

Abstract
Defining the shape, conformation, or assembly state of an RNA in solution often requires multiple investigative tools ranging from nucleotide analog interference mapping to X-ray crystallography. A key addition to this toolbox is small-angle X-ray scattering (SAXS). SAXS provides direct structural information regarding the size, shape, and flexibility of the particle in solution and has proven powerful for analyses of RNA structures with minimal requirements for sample concentration and volumes. In principle, SAXS can provide reliable data on small and large RNA molecules. In practice, SAXS investigations of RNA samples can show inconsistencies that suggest limitations in the SAXS experimental analyses or problems with the samples. Here, we show through investigations on the SAM-I riboswitch, the Group I intron P4-P6 domain, 30S ribosomal subunit from Sulfolobus solfataricus (30S), brome mosaic virus tRNA-like structure (BMV TLS), Thermotoga maritima asd lysine riboswitch, the recombinant tRNAval, and yeast tRNAphe that many problems with SAXS experiments on RNA samples derive from heterogeneity of the folded RNA. Furthermore, we propose and test a general approach to reducing these sample limitations for accurate SAXS analyses of RNA. Together our method and results show that SAXS with synchrotron radiation has great potential to provide accurate RNA shapes, conformations, and assembly states in solution that inform RNA biological functions in fundamental ways.
Keywords: small-angle X-ray scattering (SAXS), RNA folding, modeling, misfolded
INTRODUCTION
For most noncoding functional RNAs, defining their respective shapes and conformations in solution is a critical step toward understanding their functional role in a biological assemblage. Defining this conformational space can be made difficult by intrinsic RNA flexibility, thus precluding many classical approaches to structural investigations. Nonetheless, alternative methods are available, such as small-angle X-ray scattering (SAXS), which can provide in-solution structural information of biological particle describing the size, shape, and compactness of the scattering macromolecules (Glatter and Kratky 1982). This in-solution structural technique is experiencing a revival largely due to overall improvements in data collection technologies and computational algorithms (for review, see Putnam et al. 2007). In the absence of any prior structural information, SAXS can provide the first structural insights into a biological macromolecule addressing the basic questions of oligomerization, foldedness, or flexibility of the particle (Tsutakawa et al. 2007; Hura et al. 2009). Unlike X-ray crystallography and nuclear magnetic resonance (NMR), SAXS is a technique performed under dilute conditions, thus requiring minimal amounts of sample, and has provided reliable data on particles ranging from lysozyme at 14 kDa to the 70 S ribosome at 2700 kDa (Chen et al. 1996; Svergun et al. 1997; Montelione et al. 2000). SAXS can be an invaluable tool for the structural biologist, supplementing the traditional high-resolution techniques mentioned above; yet, the method has limitations for RNA that merit attention.
Classically, SAXS has been used to determine the radius-of-gyration, Rg, and maximum dimension, dmax, of the scattering particle. Rg is a description of the particle's distribution of mass around its center of gravity. Therefore, changes in Rg often reflect a measured conformational change that may be due to a small molecule binding or perturbation of the thermodynamic state. In fact, time-resolved Mg2+-dependent changes in Rg have been used to follow folding of several large RNAs (Perez-Salas et al. 2004; Lipfert et al. 2007b). Furthermore, if the SAXS data are of high quality, they can be used to reconstruct ab initio models of the scattering particle (Chacon et al. 1998; Svergun et al. 2001; Franke and Svergun 2009). For example, Lipfert and colleagues have used rational dissection of the Varkud satellite (VS) ribozyme and SAXS to successfully build the solution model of the entire VS ribozyme (Lipfert et al. 2008). The final model was refined against the experimental SAXS profile. In this case, SAXS was used as a modeling constraint and will likely be used in the future as an experimental constraint for refining a model constructed from phylogenetic and high-resolution chemical probing methods (Le Quesne et al. 2001; Badorrek and Weeks 2006; Stormo 2006). In some cases, a high-resolution crystal structure may be available, and SAXS provides an opportunity to validate the overall shape of a crystal structure in solution, free of crystal packing forces or other solid state influences. Nonetheless, all of the above modeling approaches fundamentally require quality SAXS data on a defined RNA sample.
The production of RNA generally involves an in vitro transcription, purification by a denaturing process, concentration, and storage at a cryogenic temperature (<−20°C). Then, prior to any structural or biochemical investigation, the RNA is thermally annealed in the presence or absences of salts to produce the folded RNA. Often, minor populations of the annealed RNA sample will be misfolded, thereby contributing to a constant background noise in biochemical experiments (Uhlenbeck 1995). During crystallization, the misfolded RNA may isomerize to the correctly folded state or be selected against by the crystal due to the restrictive nature of the lattice. In contrast, SAXS data are collected over the entire ensemble of particles so any misfolded RNA will contribute directly to the observed X-ray scattering. The magnitude of the misfolded contribution is dependent on the mass, shape, and concentration of the misfolded RNA. For a misfolded dimeric species, its contribution to the observed X-ray scattering signal will scale as the square of the scattering mass, contributing an inherently fourfold greater signal than its respective monomeric form. Likewise, a misfolded monomeric species will have a fundamentally different hydrodynamic shape whose contribution to the observed SAXS signal will scale directly with concentration. Therefore, to achieve accuracy, SAXS data analyses and reconstructions must be made from homogenous samples or at the very least from well-defined samples. Here, we propose and test a general approach to reducing these sample limitations for accurate SAXS analyses of RNA. Our method demonstrates that SAXS can be greatly enhanced to provide accurate RNA shapes, conformations, and assembly states in solution.
