

Abstract
The question of which dendritic cells (DCs) respond to pulmonary antigens and cross-prime CD8+ T cells remains controversial. We show that influenza-specific CD8+ T cell priming is controlled by different DCs at different times after infection. Whereas early priming is controlled by both CD103+CD11blo and CD103-CD11bhi DCs, CD103-CD11bhi DCs dominate antigen presentation at the peak of infection. Moreover, CD103-CD11bhi DCs capture exogenous antigens in the lung and directly cross-prime CD8+ T cells in the draining lymph node without transferring antigen to CD8α+ DCs. Finally, we show that CD103-CD11bhi DCs are the only DCs to express CD70 after influenza infection and that CD70 expression on CD103-CD11bhi DCs licenses them to expand CD8+ T cells responding to both influenza and exogenous ovalbumin.
Introduction
CD8+ T cell priming to influenza requires antigen presentation by activated dendritic cells (DCs)1-4 that express costimulatory molecules, such as CD40, CD80 and CD86, which promote T cell proliferation and differentiation. Some activated DCs also express CD70 [http://www.signaling-gateway.org/molecule/query?afcsid=A000547], the ligand for CD27 [http://www.signaling-gateway.org/molecule/query?afcsid=A000546], which facilitates primary and secondary CD8+ T cell responses 5-8. However, it remains unclear which DCs express CD70 and how CD70 expression impacts their ability to promote CD8+ T cell proliferation and differentiation.
Within lymph nodes (LNs), DCs can be broadly separated into resident DCs, which differentiate locally from precursors that enter LNs from the blood, and migratory or tissue DCs (tDCs), which differentiate in peripheral tissues and, upon activation, migrate into draining LNs via afferent lymphatics3,9,10. LN resident DCs include plasmacytoid DCs (pDC), and conventional DCs (cDCs), which can be subdivided based on CD8α and CD4 expression3. tDCs can also be subdivided based on the expression of the integrin αEβ7 (CD103; http://www.signaling-gateway.org/molecule/query?afcsid=A001208)11,12. In the lung, CD103+CD11blo tDCs are located in the airway mucosa between epithelial cells, whereas CD103−CD11bhi tDCs are located in the submucosa and lung parenchyma11,13. Although trafficking of pulmonary tDCs to the mediastinal LN (mLN) is essential for the generation of adaptive immunity, the roles played by various DC subsets in CD8+ T cell responses are not fully understood12,14-18.
CD8+ T cells respond to a wide variety of antigens, some of which are not expressed by DCs. Thus, DCs must have a way of acquiring external antigens and processing these in the major histocompatibility complex (MHC) class I pathway - a process known as cross–presentation2,3,19. The current paradigm suggests that CD8α+ cDCs are responsible for cross-priming CD8+ T cells to influenza virus16,20-23 - a model inconsistent with the idea that CD8α+ cDCs are LN resident DCs and do not traffic through peripheral tissues. However, tDCs may carry antigens from the lung, home to the LN and transfer antigen to CD8α+ cDCs, which then cross-prime CD8+ T cells3,16,23-25. More recent studies challenge this model and suggest that some CD8α− migratory DCs directly participate in cross-presentation14,17,26. Thus, the ability of various DC subsets to acquire antigen in the lungs, express the appropriate costimulatory molecules and cross-prime CD8+ T cell responses remains unclear.
Here we show that, early after pulmonary influenza infection, both CD103+CD11blo and CD103−CD11bhi tDCs, but not CD8α+ cDCs, migrated from the lung to the mLNs and presented influenza nucleoprotein (NP). However, CD103−CD11bhi tDCs rapidly accumulated in the mLNs and lung, and at the peak of infection, completely dominated the presentation of NP. Moreover, CD103−CD11bhi tDCs efficiently captured exogenous antigens within the lung, processed these antigens into the MHC class I pathway, migrated to the draining LNs and cross-primed CD8+ T cells. Finally, we show that CD103−CD11bhi tDCs were the only DCs that up-regulated CD70 after influenza infection and that CD70 expression on CD103−CD11bhi tDCs was functionally important for CD8+ T cell responses to both influenza NP and exogenous ovalbumin (OVA). Together, these results establish a new paradigm of which DC subsets acquire exogenous and virus-derived antigens in the lung and cross-prime CD8+ T cell responses.
Results
Kinetics of the NP-specific CD8+ T cell response
To determine the kinetics of the CD8+ T cell response to influenza NP, we infected C57BL/6 mice with influenza A/PR8/34 (PR8) and enumerated NP-specific CD8+ T cells in the draining mLNs and lungs. We first observed NP-specific CD8+ T cells in the mLNs between days 3 and 5 after infection (Fig. 1a) - a time when NP-specific CD8+ T cells in the lung were still undetectable by flow cytometry (Fig. 1b). However, increases in NP-specific CD8+ T cells were observed in both mLNs and lungs between days 5 and 10, when NP-specific CD8+ T cells were rapidly proliferating in both organs (not shown). The number of NP-specific CD8+ T cells peaked 10 days after infection in the mLNs (Fig. 1a) and 12 days after infection in the lung (Fig. 1b). These data are consistent with the idea that influenza-specific CD8+ T cells are first primed in the draining mLNs before homing to the lung, where they continue to proliferate and perform their effector functions.
Figure 1. The NP-specific CD8+ T cell in the mLN precedes that in the lung.
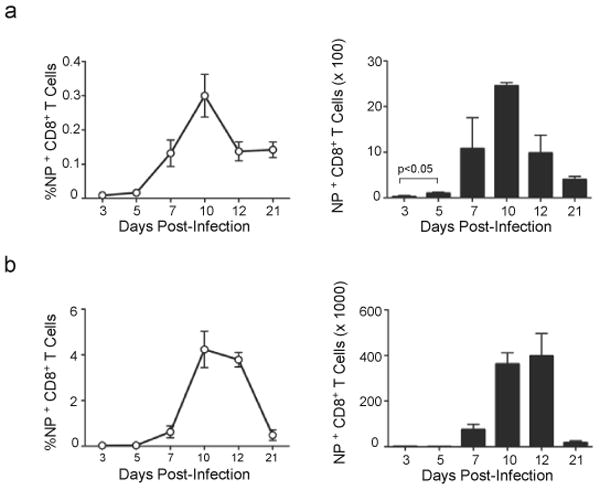
(a,b) C57BL/6 mice were infected intranasally with PR8. Frequencies of NP-specific CD8+ T cells in mLNs (a) and lungs (b) were determined by flow cytometry (left) and the total numbers (right) were calculated at the indicated times. Data are shown as the mean ± SD (n = 5 mice/point). Data are representative of three independent experiments.
DC subsets in the mLN after influenza virus infection
Since T cells are primed by DCs, we next characterized DC subsets in the mLNs. We observed three major populations of DCs in the mLNs based on the expression of CD11c and MHC class II (Fig. 2a, left). The major subset consisted of MHCIIhiCD11cmedB220− cells, which appeared to be migratory tDCs that arrive from the lung3,27. The tDCs were further divided into two subpopulations based on expression of CD103. The MHCIIhiCD11cmedB220−CD103+ tDCs expressed low amounts of CD11b and Ly6C and were designated CD103+CD11blo tDCs, whereas the MHCIIhiCD11cmedB220−CD103−tDCs expressed abundant CD11b and had intermediate Ly6C expression and were designated CD103−CD11bhi tDCs. These latter cells are similar to MHCIIhiCD11cmedCD11bhiLy6Cmed dermal monocyte-derived DCs10. Interestingly, CD8α was weakly detected on both CD103−CD11bhi and CD103+CD11blo tDCs (Supplementary Fig. 1a). We also observed MHCIImedCD11chiB220− DCs, which appeared to correspond to resident cDCs that differentiate locally in the LNs from blood-derived precursors3. This population was subdivided into three subsets; CD11bloCD4−CD8α+Ly6Clo cells that we called CD8α+ cDCs, CD11bhiCD4+CD8α−Ly6Clo cells that we called CD4+ cDCs and CD11bhiCD4−CD8α−Ly6Clo cells that we called double-negative (DN) cDCs. Whereas DN cDCs and CD4+ cDCs did not express CD103, CD8α+ cDCs could be further subdivided based on CD103 expression into CD103+ and CD103−CD8α+ cDCs (Supplementary Fig. 1b). Finally, we identified MHCIImedCD11cmedB220−CD11bhiCD4−CD8αloCD103−Ly6Chi DCs, which appear to correspond to inflammatory DCs (iDCs) that differentiate locally in the LN from blood-recruited monocytes9,10.
