

Abstract
Human c-Rel (REL) is a member of the NF-κB family of transcription factors, and one of its primary physiological roles is in the regulation of B-cell proliferation and survival. Although REL is primarily regulated by cytoplasmic-nuclear translocation through interaction with IκB inhibitors, REL also undergoes several posttranslational modifications that have been proposed to modulate its transcriptional activation activity. For example, phosphorylation of C-terminal sequences of REL has been proposed to increase its transactivation activity. In this report, we have used immune complex kinase assays to identify Ser484 and Ser494 as the primary sites of IKKα- and IKKβ-mediated in vitro phosphorylation in the C-terminal transactivation domain of REL. However, in cotransfection studies in A293 cells we have failed to detect IKKβ-mediated phosphorylation of these sites on REL in vivo, nor does IKKβ appear to interact with REL in these cells. Ser-to-Ala mutation of Ser484 and Ser494 does not affect IKK’s ability to enhance GAL4-REL transactivation in reporter gene assays in A293 cells. We also show that the previously reported effects of overexpressed IKK and tumor necrosis factor treatment on GAL4-REL transactivation are due to IKK-mediated activation of the endogenous NF-κB pathway, which increases transcription from κB sites in the promoter of a commonly used GAL4 expression vector. Taken together, these results do not support a role for IKK-mediated phosphorylation as means for regulating the activity of REL in vivo.
Key words: c-Rel, Phosphorylation, IKK, Transactivation, GAL4 reporter assay, NF-κB
INTRODUCTION
The human c-rel proto-oncogene (REL) encodes a transcription factor that is a member of the NF-κB family of proteins, which also includes p65/RelA, RelB, p52/p100, and p50/p105 (3). The expression of c-rel is important for normal and malignant B-cell proliferation and survival. c-rel knockout mice develop normally, but these mice have immune defects because their B cells cannot proliferate in response to mitogenic stimulation. Moreover, the REL gene is amplified in several types of human B-cell lymphoma, including Hodgkin’s lymphomas and diffuse large B-cell lymphomas (3).
REL is a 587 amino acid (aa) polypeptide that can bind DNA as a homodimer or as a dimer with other NF-κB family members (3). The N-terminal approximately 300 aa of REL largely comprise a conserved domain called the Rel homology domain (RHD), which mediates dimerization, DNA binding, nuclear localization, and binding to its inhibitor, IκB. The C-terminal half of REL (aa 296–587) contains an inhibitory linker region (10) followed by a transactivation domain that contains two subdomains: subdomain I (aa 422–497) and subdomain II (aa 518–587) (14,21) (Fig. 1A).
Figure 1.
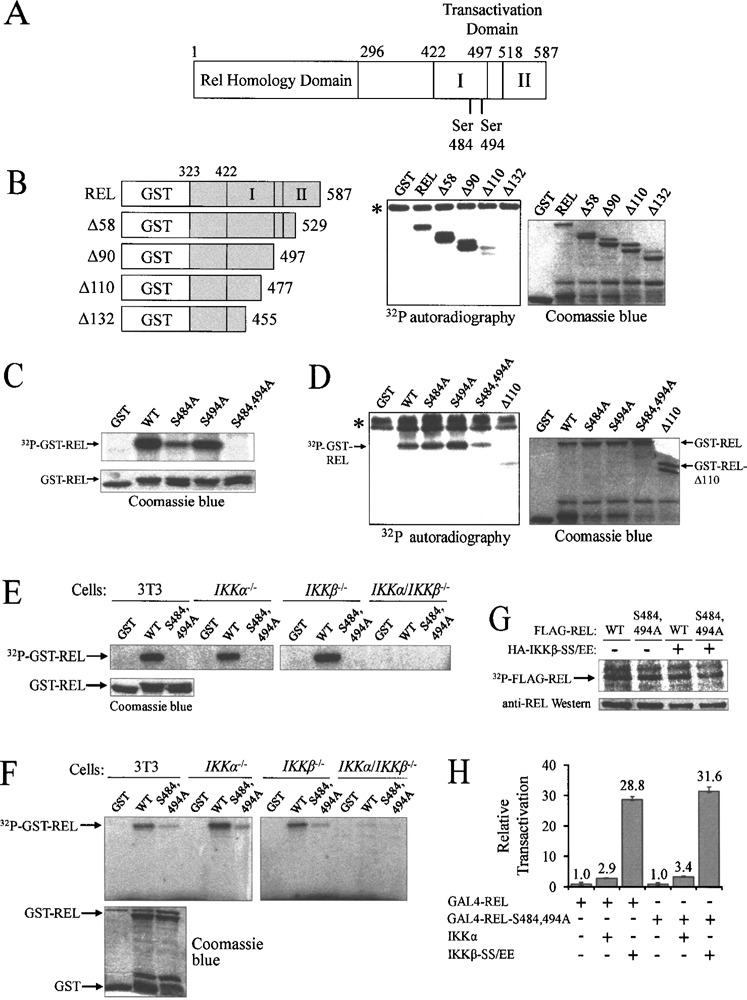
