

Abstract
Tertiary interactions between loops and helical stems play critical roles in the biological function of many RNA pseudoknots. However, quantitative predictions for RNA tertiary interactions remain elusive. Here we report a statistical mechanical model for the prediction of noncanonical loop–stem base-pairing interactions in RNA pseudoknots. Central to the model is the evaluation of the conformational entropy for the pseudoknotted folds with defined loop–stem tertiary structural contacts. We develop an RNA virtual bond-based conformational model (Vfold model), which permits a rigorous computation of the conformational entropy for a given fold that contains loop–stem tertiary contacts. With the entropy parameters predicted from the Vfold model and the energy parameters for the tertiary contacts as inserted parameters, we can then predict the RNA folding thermodynamics, from which we can extract the tertiary contact thermodynamic parameters from theory–experimental comparisons. These comparisons reveal a contact enthalpy (ΔH) of −14 kcal/mol and a contact entropy (ΔS) of −38 cal/mol/K for a protonated C+•(G–C) base triple at pH 7.0, and (ΔH = −7 kcal/mol, ΔS = −19 cal/mol/K) for an unprotonated base triple. Tests of the model for a series of pseudoknots show good theory–experiment agreement. Based on the extracted energy parameters for the tertiary structural contacts, the model enables predictions for the structure, stability, and folding pathways for RNA pseudoknots with known or postulated loop–stem tertiary contacts from the nucleotide sequence alone.
Keywords: tertiary interaction, RNA pseudoknot, folding thermodynamics, RNA structure prediction
INTRODUCTION
Pseudoknots are tied to many biological functions from ribosomal frameshifting (Draper 1990; Gesteland and Atkins 1996; Giedroc et al. 2000; Staple and Butcher 2005; Giedroc and Cornish 2009) to human/yeast telomerase RNA (hTR) activity (Comolli et al. 2002; Theimer et al. 2003, 2005; Chen and Greider 2005; Cao and Chen 2007; Shefer et al. 2007; Qiao and Cech 2008) to metabolite-sensing riboswitches (Kang et al. 2009; Klein et al. 2009; Spitale et al. 2009). In a pseudoknot, the close physical proximity between the loop and stem often induces loop–stem tertiary structural interactions or contacts (Kim et al. 1999; Su et al. 1999; Cornish et al. 2005; Theimer et al. 2005; Cornish and Giedroc 2006; Chen et al. 2007; Ulyanov et al. 2007). Mutations that disrupt loop–stem tertiary contacts have been found to dramatically reduce biological activity, often due to a measurable reduction in the thermodynamic stability of the native pseudoknot relative to other partially folded conformations (Nixon et al. 2002a; Theimer et al. 2005).
In hairpin-type (H-type) pseudoknots, loops L1 and L2 (L2 is alternatively denoted L3 with L2 the short loop between the two stems at the helical junction) lie in major and minor grooves of stems S2 and S1, respectively (Giedroc et al. 2000). The most frequently occurring loop–stem tertiary interaction is a base triple interaction, wherein a nucleotide in one of the loops forms specific hydrogen bonds with a Watson–Crick base pair in the adjacent helical stem. In general, loop nucleotides can approach the base pair either from the major groove side or the minor groove side of the base pair. Although a wide range of interactions is possible, most often, loop pyrimidines (C or U) form L1–S2 major groove Hoogsteen base pairs with purine (G or A, respectively) N7–N6/O6 group to create U•(A–U) and C+•(G–C) base triples; in contrast, nucleotides from the minor groove side are in close proximity to the 2′-OH group and N3/O2–N2 side of one of the bases in the base pair and form L2–S1 base triples.
Not surprisingly, experimental studies reveal that specific tertiary structural hydrogen-bonding interactions depend strongly on nucleotide sequence conservation and solution conditions, notably pH, monovalent salt, and/or divalent salt concentration (Soto et al. 2007). For example, protonation of the cytidine N3 strongly stabilizes a major groove C+•G base pair in the context of a C+•(G–C) base triple by 2–3 kcal/mol to the overall pseudoknot stability. This value can be dependent on the context of the interaction and may or may not be linked to Mg2+ binding to the folded RNA (Nixon and Giedroc 2000; Nixon et al. 2002a; Nakano and Bevilacqua 2007). Minor groove-derived adenosine or cytidine-based hydrogen-bonding interactions to 2′-OH donors are also stabilizing and may function cooperatively with other base-pairing interactions (Cornish et al. 2005; Cornish and Giedroc, 2006). Finally, these loop–stem interactions may be augmented by pseudoknot L1 loop–L2 loop (typically U–A) Hoogsteen base pairs (Nixon et al. 2002b; Theimer et al. 2005; Giedroc and Cornish 2009; Kang et al. 2009; Klein et al. 2009; Spitale et al. 2009).
Despite the extensive experimental studies on the loop–stem tertiary interaction energetics, quantitative predictions for the structures and folding stabilities for loop–stem tertiary contacts from the nucleotide sequence remain an unsolved problem (Batey et al. 1999; Hermann and Patel 1999; Leontis et al. 2006; Mathews and Turner 2006; Das and Baker 2007; Shapiro et al. 2007; Chauhan and Woodson 2008; Ding et al. 2008; Parisien and Major 2008; Xin et al. 2008; Zhang et al. 2008). A major complication is the strong coupling of tertiary structural folding to divalent ion concentration; however, for the simple H-type pseudoknots discussed here, the folded state is achieved in the absence of Mg2+ and can be stabilized fully by monovalent salt (Soto et al. 2007). Even in these systems, however, another difficulty is a lack of energetics parameters for the tertiary contacts and the inability to accurately compute the conformational entropy for a pseudoknotted tertiary structural fold. Previous quantum mechanical computations for the tertiary contact energies provide useful estimations for the base triple energies (Oliva et al. 2006). However, the explicit nucleotide-solvent interactions, which can be important both enthalpically and entropically, were not considered in these calculations. An alternative approach is to extract the parameters from the experimental thermodynamic unfolding data. To do so requires a statistical mechanical model that bridges the macroscopic thermodynamics and the site-specific energetics parameters for individual microscopic interactions. In this paper we report the development of such a model for loop–stem tertiary interactions in pseudoknotted RNAs.
Previous molecular dynamics (MD) simulations on the BWYV (Saszar et al. 2001) and hTR pseudoknots (Yingling and Shapiro 2006) have uncovered local structural perturbations associated with the folded state. The predictions for the structure and thermal stability, however, involve large conformational changes and a large conformational ensemble, from fully unfolded, to partially folded intermediates to fully folded structures. In addition, unlike protein folding, the RNA folding energy landscape may well be rugged and may involve multiple structural rearrangements in the folding process (Chen and Dill 2000; Chen 2008; Hyeon and Thirmulai 2008). Tertiary structural contacts can sometimes drive a rearrangement of the secondary structure for RNAs such as the P5abc domain of the group I intron (Wu and Tinoco 1994; Thirumalai and Woodson 1996). It is therefore necessary to consider the complete conformational ensemble, including structures with the formation of various tertiary contacts for different secondary structures. Herein, we develop such a statistical mechanical approach that allows us to consider the complete conformational ensemble.
Existing theories for the stability of RNA secondary structure are not applicable to the tertiary interactions in pseudoknotted RNA structures. A notable difference in the energetics for the secondary versus tertiary structure derives from the additivity of the free energy. For secondary structures, the nearest-neighbor model (Serra and Turner 1995) posits that the total free energy, enthalpy, and entropy of the structure is equal to the sum of free energies of individual substructural units (base stacks, loops, etc.). For tertiary folds, however, the additivity principle fails because different parts of the structure are brought together by long-range tertiary contacts that essentially cross-link the structure; as a result, the total free energy and entropy of the folded structure is changed in a way that will not be equal to the sum of the free energy and entropy for each individual substructure. Therefore, a method is required that can explicitly treat how folding different substructures influences the energetics of the tertiary structural fold.
In an RNA pseudoknot, a minimum of two loops (L1 and L2) span the corresponding helical stem (S2 and S1, respectively). A loop and a helix can become correlated through, for example, the excluded volume interactions, that is, a loop and helix cannot occupy the same space. The structure of the helix, including the major and minor grooves, can significantly influence the loop conformational variability simply through the loop–stem excluded volume interactions, imposing a strong restriction on the conformational space and thus the entropy of the loop. As a result, the total free energy of a pseudoknot cannot be computed from the sum of the free energy of helices and individual loops; instead, the helix and loop together must be considered as an integral unit when the loop entropy is calculated.
Loop–stem tertiary structural interactions would add additional significant restrictions on the conformational entropy of a loop, with the resultant entropic decrease expected to destabilize the pseudoknotted structure. However, the hydrogen bonds associated with loop–stem contacts will tend to “stick” the loop into the stem, which results in net stabilization of the tertiary structural contacts. The hydrogen bond-induced stabilization competes with the entropy-induced destabilization, leading to a complex stability of the structures and the free energy landscape for the structures.