Figure 2. DC subsets in mLN after influenza infection.
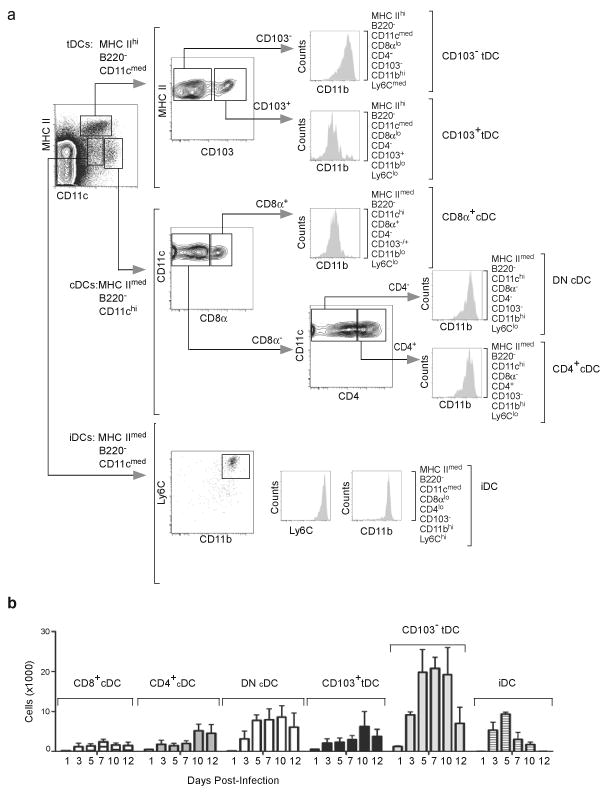
(a) Mice were infected intranasally with PR8 and DC subsets were analyzed by flow cytometry on day 7 after infection. Plasmacytoid DCs were initially excluded based on B220 expression and the three major subsets that remained were defined as conventional DCs (cDCs: MHCIImedCD11chi), tissue DCs (tDCs: MHCIIhiCD11cmed) and inflammatory DCs (iDCs: MHCIImedCD11cmedLy6chi). Each of these populations was further characterized based on the expression of CD8α, CD4, CD103, CD11b and Ly6C.
(b) C57BL/6 mice were infected intranasally with PR8 and CD8α+ cDCs, CD4+CD8α−cDCs, CD8−CD4−cDCs, CD103+CD11blo tDCs, CD103−CD11bhi tDCs and iDCs were enumerated by flow cytometry as described in a. Values represent the total numbers (mean ± SD) of each subset calculated at each time point (n = 4–5 mice / time point). Data are representative of three independent experiments.
As expected, the total number of DCs in the mLNs increased after influenza infection, peaking between day 5 and 10 after infection (Fig. 2b), the same time that NP-specific CD8+ T cells were proliferating in the mLNs. However, the accumulation of various cDC subsets was not equitable. In particular, CD103−CD11bhi tDCs were the most numerous after influenza infection, whereas CD8α+ cDCs and CD103+CD11blo tDCs were minor populations (Fig. 2b).
CD103−CD11bhi tDCs migrate to the mLN
Migration of DCs from peripheral tissues to draining LNs is important for priming T cell responses. To determine which DC subsets were recruited from lung to the mLNs after influenza infection, we intranasally instilled carboxyfluorescein succinimidyl ester (CFSE) into the lungs of influenza-infected mice and enumerated CFSE+CD11c+ DCs in the mLN 16 h later. Our results show that, although virus infection quickly enhanced DC recruitment from the lung to the mLNs (Fig. 3a), maximal migratory flux was observed on day 5 after infection. The majority of migratory DCs were MHCIIhiCD11cmed tDCs (Fig. 3b) and relatively few were MHCIImedCD11chi cDCs or MHCIImedCD11cmed iDCs.
Figure 3. Influenza infection triggers recruitment of CD103−CD11bhi tDCs to the mLN.
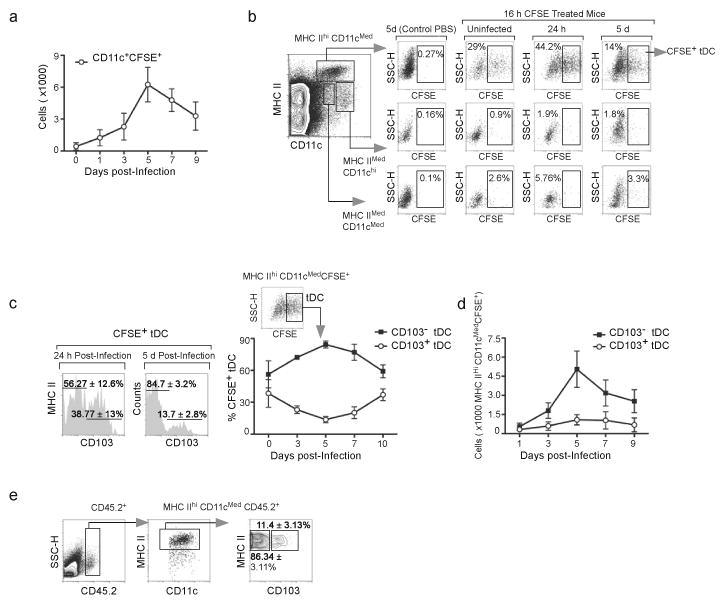
(a) C57BL/6 mice were infected with PR8 and 16 h prior to sacrifice, cells in the lung were labeled with CFSE by intranasal instillation. CFSE-labeled CD11c+ cells in the mLNs were enumerated by flow cytometry at the indicated times. Values represent the number (mean ± SD) of CFSE-labeled CD11c+ cells calculated for each time point (n = 4–5 mice/time point). Data are representative of three independent experiments.
(b) Influenza-infected mice were treated with CFSE or PBS as indicated in a and the phenotypes of CFSE-labeled DCs were determined by flow cytometry at the indicated times. Percentage of CFSE-labeled cells within tDC, cDC and iDC subsets are shown.
(c,d) Influenza-infected mice were treated with CFSE as indicated in a and the frequencies (c) and the total numbers (d) of CFSE-labeled CD103+CD11blo and CD103−CD11bhi tDCs were determined by flow cytometry. Data are shown as the mean ± SD (n = 4–5 mice/time point). Data are representative of three independent experiments.
(e) C57BL/6 donor mice (CD45.2) and CD45.1 recipient mice were infected intranasally with PR8. CD11c+ cells were isolated from donors lungs 5 days after infection and intranasally transferred to recipients (also 5 days after infection). Frequencies of CD103+CD11blo and CD103−CD11bhi cells with the donor marker (CD45.2) in the mLNs were determined 24 h later by flow cytometry. Results are representative of five experiments.
We next examined whether CD103 was expressed on the tDCs that migrated from the lungs to the mLNs. We found that CD103+CD11blo tDCs comprised around 40% of tDCs recruited into the mLNs immediately after infection (Fig. 3c). However, the migration of CD103−CD11bhi tDCs progressively outpaced that of the CD103+CD11blo tDCs cells and by day 5, the CD103−CD11bhi tDCs comprised 80–85% of the CFSE-labeled DCs recruited from the lung (Fig. 3c). In fact, CD103−CD11bhi tDCs were the dominant DC population recruited to the mLN from the lung after influenza infection (Fig. 3d).