IKKβ in vitro phosphorylation sites on REL are not required for IKKβ-enhanced transactivation by GAL4-REL. (A) Schematic of the REL protein showing the location of IKKβ phosphorylation sites in the C-terminal domain. Transactivation subdomains I and II are indicated. (B) FLAG-IKKβ protein was immunoprecipitated from transfected A293 cells and immune complex kinase assays were performed with GST alone and each of the depicted GST-REL deletion mutants (left panel). Coomassie blue staining shows protein loading (right panel). Asterisk (*) denotes IKKβ autophosphorylation. (C) FLAG-IKKβ immune complex kinase assays were performed with GST and GST-REL aa 476–504 with the indicated point mutants (upper panel). Coomassie blue staining shows protein loading (lower panel). (D) FLAG-IKKβ immune complex kinase assays were performed with GST and GST-REL aa 323–587 with the indicated point mutants (left panel). Coomassie blue staining shows protein loading (right panel). Asterisk (*) denotes IKKβ autophosphorylation. (E and F) Anti-NEMO complexes were immunopreciptated from the indicated mouse fibroblast cell lines and were used in immune complex kinase assays with GST GST-REL and GST-REL-S484 494A: (E) GST-REL aa 476–504; (F) GST-REL aa 323–587 (upper panels). Coomassie blue staining shows protein loading (lower panels). (G) Cells were transfected with 1 5 μg of FLAG-REL or FLAG-REL-S484,494A and 1.5 μg of pcDNA or HA-IKKβ-SS/EE. Cells were incubated for 48 h and were then radiolabeled with [32P]orthophosphate for 3 h prior to lysis. FLAG-REL was detected by phosphorimaging (upper panel) and by an anti-REL Western blotting of the same filter (lower panel). The positions of the FLAG-REL protein are indicated. (H) Cells were transfected with 100 ng of pSG-REL or pSG-REL-S484 494A GAL4-fusion expression vector 0.5 pcDNA, IKKα, or IKKβ-SS/EE, and 0.5 μg of GAL4-luciferase plasmid. Cells were incubated for 48 h, and luciferase and β-galactosidase activites were then determined and values normalized to GAL4-REL plus the pcDNA vector (10) GAL4-REL-S484 494A plus IKK values were normalized to GAL4-REL-S484 494A alone (10) In all cases values are the averages of three experiments performed with triplicate samples
NF-κB proteins are primarily regulated at the level of cytoplasmic-nuclear localization through controlled phosphorylation and degradation of the interacting IκB inhibitors; however, the activity of the NF-κB proteins is fine-tuned through several regulatory posttranslational modifications (15). For example, the IκB kinases (IKK) IKKα and IKKβ can phosphorylate RelA on Ser536 in vitro and in vivo (18), and phosphorylation of Ser536 can affect RelA-driven transactivation and RelA stability (9,24).
IKK has also been shown to phosphorylate the C-terminal transactivation domain of REL in in vitro immune complex kinase assays (4,9,23). Moreover, overexpression of IKKβ and activation of endogenous IKK by treatment of cells with TNF-α have been reported to enhance the transactivation ability of GAL4-REL fusion proteins containing the REL C-terminal transactivation domain (13,14,21,23). However, the sites of IKK phosphorylation on REL and a role for IKK phosphorylation of REL activity in vivo have not been clearly demonstrated.
