion assays with miR-34 family and/or MET siRNA transfected SKOV-3 cells. Notably, while MET knock down was observed after MET siRNA treatment, CDK4, which is another target of miR-34 family, was not affected by MET siRNA but only after miR-34 family transfection (Figure 4C). As expected, MET siRNA, individual miR-34 reconstitution by transfection caused significant reduction in motility and invasion in the presence of the MET ligand, HGF (Figure 4D and E and Supplementary Figure 4). However, when miR-34 and MET siRNA were transfected together no further reduction was observed, demonstrating that MET downregulation by miR-34 is largely responsible for the reduced invasion.
Next, we asked whether miR-34 family reconstitution reduces cell proliferation, since miR-34 family also can target cell cycle related genes such as CDK4 (Figure 4C). Transfection of SKOV-3 cells with either 15 nM miR-34a, miR-34b* or miR-34c reduced proliferation by approximately 30% compared to control transfected cells (p=0.0044, p=0.0042 and p=0.0106, respectively) (Figure 4F). We next treated cells with a combination of 5 nM of each miR-34 family member to determine whether additional suppression could be achieved due to sequence, and presumably target, differences of miR-34 family members. Although the percentage of proliferative cells was reduced to 20.7% (p=0.0021 compared to control), the difference in reduction compared to miR-34c transfection individually was not statistically significant (p=0.0876). Additionally, we assessed the amount of apoptosis in miR-34 transfected cells by determination of cleaved caspase 3 staining (Supplementary Figure 5). No significant change in number of apoptotic cells was observed in p53 mutant SKOV-3, consistent with miR-34-induced apoptosis being p53-depenedent (31).
Discussion
Previously we showed that miR-34b-5p and miR-34c expression is reduced in a p53-dependent manner in a mouse model of EOC, while others also reported reduced miR-34 expression in a variety of cell lines and mouse models (10–15). These results led us to question the involvement of miR-34 in human EOC and we show here that miR-34 family expression is also significantly reduced in human EOC, particularly in patients with p53 mutations.
Recently, the involvement of Drosha and Dicer in EOC has been reported, linking reduced expression of these proteins to poor outcome (25). Yet while reduced Drosha/Dicer processing might be expected to lead to globally decreased miRNA expression, this does not appear to account for all miRNA expression defects, given that miR-199a* expression is modestly increased (Supplementary Figure 2). Moreover, unlike miR-34a, miR-34b*/c expression is not reduced in tumors with wild type p53. This discrepancy between miR-34a and miR-34b*/c expression suggests that, in addition to shared p53-dependent transactivation, control mechanisms unique to miR-34a are altered in tumors with wild type p53. Underlining differences between the two mir-34 loci is our observation that mir-34b*/c, but not mir-34a, is significantly associated with stage 4 distant metastatic disease (Figure 1B).
To understand the cause of these differences we investigated the role of promoter methylation and copy number variations of mir-34. mir-34 promoter methylation has been reported in several tumor types (27, 28), while megabase pair deletions at chromosome 1p36 containing the mir-34a locus have been identified in 7 low grade serous carcinomas (32) and in neuroblastoma (33). Indeed promoter methylation was observed at mir-34a and mir-34b*/c in 27% and 47% of EOC samples, respectively. Furthermore, reduced mir-34a copy number was observed in 39% of samples. However, there was no direct correlation between methylation or copy number and miR-34 expression levels. There are several possible explanations that may account for these data. Firstly, although our p53 sequencing data identified p53 mutations with high confidence, mutation in genes that regulate p53 may be involved, such as MDM2 which post-translationally silences p53 through ubiquitinylation (34, 35), while p53 may itself be epigenetically silenced (36). Secondly, many miRNAs, including miR-34a, have been shown by Chang et al. to be suppressed by c-Myc (37), an oncogene frequently over expressed in multiple tumor types, including in EOC. Additionally, it is likely that additional transcription factors regulate miR-34 expression. Very recently, Christoffersen et al. studied human primary hTERT-immortalized TIG3 fibroblasts and observed p53-independent transcription of miR-34a (38). Oncogene-induced senescence (OIS) mediated through B-RAF activation induces miR-34a expression in cells treated with a p53 siRNA or a p53 dominant negative variant. Through chromatin immunoprecipitation experiments, the authors demonstrate that the ETS oncogene family member and transcription factor ELK1 binds a conserved region in the mir-34a promoter.