To confirm that the CD103−CD11bhi tDCs cells were actually migrating from the lungs rather than simply acquiring CFSE in the mLNs, we purified total CD11c+ DCs from the lungs of CD45.2+ C57BL/6 mice that had been infected with influenza 5 days previously and intranasally transferred them into influenza-infected CD45.1+ recipient mice. We then determined the frequencies of donor CD103+CD11blo and CD103−CD11bhi tDCs within the mLNs 24 h after adoptive transfer. We found that both CD103−CD11bhi and CD103+CD11blo tDCs migrated to the mLNs of recipient mice (Fig. 3e). Although the majority (86%) of the tDCs that migrated to the mLN lacked CD103 expression, this frequency was similar to the frequency of CD103−CD11bhi tDCs found in the transferred population (not shown). Thus, both CD103+CD11blo and CD103−CD11bhi tDCs migrate from the lung to the mLNs, but the CD103−CD11bhi subset is recruited at higher numbers during the peak of infection.
CD103−CD11bhi tDCs accumulate in the lung
The preferential accumulation of CD103−CD11bhi tDCs in the mLNs after influenza infection could reflect changes in DC populations in the lung. To test this possibility, we enumerated DC subsets in the lungs following influenza infection. Lung macrophages and DCs express similar amounts of CD11c28. Therefore, we initially excluded macrophages based on autofluorescence and then gated on MHCII+CD11c+ DCs (Fig. 4a, left), which were divided into B220−DCs and B220+ pDCs. The B220− subset was further subdivided into CD103−CD11bhi tDCs and CD103+CD11blo tDCs.
Figure 4. CD103−CD11bhi tDCs accumulate in the lung after influenza infection.
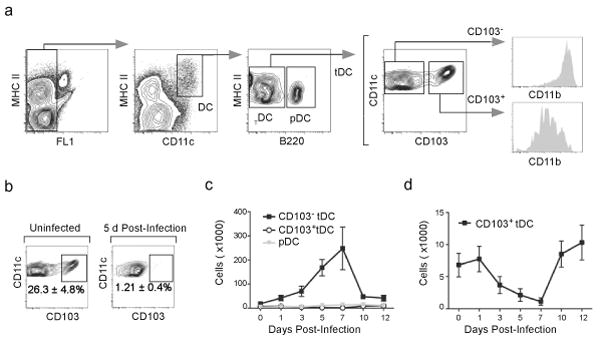
(a) C57BL/6 mice were infected with PR8 and DC subsets in the lung were analyzed by flow cytometry on day 12 after infection. Autofluorescent cells were excluded from the analysis and DCs were subsequently defined as autoloMCHII+CD11c+. pDCs were excluded based on B220 expression and the remaining DCs were divided based on the expression of CD103 and CD11b. (b) C57BL/6 mice were infected intranasally with PR8 or left uninfected and the frequencies of CD103+CD11blo and CD103−CD11bhi tDCs were determined by flow cytometry 5 days later. Percentages indicate the frequency of CD103+ cells within the total DC population. (c) C57BL/6 mice were infected intranasally with PR8 and the frequencies of pDCs as well as CD103+CD11blo and CD103−CD11bhi tDCs were determined by flow cytometry at the indicated times. Values represent the total number (mean ± SD) of DCs in each subset calculated for each time point (n = 4–5 mice/time point). Data are representative of three independent experiments. (d) Data for CD103+CD11blo DCs from panel c with expanded scale.
Under steady-state conditions, we found that the CD103+CD11blo tDCs represented about 30% of total tDCs in the lungs (Fig. 4b, left). However, infection altered the ratio of CD103+CD11blo and CD103−CD11bhi tDCs such that the CD103−CD11bhi tDCs represented more than 90% of the tDCs in the lung by day 5 after infection (Fig. 4b). Influenza virus infection triggered the accumulation of CD103−CD11bhi tDCs in the lungs (Fig. 4c), which peaked 7 days after infection. In contrast, the number of CD103+CD11blo was reduced after influenza infection (Fig. 4d), suggesting that the increased numbers of CD103−CD11bhi tDCs in the mLNs after influenza infection is a consequence of increased numbers of CD103−CD11bhi tDCs in the lung that migrate to the mLNs.
CD103−CD11bhi tDCs present NP to CD8+ T cells
The simultaneous accumulation of CD103−CD11bhi tDCs and NP-specific CD8+ T cells in the mLNs suggested a prominant role for this DC subset in antigen presentation. To test this possibility, we sorted DC subsets from the mLNs of infected mice and co-cultured them with CFSE-labeled CD8+ T cells from the mLNs of mice infected 7 days previously. We found that, prior to culture, the input CD8+ T cell population contained about 2% NP-specific T cells (Fig. 5a). When cultured for 3 days with control DCs from uninfected mice, the NP-specific CD8+ T cells failed to proliferate (Fig. 5b). When cultured with DC subsets from infected mice, we found that only tDCs (Fig. 5c,d) but not CD8α+ cDCs (Fig. 5e), CD8α− cDCs (Fig. 5f) or iDCs (Supplementary Fig. 2) induced substantial CD8+ T cell proliferation. Interestingly, whereas both CD103−CD11bhi and CD103+CD11blo tDCs equally induced NP-specific CD8+ T cell proliferation 3 days after infection (Fig. 5g), the capacity of CD103+CD11blo tDCs to expand NP-specific CD8+ T cells strongly declined by days 5 and 7 after infection (Fig. 5h,i). By contrast, CD103−CD11bhi tDCs retained their ability to present NP on days 5 and 7 after infection (Fig. 5h,i). Given that CD103−CD11bhi tDCs were markedly more abundant in the mLNs than other DC subsets (Fig. 2b) and that CD103−CD11bhi tDCs primed NP-specific CD8+ T cell proliferation more efficiently than the other DC subsets in the mLN at the peak of T cell response (Fig. 5), our results suggest that CD103−CD11bhi tDCs are central players in CD8+ T cell responses to influenza infection.
Figure 5. CD103−CD11bhi tDCs continue to present antigen at late times after infection.
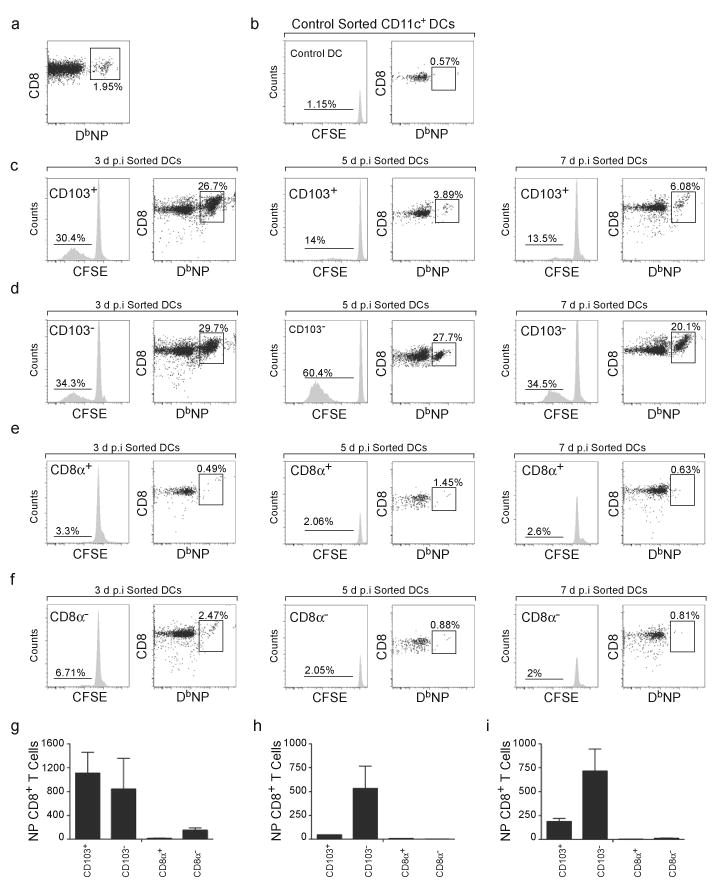
(a) C57BL/6 mice were infected with PR8 and CD8+ T cells were purified from draining mLNs 7 days later. The frequency of NP-specific CD8+ T cells in the purified population was evaluated by flow cytometry. (b) Purified CD8+ T cells from panel a were labeled with CFSE and co-cultured with CD11c+ DCs from the spleens of uninfected mice. The frequency of NP-specific CD8+ T cells that had divided (histogram) and frequency of NP-specific CD8+ T cells in the total CD8+ T cell population (dot plot) were evaluated by flow cytometry after 3 days of culture.(c–f) Purified CD8+ T cells from panel a were labeled with CFSE and co-cultured with CD103+CD11blo tDCs (c), CD103−CD11bhi tDCs (d), CD8α+ cDCs (e) and CD8α−cDCs (f) that were sorted from the mLNs of mice that had been infected with PR8 3, 5 or 7 days previously. The frequency of NP-specific CD8+ T cells that had divided (histogram) and frequency of NP-specific CD8+ T cells in the total CD8+ T cell population (dot plot) were evaluated by flow cytometry after 3 days of culture. (g–i) The number (mean ± SD) of proliferating CFSElo NP-specific CD8+ T cells was calculated for cultures initiated with day 3 DCs (g), day 5 DCs (h) and day 7 DCs (i). All values were obtained in triplicate. The results in panels a-i are representative of three independent experiments.