RESULTS
Limitations to SAXS analysis due to refolding artifacts
Multiple folding pathways are available to an RNA during in vitro refolding (Treiber et al. 1998; Silverman et al. 2000). In some cases, a pathway may lead to a kinetically trapped non-native conformation, thus producing structural heterogeneities or refolding artifacts in the sample (Rook et al. 1999). This is observed for both the SAM-I riboswitch and the Δ(C209)P4-P6 domain of the group I intron (Treiber and Williamson 2001). Each RNA was purified to a single band on a denaturing gel and refolded as described (Juneau and Cech 1999; Kieft and Batey 2004; Montange and Batey 2006). Separation of the refolded RNA samples by analytical size-exclusion chromatography (SEC) clearly shows a conformational heterogeneity described by a single predominant peak preceded by a smaller peak that constituted 5–15% of the total signal (Fig. 1A,B). Light scattering analyses determined the molecular mass of the predominant peaks to correspond to the expected molecular mass for each monomeric species at 29.9 and 52.4 kDa for SAM-I and P4-P6 RNA, respectively (Table 1). It was not possible to determine the mass of either secondary peak due to the concentration requirements of the light scattering analysis. However, the stable partitioning, characteristic 260/280 nm absorbance ratio, and earlier elution times for both secondary peaks suggests that an alternate state of the RNA is present after refolding.
FIGURE 1.

Size exclusion chromatographic (SEC) analysis of SAM-I and P4-P6 domain. Gel-filtration elution profiles for folded RNAs. Vertical axis is the mAu at 280 nm. (A) SAM-I RNA before purification (broken green line) and after SAXS of the purified RNA (red line); (B) P4-P6 domain (solid green line). Arrows mark regions of heterogeneity.
TABLE 1.
Biophysical constants and experimental SAXS parameters

The effect of the sample heterogeneity on a SAXS data set is shown by analysis of the SAM-I RNA. SAM-I is a 94-nucleotide RNA aptamer capable of binding a small molecule, S-adenosylmethionine (SAM) (McDaniel et al. 2003; Winkler et al. 2003). X-ray crystallography of the bound SAM-I complex reveals that the ligand is deeply buried in the aptamer, suggesting a macromolecular conformational change may occur during binding (McDaniel et al. 2003; Montange and Batey 2006; Heppell and Lafontaine 2008). Ligand-dependent macromolecular conformational changes are robustly detected by SAXS by simply comparing the scattering profiles of the ligand-bound and ligand-free samples. Figure 2A illustrates the SAXS profiles of the refolded SAM-I riboswitch in the presence (SAM-plus) and absence (SAM-free) of SAM. Qualitatively, the addition of the ligand causes a sharper decay in the overall SAXS profile, suggesting compaction of the RNA in the presence of the ligand. Transforming the data via a Kratky plot [q2•I(q) versus q] (Fig. 2B) emphasizes the conformational differences, and for the SAM-plus sample, the Kratky plot converges faster to baseline than SAM-free, supporting the assertion that the SAM-plus RNA is more compact (Doniach 2001). Furthermore, any compaction of the RNA should demonstrate a comparable decrease in the Rg within the Guinier region (q•Rg < 1.3) of the data (Feigin and Svergun 1987). Yet, the Guinier Rg for the SAM-free RNA (31.1 Å) is smaller than that of the SAM-plus RNA (34.4 Å), an observation inconsistent with the above Kratky analysis. For the SAM-I RNA, the presence of the misfolded RNA during SAXS data collection resulted in a comparative inconsistency between the Guinier and Kratky analyses.
FIGURE 2.

SAXS analyses of refolded SAM-I and P4-P6 domain RNA. (A) Experimental SAXS curves of SAM-plus (red) and SAM-free (black) overlaid with the theoretical scattering curve (orange) calculated from X-ray crystal structure (PDB: 2GIS). The experimental curves were placed on a relative scale in PRIMUS, and the theoretical curve was manually adjusted to illustrate the overall disagreement with the experimental curves. The arrow marks a region of disagreement between the two scattering curves. (B) Kratky plots of the SAM-plus (red) and SAM-free (black) scattering data. (C) Crystallographic and ab initio SAXS models of SAM-I. The surface model (orange) is the ab initio model calculated using the SAM-plus data and represents an average of eight independent particle reconstructions using the program DAMMIF. The SAM-plus data were transformed using a dmax of 118 Å. The X-ray crystal structure (PDB: 2GIS) of SAM-I (orange) is shown as reference with its corresponding dmax. (D) Experimental SAXS curve of the P4-P6 domain (black circles) with the theoretical SAXS profile (orange) calculated from the X-ray crystal structure (PDB: 1L8V). The data were scaled to a common low-angle scattering intensity for purposes of an overlay using PRIMUS. (E) Kratky plot of the P4-P6 domain SAXS data. (F) X-ray crystal structure (orange ribbon or solvent-exposed surface) superimposed with the surface ab initio model (gray surface or wire mesh) calculated from the P4-P6 domain SAXS data, an average of eight independent DAMMIN/F runs using data transformed with a dmax of 178 Å. Crystallographic models were aligned with SUPCOMB20.