Clearly, future studies are required to obtain a more complete understanding of the regulation of miR-34 expression in both normal development and in disease. However, the observation of B-RAF induced miR-34a expression through OIS raises an important question regarding the etiology of EOC. It has been hypothesized that low grade and high grade serous tumors have distinct precursor lesions (3, 39), with p53 mutations rarely found in low grade tumors but common in high grade. Interestingly, and further emphasizing the differing molecular defects between the two tumor types, activating B-RAF mutations are found exclusively in low grade serous EOC (40, 41). Together, this suggests that activation of miR-34a transcription by B-RAF/ELK1 and p53 in low grade serous EOC induces senescence and prevents progression to high grade disease. In contrast, a lack of B-RAF mutations combined with frequent p53 mutation in high grade serous EOC would appear to largely eliminate miR-34 expression and subsequently tumor suppression capability is lost. Additionally, while p53 mutation has no impact on miR-34b*/c methylation, miR-34a methylation is more common in samples with wild type p53, consistent with a requirement for diminished p53-miR-34 activity in order to progress to carcinoma. Together, these observations suggest that high grade tumors arise from a population of cells with mutated p53 but wild type B-RAF.
Our data demonstrating correlation of miR-34b*/c expression with metastatic disease suggests that these two miRNAs will be useful as a prognostic marker. This observation is in a good agreement with recent report that low miR-34a levels are correlated with increased probability of relapse in non-small cell lung carcinoma (42) and reinforces the importance of decreased miR-34 expression. Future studies based on complete follow-up data will determine if miR-34 expression correlates with survival of EOC patients.
Our functional studies of miR-34 reconstitution suggest therapeutic applications too. miRNAs represent attractive candidates for gene therapy approaches for several reasons. Computationally, individual miRNAs have been predicted to target tens or hundreds of mRNAs for translational repression. Indeed, one miRNA may regulate many targets. For example, in the case of miR-223 in neutrophils, hundreds of proteins are directly repressed which has a significant impact on phenotype, despite protein repression being relatively modest (43, 44). It is noteworthy that microarray experiments performed after miR-34 reconstitution in cancer cell lines revealed highly significant alterations that are clustered to important biological pathways, for example cell cycle pathway genes (12, 15). These data suggest that reconstituted expression of a downregulated miR-34 might reset or otherwise induce normal function of these gene networks.
Due to its large number of transcriptional targets p53 has also been an attractive candidate for gene therapy. It was therefore disappointing that p53 gene therapy in EOC failed in phase II/III clinical trials (45). Yet while p53 therapy may have failed due to p53 degradation through MDM2 and dominant negative tetramer formation, gene therapy with a p53-independent miR-34 transgene under control of a strong promoter would not expected to face either of these problems. Furthermore, miR-34 may be more attractive than p53 due to its small size, making it more amenable to packaging in viral and non-viral technologies. No less of importance, a number of EOCs do not harbor p53 mutations but express low levels of miR-34 which could be corrected. Thus, it would be interesting to study the consequence of miR-34 delivery to EOC cells with wild type p53. However, currently characterized EOC cell lines carrying wild type p53, such as the serous adenocarcinoma cell line A2780 and clear cell carcinoma cell line TOV-21G (15, 18), demonstrate high levels of expression of endogenous miR-34.
MET is one of few common targets for all three miR-34 family members. Taking into account that the majority of EOC express elevated levels of MET (29) we have tested the role of all miR-34 family members in repression of MET in EOC cells by using Western blotting, invasion and motility assays, finding that all three members inhibit MET. Small-molecule MET inhibitors are now under clinical trials for several cancers (46), and Sawada et al. showed that MET siRNA could reduce adhesion, invasion, metastasis and tumor burden in intraperitoneal ovarian cancer xenograft model, but did not affect proliferation (47). On the contrary, miR-34 therapeutics may have advantages compared to siRNA approach, since miR-34 is capable of regulating cell proliferation as well as invasion through targeting several target genes in addition to MET, such as MYC, E2F3 and others. This is underscored by our experiment demonstrating reduced CDK4 protein after miR-34 treatment, but not MET siRNA.
Taken together, our data demonstrates the frequent reduction of miR-34 family expression in EOC and its functional properties as an inhibitor of proliferation and invasion. This miRNA family is therefore an attractive candidate gene for further studies aimed at better understanding of disease pathogenesis and development of novel therapeutic approaches.
Supplementary Material
Acknowledgments
This work was supported by NIH grants CA112354, and RR17595 and a Marsha Rivkin Cancer Center Pilot Study Grant to AYN, Cornell Vertebrate Genomics Scholarship to DCC, a Graduate Research Assistantship of the College of Veterinary Medicine to CH and an Ovarian Cancer Research Fund Program of Excellence in Ovarian Cancer Research Postdoctoral Fellowship CUMC/POE02.06 to AM. This work was also supported in part by grants from the U. T. M. D. Anderson Cancer Center Ovarian Cancer SPORE (P50 CA083639) and Ovarian Cancer Research Fund, Inc. to AKS.