CD103−CD11bhi tDCs cross-present antigens to CD8+ T cells
Previous reports suggest that migratory DCs obtain exogenous antigens within infected tissues, migrate to the corresponding draining LNs and efficiently prime CD8+ T cells by cross-presentation14,17,26. To test whether antigens captured in the lungs could be cross-presented to CD8+ T cells within the mLNs after influenza infection, we adoptively transferred CFSE-labeled OVA-specific OT-I cells into recipient mice that had been infected with influenza 4 days earlier. We then intranasally administrated 60 μg of soluble OVA or PBS the following day and analyzed the proliferation of the transferred cells in the mLN 72 h later. We observed robust proliferation of OT-I cells in OVA-treated mice, but not in PBS-treated mice (Fig. 6a), demonstrating that exogenous antigens in the lung were efficiently cross-presented to CD8+ T cells. Since the OT-I cells used in the previous experiment were not on a Rag1−/− background, they were likely to include some antigen-experienced cells. To test whether naïve OT-I cells could proliferate in response to exogenous OVA, we purified CD44lo OT-I cells (Supplementary Fig. 3a) and repeated the above experiment. Consistent with our earlier results, we found that naïve OT-I cells could be primed by exogenous OVA delivered to the lung (Supplementary Fig. 3b).
Figure 6. CD103−CD11bhi tDCs cross-present soluble antigens captured in the lung.
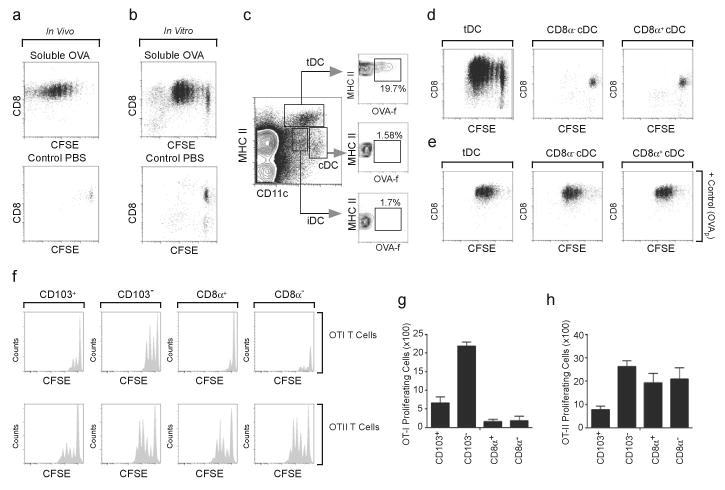
(a) CFSE-labeled OT-I cells (CD45.2) were adoptively transferred into CD45.1 mice 4 days after PR8 infection and recipient mice were treated intranasally with PBS or 60 μg of FITC-OVA 24 h later. Proliferation of transferred OT-I cells in the mLN was assessed 72 h later by flow cytometry. Plots shown are gated on CD45.2. Results are representative of five experiments. (b) C57BL/6 mice were infected with PR8 and treated intranasally with PBS or 60 μg of soluble FITC-OVA 5 days later. CD11c+ cells were purified from mLNs 16 h after OVA administration and were co-cultured with naïve CFSE-labeled OT-I cells. Proliferation of OT-I cells in culture was assessed 72 h later by flow cytometry. These results are representative of three independent experiments. (c) C57BL/6 mice were infected with PR8 and treated intranasally with 60 μg of soluble FITC-OVA 5 days after infection. The phenotype of FITC-labeled DCs was determined 16 h later by flow cytometry. Data are representative of three independent experiments (n = 4–5). (d,e) C57BL/6 mice were infected with PR8 and treated intranasally with PBS or 60 μg of soluble FITC-OVA 5 days later. DC subsets were purified from mLNs 16 h after OVA administration and were co-cultured with naïve CFSE-labeled OT-I cells without peptide (d) or with OVA257-264 (e). Proliferation of OTI cells in culture was assessed 72 h later by flow cytometry (d). These results are representative of three independent experiments. (f–h) Mice were infected with PR8 and DC subsets were sorted from mLNs 5 days later. Purified CFSE-labeled OT-I cells (top) or CFSE-labeled OTII cells (bottom) were co-cultured with sorted DC subsets pulsed with 25 μg/ml of soluble OVA for 48 h (OT-I cells) or 72 h (OT-II cells). CFSE dilution was assayed by flow cytometry after gating on T cells (f). The number (mean ± SD) of proliferating CFSElo OT-I cells (g) or OT-II cells (h) was calculated. All values were obtained in triplicate and the results shown are representative of three independent experiments.
To track the DCs that were responsible for cross-presentation, we intranasally administered soluble fluorescein isothiocyanate (FITC)-OVA to mice 5 days after virus infection, purified CD11c+ cells from the mLNs and used them as APCs for CFSE-labeled OT-I cells in vitro. We found that OT-I cells rapidly proliferated when primed with CD11c+ cells from the mLNs of mice that had been administered FITC-OVA, whereas OT-I cells did not proliferate when cultured with CD11c+ cells from PBS-treated mice (Fig. 6b). To determine what DC subset was responsible for cross-presentation, we analyzed which DCs acquired FITC-labeled OVA. We found that only tDCs and not cDCs or iDCs acquired FITC-OVA (Fig. 6c), suggesting that only the DCs that migrate from the lung were capable of presenting lung-derived antigens to CD8+ T cells. However, previous studies suggest that migratory DCs do not directly cross-present antigen to CD8+ T within the draining LNs cells, but transfer those antigens to resident CD8α+ cDCs, which cross-prime CD8+ T cells16,24,25. To directly determine which DCs were responsible for cross-priming CD8+ T cells, we sorted DC subsets from the mLNs of mice that had been intranasally administered OVA 5 days after influenza infection and used these cells to prime CFSE-labeled OT-I cells in vitro. Whereas sorted tDCs induced OT-I proliferation (Fig. 6d), neither CD8α+ cDCs nor CD8α−cDCs were effective. However, failure of cDC subpopulations to prime OT-I cells was not due to an inability to stimulate CD8+ T cells, as inclusion of exogenous OVA peptide (amino acids 257-264) induced comparable proliferation of OT-I cells when cultured with any of the DC subsets (Fig. 6e). These data suggest that tDCs are the primary DC population that acquires soluble antigens in the lung and cross-primes CD8+ T cells.
Although it was clear that tDCs were the major DC population that captured and cross-presented antigens from the lung, it was not clear whether some of the cDCs also had cross-priming ability, but did not have access to antigens from the lung. To directly test whether tDCs and cDCs had similar abilities to cross-present antigens, we sorted DC subpopulations from the mLNs of mice infected with influenza 5 days earlier, pulsed them in vitro with soluble OVA and co-cultured them with CFSE-labeled OT-I cells or CFSE-labeled OT-II CD4+ T cells. We found that tDCs, particularly CD103−CD11bhi tDCs, were more efficient than either CD8α+ or CD8α−cDCs at cross-presenting soluble OVA (Fig. 6f, top, and 6g). By contrast, no differences in antigen presenting ability were detected when DCs subsets were pulsed with OVA257-264 (not shown). Moreover, CD8α+cDCs, CD8α−cDCs and CD103−CD11bhi tDCs efficiently presented soluble antigen to OT-II cells, whereas CD103+CD11blo tDCs were only modestly effective (Fig. 6f, bottom, and 6h). We also performed this same experiment using sorted CD44lo naïve OT-I cells (Supplementary Fig. 3c) and again found that CD103−CD11bhi tDCs most efficiently primed naïve OT-I cells (Supplementary Fig. 3d,e). CD103+CD11blo tDCs could also prime naïve OT-I cells, albeit less efficiently, but neither CD8α+ nor CD8α−cDCs were able to prime naïve OT-I cells (Supplementary Fig. 3d,e). Taken together, our results demonstrate that CD103−CD11bhi tDCs efficiently capture antigens from the lung, migrate to the draining mLNs and cross-prime CD8+ T cells.