In addition, comparison of the SAM-plus SAXS profile with the theoretical scattering profile calculated from the X-ray crystal structure (Fig. 2A) reveals a significant disagreement between the putative in-solution and X-ray crystal structures. The χ2 for the fit of the X-ray structure to the SAXS curve is 5.3 and strongly suggests the shape of SAM-I in solution is fundamentally different from its X-ray crystal structure. Furthermore, the ab initio model calculated from the experimental SAM-plus SAXS data shows an elongated shape with a dmax (118 Å) that is almost twice the size of the X-ray crystal structure (63 Å) (Fig. 2C). These observations suggest that the SAXS experiment is inaccurate, the sample is corrupted, or the structure of SAM-I in solution is fundamentally different from its crystal form.
A similar disagreement between the solution scattering data and the X-ray crystal structure was observed with the (ΔC209)P4-P6 domain of the group I intron (Fig. 2D). The P4-P6 domain is a well-studied structured RNA known to fold into a compact structure in solution. The χ2 for the fit of the X-ray crystal structure (Protein Data Bank [PDB] no. 1L8V) to the experimental SAXS curve was 4.7. The Rg for the X-ray crystal structure (29.58 Å) was significantly smaller than that of the experimental Guinier Rg (32.2 ± 0.1 Å), suggesting the P4-P6 domain is less compact in solution. However, transforming the data in a Kratky plot (Fig. 2E) suggested the RNA is folded and not a random coil. The ab initio model based on the SAXS data is elongated and similar to a previously published ab initio reconstruction for the P4-P6 domain (Lipfert et al. 2007a); however, this model is inconsistent with the overall shape suggested by the X-ray crystal structure.
In each of these two representative cases, the derived ab initio SAXS models from the SAM-I and P4-P6 domain samples were inconsistent with the X-ray crystal structures. Our characterizations determined that these problems are characteristic of SAXS data collected from samples that contain a minor population of RNA in an alternate hydrodynamic state. Consequently, as we show below, significant improvements in SAXS data and interpretation can be robustly achieved by a SEC purification of the refolded RNA prior to data collection.
Improving SAXS data by SEC purification
SEC purification of an RNA sample prior to SAXS data collection offers two main advantages: First, the SEC profile itself assesses the sample heterogeneity; and second, the SEC purification ensures the RNA is in a defined buffer for recovering the SAXS curve. It is established that the purification of RNA via denaturing PAGE will contain contamination from acrylamide oligomers that will consequently interfere with the SAXS curve in the high q region (q >0.15 Å) (Lukavsky and Puglisi 2004). As shown in Figure 1, A and B, the SEC profiles are due to a folding protocol optimized for each respective RNA. We observed that the application of different folding protocols, i.e., snap-cooling versus slow-cooling and in the presence or absence of Mg2+, produced wildly different SEC profiles (data not shown). Nonetheless, the optimized folding protocol for each RNA tested above still resulted in a conformational heterogeneity that was stable to partitioning by SEC. We suggest that any RNA sample for SAXS be reduced to a single peak where the putative ensemble consists of a population of RNA that can freely interconvert. Such a population is characterized by a symmetric elution peak in a typical size-exclusion chromatogram.
To optimally achieve SEC separation of folded RNA samples for SAXS, we used a 2.4-mL gel-filtration column with sample volumes of 25–50 μL. The column is operated at 40 μL per minute. For a particular RNA in a specified buffer condition, an analytical scale SEC run is performed where a 25-μL annealed RNA sample is injected at 0.2–1 mg/mL. This preliminary run is necessary as it defines the sample complexity, elution time of the peak of interest, and approximate absorbance at 260 nm. In our laboratory, a multi-angle light scattering (MALS) detector attached to the SEC run allows for a direct determination of the absolute molecular weight of the eluting peak. In this case, RNA samples should be injected at 2–5 mg/mL to ensure a reliable light scattering signal. More importantly, the integration of a MALS instrument in the purification pipeline is necessary for RNA samples as suitable mass standards for accurate mass determination by I(0) measurements are not readily available commercially.
Following the analytical run, a 50-μL preparative scale purification is performed at ∼10 mg/mL of RNA. For the SAM-I and P4-P6 RNAs, the refolding at the higher concentration did not produce any additional artifacts; however, it may be necessary to refold the RNA under dilute conditions and concentrated to the requisite 50-μL volume using a spin-concentrator (10,000 MWCO Sartorius Vivaspin 500). During the separation, a 60–100-μL fraction (2–3 drops) corresponding to the predominant peak identified from the analytical run is taken for SAXS analysis. This micropurification scheme generally results in an approximate fivefold dilution of the sample. Most importantly, a buffer sample (blank) must be collected after the SEC run for dilution of the purified RNA sample and buffer subtraction of the collected SAXS data. Notably, a size-exclusion column will release resin particles during the initial pressurization of the system, and any buffer taken for X-ray scattering should be taken from a SEC column that has been running for at least one column volume to avoid resin contamination.
For the SAM-I and P4-P6 domain samples, purification of the RNAs prior to SAXS analysis resulted in a marked improvement in their respective experimental SAXS profiles (Fig. 3A,B). In the case of the SAM-I RNA, the RNA was refolded and purified in the presence of 100 μM SAM. The purification improves the χ2 of the X-ray crystal structure fit to the experimental SAXS data significantly from 5.3 to 1.6 (Fig. 3B). Likewise, for the P4-P6 domain, the fit of the experimental SAXS data to the crystal structure improves from 4.7 to 2.3 (Fig. 3A). These improvements highlight the high sensitivity of SAXS to the presence of misfolded or unaccounted for particles. The micropurification system requires minimal volume for several analytical runs and system equilibration, a total of ∼25 mL of buffer. Thus, it is possible to purify RNA samples in the presence of small molecules such as SAM without extraordinary material costs.