Footnotes
Translational Relevance
Diagnosis and treatment of EOC is particularly difficult due to poor understanding of disease pathogenesis. We show that miR-34 expression is frequently decreased in EOC due to several mechanisms, but mainly due to p53 mutation. Importantly, reconstitution of miR-34 in human ovarian cancer cells results in decreased proliferation and invasion, at least partially by inhibition of the MET oncogene. Taken together, these data suggest that miR-34 family is important for EOC development and may be an attractive candidate for development of novel therapeutic approaches.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Bast RC, Jr, Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer. 2009;9:415–28. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corney DC, Flesken-Nikitin A, Choi J, Nikitin AY. Role of p53 and Rb in ovarian cancer. Adv Exp Med Biol. 2008;622:99–117. doi: 10.1007/978-0-387-68969-2_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flesken-Nikitin A, Choi KC, Eng JP, Shmidt EN, Nikitin AY. Induction of carcinogenesis by concurrent inactivation of p53 and Rb1 in the mouse ovarian surface epithelium. Cancer Research. 2003;63:3459–63. [PubMed] [Google Scholar]
- 5.Dinulescu DM, Ince TA, Quade BJ, Shafer SA, Crowley D, Jacks T. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nature Medicine. 2005;11:63–70. doi: 10.1038/nm1173. [DOI] [PubMed] [Google Scholar]
- 6.Wu R, Hendrix-Lucas N, Kuick R, et al. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/beta-catenin and PI3K/Pten signaling pathways. Cancer Cell. 2007;11:321–33. doi: 10.1016/j.ccr.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 7.Iorio MV, Visone R, Di Leva G, et al. MicroRNA Signatures in Human Ovarian Cancer. Cancer Research. 2007;67:8699–707. doi: 10.1158/0008-5472.CAN-07-1936. [DOI] [PubMed] [Google Scholar]
- 8.Zhang L, Volinia S, Bonome T, et al. Genomic and epigenetic alterations deregulate microRNA expression in human epithelial ovarian cancer. Proc Natl Acad Sci U S A. 2008;105:7004–9. doi: 10.1073/pnas.0801615105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nature Reviews: Cancer. 2006;6:259–69. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 10.Corney DC, Flesken-Nikitin A, Godwin AK, Wang W, Nikitin AY. MicroRNA-34b and MicroRNA-34c Are Targets of p53 and Cooperate in Control of Cell Proliferation and Adhesion-Independent Growth. Cancer Research. 2007;67:8433–8. doi: 10.1158/0008-5472.CAN-07-1585. [DOI] [PubMed] [Google Scholar]
- 11.Tarasov V, Jung P, Verdoodt B, et al. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle. 2007;6:1586–93. doi: 10.4161/cc.6.13.4436. [DOI] [PubMed] [Google Scholar]
- 12.Bommer GT, Gerin I, Feng Y, et al. p53-Mediated Activation of miRNA34 Candidate Tumor-Suppressor Genes. Current Biology. 2007;17:1298–307. doi: 10.1016/j.cub.2007.06.068. [DOI] [PubMed] [Google Scholar]
- 13.Chang TC, Wentzel EA, Kent OA, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Molecular Cell. 2007;26:745–52. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raver-Shapira N, Marciano E, Meiri E, et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Molecular Cell. 2007;26:731–43. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 15.He L, He X, Lim LP, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–4. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2009 doi: 10.1038/cdd.2009.56. [DOI] [PubMed] [Google Scholar]
- 17.Landgraf P, Rusu M, Sheridan R, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–14. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Migliore C, Petrelli A, Ghiso E, et al. MicroRNAs impair MET-mediated invasive growth. Cancer Res. 2008;68:10128–36. doi: 10.1158/0008-5472.CAN-08-2148. [DOI] [PubMed] [Google Scholar]
- 19.Stark A, Kheradpour P, Parts L, et al. Systematic discovery and characterization of fly microRNAs using 12 Drosophila genomes. Genome Res. 2007;17:1865–79. doi: 10.1101/gr.6593807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen C, Ridzon DA, Broomer AJ, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Research. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 22.Nikitin A, Lee WH. Early loss of the retinoblastoma gene is associated with impaired growth inhibitory innervation during melanotroph carcinogenesis in Rb+/− mice. Genes & Development. 1996;10:1870–9. doi: 10.1101/gad.10.15.1870. [DOI] [PubMed] [Google Scholar]