The theory developed here aims to predict the native structure, conformational ensemble, and folding stability from the nucleotide sequence alone. To do this, we employ a virtual bond-based conformational model (Vfold model) to describe an RNA tertiary structural fold. In order to provide a precise description for tertiary interactions, the model uses the sugar-base orientations in addition to the traditional backbone conformations (Leontis and Westhof 2001; Walberer et al. 2003) to describe the structure. This study focuses on the loop–stem base triple interactions, the most frequently occurring tertiary contacts in an RNA pseudoknot. In order to predict the tertiary interactions from the nucleotide sequence, we first use the Vfold model (Cao and Chen 2005, 2006) to compute the entropy for the different structures that contain various loop–stem contacts. Then, for a given sequence, we enumerate all the possible conformations, including component S1 and S2 hairpin structures and the pseudoknots, with or without loop–stem tertiary contacts. For each structure, we compute the free energy based on the pre-calculated entropy from the Vfold model. Summing over all the possible structures gives the partition function, from which we predict the folding stability and the structural distributions, with the energy parameters for the tertiary structural contacts as input parameters. Through theory–experiment comparison of the folding thermodynamics, we can fit the enthalpy and entropy parameters for the tertiary contacts. As an application of this tertiary folding model, we investigate the stable structures, folding stabilities, and equilibrium unfolding pathways for a series of experimentally studied pseudoknots. Theory–experiment tests have shown good agreements. As a further application of the model, we predict the stability for several telomerase RNA pseudoknots and find a wealth of tertiary interactions that can significantly alter the pseudoknot stability.
THEORY AND MODEL
Structural model for tertiary interaction
Because the C–O bonds in the polynucleotide backbone tend to adopt a fixed conformational state, the conformational degree of freedom can be approximately represented by two virtual bonds, 5′P–C4′3′ and 5′C4′–P3′, respectively (Olson 1980). Statistics of the known RNA three-dimensional (3D) structure show that the torsion angles β and ɛ (Fig. 1A) are close to the trans rotameric state (Murthy et al. 1999; Murray et al. 2003; Richardson et al. 2008). Note that the distribution of the ɛ torsion is centered at −150° with a broad shoulder and the β torsion is not exactly centered at 180° (Murthy et al. 1999). Therefore, the two-bond representation of the backbone conformation is a coarse approximation to the 46 discrete conformers of the RNA backbone (Richardson et al. 2008). Our previous studies indicate that the virtual-bond model can give reliable estimations for the conformational entropy. For instance, the calculated entropies for the hairpin and internal and bulge loops are quite close to the experimental measurements (Cao and Chen 2005) and the predicted loop entropy parameters for pseudoknots lead to accurate predictions for pseudoknot stabilities. Because the exhaustive enumeration of the conformations using the 46 conformers per nucleotide backbone or the all-atom sampling for the full conformational ensemble is computationally infeasible, the simplified virtual bond approach may provide a viable approach to the computation of the conformational entropy. The underpinning idea is that the all-atom free energy landscape can have a discrete number of local minima and the different structures at the different local minima may be approximately characterized by the ensemble of virtual bond conformations. Therefore, we can estimate the all-atom conformational entropy from the conformational ensemble of the virtual bond structures. Based on the virtual bond representation of RNA conformations, we recently developed an RNA folding model (Vfold model). The development of the Vfold model is motivated by the need to have a simplified conformational model for RNAs that can capture the realism of the key features of the structures, such as the major and minor grooves of the helices and the atomistic conformations of the tertiary contacts. Surveys of the experimentally determined structures suggest that the conformations of the virtual bonds are rotameric (Olson and Flory 1972; Olson 1980; Duarte and Pyle 1998; Wadley et al. 2007). Such discrete nature of the conformations leads to significant simplifications for modeling RNA structures (Wadley et al. 2007; Cao and Chen 2009).
FIGURE 1.

(A) Virtual bonds for a nucleotide: 5′P–C4′3′, 5′C4′–P3′, and C4′–N1 (primidine) or C4′–N9 (purine). (B) The secondary structure for hTR pseudoknot (Theimer et al. 2005). (C) The three-dimensional structure of the hTR pseudoknot (PDB code: 1YMO) (Theimer et al. 2005). Loops L1 and L2 span across the major and minor grooves of helices S2 and S1, respectively.
A traditional virtual bond model for nucleic structure employs two virtual bonds and accounts for the conformation of the backbone only (Olson and Flory 1972; Olson 1980). Recently, motivated by the importance of the sugar-base orientation in tertiary interactions, we developed a new virtual bond-based RNA folding model (Vfold model) by considering the orientation of the base. Specifically, besides the original two backbone virtual bonds, we include a third bond C4′–N1 (pyrimidine) or C4′–N9 (purine) to describe the sugar-base configuration (see Fig. 1A). From the known PDB database (Michiels et al. 2001; Theimer et al. 2005) for RNA pseudoknots, we find that the distance (DCN) between N1 (N9) and C4′ atoms is close to a fixed value 3.9 Å (the average value of DCN is 3.4 Å). In Supplemental Figure S1, we test the sensitivity of the entropy parameter on DCN and find that the difference in the conformational entropies based on the two different distances is not significant. In addition, we also find that the torsion angle between plane Pi–C4′–Pi + 1 and plane Pi–C4′–N1 (N9) is close to the fixed g−1 isomeric state (Cao and Chen 2009). Therefore, for each Pi–C4′–Pi + 1 backbone conformation, we can fix the N1 (N9) position. A previous statistical study on the RNA molecule also supports a rigid base orientation for a given backbone conformation (Olson and Flory 1972).
A simple H-type RNA pseudoknot consists of two loops (L1 and L2) that span the major and minor grooves, respectively, of S2 and S1 helical stems (Fig. 1B,C). Based on the atomic coordinates for an A-form RNA helix, we can reduce an all-atom helix structure into a reduced virtual bond structure. For loops, which are less rigid than helices, we use the usual gauche+ (g+), trans (t), and gauche−1 (g−1) rotational isomeric states for a polymer (Flory 1969) to represent the torsions of the backbone virtual bonds. A great simplification of the virtual bond-based modeling comes from the fact that the above rotameric virtual bond conformations can be conveniently configured in a diamond lattice by treating a lattice bond as virtual bond (Cao and Chen 2005, 2006). As a result, we can generate flexible loop conformations through self-avoiding random walks in the diamond lattice.
Helix stems
For a generic H-type RNA pseudoknot, the intervening junction region between the two helical stems (see Fig. 1) is often bent and/or over-rotated and may contain zero, one, or many intervening nucleotides (Giedroc et al. 2000; Pfingsten et al. 2007; Klein et al. 2009). The deviation of the two-stem structure away from the coaxially stacked continuous A-form helix is due to the conformational propensity of the intervening junction/loop, the loop–stem tertiary contacts, and the conformational viability (chain connectivity) of the loops that span across the respective helices. As a result, it will not, in most cases, be appropriate to model the two-stem system simply as a rigid continuous A-form helix. For example, as shown in Figure 1B, the hTR pseudoknot contains two helices (S1 and S2) of lengths equal to 6 base pairs (bp) (S1) and 9 bp (S2), respectively, and an array of loop–stem tertiary interactions as defined by the solution structure (Theimer et al. 2005). For the hTR, the two stems are bent by ∼30° (Theimer et al. 2005). In addition, the loop–helix interaction can also cause minor distortion of the helices away from perfect A-form RNA helical geometry. As a result, replacing the helices by a continuous A-form helix would cause a large distortion of the structure with a root-mean-square deviation (RMSD) of 6.2 Å, predominantly due to the noncoaxial orientation of the two helices. In the calculation, we use the best molecular fit program (Ferro and Hermans 1971) to calculate the RMSD.
We use the two helical stems in the hTR pseudoknot as a template for modeling the two-helix structure for a general pseudoknot that contains loop–stem tertiary interactions. Specifically, we use the P, C4′, and N1 (N9) coordinates in the solution structure of the hTR pseudoknot (Theimer et al. 2005) to generate the virtual bond representation for the two helices as a unit. The major motivation here is to capture any noncoaxial orientations of the two helices. We model each of the two stems by an A-form RNA helix, which causes small RMSDs equal to 1.6 and 1.4 Å for the 6-bp stem 1 and the 9-bp stem 2, respectively. To develop a versatile template system for modeling pseudoknots of various helix lengths, we elongate each helix to 10 bp to generate a two-stem structure with each helix length equal to 10 bp. Using this two-stem structure as a template, we can construct the 3D structure for pseudoknot helices of any stem lengths ≤10 bp. To test the validity of the approach, we computed the RMSD between the template and two other pseudoknots, including the sugarcane yellow leaf virus (ScYLV) (Cornish et al. 2005) and beet western yellows virus (BWYV) P1-P2 plant viral frameshifting pseudoknots (Su et al. 1999), each of which contains different loop–stem base triple interactions. The resultant RMSDs are 3.8 Å and 3.6 Å for ScYLV and BWYV pseudoknots, respectively. This suggests that the template structure may provide an approximate description for the two-helix configurations for a general pseudoknot that contains loop–stem tertiary interactions. Here the RMSDs are calculated over all the P, C4′, and N1 (N9) atoms that define the virtual bonds.