FIGURE 3.

SAXS data and models from SEC purified refolded SAM-I and P4-P6 domain. Experimental SAXS profiles of purified SAM-plus (A) and P4-P6 domain (B). In both cases, the orange curve represents the theoretical SAXS profile calculated from X-ray crystal structures of SAM-I (PDB: 2GIS) and P4-P6 domain (PDB: 1L8V). DAMMIN/F ab initio models in two orientations calculated from the SEC purified SAXS data SAM-I RNA (C) and P4-P6 domain (D). Orange backbone models represent the X-ray crystal structures for SAM-I and P4-P6 domain superposed with the program SUPCOMB.
In addition, the SAXS measurements on SAM-free RNA purified in the absence of SAM provides a clearer picture regarding the unliganded state of SAM-I. Comparatively, the data between the SAM-free and SAM-plus states and their respective transformations in the Kratky and P(r) plots are now consistent with a ligand-induced conformational change (data not shown). In the presence of SAM, the Guinier Rg and dmax decrease from 24.2 to 22.4 Å and from 81 to 76 Å, respectively (Table 1). The SEC purification helps to precisely define the sample such that the SAXS data are rendered more reliable and useful toward structural interpretation.
SEC profile and SAXS data on additional representative RNAs
We expected that the SEC separation prior to SAXS may be applicable to RNAs of widely differing sizes (Table 1). To test this, we therefore applied the chromatographic separation technique to the 30S ribosomal subunit from Sulfolobus solfataricus (30S), the brome mosaic virus tRNA-like structure (BMV TLS), the Thermotoga maritima asd lysine riboswitch (Lys), recombinant tRNAval, and yeast tRNAphe.
In all cases, the native or folded samples contained significant heterogeneity as judged by their respective SEC profiles (Fig. 4A). The heterogeneity constituted 5, 11, 30, and 50% of the signal for the 30S, BMV TLS, Lys, and tRNAs samples, respectively. Any SAXS data collected directly from these samples would be of a mixed population, thereby confounding structural analyses and any further structural modeling. Therefore, samples for SAXS were taken from fractions corresponding to the top of the main peaks (Fig. 4B–E).
FIGURE 4.

SEC profile and SAXS curves of the 30S subunit, BMV TLS, lysine riboswitch, and tRNAphe. (A) Elution profile of studied RNAs. The signal for the 30S subunit (red) was measured using a refractive index detector and scaled for the figure. Each elution profile was shifted for clarification by 0.3, 0.6, 0.9, and 1.2 mL for BMV TLS (BMV) (green), lysine riboswitch (Lys) (cyan), tRNAVal (Val) (gray), and tRNAphe (Phe) (orange), respectively. SAXS profiles for 30S subunit (B), BMV TLS RNA (C), lysine riboswitch (D) in the presence (cyan) and absence (gray) of lysine, and tRNAphe (orange) and tRNAval (gray) (E). Data for tRNAval were collected with a larger beamstop mask truncating the data at lower scattering angles.
The lysine riboswitch is a metabolite sensing element known to bind lysine (Sudarsan et al. 2003); therefore, two separate SEC purifications were performed in the presence (+) and absence (−) of lysine (Garst et al. 2008). The corresponding SAXS profiles (Fig. 4D) parallel each other in the low q region (q <0.15 Å), indicating that the riboswitch does not undergo significant global changes in solution upon metabolite binding. In fact, the transformed P(r) functions superimpose (Garst et al. 2008).
Interestingly, tRNAval had the highest amount of heterogeneity that preceded the main elution peak. The tRNA was purified to a single band on a denaturing gel. However, the SEC profile suggests the refolding procedure of the tRNA created many misfolded species of varying sizes. The molecular mass of the main peak was determined to be 24.5 kDa ± 10% by MALS (Table 1). Likewise, tRNAphe broadened on the latter side of the main peak, suggesting additional species are present with smaller hydrodynamic properties or mass. The SAXS data are similar and parallel each other through most of the scattering angles, suggesting the two tRNAs share structural features in solution (Fig. 4E).
Ab initio modeling
For a homogeneous particle in solution, the SAXS profile is determined primarily by the shape of the particle (Svergun and Stuhrmann 1970a). There are multiple effective algorithms available for reconstructing shapes from SAXS data, notably DAMMIN/F, GASBOR, and DALAI_GA (Chacon et al. 1998; Svergun 1999; Svergun et al. 2001). The success in generating a set of useable models from SAXS data will ultimately rely on the quality of the data collected and the state of the macromolecule in solution, i.e., is it fully folded? To address the foldedness question, the SAXS data can be transformed into a Kratky plot (see Materials and Methods), where a well-folded homogenous particle will demonstrate a parabolic curve and an unfolded particle will demonstrate a hyperbolic curve.