CD70 expression on CD103−CD11bhi tDCs
To better understand the role of lung migratory DCs in immunity to influenza, we evaluated the maturation of CD103−CD11bhi and CD103+CD11blo tDCs within the mLNs after influenza infection. We found that the kinetics of CD40 expression was almost identical on both CD103+CD11blo and CD103−CD11bhi tDC populations, with the peak of CD40 expression occurring between days 3 and 7 after infection (Fig. 7a). Although CD86 expression peaked earlier than CD40, its expression was also nearly identical on both CD103+CD11blo and CD103−CD11bhi tDCs (Fig. 7b). In contrast, the expression of CD70, the ligand for CD27 (ref.29), was increased on CD103−CD11bhi tDCs and not on CD103+CD11blo tDC (Fig. 7c), with maximal expression of CD70 observed at 5 days after infection (Figure 7c,d). Unlike previous studies17 we did not observe CD70 on either CD8α+ or CD8α−cDCs at any time point after infection (Supplementary Fig. 4). Thus, CD103−CD11bhi tDCs appear to be the primary migratory DC subset that expresses the costimulatory molecule, CD70, after influenza infection.
Figure 7. CD70 expression is a hallmark of CD103−CD11bhi tDCs.
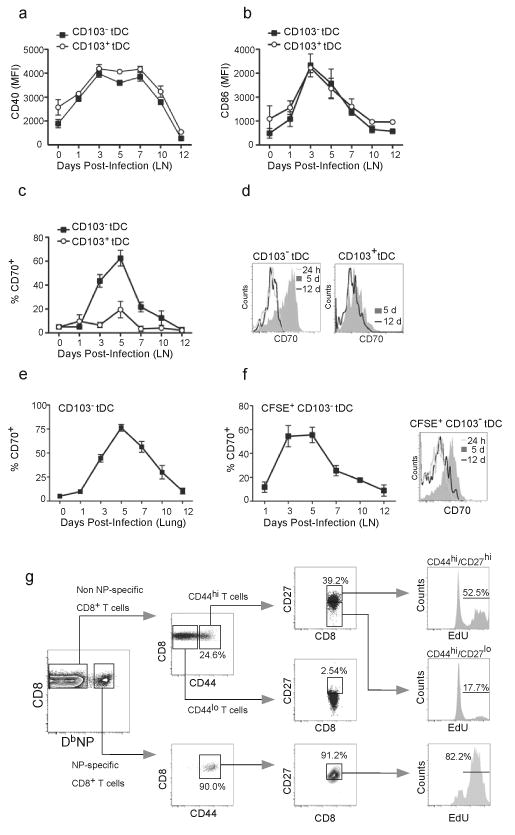
(a–c) C57BL/6 mice were infected intranasally with PR8 and the expression of CD40 (a), CD86 (b) and CD70 (c) on CD103+CD11blo and CD103−CD11bhi tDCs was determined by flow cytometry. Values represent the mean ± SD of the mean fluorescence intensity (MFI) or percentage of positive cells (n = 4–5 mice/time point). Data are representative of three independent experiments. (d) C57BL/6 mice were infected intranasally with PR8 and the expression of CD70 on CD103+CD11blo and CD103−CD11bhi tDCs in the mLNs was compared by flow cytometry at 24 h, 5 days and 12 days after infection. Data are representative of three independent experiments. (e) C57BL/6 mice were infected intranasally with PR8 and the frequency of CD70-expressing CD103−CD11bhi tDCs in the lungs was determined by flow cytometry. Data are representative of three independent experiments. (f) C57BL/6 mice were infected intranasally with PR8 and 16 h prior to sacrifice, cells in the lung were labeled with CFSE by intranasal instillation. The frequency of CD70-expressing, CFSE-labeled CD103−CD11bhi cells in the mLNs was determined by flow cytometry at the indicated times. Values represent the percentage (mean ± SD) of CD70-expressing, CFSE-labeled, CD103−CD11bhi cells calculated for each time point (n = 4–5 mice / time point). Data are representative of three independent experiments. (g) C57BL/6 mice were infected intranasally with PR8 and injected with EdU 3 times, 6 h apart on day 6 after infection. The frequency of EdU-labeled cells in various CD8+ T cell subsets in the mLN was determined by flow cytometry 24 h after the first EdU injection. Plots are representative of two independent experiments (n = 5).
Although it was clear that CD70 was uniquely expressed on CD103−CD11bhi cells in the draining LN, it was not clear whether these cells were derived from CD70-expressing cells in the lung or whether CD70 expression was acquired in the LN or in transit from the lung to the LN. Therefore, we next analyzed CD70 expression on CD103−CD11bhi cells in the lung. We found that CD70 was expressed with similar kinetics and on similar frequencies of CD103−CD11bhi cells in the lungs and mLNs (Fig. 7e), suggesting that CD70 was initially expressed in the lung on CD103−CD11bhi cells prior to their migration to the mLN. To demonstrate that CD70 cells in the draining LN were recently derived from the lung, we intranasally administered CFSE to the lungs of influenza infected mice and then determined the frequency of CD70-expressing cells in the CFSE-labeled CD103−CD11bhi tDC population. We found that nearly 60% of the CFSE-labeled CD103−CD11bhi cells in the LN expressed CD70 on day 5 (Fig. 7f), again suggesting that these cells had recently migrated from the lung.
CD70–CD27 interactions play a central role in both primary and secondary CD8+ T cell responses to influenza5,6. To test whether CD27 expression correlated with proliferation of CD8+ T cells, we assayed 5-ethynyl-2′-deoxyuridine (EdU) incorporation and CD27 expression on NP-specific CD8+ T cells. We found that the majority of NP-specific CD8+ T cells co-expressed abundant CD44 and CD27 and that more than 80% of these cells incorporated EdU during a 16-h pulse (Fig. 7g). We also found relatively high expression of CD27 on CD44hi CD8+ T cells that were not specific for NP. However, CD27 expression was substantially lower on CD44lo CD8+ T cells (Fig. 7g), probably because many of these cells were not responding to influenza. Interestingly, of the CD44hi NP-non-specific CD8+ T cells, those that expressed high amounts of CD27 incorporated EdU at a much higher frequency than those that expressed less CD27 (Fig. 7g). We obtained similar results when we examined NP-specific CD8+ T cells in the lung on day 7 after influenza infection (Supplementary Fig. 5). Thus, CD27 expression appears to correlate with the proliferation of effector CD8+ T cells.
CD70 promotes expansion of NP-specific CD8+ T cells
To address whether CD70–CD27 interactions were required for NP-specific CD8+ T cell proliferation, we first blocked CD70–CD27 in vitro. We found that the ability of CD103−CD11bhi tDCs to promote the proliferation of NP-specific CD8+ T cells was markedly inhibited by anti-CD70 (Fig. 8a). Next, we analyzed whether cross-presentation of soluble antigens by CD103−CD11bhi tDCs required CD70 expression. We found that CD103−CD11bhi tDCs pulsed with soluble OVA strongly promoted OTI proliferation in the presence of control antibody, but not with anti-CD70 (Fig. 8b). To address whether the late expansion of NP-specific CD8+ T cells was dependent on CD70 expression in vivo, influenza infected mice were treated with anti-CD70 4 days after primary infection and frequencies of NP-specific CD8+ T cells were determined 6 days after antibody administration (day 10 after infection). We observed a clear reduction of NP-specific CD8+ T cells in mice that were treated with anti-CD70 compared to isotype control-treated mice (Fig. 8c), confirming that in vivo expansion of influenza NP-specific CD8+ T cells was dependent on CD70–CD27 interactions.