Loops
We model the loop conformations by self-avoiding walks in diamond lattice (Cao and Chen 2005, 2006), where each virtual bond is represented by a lattice bond. For a given two-stem structure, we count the number of loop conformations Ωloop through exhaustive computer enumeration and calculate the loop entropy as ΔSloop = kB ln Ωloop. Ignoring correlation between the two loops, we then compute the total loop entropy as the sum of each individual loop entropy. For each loop, the conformational entropy is determined by three factors: the volume exclusion between the different nucleotides within the loop, between the loop and helix nucleotides, and the formation of the base triple contacts between loop and helix nucleotides. In the enumeration of loop conformations, we take into account all three of these factors, which, in addition to the chain connectivity of whole molecule, impose rather strong restrictions on the loop conformation.
Because our template for the two-helix structure is derived from a specific pseudoknot (the hTR pseudoknot), we next determined the degree to which the template leads to reliable loop entropies for other pseudoknots. As a first test, we replaced the structure for the two helices in the hTR pseudoknot with the corresponding two-helix structure of the T2 gene 32 pseudoknot (Holland et al. 1999) and then recalculated the loop entropy of the chimeric structure. Our results for the loop entropy show that altering the helix structures leads to a minor error of 10% in total loop entropy. Therefore, the hTR pseudoknot-derived helix template would appear to return reliable estimations of the loop entropy that could be used for structure predictions.
As discussed above, different base triples can be formed between the loop and the stem. A given S1–L2 or S2–L1 helix–loop pair often contains multiple base triples. We denote a set of (multiple) loop–stem base triples as a “tertiary contact mode.” For a given helix and loop, we enumerate all possible modes and compute the loop entropy for each given mode using the Vfold model. We then tabulate the loop entropy parameters as a function of the helix length, loop length, and the mode. Because the L1 and L2 loops are nonsymmetric and span the major and minor helix grooves of the S2 and S1 stems, respectively, we make two separate tables for the two loops. With such loop entropy tables for individual tertiary structural folds, we can readily compute the entropy for any pseudoknot with a given loop–stem base triple tertiary contact architecture.
Base triples
A base triple is formed when a loop residue forms hydrogen bonds, for example, Hoogsteen hydrogen bond into the major groove, with a base pair in the helix (see Figs. 1B, 2). Solution NMR and crystallographic structures for a variety of RNAs, including the sugarcane yellow leaf virus (ScYLV) (Cornish et al. 2005) and BWYV (Su et al. 1999) plant frameshifting pseudoknots and the hTR (Theimer et al. 2005) pseudoknot, reveal eight types of frequently occurring loop–stem base triples, which can be designated A•(G–C), C•(G–C), A•(C–G), C•(C–G), A•(A–U), U•(A–U), A•(U–A), and U•(U–A) (Fig. 2A–G for seven base triples). In the current coarse-grained model, no distinction is made as to whether an interaction occurs in the minor or major groove of the helical stem, i.e., we allow these base triples to be formed in both minor and major grooves. Furthermore, the model makes no effort to distinguish base-pairing patterns in two similar base triples such as C•(G–C) or C•(C–G) (Nixon et al. 2002b).
FIGURE 2.

(A–G) The configurations for seven base triples selected from the BWYV (Su et al. 1999), ScYLV (Cornish et al. 2005), and hTR pseudoknots (Theimer et al. 2005). (H) A simplified configuration for the base triple U•(A–U). Nucleotide 3 (U) forms a base triple interaction with base pair 2 (A)–1 (U), and we use three atoms (P, C4′, and N1 or N9) to represent a nucleotide. DN12 or DN13 denotes the N1 or 9–N1 or 9 distance between two nucleotides. γ is the torsion angle C4′–N1 or 9–C4′–N1 or 9. α is the bond angle for C4′–N1 or 9–C4′.
Since RNA conformations are represented by virtual bonds in the Vfold model, a method is required to identify base triples at the level of virtual bonds. For illustration, we use the U•(A–U) major groove Hoogsteen base triple of the hTR pseudoknot (Fig. 2G). The hydrogen bonds restrict the flexibility of the nucleotides in the loops. Our survey of the known pseudoknot structures leads to the following operationally defined rules for the virtual bond configurations for the formation of a base triple (see Fig. 2H; Table 1):
The close distance between the nucleotide in the loop and the nucleotides in the helix: 5.0 Å ≤ DN12 or DN13 ≤ 14.0 Å;
The relative orientations of the sugar-base virtual bonds: (α12, α13, α21, and α31) ≤ 120°;
The torsion angle |γ12 or γ13| ≤ 120°.
TABLE 1.
Distances, torsion angles, and bond angles for the known triple bases

Using these rules, we can identify the base triples for a given virtual bond structure of RNA pseudoknot.
Partition function
Central to the prediction of the structure and folding stability from the nucleotide sequence is the partition function Q, which is the weighted sum over all the possible structures:
where ΔGs is the free energy of a given structure s. To compute the partition function from the nucleotide sequence, we enumerate all the possible secondary structures and pseudoknotted structures with and without loop–stem base triple contacts. For each pseudoknot, we exhaustively enumerate all the possible tertiary contact modes (combinations of different base triples) between stem S1 and loop L2 and between stem S2 and loop L1, respectively. The number of tertiary contact modes Nmode for the base triples between an m-nucleotide (nt) loop and an n-nt helix can be estimated from the following formula:
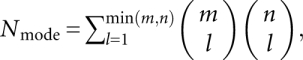 |
where l is the number of the base triples in the mode. The (combinatorial) number of modes and hence the computational time grows exponentially with the loop and stem lengths. Our benchmark test for the computational time (Fig. 3) for the different loop lengths shows that it requires ∼20 h to enumerate the loop conformations for all the tertiary modes for a pseudoknot with S1 = 9 bp and L2 = 9 nt. Thus, in order to efficiently enumerate all the tertiary modes for pseudoknots, we restrict the stem length to ≤9 bp and loop length ≤9 nt in the conformational ensemble for each partition function calculation.
FIGURE 3.

Plot of the CPU time for the enumeration of the complete ensemble of tertiary contacts for different loop length (L2) and fixed stem length (S1 = 9 bp). Note that the y-axis is in log scale.
For a given pseudoknot, we use the nearest-neighbor model and the empirical thermodynamic parameters (Turner rules) (Serra and Turner 1995) to calculate the free energy ΔGstacks for the stems as the sum of the free energies for each base stack. Using the pre-calculated loop entropy tables, we obtain the loop entropy from the loop conformational count Ωm for a tertiary contact mode m. We compute the partition function QPK from the sum over all the different tertiary contact modes (denoted by m1 for contacts between L2 and S1 and m2 for contacts between L1 and S2):
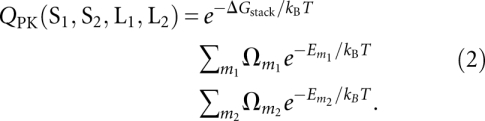 |
Here the tertiary contact free energy Em for a given tertiary contact mode m accounts for the free energy change, excluding the loop entropy change, upon the formation of the base triples. As the lowest-order approximation, we assume that the total free energy of the tertiary contacts for a given mode m is the sum of the free energy for each individual contact:
where Δhi and Δsi are the enthalpy and entropy, respectively, for the formation of the i-th base triple. The enthalpy is mainly derived from the hydrogen-bonding interactions, while the entropy comes from the reduced conformational freedom for the bases and the change of the solvent entropy associated with the formation of the base triple. As shown below, Equation 2 allows us to optimize (fit) the values of the enthalpy and entropy for different types of base triples from the experimental thermodynamic data for a training set of selected pseudoknots. We do not take into account the coaxial stacking in the prediction due to the bent configuration of the two stems, nor do we explicitly consider any potential loop L1–loop L2 base-pairing interactions.
Using a recursive algorithm, we can efficiently calculate the total partition function (Qtot) for a given sequence (Cao and Chen 2006). From the conditional partition function (Qij) with the condition that nucleotides i and j form a base pair, we calculate the probability for nucleotides i and j to form a base pair:
From Pij for all the possible (i, j) pairs, we predict the structure and the equilibrium folding pathway as a function of temperature as well as a heat capacity melting curve C(T), or melting profile, directly from the partition function Qtot:
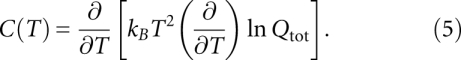 |
We can evaluate our theory through theory-experiment comparisons of the calculated and experimentally determined heat capacity thermal unfolding curves for different RNA sequences.