For the purified SAM-I, P4-P6 domain, 30S, BMV TLS, Lys, and tRNAphe samples, Kratky plots suggested the samples were well-folded RNAs (data not shown). Therefore, ab initio reconstructions were performed with the program DAMMIN/F (Svergun et al. 2001). For each data set, a total of eight reconstructions were performed and averaged together to generate the final model (Figs. 3C,D, 5). The 30S ribosomal particle is a large mega-Da ribonucleoprotein complex with a dmax of 231 Å. The reconstruction (Fig. 5A) shows a particle that is asymmetric with two lobes; its overall shape is consistent with the known X-ray crystal structure (PDB: 1FJG) from Thermus thermophilus (Carter et al. 2000). The BMV TLS RNA is a structured RNA that lacks a previously determined X-ray crystal structure (Hammond et al. 2009). Nonetheless, the SAXS ab initio reconstructions reveal a shape with unique features that are largely dictated by a 22 Å measure (Fig. 5B). Importantly, this measure is roughly the width of an RNA helix, and at the resolution of SAXS, a helix is an easily discernable secondary structural element found in folded RNAs. Such helical features are well demonstrated in the SAXS Lys and tRNAphe reconstructions for which there are known X-ray crystal structures (Fig. 5C,D). The lysine riboswitch folds into a structure with three helical regions tightly packed into a core. As evidenced by the superposition of the X-ray crystal structure (PDB: 3D0U) with the ab initio bead model (Fig. 5D), the helical elements forming the core and extension are easily discernable.
FIGURE 5.

ab initio models of the 30S subunit, BMV TLS, lysine riboswitch, and tRNAphe. (A) S. solfataricus 30S ribosomal subunit; (B) BMV TLS RNA; (C) bound lysine riboswitch; (D) tRNAphe. All ab initio models were created with DAMMIF and averaged from eight independent runs with DAMAVER using SAXS data collected from. SEC purified samples. Two contoured surfaces are used for models where no prior PDB entry is available. For A and B, the two surfaces represented the model at relative contours of 0.5 (wireframe) and 1 (solid). For C and D, the bead models (white) are aligned with the respective crystal structures (orange).
DISCUSSION
Here, we examined SAXS results on RNA samples covering a wide range of sizes, shapes, and particle composition (protein/RNA). For SAXS and NMR analyses of samples in solution, data can be collected from every sample, so guidelines for assessing sample and data quality are important (Lukavsky and Puglisi 2004). In the absence of suitable RNA standards, we showed that MALS experiments provide key independent assessments of sample mass and homogeneity. For each set of data comprising the concentration series, consistency among Rg, dmax, and the Kratky analysis provided a useful internal check of data quality.
Conventional methods for preparing folded RNA samples, i.e., snap- or slow-cooling, are prone to producing heterogeneities of varying degrees (Figs. 1, 4A). The consequence of these heterogeneities will largely limit the scope of the SAXS experiment unless they are appropriately addressed as shown here. Experiments with SAM-I showed that the presence of the heterogeneities grossly distorted the low scattering angle region (q <0.1 Å) such that changes in Rg were opposite of a ligand-induced compaction of the RNA (Fig. 2). In this circumstance, the utility of SAXS is limited to making a qualitative statement that a conformational change is occurring in the presence of ligand. Any further assessment regarding the pair-distribution function, Rg, or modeling is rendered unreliable by the misfolded species.
Importantly, restoring the quantitative utility of SAXS was readily achieved by focusing on the purification of the SAXS sample. The SAXS data for the SAM-I RNA improved where the observed changes to the Rg, P(r) distribution, and Kratky plot were consistent between the SAM-free and SAM-plus measurements. The benefits of a hydrodynamic purification were further strengthened by SAXS experiments on the P4-P6 domain. Here, the ab initio modeling became more consistent with the global shape of the RNA (Fig. 3D), arguing the possibility that these ab initio bead models could provide reliable scaffolds for model building. Considering that advances in chemical probing techniques of RNA identifies helical and flexible regions in a secondary structure map, SAXS can provide a powerful tool in constraining models built in the absence of atomic level information (Grishaev et al. 2008). These models should be consistent with the respective secondary structures and provide specific models to evaluate experimentally. Such experimentally tested models promise to provide useful and reliable frameworks for subsequent biochemical and biological investigations. RNA antibodies have proven valuable tools and can aid crystallization and structural studies (Ye et al. 2008). As the reactivity of antipeptide antibodies with the intact protein is a function of the mobility of protein regions (Tainer et al. 1984), SAXS analyses methods that distinguish solution flexibility from sample heterogeneity, as presented here, may prove useful for choosing RNA regions for making antibodies. Our method demonstrates that the direct application of a size-exclusion column to an RNA sample is an effective method for making high-quality SAXS samples for structural interpretation, inferring dynamic assemblies, and studying conformational states in solution.
MATERIALS AND METHODS
RNA preparation
The lysine riboswitch RNA sample and plasmids encoding the SAM-1 riboswitch and P4-P6 domain were kindly provided by Dr. Robert T. Batey (University of Colorado at Boulder). The BMV RNA sample was kindly provided by Dr. Jeff Kieft (University of Colorado Health Sciences Center at Aurora). One milligram of tRNAphe (Sigma-Aldrich) was resuspended in 200 μL of RNA storage buffer containing 10 mM HEPES at pH 6.5 and 0.2 mM ethylenediaminetetraacetic acid (EDTA).
The SAM-I and (ΔC208)P4-P6 domain RNA samples were prepared from in vitro T7 polymerase transcriptions reactions using PCR-generated DNA templates. The crude RNA was purified by 10%–12% denaturing gel electrophoresis as described (Doudna 1997). RNA samples were eluted from crushed gel slices overnight at 4°C into RNA storage buffer as described above.
The Sulfolobus solfataricus 30S ribosomal subunit was kindly provided by Steve Yannone at the Lawrence Berkeley National Laboratory and purified as described (RP Rambo, K Frankel, JA Tainer, and SM Yannone, in prep.). SEC purifications were performed in native buffer containing 40 mM imidazole at pH 5.4, 75 mM KCl, 75 mM NaCl, 5 mM MgCl2, 2 mM [tris(2-carboxyethyl)phosphine] (Sigma-Aldrich), and 0.2% NaAzide.