In this report, we have identified Ser484 and Ser494 as the primary sites of IKK-mediated in vitro phosphorylation in the C-terminal transactivation domain of REL. However, these sites do not appear to be sites of IKK phosphorylation on REL in vivo, nor do mutations at these sites affect IKK’s ability to enhance GAL4-REL transactivation. Finally, we show that the previously reported effects of IKK on GAL4-REL transactivation are due to IKK-mediated activation of the endogenous NF-κB pathway, which increases expression from κB sites in the promoter of a commonly used GAL4 expression vector. As such, these results do not support an in vivo role for IKK phosphorylation affecting the activity of REL.
MATERIALS AND METHODS
Plasmids
DNA manipulations were carried out by standard methods (19). Further details of all subclones and primers used in this study can be obtained at www.nf-kb.org.
GST bacterial expression plasmids pGEX-KG and pGEX-REL (REL aa 323–587) have been described previously (23). pGEX-REL-S484,494A was created by standard overlapping PCR mutagenesis techniques. GST-REL deletion mutants Δ58, Δ90, Δ110, and Δ132 were created by subcloning EcoRI-HindIII PCR fragments from plasmids containing these deletions (21) into pGEX-KG. GST-REL aa 476–504 plasmids containing wild-type, S484A, S494A, or S484,494A sequences were created by PCR mutagenesis techniques and were subcloned into pGEX-KG using EcoRI and HindIII.
pcDNA3-FLAG was a kind gift of Bakary Sylla (International Agency for Research on Cancer, Lyon, France). pcDNA-FLAG-REL was created by sub-cloning an EcoRI-BamHI PCR fragment containing the N-terminal half of REL into pcDNA3-FLAG, followed by subcloning an EcoRV-XhoI fragment containing the C-terminal half of REL into that vector. pcDNA-FLAG-REL-S484,494A was created by subcloning an EcoRV-XhoI fragment containing the S484,494A double mutant to replace the corresponding sequences in pcDNA-FLAG-REL.
pcDNA-FLAG-IKKα, pcDNA-FLAG-IKKβ, pc-DNA-FLAG-IKKβ-S177,181E, pcDNA-mouse-RelA, and pcDNA-REL have been described previously (10–12,23). pcDNA-FLAG-IκBα-S32,36A super-repressor was a kind gift of Susan Kandarian (Boston University). pcDNA-HA-IKKβ-S177,181E was a kind gift of Sankar Ghosh (Yale University).
GAL4 expression plasmids pSG424 (aa 1–147 of GAL4) and pSG-REL (REL aa 278–587), reporter plasmid GAL4-site luciferase, and transfection normalization plasmid RSV-βgal have been described previously (21). pSG-REL-S484,494A was created by subcloning an EcoRV-NdeI REL fragment containing the S484,494A double mutant into pSG-REL digested with EcoRV and NdeI to replace the wild-type REL sequences. pcDNA-GAL4-REL was created by subcloning a HindIII-XbaI fragment from pSG-REL into pcDNA 3.1 (−).
The reporter plasmid SV40-luciferase was created by subcloning a PvuII-HindIII fragment from pSG424 containing the SV40 promoter into pGL3 Promoter Vector (Promega, Madison, WI) that had been digested with SmaI and HindIII. The reporter plasmid RSV-luciferase was created by subcloning an NdeI/Klenow-treated to HindIII fragment containing the Rous sarcoma virus LTR into pGL3 Promoter Vector that had been digested with SmaI and HindIII.
Cell Culture and Transfection
A293 human embryonic kidney cells and 3T3, IKKα −/−, IKKβ −/−, and IKKβ −/− mouse embryonic fibroblasts were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Biologos, Montgomery, IL) as previously described (22).