RESULTS AND DISCUSSION
Energetic parameters for base triples
The Vfold model developed in the present study allows us to extract the tertiary contact energetic parameters from experimental data reported for a training set of RNA pseudoknots. We note that the approach is different from a simple curve fitting exercise because the theory is based on the rigorous statistical mechanical properties of the system. As a first-order approximation, we classify the base triples into two types: protonated [C+•(C–G) or C+•(G–C)] and unprotonated base triples [A•(A–U), A•(U–A), U•(A–U), U•(U–A), A•(C–G), A•(G–C)]. The classification is based on the protonation state of the cytosine N3 at pH 7. A protonated base triple can have a higher stability than an unprotonated base triple (Nixon et al. 2002b; Giedroc and Cornish 2009). We note that the base-pairing structure of the protonated C+•G Hoogsteen base pair is identical in both the C+•(C–G) and C+•(G–C) base triples, with the C+•(C–G) base triple formed by an S-turn in the polynucleotide chain (Nixon et al. 2002b; Giedroc and Cornish 2009).
We next optimized the (Δh, Δs) parameters (Eq. 3) from direct theory–experiment comparisons for the melting thermodynamics for four different RNAs in the training set. These include the PEMV-1 (Nixon et al. 2002a), BWYV wild-type, and BWYV U8 frameshifting plant viral pseudoknots (Nixon and Giedroc 2000; Soto et al. 2007), and the ΔU177 hTR pseudoknot (Theimer et al. 2005). These systems were selected because they contain a variety of loop–stem base triples and their folding thermodynamics have been studied experimentally. Thermodynamic measurements were performed at different solution conditions. For example, 0.5 M KCl was used for plant viral frameshifting RNAs, and 0.2 M NaCl for the ΔU177 hTR mutant, with the empirical base-stacking energy parameters used to evaluate the free energies for the formation of helical stems compiled at 1 M NaCl “standard” conditions (Serra and Turner 1995). To account for the effect of the different salt concentrations, we employed the following empirical correction (SantaLucia 1998; Tan and Chen 2006) for the enthalpy and entropy of individual base-pair stacks: ΔH(Na+) = ΔH(1M); ΔS(Na+) = ΔS(1M) + 0.368 ln [Na+], where ΔH(1 M) and ΔS(1 M) are the empirical enthalpy and entropy for a base-pair stack at 1 M NaCl (Serra and Turner 1995). Previous tests on RNA hairpins suggest that the above correction is quite reliable for salt concentrations that are not too low (≥millimolar) (Tan and Chen 2006; Zhang and Chen 2006).
The experimental melting profiles acquired at pH 7.0 show multiple melting transitions (Fig. 4). For the plant viral RNAs, previous studies reveal three partially overlapping, sequential unfolding transitions that were assigned to the disruption of the tertiary loop–stem interactions, followed by successive unfolding of each of the helical stems to the unfolded state (Nixon and Giedroc 2000; Nixon et al. 2002a; Soto et al. 2007). However, for purposes of extracting best-fit thermodynamic parameters, these melting profiles were simply “fit” to two sequential unfolding steps defined by Tm1 and Tm2 (Fig. 4A–C). For the hTR ΔU177 pseudoknot (see Fig. 1) three discrete unfolding steps are clearly observed in the unfolding profiles, and thus it was modeled as such (Fig. 4D). The optimal fitting for the melting temperatures for the two transitions Tm1 and Tm2 of BWYV, U8 and PEMV, shown in Figure 4, leads to the following results for the tertiary contact parameters:
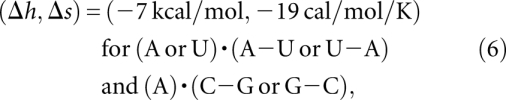 |
and
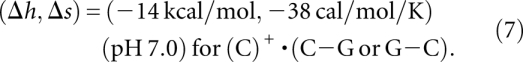 |
FIGURE 4.

Comparisons between the calculated (dashed lines) and the experimental (solid lines) thermal unfolding curves for (A) the BWYV wild-type pseudoknot (Nixon and Giedroc 2000), (B) the C8U BWYV pseudoknot (Nixon and Giedroc 2000), (C) the PEMV frameshifting pseudoknot (Nixon et al. 2002a), and (D) the ΔU177 hTR RNA pseudoknot (Theimer et al. 2005). The calculated and experimental melting curves for PEMV, BWYV wild-type, and BWYV U8 mutant are measured at 0.5 KCl (pH 7.0), while for the ΔU177 hTR RNA, the salt concentration is 0.2 NaCl (pH 7.0).
The uncertainties for (Δh, Δs) are about (±0.5 kcal/mol, ±1 cal/mol/K). The above parameters are derived from a simultaneous fit to BWYV, U8, and PEMV-1 data sets. As shown in Figure 5, the melting temperature is sensitive to the (Δh, Δs). Due to the nature of the Vfold model, the parameters are nucleotide-based instead of atom-based; as a result, the parameters are not specific to the chemical structure or atomistic details. In addition, to estimate the use of the coarse-grained conformational model may also contribute to the uncertainty of the fitted (Δh, Δs). How sensitive is the fitted (Δh, Δs) to the conformational entropy model? Our test results show that a change of the conformational entropy by 1 kB would lead to the new best-fit parameters (Δh, Δs) = (−6 kcal/mol/K, −15 cal/mol/K) for the unprotonated base triples and (−13 kcal/mol/K, −35 cal/mol/K) for the protonated base triples. The small change in the conformational entropy does not cause significant changes in the melting curve profile (see Supplemental Fig. S2).
FIGURE 5.

The predicted melting temperature (Tm1) for the disruption of the tertiary contacts in the BWYV pseudoknot with different enthalpy and entropy parameters. (A,B) We fix the energy parameter for the nonprotonated base triples at (enthalpy, entropy) = (−7 kcal/mol, −19 cal/mol/K) and vary the entropy and enthalpy parameters for the protonated base triples. (C,D) We fix the energy parameter for the protonated base triples at (enthalpy, entropy) = (−14 kcal/mol, −38 cal/mol/K) and vary the entropy and enthalpy parameters for nonprotonated base triples. We find that the melting temperature for the disruption of the tertiary interactions is sensitive to the enthalpy and entropy parameters. The experimentally determined low-temperature maximum in the melting profile under these conditions is Tm1 = 63°C (Nixon and Giedroc 2000).
The above parameter set is consistent with the experimental finding that turning off the protonation would lead to an enthalpic increase of ∼7 kcal/mol and a free energy increase of 1.5 kcal/mol (Cornish et al. 2005). Our above fitted parameter Δh = 7 kcal/mol is also close to the previous experimental result Δh = 5 kcal/mol for a U•(A–U) base triple (Krakauer and Sturtevant 1968). The 2.0 kcal/mol−1 theory–experiment discrepancy (Δh = 7 kcal/mol versus 5 kcal/mol) is probably caused by the simplifying assumption that distinct nonprotonated base triples irrespective of structure are forced to be characterized by the same thermodynamic parameters.
In general, the predicted melting curves agree quite well with the experimental melting profiles. For example, both theoretical predictions and experimental measurements give two peaks in the melting curves for PEMV-1, BWYV, and U8 pseudoknots, which correspond, in this simplified model, to the disruption of the tertiary interactions following helical unfolding structures, respectively. For the BWYV U8 mutant RNA, the C+•(G–C) base triple in the wild type is disrupted by the C8-to-U8 mutant. The melting temperature for the first unfolding transition, which is derived from the nearly coincident disruption of tertiary structural interaction as well as stem S2 (Nixon and Giedroc 2000), is ∼10°C lower for the U8 mutant than for the BWYV wild-type RNA.
To test the robustness of the extracted parameters, we examined the sensitivity of the melting temperature (Tm1) to the tertiary energy parameters (Δh, Δs). As shown in Figure 5, the melting temperatures are quite sensitive to the magnitude of (Δh, Δs), with Tm1 rapidly decreasing with a decreased enthalpy |Δh| or increased entropy |Δs|. Therefore, the fact that the single set of the fitted (Δh, Δs) values is sufficient to predict the unfolding behavior of four different RNA systems (Fig. 4) suggests that the extracted parameters in Equations 6 and 7 are robust and thus could be used for predictive purposes.
For the ΔU177 hTR RNA, a loop–loop U7•A37 Hoogsteen base pair (see Figs. 1B; 2A) is found in the solution structure (Theimer et al. 2005); an exactly analogous U9•A27 base pair (Nixon et al. 2002a,b; Giedroc and Cornish 2009) is found in the PEMV-1 RNA structure as well. In the hTR pseudoknot, the loop–loop base pair plays a significant role in the stabilization of the pseudoknot, causing the elevation of the melting temperature for the tertiary interactions by nearly 20°C (Theimer et al. 2005). In the present form of the model, loop–loop contacts are not explicitly considered; as a result, our model cannot predict the U7•A37 Hoogsteen base pair. In order to account for such loop–loop contact, we simply assign an interaction energy for a conventional Watson–Crick U7–A37 base pair. Using a Δh of −8 kcal/mol (Krakauer and Sturtevant 1968), the fitted Δs from the experimental melting curve (Fig. 4D) is −16 cal/mol/K.