Size-exclusion chromatography
Chromatographic separations were performed with the Ettan LC liquid chromatography system (GE Healthcare) configured with a 3-wavelength ultraviolet-visual (UV-VIS) detector. Purifications were performed with either a Superose 6 (for 30S, BMV, and lysine riboswitch), Superdex 75 (tRNA-phe and val), or Superdex 200 (SAM riboswitch and P4P6 domain) PC 3.2/2.4 mL column (GE Healthcare). BMV TLS, SAM-I, P4-P6 domain RNAs were purified in running buffer containing 20 mM MOPS at pH 6.5, 50 mM KCl, and 7.6 mM MgCl2. An additional purification of SAM-I was performed where the running buffer was supplemented with 100 μM SAM (Sigma-Aldrich). The lysine riboswitch was purified in buffer containing 20 mM HEPES at pH 6.5, 50 mM KCl, and either 5 mM MgCl2, ±2 mM lysine, or 2 mM EDTA. Flowthrough from each of the respective buffers was kept for buffer subtraction during SAXS analysis. The SAM-I and P4-P6 domain RNA purifications and experiments were performed as five independent sets of experiments. The lysine riboswitch and BMV RNA were purified through three independent trials.
Multi-angle laser light scattering
MALS experiments were performed using an 18-angle DAWN HELEOS light scattering detector connected in tandem to an Optilab refractive index concentration detector (Wyatt Technology). Detector 12 of the DAWN HELEOS was replaced with a DynaPro quasielastic light scattering (QELS) detector (Wyatt Technology). System normalization and calibration was performed with BSA using a 50-μL sample at 10 mg/mL in SEC running buffer and a dn/dc value of 0.18–0.185. The light scattering experiments were used to perform analytical scale chromatographic separations for mass determination of the principle peaks in the SEC analysis. A refractive index of 0.17 (Rambo and Doudna 2004) and 0.178 was used for all RNAs and the 30S subunit, respectively. The MALS data in coordination with the QELS results were used to monitor the monodispersity throughout the SEC profile.
Small-angle X-ray scattering
SAXS experiments were performed at beamline 12.3.1 of the Advanced Light Source, Lawrence Berkeley National Laboratory. Twenty microliters of purified RNA samples and corresponding matching buffers were loaded into a 96-well plate (Nunc) and covered with protective film. RNA samples were collected at 2–3 mg/mL and diluted serial in the plate. Automated loading of the SAXS samples into the sample cuvette was achieved using a Hamilton syringe robot as described (Hura et al. 2009). For each sample, several exposures were taken using the following time sequence of 5, 50, and 5 sec or 6, 60, and 6 sec. All data collections were performed at room temperature. The first and last exposures of each sequence were compared to assess for radiation damage. Integration, scaling, and buffer subtraction were accomplished using the program Ogre (Greg Hura, Lawrence Berkeley National Laboratory). The scattering angle, q, was calculated as 4 π sin(θ/2)/λ where θ/2 is the scattering angle measured from the Bragg plane and λ is the wavelength in Å. A fixed detector distance was maintained at 1.486 m and with a wavelength of 1.03 Å (12,000 eV), and data were collected over the q range of 0.0088–0.322 Å−1.
The merging of the short and long exposures was achieved using the program PRIMUS (Konarev et al. 2003). Overlays of the merged data sets were used to detect concentration dependent scattering in the lowest q region. Indirect Fourier transforms of the SAXS scattering curve was performed with Gnom (Svergun 1992).
For well-folded particles, the SAXS profiles with the lowest noise and free of interparticle interference were subsequently used for ab initio modeling with the program DAMMIN/F. The determination of a well-folded particle was made using a Kratky plot. The Kratky plot is based on a derivation by Porod (Glatter and Kratky 1982), who noted that the integral of the scattered intensity, I(q), times the square of the scattering angle, q, approaches a constant value, Q:
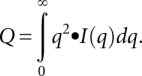 |
Stated simply, the area under the curve in a Kratky plot [q2•I(q) versus q] should approach a constant value. This is easily visualized in the plot by recognizing that a parabolic curve will capture an area whereas a hyperbolic curve does not. Empirically, folded proteins and RNAs demonstrate similar parabolic features in a Kratky plot. For each refined and merged SAXS curve, multiple independent DAMMIN/F runs were performed, superimposed, and averaged with the program DAMAVER (Volkov and Svergun 2003).
Where available, calculated biophysical parameters for each RNA were calculated from X-ray crystal structures 2GIS, 1L8V, 3D0U, 1EVV, and 1FJG for SAM-I, P4-P6 domain, lysine riboswitch, tRNAphe, and 30S ribosomal subunit as well as theoretical scattering curves using CRYSOL (Svergun et al. 1995). Molecular weights were calculated from RNA primary sequence using http://www.basic.northwestern.edu/biotools/oligocalc.html (Kibbe 2007). Graphs were prepared with Kaliedegraph. Surface models were prepared with pdb2vol from SITUS (Wriggers et al. 1999). Figures with structural models were oriented with VMD and rendered with Povray.
SAXS data and models are deposited in the SAXS biologically integrated structures in solution (Bioisis) database (http://www.bioisis.net).