Figure 8. CD70 expression on CD103−CD11bhi tDCs promotes the expansion of NP-specific CD8+ T cells.
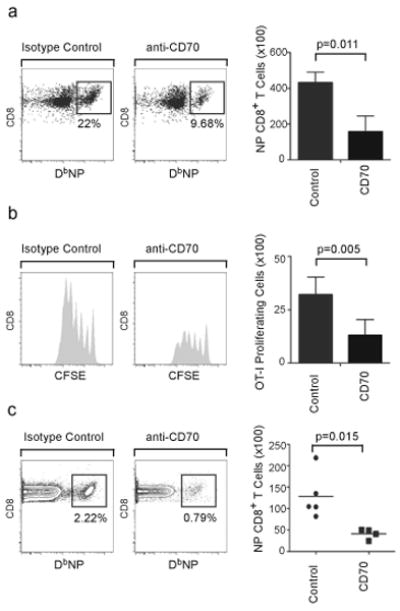
(a) C57BL/6 mice were infected with PR8 for 7 days, when CD103−CD11bhi tDCs as well as CD8+ T cells were purified from mLNs. Purified CD8+ T cells were cocultured with sorted CD103−CD11bhi tDC with 20 μg/ml of either CD70-specific blocking antibody (anti-CD70) or isotype control. The number (mean ± SD) of NP-specific CD8+ T cells was determined 96 h later by flow cytometry. All values were obtained in triplicate. The results shown are representative of three independent experiments. (b) C57BL/6 mice were infected with PR8 for 7 days, when CD103−CD11bhi tDCs were sorted from mLNs, pulsed with 25 μg/ml of soluble OVA and co-cultured for 48 h with naïve CFSE-labeled OT-I cells with 20 μg/ml of either anti-CD70 or isotype control. The number (mean ± SD) of proliferating CFSElo T cells was determined by flow cytometry. All values were obtained in triplicate. The results shown are representative of two independent experiments. (c) C57BL/6 mice were infected with PR8 and starting on day 4, mice were treated with 500 μg of either anti-CD70 or control antibody. The frequency of NP-specific CD8+ T cells in the mLN was determined by flow cytometry on day 10 after infection and the total numbers of NP-specific CD8+ T cells were calculated. Data are representative of two independent experiments.
DISCUSSION
Our results demonstrate that CD103−CD11bhi tDCs play a critical role in NP-specific CD8+ T cell expansion after pulmonary influenza infection. These results differ from previous findings in several respects. First, we find that DC migration from the lung to the mLN continues to increase through day 5 after influenza infection, rather than peaking in the first day or two30. Interestingly, the DCs migrating from the lung to the mLN during this time include both CD103+CD11blo and CD103−CD11bhi tDCs, both of which are potent APCs. These results are consistent with other reports showing that tissue-derived CD103+ DCs present antigen in draining LNs immediately after infection or immunization14,18. However, we find that the CD103−CD11bhi tDCs migrating from the lung to the LN vastly outnumber the CD103+CD11blo cells by day 5 after infection and are more potent APCs than CD103+CD11blo tDCs on a per cell basis. Thus, the CD103−CD11bhi tDCs take over antigen presentation in the mLN during the peak of the response.
Although we did not explicitly examine the cellular origin of CD103−CD11bhi tDCs in the mLN, they are likely to be similar to the MHCIIhiCD11cmedCD11bhiLy6Cmed DCs described after Leishmania major infection10. These dermal monocyte-derived DCs are recruited to the dermis and locally differentiate into DCs. Upon activation, they migrate to the draining LN and present antigen to T cells. In a parallel way, we suggest that influenza infection leads to the recruitment of monocyte precursors to the lung, which differentiate locally into CD103−CD11bhi tDCs. Once activated, the CD103−CD11bhi tDCs in the lung then migrate to the draining mLN, where they present antigen to CD8+ T cells.
Interestingly, CD103+CD11blo and CD103−CD11bhi tDCs develop from different monocyte precursors31,32 that appear to respond to different growth factors. For example, Flt3 ligand promotes the expansion of CD11bhi but not CD11blo DCs in the lung33, whereas epithelial-derived factors, like TGFβ and retinoic acid promote the differentiation of CD103+ tDCs34,35. Given that influenza infection destroys the respiratory epithelium, it is possible that alterations in pulmonary cytokine expression after influenza infection promotes the accumulation of CD103−CD11bhi tDCs at the expense of CD103+CD11blo tDCs. Selective exposure of CD103+CD11blo tDCs to influenza in the epithelium11 is also likely to make them highly susceptible to infection, whereas the CD103−CD11bhi tDCs in the lung parenchyma are likely to remain uninfected36. The placement of CD103+CD11blo tDCs in the epithelium allows them to effectively acquire antigen and prime CD8+ T cells when the epithelium is intact, such as after exposure to inert antigens37 or perhaps after infection with non-lytic viruses14,18 or very early after influenza infection14. In contrast, CD103−CD11bhi tDCs are thought to promote tolerance or preferentially prime CD4+ T cells under steady state conditions37. However, CD103−CD11bhi tDCs are likely to progressively gain access to influenza antigens once the epithelium is disrupted. Thus, temporal changes in growth-promoting cytokines, susceptibility to infection and antigen availability allow CD103−CD11bhi tDCs to dominate the late stages of antigen presentation to CD8+ T cells after influenza infection.
CD8α+ cDC are often implicated as the major DC subset able to cross-prime CD8+ T cells16,20-23,38. However, we find that early after influenza infection, both CD103+CD11blo tDCs and CD103−CD11bhi tDCs, but not CD8α+ cDCs, acquire soluble antigen in the lungs, migrate to the mLN and cross-prime CD8+ T cells directly, without handing off antigen to other DCs. Although these results contradict the prevailing dogma, the role of DC subsets in cross-priming CD8+ T cell responses depends on how antigen is delivered and how DCs are activated. For example, pDCs39,40, CD8α−cDC41,42 and monocyte-derived migratory DCs14,17,26, can efficiently cross-present exogenous antigens under some circumstances. Moreover, although CD8α+ cDCs are the only DCs to cross-prime influenza-specific CD8+ T cells after intravenous or subcutaneous infection20, both CD8α+ DCs and airway-derived CD8α−CD11blo DCs prime CD8+ T cells after intranasal infection16. Even the ability of CD8α+ DCs to cross-prime CD8+ T cells to soluble OVA depends on the lymphoid organ from which they are isolated43. Thus, cross-priming by DCs can be strongly influenced by how DCs acquire antigen and the type of antigen they encounter.
The contradictory results concerning the role of CD8α+ DCs in cross-priming may also be partly attributed to the methods used to phenotype DC subsets. Many authors sort bulk CD8α+CD11c+ DCs or deplete CD8α+ cells from total CD11c+ cells and then test cross-priming activity. However, we find that CD103+CD11blo tDCs, CD103−CD11bhi tDCs and even iDCs all express moderate levels of CD8α14,41. As a result, purification of bulk CD8α+CD11c+ cells without additional characterization using other markers will likely result in a mixed population of CD8α+ DCs and tDCs. Similarly, the expression of CD11b does not distinguish many of the DC subsets described here without the use of additional markers, like MHCII and CD11c. Using a multi-parameter sorting strategy, we and others14 separate true CD8α+ cDCs from other DC subsets that may express low levels of CD8α.
Taken together, our results suggest a new paradigm for how DC subsets promote pulmonary CD8+ T cell responses. Initial immune responses to lytic viruses, such as influenza, are initiated by both CD103+CD11blo tDCs and CD103−CD11bhi tDCs, which acquire antigen in the lung, migrate to the mLN and prime CD8+ T cells. CD103+CD11blo tDCs are soon depleted from the lung and are poorly replenished. As a result, CD103−CD11bhi tDCs rapidly become the primary DC subset that acquires virus-derived and exogenous antigens and cross-primes CD8+ T cells directly, without handing off antigen to CD8α+ cDCs. Once virus infection is cleared and the epithelium is repaired, CD103+CD11blo tDCs are restored to the lung and homeostasis is re-established.