Equilibrium unfolding pathways
BWYV pseudoknot
Previous experimental studies (Nixon and Giedroc 2000; Soto et al. 2007) suggested a three-step equilibrium folding pathway for the wild-type BWYV RNA: the native folded state (denoted F), to a pseudoknotted state with the tertiary interactions disrupted (PK), to the most stable component hairpin (hp or S) intermediate, to the fully unfolded state (U). To use our model to predict the folding pathways from the sequence, we first compute the partition functions (Eqs. 1 and 2) at different temperatures, from which we predict the melting curve (Eq. 5). From the melting curve, we identify the melting temperatures for each structural transition. In each different temperature regime, we predict the dominant (most probable) structure (s) from the base-pairing probability (Eq. 4). The change of the dominant structures as temperature changes gives the predicted equilibrium unfolding pathway.
Figure 6A shows the theoretically predicted folding pathway for the wild-type BWYV pseudoknot. We find that at low temperature (T ∼20°C), both loops are involved in base triple interactions, with the model predicting that the protonated C8+•G12 base pair contributes about −3.0 kcal/mol to the overall folding stability at T ∼20°C as dictated by the fitted entropy and enthalpy parameters for a protonated base triple (see Eq. 7). We note that the fact that this value is very close to the experimentally determined ΔΔG for protonation is not unexpected since the BWYV RNA was one of the RNAs in the training set. Moreover, we find that loop residues A24 and C22 form base triples with base pairs G7–C14 and G6–C15, respectively. These predicted base triples are consistent with the crystallographic structure of this RNA (Su et al. 1999). This structure reveals that C22 is flipped toward the base pairs G6–C15 and C5–G16 and A20 also forms a base triple with G4–C17 and C5–G16. Our calculation also predicts that A20 forms a base triple only with the C5–G16 base pair. This minor difference does not alter the global stability of the pseudoknot to any measurable degree. As the temperature increases, the tertiary interactions are disrupted first. This is because breaking the tertiary interactions causes a dramatic increase in the entropy due to the change in the conformational entropy for the backbone, bases, and solvent molecules. In addition, we find that stem S1 is the most stable structural component, which is the last structural component to be disrupted. Unfolding of stem 1 results in a sharp peak at T ∼90°C in the melting curve (Fig. 4A). The three-step sequential unfolding pathway for wild-type BWYV pseudoknot is consistent with that proposed previously (Nixon and Giedroc 2000; Soto et al. 2007), despite the fact that Δh and Δs for tertiary structural contacts were optimized assuming a two-step unfolding curve for this RNA.
FIGURE 6.

The predicted equilibrium unfolding pathways for (A–D) BWYV and (E–H) ΔU177 hTR RNAs. We also show the experimentally observed loop–stem base triples between the loops and stems. The results support the F to PK to hp to U folding pathway (Nixon and Giedroc 2000), where F is the folded state with the tertiary interactions, PK is the pseudoknot structure without the tertiary interactions, hp is a folding intermediate (usually a stem–loop structure), and U is the unfolded state. In the density plots, the x- and y-axes denote the indices of the nucleotides i and j, and the darkness of the points denote the base-pairing probability Pij (from Eq. 4).
ΔU177 hTR pseudoknot
Thermodynamic unfolding experiments show that the tertiary interactions are disrupted at T ∼60°C. NMR experiments (Theimer et al. 2005) indicate three Hoogsteen base pairs are found in the U•(U–A) base triple between loop L1 and the major groove of stem S2. Our model correctly predicts that U9 and U10 form tertiary contacts with U22–A39 and U21–A40, respectively, at low temperature (see Fig. 6E–H). The previous molecular dynamics simulation failed to predict the correct tertiary interactions for the hTR pseudoknot (Yingling and Shapiro 2006). In contrast, our Vfold model appears to give much improved predictions for these tertiary interactions. However, the Vfold model cannot account for the all-atom details, relying instead on discrete rotameric states for the backbone conformation. As a result, the model cannot predict base triples with atomistic details not considered in the Vfold model. For instance, the model does not predict the U8•(U21–A40) and A35•(U5–A25) base triples in the ΔU177 hTR pseudoknot (Theimer et al. 2005).
From the temperature dependence of the ensemble of structures, we are able to predict the unfolding pathway for the ΔU177 hTR RNA. We find that the tertiary interactions in ΔU177 are completely disrupted as the temperature approaches 70°C (Fig. 4D). In accordance with the experimental findings, our model predicts that a shoulder at ∼60°C in the melting curve in Figure 4D corresponds to the disruption of the tertiary interactions. Further unfolding steps involve the unfolding of stem S1, followed by unfolding of stem S2. The above two transitions result in two peaks at temperatures 70°C and 80°C, respectively. The resultant ΔU177 supports the general pathway.
Predicting loop–helix tertiary interactions
Using the Vfold model, we can also predict the tertiary structural interactions for RNA pseudoknots that are not in the training set. We first tested our Vfold model for the prediction of simple nonpseudoknot RNA conformations such as hairpins. Our test results (Supplemental Table S1) indicate that the model can accurately predict the base pairs for the tested sequences. The accuracy of the theoretical predictions is measured by two parameters: sensitivity SE and specificity SP: SE = Np(c)/Ne, SP = Np(c)/Np, where Np(c) is the number of correctly predicted canonical base pairs, and Ne and Np are the total numbers of canonical base pairs in the experimentally determined and theoretically predicted structures, respectively.
We then tested the theory for the predictions of pseudoknotted structures. Specifically, we investigated the structures, stabilities, and unfolding pathways for three additional plant viral frameshifting RNA pseudoknots from sugarcane yellow leaf virus (ScYLV) and potato leaf roll virus (PLRV), which are of known structure (Cornish et al. 2005; Pallan et al. 2005), and cucurbit aphid-borne yellows virus (CABYV) (of unknown structure) and compared those predictions for the refined structure of the PEMV-1 RNA, which is one of the RNAs in the training set.
For the ScYLV RNA, the model correctly predicts that the C8+ and C27 form major- and minor-groove-derived tertiary structural contacts with G12–C28 and G7–C14 base pairs, respectively. In addition, the model predicts that nucleotides A20, A21, and A22 from L2 are capable of forming minor groove tertiary contacts with the G4–C17 base pair, while A22, A23, and A24 can form tertiary contacts with the U5–A16 base pair. Inspection of the solution structure reveals that A21 and A22 form base triple interactions with G4–C17 and U5–A16, respectively (Cornish et al. 2005). Thus, although the prediction is unable to pinpoint the precise hydrogen bonding interactions with certainty, the model predicts two L2–S1 interactions at the top of the molecule, in agreement with the solution structure (Cornish et al. 2005).
For the CABYV RNA, Figure 7B shows that we can correctly predict the pseudoknot structure only if we consider the pseudoknot stem–loop interactions since a model (Cao and Chen 2006) that ignores the tertiary structural interactions predicts the S1 hairpin rather than the pseudoknot. This finding reveals that tertiary structural interactions play a vital role for the structure and stability of the CABYV pseudoknot, as previously documented experimentally for the BWYV pseudoknot (Nixon et al. 2002a). For the refined solution structural model of the PEMV-1 pseudoknot (Giedroc and Cornish 2009), we predict that the C10+, C24, and A26 will form tertiary structural contacts with the C13–G28, G7–C16, and G8–C15 base pairs, respectively, each of which is found in the refined solution structure of this RNA (Giedroc and Cornish 2009).
FIGURE 7.

The predicted tertiary interactions and the density plots for the base-pairing probability for three pseudoknot-forming sequences: (A) ScYLV RNA (Cornish et al. 2005), (B) CABYV RNA, (C) PEMV-1 RNA, and (D) PLRV-M (Pallan et al. 2005) at 20°C. In C we number the PEMV-1 sequence from G0 to A32 in order to be consistent with the experiment (Nixon et al. 2002b; Giedroc and Cornish 2009).
We next used our methods to predict tertiary structural loop–stem interactions in a mutant luteoviral PLRV pseudoknot, designated PLRV-M, whose crystallographic structure is known to high resolution (1.35 Å) (Pallan et al. 2005). The Vfold model (Fig. 7D) correctly predicts that nucleotide C7 and A25/A24 (A25 is involved in two H bonds; A24 is involved in just one) form base triple interactions with G11–C26 and G6–C13 base pairs, respectively. However, the model does not predict the hydrogen bonding interactions between A23 and the G5–C14 base pair. In addition, our model cannot predict the detailed tertiary interaction between A20 and the two base pairs G3–C16 and C4–G15 (Pallan et al. 2005) because such tertiary interactions are not considered in the theory.