ACKNOWLEDGMENTS
We acknowledge Robert Batey and Jeff Kieft for providing RNA samples and Steve Yannone for the 30S ribosomal subunit from Sulfolobus solfataricus. We thank John Hammond, Robert Batey, Jeff Kieft, Gareth Williams, Greg Hura, Michal Hammel, Ken Frankel, Kevin Dyer, Jane Tanamachi, and Elisa Lutzer for insightful discussions. Support for this work and data collection at the Lawrence Berkeley National Laboratory SIBYLS beamline of the Advanced Light Source came from the DOE program Integrated Diffraction Analysis Technologies (IDAT) under Contract DE-AC02-05CH11231 with the U.S. Department of Energy.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.1946310.
REFERENCES
- Badorrek CS, Weeks KM. Architecture of a γ retroviral genomic RNA dimer. Biochemistry. 2006;45:12664–12672. doi: 10.1021/bi060521k. [DOI] [PubMed] [Google Scholar]
- Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature. 2000;407:340–348. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- Chacon P, Moran F, Diaz JF, Pantos E, Andreu JM. Low-resolution structures of proteins in solution retrieved from X-ray scattering with a genetic algorithm. Biophys J. 1998;74:2760–2775. doi: 10.1016/S0006-3495(98)77984-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Hodgson KO, Doniach S. A lysozyme folding intermediate revealed by solution X-ray scattering. J Mol Biol. 1996;261:658–671. doi: 10.1006/jmbi.1996.0491. [DOI] [PubMed] [Google Scholar]
- Doniach S. Changes in biomolecular conformation seen by small-angle X-ray scattering. Chem Rev. 2001;101:1763–1778. doi: 10.1021/cr990071k. [DOI] [PubMed] [Google Scholar]
- Doudna JA. Preparation of homogeneous ribozyme RNA for crystallization. Methods Mol Biol. 1997;74:365–370. doi: 10.1385/0-89603-389-9:365. [DOI] [PubMed] [Google Scholar]
- Feigin LA, Svergun DI. In: Structure analysis by small-angle x-ray and neutron scattering. Taylor GW, editor. Plenum; New York: 1987. [Google Scholar]
- Franke D, Svergun DI. DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J Appl Crystallogr. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garst AD, Heroux A, Rambo RP, Batey RT. Crystal structure of the lysine riboswitch regulatory mRNA element. J Biol Chem. 2008;283:22347–22351. doi: 10.1074/jbc.C800120200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatter O, Kratky O. Small-angle x-ray scattering. Academic; London, UK: 1982. [DOI] [PubMed] [Google Scholar]
- Grishaev A, Ying J, Marella CD, Pardi A, Bax A. Solution structure of tRNAVal from refinement of homology model against residual dipolar coupling and SAXS data. J Biomol NMR. 2008;42:99–109. doi: 10.1007/s10858-008-9267-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond JA, Rambo RP, Filbin ME, Kieft JS. Comparison and functional implications of the 3D architectures of viral tRNA-like structures. RNA. 2009;15:294–307. doi: 10.1261/rna.1360709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppell B, Lafontaine DA. Folding of the SAM aptamer is determined by the formation of a K-turn-dependent pseudoknot. Biochemistry. 2008;47:1490–1499. doi: 10.1021/bi701164y. [DOI] [PubMed] [Google Scholar]
- Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, 2nd, Tsutakawa SE, Jenney FE, Jr, Classen S, Frankel KA, Hopkins RC, et al. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6:606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juneau K, Cech TR. In vitro selection of RNAs with increased tertiary structure stability. RNA. 1999;5:1119–1129. doi: 10.1017/s135583829999074x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibbe WA. OligoCalc: An online oligonucleotide properties calculator. Nucleic Acids Res. 2007;35(Web Server issue):W43–46. doi: 10.1093/nar/gkm234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieft JS, Batey RT. A general method for rapid and nondenaturing purification of RNAs. RNA. 2004;10:988–995. doi: 10.1261/rna.7040604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: A Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr. 2003;36:1277–1282. [Google Scholar]
- Le Quesne JP, Stoneley M, Fraser GA, Willis AE. Derivation of a structural model for the c-myc IRES. J Mol Biol. 2001;310:111–126. doi: 10.1006/jmbi.2001.4745. [DOI] [PubMed] [Google Scholar]
- Lipfert J, Chu VB, Bai Y, Herschlag D, Doniach S. Low-resolution models for nucleic acids from small-angle X-ray scattering with applications to electrostatic modeling. J Appl Crystallogr. 2007a;40:S229, S234. doi: 10.1107/S0021889807001707. [DOI] [Google Scholar]