Methods
Mice, infection and in vivo cell labeling
C57BL/6, C57BL/6.OT-I (OT-I), C57BL/6.OT-II (OT-II) and C57BL/6.IgHa.Thy-1a.Ptrpca (CD45.1) mice were bred in the University of Rochester animal facility. Primary infections were performed intranasally with 500 EIU of influenza A/PR8/34 (PR8) in 100 μl of PBS. Lung DCs were labeled in situ by intranasally administering either 30 μl of 8 mM CFSE (Molecular Probes) or 60 μg FITC-OVA (Molecular Probes) in PBS 16 h before sacrifice at the indicated times. Proliferating cells were labeled in situ by intravenously injecting 0.5 mg of EdU (Invitrogen) three times every 6 h starting 24 h before sacrifice. In some experiments, mice were intraperitoneally injected with 500 μg of anti-CD70, FR70, or 500 μg of rat IgG2b isotype control (both from Bioxcell). All experimental procedures involving animals were approved by the University of Rochester University Committee on Animal Resources and were performed according to the guidelines outlined by the National Research Council.
Cell preparation and flow cytometry
Single cell suspensions were prepared from the lungs by cutting into small fragments and digesting with 0.6 mg/ml collagenase A and 30 μg/ml DNAse I (both from Sigma) in RPMI 1640 for 45 min at 37 °C. Digested lungs were mechanically disrupted by passage through a wire mesh and live mononuclear leuokocytes were enriched from lung suspensions by density gradient centrifugation using 1-Step Polymorphs (Accurate Chemical). Single cell suspensions were obtained from mLNs and spleens by mashing through 70 μM nylon cell strainer (BD-Biosciences) without enzymatic digestion. RBCs were lysed with 150 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA.
Fc receptors were blocked with 10 μg/ml 2.4G2, followed by staining with fluorochrome-conjugated antibodies or MHC class I tetramers. The H-2Db class I tetramer containing NP366-374 peptide was generated by the Trudeau Institute Molecular Biology Core Facility. Fluorochrome-labeled antibodies to CD8α (53-6.7), CD4 (RM4-5), CD27 (L6.3A10), CD40 (3/23), CD44 (IM7), CD45.1 (A20), CD45.2 (104), CD86 (GL1), CD11b (MI/70), Ly6C (AL-21), B220 (RA3-6B2) and MHC Class II (AF6-120.1) were obtained from BD-Biosciences. CD11c (N418), CD70 (FR70) and CD103 (2E7) were obtained from eBioscience. EdU-labeled cells were detected by intracellular staining using the Click-iT EdU Flow Cytometry Assay Kit (Invitrogen) according to manufacture's instructions. Flow cytometry was performed on either FACSCalibur or FACSCanto II Flow Cytometers (BD-Biosciences) available through the Flow Cytometry Core Facility at the University of Rochester.
Cell purification, CFSE labeling and adoptive transfer
CD8+ T cells from influenza-infected C57BL/6 mice and from naïve OT-I mice as well as CD4+ T cells from naïve OT-II mice were obtained by first depleting CD11c+ cells using anti-CD11c MACS beads (Miltenyi Biotec). T cells were subsequently isolated using anti-CD8 or anti-CD4 MACS beads and LS columns (Miltenyi Biotec), according to the manufacturer's instructions. In some experiments, CD44lo naïve OT-I cells were purified using the CD8+ T cell isolation kit (Miltenyi Biotec) to obtain CD8+ T cells and then an additional negative selection by MACS to eliminate CD44hi cells. Alternatively, CD8+CD44lo naïve OT-I cells were directly sorted after a positive selection using anti-CD8 MACS beads from preparations of OT-I splenocytes. All T cell preparations were more than 95% pure as determined by flow cytometry. Purified OT-I and OT-II T cells were labeled with 5 μM CFSE (Molecular Probes) for 10 min at 37 °C and 1 × 105 CFSE-labeled CD8+ T cells were intravenously injected into recipient mice.
DCs were enriched from pooled mLNs or lungs using anti-CD11c MACs beads and, after staining with fluorochrome-conjugated antibodies, were sorted using a FACSAria sorter. All sorted DC populations were more than 95% pure as determined by flow cytometry. DCs purified from CD45.2 donors were intranasally transferred (1 × 105 in 50 μl of PBS) into influenza-infected CD45.1+ recipient mice.
In vitro cultures
All in vitro cultures were performed in RPMI 1640 supplemented with sodium pyruvate, HEPES pH 7.4, non-essential amino acids, penicillin, streptomycin, 2-mercaptoethanol and 10% heat-inactivated FCS (all from GIBCO). 1 × 103 sorted DCs and 1 × 104 CFSE-labeled purified T cells were cultured in 100 μl of complete medium in round-bottomed 96-well plates for 72–96 hat 37 °C. In some experiments, soluble OVA, or OVA257-264 peptide were added at 25 μg/ml or 2 μg/ml final concentration, respectively. In some cases anti-CD70 (FR70; eBioscience) or 500 μg of RatIgG2b isotype control antibody (KLH; Bioxcell) were added to the culture at 20 μg/ml final concentration.
Statistical analysis
Statistical differences in mean values were analyzed using a two tailed Student's t-test where indicated. Values of P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
The authors would like to thank L. LaMere and K. Martin for animal husbandry and the members of the URMC Flow Cytometry Core Facility for cell sorting. This work was supported by the University of Rochester and NIH grants AI61511 and HL69409 to T.D.R. and AI06856 to F.E.L. The authors declare no financial conflicts of interest related to this work.
Footnotes
Author contribution statement. A.B.T. designed and performed the experiments, analyzed the data and wrote the paper. B.L.R. optimized the dendritic cell subsetting protocol and helped interpret the data. F.E.L. helped supervise the project and edit the paper. T.D.R. supervised the project, helped design the experiments and edited the paper.