Finally, we have applied our model to predict the probabilities for the formation of loop–stem tertiary interactions in telomerase pseudoknots in species other than human, each of which are of unknown high-resolution structure (Ulyanov et al. 2007). Figure 8 shows our calculated total partition functions Qtot for all the possible structures as well as the conditional partition function Q2 for all the structures without loop–stem tertiary interactions. A large Qtot over Q2 is indicative of significantly stabilizing tertiary structural interactions between loop and stem in the RNA. From Figure 8, we conclude that there exist significant tertiary structural interactions in four of the six telomerase RNAs, these from Colpidium colpoda, Tetrahymena thermophila, Glaucoma chattoni, and Euplotes aediculatus. Similar to the human telomerase RNA pseudoknot (Theimer et al. 2005), loop–stem tertiary interactions may play important functional roles in the telomerase activity of these species (Ulyanov et al. 2007). The Vfold model can predict the loop–stem tertiary contacts (base triples) for the four telomerase sequences (see Fig. 9). The model predicts a conserved major-groove tertiary interaction between nucleotide A or U and a U–A base pair for the four telomerase sequences. The result is consistent with the predictions from molecular dynamics simulation (Ulyanov et al. 2007). In addition, we also predicted the minor-groove tertiary interaction between loop L2 and stem S1 in sequences G. chattoni, T. thermophila, and E. aediculatus (Fig. 9).
FIGURE 8.

The partition functions Qtot for all the possible structures (filled square and solid thick line) and Q2 for structures without loop–stem tertiary contacts (open squares and thin line) for the telomerase RNA pseudoknots in six different organisms: Paramecium tetraurelia, Tetrahymena paravorax, C. colpoda, G. chattoni, T. thermophila, and E. aediculatus at T = 20°C.
FIGURE 9.

The predicted base triples interactions for C. colpoda, G. chattoni, T. thermophila, and E. aediculatus at T = 20°C.
Predicting all-atom three-dimensional structures for pseudoknots
Predicting the high-resolution 3D RNA structure from sequence is not a solved problem (Major et al. 1991; Macke and Case 1998; Burks et al. 2005; Jossinet and Westhof 2005; Tan et al. 2006; Das and Baker 2007; Shapiro et al. 2007; Ding et al. 2008; Martinez et al. 2008; Parisien and Major 2008; Jonikas et al. 2009). One of the bottlenecks is the calculation of the free energy for a tertiary fold. The Vfold model allows us to compute the entropies and free energies for pseudoknotted structures and structures containing loop–stem tertiary contacts. From the predicted free energy landscape, the model gives the minimum-free-energy structure. The Vfold-predicted structure is a two-dimensional (2D) structure (i.e., structure defined by base pairs and loop–stem contacts). The 2D structure serves as the scaffold for the all-atom 3D structure. Here, using the BWYV pseudoknot as an illustrative case, we show the computational procedure for the prediction of the all-atom 3D structure from the Vfold-predicted 2D structure:
Predict the 2D structure (including the loop–stem tertiary contacts) of the RNA pseudoknot by using the Vfold model. As described in the above sections, the Vfold model can give reliable predictions for the pseudoknot 2D structures. Figure 10A gives the predicted 2D structure of the BWYV pseudoknot.
Build a scaffold 3D structure based on the virtual-bond representation (see Fig. 10B). The coordinates of P, C4, and (N1 or N9) atoms are obtained from a combined fragment-based and diamond lattice-based method as described below. The coordinates of the red nucleotides are adopted from the PEMV-1 fragment (PDB ID, 1KPZ), which has the same junction G7–C14/U13–A25 as that of the BWYV pseudoknot. The coordinates of the two helices (green) are obtained from an A-form helix. The coordinates of the remaining two nucleotides (blue) are generated by self-avoiding walks of the virtual bonds in the diamond lattice.
Construct the all-atom structure: we first extract the all-atom coordinates for A, U, G, and C nucleotides from the coordinates of the A-form helix, then append the atoms to the scaffold structure in Figure 10B. This results in the all-atom structure (Fig. 10C).
Refine the all-atom structure using Amber minimization (Case et al. 2005, 2006). First, we perform 2000 steps minimization with 500.0-kcal/mol restraints. Following the 2000 steps minimization, we run another 2000 steps minimization without restraints. In the minimization process, the negative charge in phosphate is neutralized by cation Na+. The nonbonded interactions are cut at 12 Å. The force field we use is ff77 for RNA (Cornell et al. 1995; Wang et al. 2000). Figure 10D shows the refined all-atom structure (purple-blue). The RMSD over all heavy atoms is 2.7 Å with the experimental structure (PDB code: 473d; in sand color) (Su et al. 1999).
FIGURE 10.

(A) The predicted 2D structure for the BWYV pseudoknot; (B) the predicted virtual-bond level 3D structure; (C) the all-atom structure constructed from the virtual bond structure; and (D) the refined all-atom structure using Amber 9.
Our predicted BWYV 3D structure agrees with the experimental structure quite well. Future development of the model should address the issue about how to construct the junction/loop conformation if homologous conformations are not found in the PDB database.
Summary
We have developed a new generation Vfold model that enables the prediction of loop–stem tertiary structural contacts in simple H-type RNA pseudoknots. Based on the conformational entropy parameters calculated from the Vfold model, we are able to compute the structure, stability, and structural changes that occur during temperature-induced unfolding experiments from the nucleotide sequence alone. The model leads to several useful conclusions:
We obtain rough estimates for triple base-pair enthalpy and entropy that by design are insensitive to the precise chemical nature and structural context of the specific interaction.
In general, examination of predicted equilibrium unfolding pathways reveals that the tertiary contacts are disrupted at low temperature prior to the global unfolding of the pseudoknot stems at higher temperature. Depending on the interplay between the loop and helix stabilities, a pseudoknot can unfold either through a two-state cooperative process or a noncooperative multistep process.
Loop–stem tertiary contacts are known to be crucial for the stabilization of pseudoknots (Nixon et al. 2002a). Our calculations reveal that destabilizing loop–stem base triples, particularly in the context of pseudoknots with short S2 stems, may lead to complete change of the structure of the RNA, to a partially folded hairpin loop.
We predict that there exist significant loop–stem tertiary interactions in the telomerase RNA pseudoknots of C. colpoda, T. thermophila, G. chattoni, and E. aediculatus. This implies that the tertiary structural interactions are significantly conserved and may therefore play a functional role in the activity of these telomerase RNAs (Theimer et al. 2005).
Development of the theory described here provides a starting point with which to further refine the thermodynamic parameters for the chemically different noncanonical base pairs within loop–stem base triples. Moreover, the current model assumes the additivity of the base triple interaction energies, for which there is no a priori justification. Improvement of the theory should account for the possible interferences (such as stacking) between neighboring base triples like that which has been documented to occur in many RNA pseudoknots (Cornish et al. 2005; Klein et al. 2009). Furthermore, an accurate prediction for the base triple interactions obviously requires an atomistic description of the structure. One plausible approach to enhance the utility of the current algorithm might be to use the Vfold model-predicted structure as a starting scaffold onto which one could add atomic details to further refine the structure. Finally, although we only treat the tertiary contacts in RNA pseudoknots in this study, the same methodology can be systematically extended to other more complex tertiary structures that incorporate other loop–stem base pairs, such as in the HDV pseudoknot (Ferré-D'Amaré et al. 1998), and other A-minor motifs (Nissen et al. 2001), as well as the influence of base pairing to exogenous nucleobase metabolites, like that present in the adenine, guanine, and preQ1 sensing riboswitches (Gilbert et al. 2009; Kang et al. 2009; Klein et al. 2009; Spitale et al. 2009).
SUPPLEMENTAL MATERIAL
Supplemental material can be found at http://www.rnajournal.org.
ACKNOWLEDGMENTS
We acknowledge two anonymous reviewers for careful readings and constructive suggestions. This research was supported by NIH grants GM063732 (to S.-J.C.) and AI067416 (to D.P.G.). Most of the numerical calculations were performed on the HPC resources at the University of Missouri Bioinformatics Consortium (UMBC).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.1800210.