For transfections, cells were seeded such that they were 60% confluent on the following day when transfections were performed using polyethylenimine (PEI) (Polysciences, Warrington, PA) as described previously (6,10). On the day of transfection, DNA/ PEI was incubated at a per microgram ratio of 1:3 in serum-free DMEM (200 μl for a 35-mm plate) for 15 min at room temperature. The DNA/PEI mixture was then added to 2 ml of DMEM containing 10% FBS and the final mixture was added to the cells. The next day, the medium was replaced with 2 ml of DMEM containing 10% FBS. Cells were harvested 24 h later. Where indicated, for cells treated with TNF-α (R&D Systems, Minneapolis, MN), cells were starved for 16 h in 2 ml of DMEM containing 0.5% FBS. The next day, cells were treated with 20 ng/ml TNF-α for 6 h and cells were then lysed and processed.
Immune Complex Kinase Assays
IKK immune complex kinase assays were performed essentially as described previously (11,23). For kinase assays performed with transfected FLAG-IKK, kinases were immunoprecipitated with anti-FLAG beads (Sigma, St. Louis, MO). For kinase assays performed with endogenous IKK, serum-starved cells were treated for 7.5 min with 20 ng/ml of TNF-α before harvesting in AT lysis buffer. The IKK complex was immunoprecipitated with a polyclonal antiserum against NEMO (BD Pharmingen, San Jose, CA) and protein G sepharose beads (Zymed, San Francisco, CA). Immunoprecipitates were then incubated with 5 μg GST or GST-REL and 5 (μCi [γ-32P]ATP (Amersham Biosciences, Piscataway, NJ) in kinase reaction buffer for 30 min at 30°C. Denatured samples were electrophoresed on an SDS-polyacrylamide gel, and 32P-labeled GST-REL proteins were detected by phosphorimaging. In parallel, 5 μg of GST and the relevant GST-REL proteins were electrophoresed on 10–12.5% SDS-polyacrylamide gels, and proteins were detected by staining with Coomassie blue (Bio-Rad, Hercules, CA).
Metabolic Labeling and Immunoprecipitation
A293 cells in 35-mm plates were cotransfected with 1.5 μg FLAG-REL or 1.5 μg FLAG-REL-S484,494A and with 1.5 μg HA-IKKβ-S177,181E or 1.5 μg pcDNA 3.1 (−). The next day, medium was replaced with 2 ml of DMEM containing 10% FBS. Twenty-four hours later, medium was replaced with 1 ml of DMEM without phosphate (MP Biomedicals, Solon, OH) containing 10% dialyzed FBS (Gibco, Grand Island, NY) and 0.2 mCi [32P]orthophosphate (MP Biomedicals). After 3 h, cells were harvested in lysis buffer (20 mM Tris, pH 7.5, 100 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 10 mM NaF, 1 mM Na4P2O7, 1% Triton X-100, 0.25% SDS, 1% aprotinin, 1 mM PMSF, 1 μg/ml pepstatin, 1 μg/ml leupep-tin), and FLAG-REL proteins were immunoprecipitated with anti-FLAG beads. Denatured samples were electrophoresed on a 10% SDS-polyacrylamide gel, transferred to a nitrocellulose membrane, and 32P-labeled FLAG-REL proteins were detected by phosphorimaging. The membrane was then subjected to anti-REL Western blotting as described below.
Luciferase Reporter Assays
For GAL4 reporter assays, A293 cells in 35-mm plates were transfected with 0.5 μg of reporter plasmid GAL4 site luciferase and 0.5 μg of normalization plasmid RSV-βgal. Cells were cotransfected with 100 ng of the given pSG424-based GAL4 expression plasmid, 10 ng of the given pRG424-based GAL4 expression plasmid, or 100 ng of pcDNA-GAL4-REL. For coexpression studies of GAL4-REL with IKK, IκBα-SR, REL, or RelA, 0.5 μg of the given pcDNA plasmids was used. Quantities of DNA were brought up to 2.0 μg per 35-mm plate with pcDNA.
For pSV40-luciferase and pRSV-luciferase reporter assays, A293 cells in 35-mm plates were transfected with 0.5 μg of reporter plasmid and 0.5 μg of normalization plasmid RSV-βgal. Cells were cotransfected with 1 μg of the given pcDNA-based expression plasmid.