- 23.Nelson PT, Baldwin DA, Kloosterman WP, Kauppinen S, Plasterk RH, Mourelatos Z. RAKE and LNA-ISH reveal microRNA expression and localization in archival human brain. Rna. 2006;12:187–91. doi: 10.1261/rna.2258506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pena JT, Sohn-Lee C, Rouhanifard SH, et al. miRNA in situ hybridization in formaldehyde and EDC-fixed tissues. Nat Methods. 2009;6:139–41. doi: 10.1038/nmeth.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merritt WM, Lin YG, Han LY, et al. Dicer, Drosha, and outcomes in patients with ovarian cancer. N Engl J Med. 2008;359:2641–50. doi: 10.1056/NEJMoa0803785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang H, Kong W, He L, et al. MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res. 2008;68:425–33. doi: 10.1158/0008-5472.CAN-07-2488. [DOI] [PubMed] [Google Scholar]
- 27.Lodygin D, Tarasov V, Epanchintsev A, et al. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle. 2008;7:2591–600. doi: 10.4161/cc.7.16.6533. [DOI] [PubMed] [Google Scholar]
- 28.Toyota M, Suzuki H, Sasaki Y, et al. Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res. 2008;68:4123–32. doi: 10.1158/0008-5472.CAN-08-0325. [DOI] [PubMed] [Google Scholar]
- 29.Auersperg N, Wong AS, Choi KC, Kang SK, Leung PC. Ovarian surface epithelium: biology, endocrinology, and pathology. Endocrine Reviews. 2001;22:255–88. doi: 10.1210/edrv.22.2.0422. [DOI] [PubMed] [Google Scholar]
- 30.Li N, Fu H, Tie Y, et al. miR-34a inhibits migration and invasion by down-regulation of c-Met expression in human hepatocellular carcinoma cells. Cancer Lett. 2009;275:44–53. doi: 10.1016/j.canlet.2008.09.035. [DOI] [PubMed] [Google Scholar]
- 31.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A. 2008;105:13421–6. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuo KT, Guan B, Feng Y, et al. Analysis of DNA copy number alterations in ovarian serous tumors identifies new molecular genetic changes in low-grade and high-grade carcinomas. Cancer Res. 2009;69:4036–42. doi: 10.1158/0008-5472.CAN-08-3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Welch C, Chen Y, Stallings RL. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene. 2007;26:5017–22. doi: 10.1038/sj.onc.1210293. [DOI] [PubMed] [Google Scholar]
- 34.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–9. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 35.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 36.Kang JH, Kim SJ, Noh DY, et al. Methylation in the p53 promoter is a supplementary route to breast carcinogenesis: correlation between CpG methylation in the p53 promoter and the mutation of the p53 gene in the progression from ductal carcinoma in situ to invasive ductal carcinoma. Lab Invest. 2001;81:573–9. doi: 10.1038/labinvest.3780266. [DOI] [PubMed] [Google Scholar]
- 37.Chang TC, Yu D, Lee YS, et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40:43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Christoffersen NR, Shalgi R, Frankel LB, et al. p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ. 2009 doi: 10.1038/cdd.2009.109. [DOI] [PubMed] [Google Scholar]
- 39.Singer G, Stohr R, Cope L, et al. Patterns of p53 mutations separate ovarian serous borderline tumors and low- and high-grade carcinomas and provide support for a new model of ovarian carcinogenesis: a mutational analysis with immunohistochemical correlation. Am J Surg Pathol. 2005;29:218–24. doi: 10.1097/01.pas.0000146025.91953.8d. [DOI] [PubMed] [Google Scholar]
- 40.Singer G, Oldt R, 3rd, Cohen Y, et al. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. Journal of the National Cancer Institute. 2003;95:484–6. doi: 10.1093/jnci/95.6.484. [DOI] [PubMed] [Google Scholar]
- 41.Shih Ie M, Kurman RJ. Ovarian tumorigenesis: a proposed model based on morphological and molecular genetic analysis. Am J Pathol. 2004;164:1511–8. doi: 10.1016/s0002-9440(10)63708-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gallardo E, Navarro A, Vinolas N, et al. miR-34a as a prognostic marker of relapse in surgically resected non-small-cell lung cancer. Carcinogenesis. 2009;30:1903–9. doi: 10.1093/carcin/bgp219. [DOI] [PubMed] [Google Scholar]
- 43.Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnnidis JB, Harris MH, Wheeler RT, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 2008;451:1125–9. doi: 10.1038/nature06607. [DOI] [PubMed] [Google Scholar]
- 45.Zeimet AG, Marth C. Why did p53 gene therapy fail in ovarian cancer? Lancet Oncol. 2003;4:415–22. doi: 10.1016/s1470-2045(03)01139-2. [DOI] [PubMed] [Google Scholar]
- 46.Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7:504–16. doi: 10.1038/nrd2530. [DOI] [PubMed] [Google Scholar]
- 47.Sawada K, Radjabi AR, Shinomiya N, et al. c-Met overexpression is a prognostic factor in ovarian cancer and an effective target for inhibition of peritoneal dissemination and invasion. Cancer Res. 2007;67:1670–9. doi: 10.1158/0008-5472.CAN-06-1147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.