REFERENCES
- Batey RT, Rambo RP, Doudna JA. Tertiary motifs in RNA structure and folding. Angew Chem Int Ed. 1999;38:2326–2343. doi: 10.1002/(sici)1521-3773(19990816)38:16<2326::aid-anie2326>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Burks J, Zwieb C, Mller F, Wower I, Wower J. Comparative 3-D modeling of tmRNA. BMC Mol Biol. 2005;6:14. doi: 10.1186/1471-2199-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S, Chen S-J. Predicting RNA folding thermodynamics with a reduced chain representation model. RNA. 2005;11:1884–1897. doi: 10.1261/rna.2109105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S, Chen S-J. Predicting RNA pseudoknot folding thermodynamics. Nucleic Acids Res. 2006;34:2634–2652. doi: 10.1093/nar/gkl346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S, Chen S-J. Biphasic folding kinetics of RNA pseudoknots and telomerase RNA activity. J Mol Biol. 2007;367:909–924. doi: 10.1016/j.jmb.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S, Chen S-J.2009Predicting the folding thermodynamics for the pseudoknot with interstem loops RNA 15696–706.19237463 [Google Scholar]
- Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Pearlman DA, Crowley M, et al. AMBER 9. University of California; San Francisco: 2006. [Google Scholar]
- Chauhan S, Woodson SA. Tertiary interactions determine the accuracy of RNA folding. J Am Chem Soc. 2008;130:1296–1303. doi: 10.1021/ja076166i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S-J. RNA folding: Conformational statistics, folding kinetics, and ion electrostatics. Annu Rev Biophys. 2008;37:197–214. doi: 10.1146/annurev.biophys.37.032807.125957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S-J, Dill KA. RNA folding energy landscapes. Proc Natl Acad Sci. 2000;97:646–651. doi: 10.1073/pnas.97.2.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JL, Greider CW. Functional analysis of the pseudoknot structure in human telomerase RNA. Proc Natl Acad Sci. 2005;102:8080–8085. doi: 10.1073/pnas.0502259102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Wen JD, Tinoco I., Jr Single-molecule mechanical unfolding and folding of a pseudoknot in human telomerase RNA. RNA. 2007;13:2175–2188. doi: 10.1261/rna.676707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comolli LR, Smirnov I, Xu L, Blackburn EH, James TL. A molecular switch underlies a human telomerase disease. Proc Natl Acad Sci. 2002;99:16998–17003. doi: 10.1073/pnas.262663599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A 2nd generation force-field for the simulation of proteins, nucleic-acids, and organic molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- Cornish PV, Giedroc DP. Pairwise coupling analysis of helical junction hydrogen bonding interactions in luteoviral RNA pseudoknots. Biochemistry. 2006;45:11162–11171. doi: 10.1021/bi060430n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish PV, Hennig M, Giedroc DP. A loop 2 cytidine-stem 1 minor groove interaction as a positive determinant for pseudoknot-stimulated −1 ribosomal frameshifting. Proc Natl Acad Sci. 2005;102:12694–12699. doi: 10.1073/pnas.0506166102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Baker D. Automated de novo prediction of native-like RNA tertiary structures. Proc Natl Acad Sci. 2007;104:14664–14669. doi: 10.1073/pnas.0703836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding F, Sharma S, Chalasani P, Demidov VV, Broude NE, Dokholyan NV. Ab initio RNA folding by discrete molecular dynamics: From structure prediction to folding mechanisms. RNA. 2008;14:1164–1173. doi: 10.1261/rna.894608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper DE. Pseudoknots and the control of protein synthesis. Curr Opin Cell Biol. 1990;2:1099–1103. doi: 10.1016/0955-0674(90)90162-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte CM, Pyle AM. Stepping through an RNA structure: A novel approach to conformational analysis. J Mol Biol. 1998;284:1465–1478. doi: 10.1006/jmbi.1998.2233. [DOI] [PubMed] [Google Scholar]
- Ferré-D'Amaré AR, Zhou KH, Doudna JA. Crystal structure of a hepatitis delta virus ribozyme. Nature. 1998;395:567–574. doi: 10.1038/26912. [DOI] [PubMed] [Google Scholar]
- Ferro DR, Hermans J. A different best rigid-body molecular fit routine. Acta Crystallogr A. 1971;33:345–347. [Google Scholar]
- Flory PJ. Statistical mechanics of chain molecules. Wiley; New York: 1969. [Google Scholar]
- Gesteland RF, Atkins JF. Recoding: Dynamic reprogramming of translation. Annu Rev Biochem. 1996;65:741–768. doi: 10.1146/annurev.bi.65.070196.003521. [DOI] [PubMed] [Google Scholar]
- Giedroc DP, Cornish PV. Frameshifting RNA pseudoknots: Structure and mechanism. Virus Res. 2009;139:193–208. doi: 10.1016/j.virusres.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giedroc DP, Theimer CA, Nixon PL. Structure, stability and function of RNA pseudoknots involved in stimulating ribosomal frameshifting. J Mol Biol. 2000;298:167–185. doi: 10.1006/jmbi.2000.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert SD, Reyes FE, Edwards AL, Batey RT. Adaptive ligand binding by the purine riboswitch in the recognition of guanine and adenine analogs. Structure. 2009;17:857–868. doi: 10.1016/j.str.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann T, Patel DJ. Stitching together RNA tertiary architectures. J. Mol. Biol. 1999;294:829–849. doi: 10.1006/jmbi.1999.3312. [DOI] [PubMed] [Google Scholar]
- Holland JA, Hansen MR, Du ZH, Hoffman DW. An examination of coaxial stacking of helical stems in a pseudoknot motif: The gene 32 messenger RNA pseudoknot of bacteriophage T2. RNA. 1999;5:257–271. doi: 10.1017/s1355838299981360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyeon C, Thirmulai D. Multiple probes are required to explore and control the rugged energy landscapes of RNA hairpins. J Am Chem Soc. 2008;130:1538–1539. doi: 10.1021/ja0771641. [DOI] [PubMed] [Google Scholar]
- Jonikas MA, Radmer RJ, Laederach A, Das R, Pearlman S, Herschlag D, Altman RB. Coarse-grained modeling of large RNA molecules with knowledge-based potentials and structural filters. RNA. 2009;15:189–199. doi: 10.1261/rna.1270809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jossinet F, Westhof E. Sequence to structure (S2S): Display, manipulate, and interconnect RNA data from sequence to structure. Bioinformatics. 2005;21:3320–3321. doi: 10.1093/bioinformatics/bti504. [DOI] [PubMed] [Google Scholar]
- Kang M, Peterson R, Feigon J. Structural insights into riboswitch control of the biosynthesis of queuosine, a modified nucleotide found in the anticodon of tRNA. Mol Cell. 2009;33:784–790. doi: 10.1016/j.molcel.2009.02.019. [DOI] [PubMed] [Google Scholar]
- Kim YG, Su L, Maas S, O'Neill A, Rich A. Specific mutations in a viral RNA pseudoknot drastically change ribosomal frameshifting efficiency. Proc Natl Acad Sci. 1999;96:14234–14239. doi: 10.1073/pnas.96.25.14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein DJ, Edwards TE, Ferré-D'Amaré AR. Cocrystal structure of a class I preQ1 riboswitch reveals a pseudoknot recognizing an essential hypermodified nucleobase. Nat Struct Mol Biol. 2009;16:343–344. doi: 10.1038/nsmb.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakauer H, Sturtevant JM. Heats of the helix–coil transitions of the poly A–poly U complexes. Biopolymers. 1968;6:491–512. doi: 10.1002/bip.1968.360060406. [DOI] [PubMed] [Google Scholar]
- Leontis NB, Westhof E. Geometric nomenclature and classification of RNA base pairs. RNA. 2001;7:499–512. doi: 10.1017/s1355838201002515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leontis NB, Lescoute A, Westhof E. The building blocks and motifs of RNA architecture. Curr Opin Struct Biol. 2006;16:279–287. doi: 10.1016/j.sbi.2006.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macke T, Case DA. Modeling unusual nucleic acid structures. In: Leontes NB, SantaLucia J Jr, editors. Molecular modeling of nucleic acids. American Chemical Society; Washington, DC: 1998. pp. 379–393. [Google Scholar]
- Major F, Turcotte M, Gautheret D, Lapalme G, Fillion E, Cedergren R. The combination of symbolic and numerical computation for three-dimensional modeling of RNA. Science. 1991;253:1255–1260. doi: 10.1126/science.1716375. [DOI] [PubMed] [Google Scholar]
- Martinez HM, Maizel JV, Jr, Shapiro BA. RNA2D3D: A program for generating, viewing and comparing three-dimensional models of RNA. J Biomol Struct Dyn. 2008;25:669–683. doi: 10.1080/07391102.2008.10531240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews DH, Turner DH. Prediction of RNA secondary structure by free energy minimization. Curr Opin Struct Biol. 2006;16:270–278. doi: 10.1016/j.sbi.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Michiels PJA, Versleijen AAM, Verlaan PW, Pleij CWA, Hilbers CW, Heus HA. Solution structure of the pseudoknot of SRV-1 RNA, involved in ribosomal frameshifting. J Mol Biol. 2001;310:1109–1123. doi: 10.1006/jmbi.2001.4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray LJ, Arendall WB, III, Richardson DC, Richardson JS. RNA backbone is rotameric. Proc Natl Acad Sci. 2003;100:13904–13909. doi: 10.1073/pnas.1835769100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy VL, Srinivasan R, Draper DE, Rose GD. A complete conformational map for RNA. J Mol Biol. 1999;291:313–327. doi: 10.1006/jmbi.1999.2958. [DOI] [PubMed] [Google Scholar]