- Lipfert J, Das R, Chu VB, Kudaravalli M, Boyd N, Herschlag D, Doniach S. Structural transitions and thermodynamics of a glycine-dependent riboswitch from Vibrio cholerae. J Mol Biol. 2007b;365:1393–1406. doi: 10.1016/j.jmb.2006.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipfert J, Ouellet J, Norman DG, Doniach S, Lilley DM. The complete VS ribozyme in solution studied by small-angle X-ray scattering. Structure. 2008;16:1357–1367. doi: 10.1016/j.str.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukavsky PJ, Puglisi JD. Large-scale preparation and purification of polyacrylamide-free RNA oligonucleotides. RNA. 2004;10:889–893. doi: 10.1261/rna.5264804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel BA, Grundy FJ, Artsimovitch I, Henkin TM. Transcription termination control of the S box system: Direct measurement of S-adenosylmethionine by the leader RNA. Proc Natl Acad Sci. 2003;100:3083–3088. doi: 10.1073/pnas.0630422100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montange RK, Batey RT. Structure of the S-adenosylmethionine riboswitch regulatory mRNA element. Nature. 2006;441:1172–1175. doi: 10.1038/nature04819. [DOI] [PubMed] [Google Scholar]
- Montelione GT, Zheng D, Huang YJ, Gunsalus KC, Szyperski T. Protein NMR spectroscopy in structural genomics. Nat Struct Biol. 2000;7(Suppl):982–985. doi: 10.1038/80768. [DOI] [PubMed] [Google Scholar]
- Perez-Salas UA, Rangan P, Krueger S, Briber RM, Thirumalai D, Woodson SA. Compaction of a bacterial group I ribozyme coincides with the assembly of core helices. Biochemistry. 2004;43:1746–1753. doi: 10.1021/bi035642o. [DOI] [PubMed] [Google Scholar]
- Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: Defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- Rambo RP, Doudna JA. Assembly of an active group II intron-maturase complex by protein dimerization. Biochemistry. 2004;43:6486–6497. doi: 10.1021/bi049912u. [DOI] [PubMed] [Google Scholar]
- Rook MS, Treiber DK, Williamson JR. An optimal Mg2+ concentration for kinetic folding of the Tetrahymena ribozyme. Proc Natl Acad Sci. 1999;96:12471–12476. doi: 10.1073/pnas.96.22.12471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman SK, Deras ML, Woodson SA, Scaringe SA, Cech TR. Multiple folding pathways for the P4-P6 RNA domain. Biochemistry. 2000;39:12465–12475. doi: 10.1021/bi000828y. [DOI] [PubMed] [Google Scholar]
- Stormo GD. Current protocols in bioinformatics. Chap.12, Unit 1. Wiley; New York: 2006. An overview of RNA structure prediction and applications to RNA gene prediction and RNAi design; pp. 1–3. [DOI] [PubMed] [Google Scholar]
- Sudarsan N, Wickiser JK, Nakamura S, Ebert MS, Breaker RR. An mRNA structure in bacteria that controls gene expression by binding lysine. Genes & Dev. 2003;17:2688–2697. doi: 10.1101/gad.1140003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr. 1992;25:495–503. [Google Scholar]
- Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun DI, Stuhrmann HB. New developments in direct shape determination from small-angle scattering. 1. Theory and model calculations. Acta Crystallogr A. 1970a;47:736–744. [Google Scholar]
- Svergun D, Barberato C, Koch MHJ. CRYSOL—a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr. 1995;28:768–773. [Google Scholar]
- Svergun DI, Burkhardt N, Pedersen JS, Koch MH, Volkov VV, Kozin MB, Meerwink W, Stuhrmann HB, Diedrich G, Nierhaus KH. Solution scattering structural analysis of the 70 S Escherichia coli ribosome by contrast variation. II. A model of the ribosome and its RNA at 3.5 nm resolution. J Mol Biol. 1997;271:602–618. doi: 10.1006/jmbi.1997.1191. [DOI] [PubMed] [Google Scholar]
- Svergun DI, Petoukhov MV, Koch MH. Determination of domain structure of proteins from X-ray solution scattering. Biophys J. 2001;80:2946–2953. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tainer JA, Getzoff ED, Alexander H, Houghten RA, Olson AJ, Lerner RA, Hendrickson WA. The reactivity of anti-peptide antibodies is a function of the atomic mobility of sites in a protein. Nature. 1984;312:127–134. doi: 10.1038/312127a0. [DOI] [PubMed] [Google Scholar]
- Treiber DK, Williamson JR. Beyond kinetic traps in RNA folding. Curr Opin Struct Biol. 2001;11:309–314. doi: 10.1016/s0959-440x(00)00206-2. [DOI] [PubMed] [Google Scholar]
- Treiber DK, Rook MS, Zarrinkar PP, Williamson JR. Kinetic intermediates trapped by native interactions in RNA folding. Science. 1998;279:1943–1946. doi: 10.1126/science.279.5358.1943. [DOI] [PubMed] [Google Scholar]
- Tsutakawa SE, Hura GL, Frankel KA, Cooper PK, Tainer JA. Structural analysis of flexible proteins in solution by small-angle X-ray scattering combined with crystallography. J Struct Biol. 2007;158:214–223. doi: 10.1016/j.jsb.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Uhlenbeck OC. Keeping RNA happy. RNA. 1995;1:4–6. [PMC free article] [PubMed] [Google Scholar]
- Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler WC, Nahvi A, Sudarsan N, Barrick JE, Breaker RR. An mRNA structure that controls gene expression by binding S-adenosylmethionine. Nat Struct Biol. 2003;10:701–707. doi: 10.1038/nsb967. [DOI] [PubMed] [Google Scholar]
- Wriggers W, Milligan RA, McCammon JA. Situs: A package for docking crystal structures into low-resolution maps from electron microscopy. J Struct Biol. 1999;125:185–195. doi: 10.1006/jsbi.1998.4080. [DOI] [PubMed] [Google Scholar]
- Ye JD, Tereshko V, Frederiksen JK, Koide A, Fellouse FA, Sidhu SS, Koide S, Kossiakoff AA, Piccirilli JA. Synthetic antibodies for specific recognition and crystallization of structured RNA. Proc Natl Acad Sci. 2008;105:82–87. doi: 10.1073/pnas.0709082105. [DOI] [PMC free article] [PubMed] [Google Scholar]