References
- 1.Jung S, et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heath WR, et al. Cross-presentation, dendritic cell subsets, and the generation of immunity to cellular antigens. Immunol Rev. 2004;199:9–26. doi: 10.1111/j.0105-2896.2004.00142.x. [DOI] [PubMed] [Google Scholar]
- 3.Villadangos JA, Schnorrer P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat Rev Immunol. 2007;7:543–555. doi: 10.1038/nri2103. [DOI] [PubMed] [Google Scholar]
- 4.Palm NW, Medzhitov R. Pattern recognition receptors and control of adaptive immunity. Immunol Rev. 2009;227:221–233. doi: 10.1111/j.1600-065X.2008.00731.x. [DOI] [PubMed] [Google Scholar]
- 5.Hendriks J, et al. CD27 is required for generation and long-term maintenance of T cell immunity. Nat Immunol. 2000;1:433–440. doi: 10.1038/80877. [DOI] [PubMed] [Google Scholar]
- 6.Keller AM, Schildknecht A, Xiao Y, van den Broek M, Borst J. Expression of costimulatory ligand CD70 on steady-state dendritic cells breaks CD8+ T cell tolerance and permits effective immunity. Immunity. 2008;29:934–946. doi: 10.1016/j.immuni.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 7.Dolfi DV, et al. Late signals from CD27 prevent Fas-dependent apoptosis of primary CD8+ T cells. J Immunol. 2008;180:2912–2921. doi: 10.4049/jimmunol.180.5.2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keller AM, Xiao Y, Peperzak V, Naik SH, Borst J. Costimulatory ligand CD70 allows induction of CD8+ T-cell immunity by immature dendritic cells in a vaccination setting. Blood. 2009;113:5167–5175. doi: 10.1182/blood-2008-03-148007. [DOI] [PubMed] [Google Scholar]
- 9.Nakano H, et al. Blood-derived inflammatory dendritic cells in lymph nodes stimulate acute T helper type 1 immune responses. Nat Immunol. 2009;10:394–402. doi: 10.1038/ni.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leon B, Lopez-Bravo M, Ardavin C. Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against Leishmania. Immunity. 2007;26:519–531. doi: 10.1016/j.immuni.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 11.Sung SS, et al. A major lung CD103 (αE)-β7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J Immunol. 2006;176:2161–2172. doi: 10.4049/jimmunol.176.4.2161. [DOI] [PubMed] [Google Scholar]
- 12.GeurtsvanKessel CH, Lambrecht BN. Division of labor between dendritic cell subsets of the lung. Mucosal Immunol. 2008;1:442–450. doi: 10.1038/mi.2008.39. [DOI] [PubMed] [Google Scholar]
- 13.Hammad H, Lambrecht BN. Dendritic cells and epithelial cells: linking innate and adaptive immunity in asthma. Nat Rev Immunol. 2008;8:193–204. doi: 10.1038/nri2275. [DOI] [PubMed] [Google Scholar]
- 14.Kim TS, Braciale TJ. Respiratory dendritic cell subsets differ in their capacity to support the induction of virus-specific cytotoxic CD8+ T cell responses. PLoS One. 2009;4:e4204. doi: 10.1371/journal.pone.0004204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brimnes MK, Bonifaz L, Steinman RM, Moran TM. Influenza virus-induced dendritic cell maturation is associated with the induction of strong T cell immunity to a coadministered, normally nonimmunogenic protein. J Exp Med. 2003;198:133–144. doi: 10.1084/jem.20030266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belz GT, et al. Distinct migrating and nonmigrating dendritic cell populations are involved in MHC class I-restricted antigen presentation after lung infection with virus. Proc Natl Acad Sci U S A. 2004;101:8670–8675. doi: 10.1073/pnas.0402644101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bedoui S, et al. Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat Immunol. 2009;10:488–495. doi: 10.1038/ni.1724. [DOI] [PubMed] [Google Scholar]
- 18.Lukens MV, Kruijsen D, Coenjaerts FE, Kimpen JL, van Bleek GM. Respiratory syncytial virus-induced activation and migration of respiratory dendritic cells and subsequent antigen presentation in the lung-draining lymph node. J Virol. 2009;83:7235–7243. doi: 10.1128/JVI.00452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vyas JM, Van der Veen AG, Ploegh HL. The known unknowns of antigen processing and presentation. Nat Rev Immunol. 2008;8:607–618. doi: 10.1038/nri2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Belz GT, et al. Cutting edge: conventional CD8α+ dendritic cells are generally involved in priming CTL immunity to viruses. J Immunol. 2004;172:1996–2000. doi: 10.4049/jimmunol.172.4.1996. [DOI] [PubMed] [Google Scholar]
- 21.Belz GT, Bedoui S, Kupresanin F, Carbone FR, Heath WR. Minimal activation of memory CD8+ T cell by tissue-derived dendritic cells favors the stimulation of naive CD8+ T cells. Nat Immunol. 2007;8:1060–1066. doi: 10.1038/ni1505. [DOI] [PubMed] [Google Scholar]
- 22.den Haan JM, Lehar SM, Bevan MJ. CD8+ but not CD8− dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med. 2000;192:1685–1696. doi: 10.1084/jem.192.12.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopez-Bravo M, Ardavin C. In vivo induction of immune responses to pathogens by conventional dendritic cells. Immunity. 2008;29:343–351. doi: 10.1016/j.immuni.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 24.Allan RS, et al. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity. 2006;25:153–162. doi: 10.1016/j.immuni.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 25.Carbone FR, Belz GT, Heath WR. Transfer of antigen between migrating and lymph node-resident DCs in peripheral T-cell tolerance and immunity. Trends Immunol. 2004;25:655–658. doi: 10.1016/j.it.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 26.Le Borgne M, et al. Dendritic cells rapidly recruited into epithelial tissues via CCR6/CCL20 are responsible for CD8+ T cell crosspriming in vivo. Immunity. 2006;24:191–201. doi: 10.1016/j.immuni.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 27.Vermaelen KY, Carro-Muino I, Lambrecht BN, Pauwels RA. Specific migratory dendritic cells rapidly transport antigen from the airways to the thoracic lymph nodes. J Exp Med. 2001;193:51–60. doi: 10.1084/jem.193.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vermaelen K, Pauwels R. Accurate and simple discrimination of mouse pulmonary dendritic cell and macrophage populations by flow cytometry: methodology and new insights. Cytometry A. 2004;61:170–177. doi: 10.1002/cyto.a.20064. [DOI] [PubMed] [Google Scholar]
- 29.Nolte MA, van Olffen RW, van Gisbergen KP, van Lier RA. Timing and tuning of CD27-CD70 interactions: the impact of signal strength in setting the balance between adaptive responses and immunopathology. Immunol Rev. 2009;229:216–231. doi: 10.1111/j.1600-065X.2009.00774.x. [DOI] [PubMed] [Google Scholar]
- 30.Legge KL, Braciale TJ. Accelerated migration of respiratory dendritic cells to the regional lymph nodes is limited to the early phase of pulmonary infection. Immunity. 2003;18:265–277. doi: 10.1016/s1074-7613(03)00023-2. [DOI] [PubMed] [Google Scholar]
- 31.Jakubzick C, et al. Blood monocyte subsets differentially give rise to CD103+ and CD103− pulmonary dendritic cell populations. J Immunol. 2008;180:3019–3027. doi: 10.4049/jimmunol.180.5.3019. [DOI] [PubMed] [Google Scholar]
- 32.Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J Immunol. 2008;180:2562–2572. doi: 10.4049/jimmunol.180.4.2562. [DOI] [PubMed] [Google Scholar]
- 33.Masten BJ, Olson GK, Kusewitt DF, Lipscomb MF. Flt3 ligand preferentially increases the number of functionally active myeloid dendritic cells in the lungs of mice. J Immunol. 2004;172:4077–4083. doi: 10.4049/jimmunol.172.7.4077. [DOI] [PubMed] [Google Scholar]
- 34.Iliev ID, Mileti E, Matteoli G, Chieppa M, Rescigno M. Intestinal epithelial cells promote colitis-protective regulatory T-cell differentiation through dendritic cell conditioning. Mucosal Immunol. 2009;2:340–350. doi: 10.1038/mi.2009.13. [DOI] [PubMed] [Google Scholar]
- 35.Iliev ID, et al. Human intestinal epithelial cells promote the differentiation of tolerogenic dendritic cells. Gut. 2009;58:1481–1489. doi: 10.1136/gut.2008.175166. [DOI] [PubMed] [Google Scholar]
- 36.Hao X, Kim TS, Bracial TJ. Differential response of respiratory dendritic cell subsets to influenza virus infection. J Virol. 2008;82:4908–4919. doi: 10.1128/JVI.02367-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.del Rio ML, Rodriguez-Barbosa JI, Kremmer E, Forster R. CD103− and CD103+ bronchial lymph node dendritic cells are specialized in presenting and cross-presenting innocuous antigen to CD4+ and CD8+ T cells. J Immunol. 2007;178:6861–6866. doi: 10.4049/jimmunol.178.11.6861. [DOI] [PubMed] [Google Scholar]
- 38.Belz GT, Shortman K, Bevan MJ, Heath WR. CD8α+ dendritic cells selectively present MHC class I-restricted noncytolytic viral and intracellular bacterial antigens in vivo. J Immunol. 2005;175:196–200. doi: 10.4049/jimmunol.175.1.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Di Pucchio T, et al. Direct proteasome-independent cross-presentation of viral antigen by plasmacytoid dendritic cells on major histocompatibility complex class I. Nat Immunol. 2008;9:551–557. doi: 10.1038/ni.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mouries J, et al. Plasmacytoid dendritic cells efficiently cross-prime naive T cells in vivo after TLR activation. Blood. 2008;112:3713–3722. doi: 10.1182/blood-2008-03-146290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moron G, Rueda P, Casal I, Leclerc C. CD8α−CD11b+ dendritic cells present exogenous virus-like particles to CD8+ T cells and subsequently express CD8α and CD205 molecules. J Exp Med. 2002;195:1233–1245. doi: 10.1084/jem.20011930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.den Haan JM, Bevan MJ. Constitutive versus activation-dependent cross-presentation of immune complexes by CD8+ and CD8− dendritic cells in vivo. J Exp Med. 2002;196:817–827. doi: 10.1084/jem.20020295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chung Y, et al. Anatomic location defines antigen presentation by dendritic cells to T cells in response to intravenous soluble antigens. Eur J Immunol. 2007;37:1453–1462. doi: 10.1002/eji.200636544. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.