- Nakano S, Bevilacqua PC. Mechanistic characterization of the HDV genomic ribozyme: A mutant of the C41 motif provides insight into the positioning and thermodynamic linkage of metal ions and protons. Biochemistry. 2007;46:3001–3012. doi: 10.1021/bi061732s. [DOI] [PubMed] [Google Scholar]
- Nissen P, Ippolito JA, Ban N, Moore PB, Steitz TA. RNA tertiary interactions in the large ribosomal subunit: The A-minor motif. Proc Natl Acad Sci. 2001;98:4899–4903. doi: 10.1073/pnas.081082398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon PL, Giedroc DP. Energetics of a strongly pH dependent RNA tertiary structure in a frameshifting pseudoknot. J Mol Biol. 2000;296:659–671. doi: 10.1006/jmbi.1999.3464. [DOI] [PubMed] [Google Scholar]
- Nixon PL, Cornish PV, Suram SV, Giedroc DP. Thermodynamic analysis of conserved loop–stem interactions in P1-P2 frameshifting RNA pseudoknots from plant Luteoviridae. Biochemistry. 2002a;41:10665–10674. doi: 10.1021/bi025843c. [DOI] [PubMed] [Google Scholar]
- Nixon PL, Rangan A, Kim YG, Rich A, Hoffman DW, Hennig M, Giedroc DP. Solution structure of a luteoviral P1-P2 frameshifting mRNA pseudoknot. J Mol Biol. 2002b;322:621–633. doi: 10.1016/s0022-2836(02)00779-9. [DOI] [PubMed] [Google Scholar]
- Oliva R, Cavallo L, Tramontano A. Accurate energies of hydrogen bonded nucleic acid base pairs and triplets in tRNA tertiary interactions. Nucleic Acids Res. 2006;34:865–879. doi: 10.1093/nar/gkj491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson WK. Configurational statistics of polynucleotide chains: An updated virtual bond model to treat effects of base stacking. Macromolecules. 1980;13:721–728. doi: 10.1021/ma60045a006. [DOI] [PubMed] [Google Scholar]
- Olson WK, Flory PJ. Spatial configurations of polynucleotide chains. I, Steric interactions in polyribonucleotides: A virtual bond model. Biopolymers. 1972;11:1–23. doi: 10.1002/bip.1972.360110102. [DOI] [PubMed] [Google Scholar]
- Pallan PS, Marshall WS, Harp J, Jewett FC, III, Wawrzak Z, Brown BA, II, Rich A, Egli M. Crystal structure of a luteoviral RNA pseudoknot and model for a minimal ribosomal frameshifting motif. Biochemistry. 2005;44:11315–11322. doi: 10.1021/bi051061i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisien M, Major F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008;452:51–55. doi: 10.1038/nature06684. [DOI] [PubMed] [Google Scholar]
- Pfingsten JS, Costantino DA, Kieft JS. Conservation and diversity among the three-dimensional folds of the Dicistroviridae Intergenic Region IRESes. J Mol Biol. 2007;370:856–869. doi: 10.1016/j.jmb.2007.04.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao F, Cech TR. Triple-helix structure in telomerase RNA contributes to catalysis. Nat Struct Mol Biol. 2008;15:634–640. doi: 10.1038/nsmb.1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson JS, Schneider B, Murray LW, Kapral GJ, Immormino RM, Headd JJ, Richardson DC, Ham D, Hershkovits E, Williams LD, et al. RNA backbone: Consensus all-angle conformers and modular string nomenclature (an RNA Ontology Consortium contribution) RNA. 2008;14:465–481. doi: 10.1261/rna.657708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SantaLucia J., Jr A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc Natl Acad Sci. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saszar K, Spackova N, Stefl R, Sponer J, Leontis NB. Molecular dynamics of the frame-shifting pseudoknot from beet western yellow virus: The role of non-Watson–Crick base-pairing, ordered hydration, cation binding and base mutations on stability and unfolding. J Mol Biol. 2001;313:1073–1091. doi: 10.1006/jmbi.2001.5100. [DOI] [PubMed] [Google Scholar]
- Serra MJ, Turner DH. Predicting thermodynamic properties of RNA. Methods Enzymol. 1995;259:242–261. doi: 10.1016/0076-6879(95)59047-1. [DOI] [PubMed] [Google Scholar]
- Shapiro BA, Yingling YG, Kasprzak W, Bindewald E. Bridging the gap in RNA structure prediction. Curr Opin Struct Biol. 2007;17:157–165. doi: 10.1016/j.sbi.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Shefer K, Brown Y, Gorkovoy V, Nussbaum T, Ulyanov NB, Tzfati Y. A triple helix within a pseudoknot is a conserved and essential element of telomerase RNA. Mol Cell Biol. 2007;27:2130–2143. doi: 10.1128/MCB.01826-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto AM, Misra V, Draper DE. Tertiary structure of an RNA pseudoknot is stabilized by ‘diffuse’ Mg2+ ions. Biochemistry. 2007;46:2973–2983. doi: 10.1021/bi0616753. [DOI] [PubMed] [Google Scholar]
- Spitale RC, Torelli AT, Krucinska J, Bandarian V, Wedekind JE. The structural basis for recognition of the PreQ0 metabolite by an unusually small riboswitch aptamer domain. J Biol Chem. 2009;284:11012–11016. doi: 10.1074/jbc.C900024200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staple DW, Butcher SE. Pseudoknots: RNA structures with diverse functions. PLoS Biol. 2005;3:956–959. doi: 10.1371/journal.pbio.0030213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su L, Chen LQ, Egli M, Berger JM, Rich A. Minor groove RNA triplex in the crystal structure of a ribosomal frameshifting viral pseudoknot. Nat Struct Biol. 1999;6:285–292. doi: 10.1038/6722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Z-J, Chen S-J. Nucleic acid helix stability: Effects of salt concentration, cation valence and size, and chain length. Biophys J. 2006;90:1175–1190. doi: 10.1529/biophysj.105.070904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan RKZ, Petrov AS, Harvey SC. YUP: A molecular simulation program for coarse-grained and multiscaled models. J Chem Theory Comput. 2006;2:529–540. doi: 10.1021/ct050323r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theimer CA, Finger LD, Trantirek L, Feigon J. Mutations linked to dyskeratosis congenita cause changes in the structural equilibrium in telomerase RNA. Proc Natl Acad Sci. 2003;100:449–454. doi: 10.1073/pnas.242720799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theimer CA, Blois CA, Feigon J. Structure of the human telomerase RNA pseudoknot reveals conserved tertiary interactions essential for function. Mol Cell. 2005;17:671–682. doi: 10.1016/j.molcel.2005.01.017. [DOI] [PubMed] [Google Scholar]
- Thirumalai D, Woodson SA. Kinetics of folding of proteins and RNA. Acc Chem Res. 1996;29:433–439. [Google Scholar]
- Ulyanov NB, Shefer K, James TL, Tzfati Y. Pseudoknot structures with conserved base triples in telomerase RNAs of ciliates. Nucleic Acids Res. 2007;18:6150–6160. doi: 10.1093/nar/gkm660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadley LM, Keating KS, Duarte CM, Plye AM. Evaluating and learning from RNA pseudotorsional space: Quantitative validation of a reduced representation for RNA structure. J Mol Biol. 2007;372:942–957. doi: 10.1016/j.jmb.2007.06.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walberer BJ, Cheng AC, Frankel AD. Structural diversity and isomorphism of hydrogen-bonded base interactions in nucleic acids. J Mol Biol. 2003;327:767–780. doi: 10.1016/s0022-2836(03)00090-1. [DOI] [PubMed] [Google Scholar]
- Wang JM, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J Comput Chem. 2000;21:1049–1074. [Google Scholar]
- Wu M, Tinoco I., Jr RNA folding causes secondary structure rearrangement. Proc Natl Acad Sci. 1994;95:11555–11560. doi: 10.1073/pnas.95.20.11555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin Y, Laing C, Leontis NB, Schlick T. Annotation of tertiary interactions in RNA structures reveals variations and correlations. RNA. 2008;14:2465–2477. doi: 10.1261/rna.1249208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yingling YG, Shapiro BA. The prediction of the wild-type telomerase RNA pseudoknot structure and the pivotal role of the bulge in its formation. J Mol Graph Model. 2006;25:261–274. doi: 10.1016/j.jmgm.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Zhang WB, Chen S-J. Exploring the complex folding kinetics of RNA hairpins. 1. General folding kinetics analysis. Biophys J. 2006;90:765–777. doi: 10.1529/biophysj.105.062935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Lin M, Chen R, Wang W, Liang J. Discrete state model and accurate estimation of loop entropy of RNA secondary structures. J Chem Phys. 2008;128:125107. doi: 10.1063/1.2895050. [DOI] [PMC free article] [PubMed] [Google Scholar]