Luciferase activity was measured using the Luciferase Assay System (Promega, Madison, WI). Luciferase values were normalized to βgal values in all assays (7,22).
Western Blotting
Nuclear extracts were prepared (11), and anti-REL Western blotting was performed as described previously (21,22). The rabbit anti-human REL antiserum (a kind gift of Nancy Rice, National Cancer Institute, Bethesda, MD) is directed against the REL C-terminal 15 aa, and was used at a 1:10,000 dilution.
RESULTS
The Major Sites of IKKβ-Mediated In Vitro Phosphorylation on REL Are Not Required for IKKβ-Enhanced Transactivation by GAL4-REL
Several groups have shown that sequences C-terminal to the RHD in REL can be phosphorylated by IKK in vitro; however, the individual phosphorylation sites have not been precisely identified. Namely, REL can be phosphorylated between residues 422 and 540 by IKKα and between residues 473 and 531 by IKKβ (4,9). Starczynowski et al. (23) showed that REL can be weakly phosphorylated by IKKβ on Ser525, but showed that there are additional sites of phosphorylation.
To identify the major residues in the C-terminal half of REL that are phosphorylated by IKK, immune complex kinase assays were performed using GST-REL fusion proteins containing aa 323–587 and a series of deletions of residues from the REL C-terminus (Fig. 1B). FLAG-tagged IKKβ was immunoprecipitated from transfected A293 cells and incubated with these bacterially expressed GST-REL substrates. GST-REL aa 323–587, aa 323–529 (A58), and aa 323–497 (A90) were strongly phosphorylated by IKKβ, whereas GST-REL aa 323–477 (Δ110) and aa 323–455 (A132) were not appreciably phosphorylated above the level seen with GST alone. Coomassie blue staining shows approximately equal loading of all substrates. These results indicate that the major IKKβ phosphorylation sites on REL are between residues 477 and 497.
IKK is almost exclusively a serine-specific protein kinase (20). Between aa 477 and 497, there are three serine residues, at residues 484, 491, and 494. To determine which specific sites are phosphorylated by IKKβ, immune complex kinase assays were performed using a GST-REL substrate containing aa 476–504 and single mutants S484A, S494A, or double mutant S484,494A (Fig. 1C). The wild-type REL peptide and both single mutants were readily phosphorylated, whereas the GST-REL-S484,494A double mutant was not. This result shows that Ser484 and Ser494 are IKKβ phosphorylation sites in vitro and rules out Ser491 as a site of IKKβ phosphorylation.
An IKKβ immune complex kinase assay was next performed using these same mutants in the context of REL aa 323–587, containing the entire C-terminal transactivation domain of REL (Fig. 1D). GST-REL and both single mutants (S484A and S494A) were phosphorylated strongly, whereas mutant S484,494A showed greatly reduced phosphorylation (GST-REL-A110 was included as a negative control). We conclude that Ser484 and Ser494 are the major sites of in vitro phosphorylation by IKKβ in the C-terminal half of REL. We have also determined by immune complex kinase assays that Ser484 and Ser494 are sites of IKKα phosphorylation (data not shown).
To determine whether REL can be phosphorylated on Ser484 and Ser494 by endogenous IKK, kinases assays were performed using immunoprecipitated IKK complexes from 3T3 cells or from comparable IKKα −/−, IKKα −/−, and IKKα/IKKβ −/− mouse fibroblast cell lines. Cells were either untreated or were stimulated with TNF-α, and IKK complexes were immunoprecipitated with anti-NEMO antiserum. Immune complex kinase assays were then performed using GST, GST-REL, and GST-REL-S484,494A as substrates, either as contained within the aa 476–504 peptide (Fig. 1E) or within the entire C-terminal half of REL (aa 323–587) (Fig. 1F). IKK complexes immunoprecipitated from 3T3 cells and both single IKK knockout cells phosphorylated wild-type GST-REL, but not GST alone. GST-REL-S484,494A was phosphorylated much more weakly than wild-type REL in the immune complexes from 3T3 and single IKK knockout cells (Figs. 1E, F). GST-REL fusion proteins were not phosphorylated using IKK complexes immunoprecipitated from the IKKα/IKKβ −/− double knockout cells. Therefore, endogenous IKKα and IKKβ appear to show the same specificity for phosphorylating REL Ser484 and Ser494 as overexpressed IKKα and IKKβ.
To determine whether IKKβ can phosphorylate REL on Ser484 and Ser494 in vivo, we performed 32P metabolic labeling. A293 cells were transfected with FLAG-tagged REL or REL-S484,494A, with and without HA-IKKβ-S177,181E (SS/EE), a constitutively active IKKβ mutant. Cells were radiolabeled with [32P]orthophosphate and FLAG-REL proteins were immunoprecipitated. In all cases, REL and REL-S484,494A showed similar amounts of phosphate labeling (Fig. 1G). Anti-REL Western blotting confirmed that the radiolabeled bands contained approximately equal amounts of REL protein. Therefore, Ser484 and Ser494 in REL are unlikely to be sites of in vivo phosphorylation by IKKβ.
It has been shown previously that cotransfection with IKKβ can increase the transactivation ability of GAL4-REL about fourfold in A293 human embryonic kidney cells (23). We repeated this assay in A293 cells, using IKKβ-SS/EE and the same GAL4-REL fusion protein containing the REL transactivation domain (aa 278–587) and measured transactivation from a GAL4-site luciferase reporter. GAL4-REL can activate reporter gene activity by over 200-fold compared to GAL4 (1–147) alone (21); therefore, to achieve a maximal effect of IKKβ on GAL4-REL transactivation, we optimized the assay by titrating down the amount of GAL4-REL expression plasmid compared to the IKKβ-SS/EE expression plasmid (100 ng pSG-REL to 0.5 μg FLAG-IKKβ-SS/EE). Under these conditions, we observed an approximately 30-fold increase in GAL4-REL transactivation when cells were cotransfected with an expression plasmid for the constitutively active IKKβ-SS/EE protein, compared to cells cotransfected with the empty vector control (Fig. 1H).
To determine whether IKK-mediated enhancement of GAL4-REL transactivation is due to phosphorylation of REL by IKK at serine residues 484 and 494, we measured transactivation by GAL4 fusion proteins containing the C-terminal sequences from wild-type REL or REL-S484,494A, with and without IKKβ-SS/EE in A293 cells (Fig. 1H). In the absence of IKKβ-SS/EE, GAL4-REL and GAL4-REL-S484,494A activated transcription to approximately the same extent. Moreover, IKKβ-SS/EE increased transactivation by both GAL4-REL and GAL4-REL-S484,494A by approximately 30-fold (Fig. 1H). In similar experiments with (wild-type) IKKα, both GAL4-REL and GAL4-REL-S484,494A activated transcription approximately threefold higher when cotransfected with IKKα than with the vector control. We have conducted similar experiments using full-length REL and REL S484,494A and a KB-site reporter plasmid. We observed no significant differences in transactivation by wild-type REL versus REL-S484,494A (data not shown). Taken together, these results show that IKK can increase the transactivation ability of GAL4-REL, but that this effect does not require phosphorylation of Ser484 or Ser494, which are major sites of REL phosphorylation by IKK in vitro.
The IκBα Super-Repressor Abolishes IKKβ- and NF-κB-Mediated Enhancement of GAL4-REL Transactivation
Because mutation of the major IKKβ phosphorylation sites in REL does not affect IKKβ-mediated enhancement of GAL4-REL transactivation, it suggested to us that this enhancement proceeds through a mechanism other than IKKβ phosphorylation of REL. As a first step towards describing this indirect mechanism, we sought to determine whether disruption of downstream effects of IKKβ on NF-κB affected its ability to enhance GAL4-REL transactivation. Because IKKβ-SS/EE is a potent inducer of NF-κB, we determined the effect of the IκBα super-repressor (IκBα-SR) on IKKβ enhancement of GAL4-REL transactivation. The IκBα-SR does not dissociate from NF-κB dimers, and therefore blocks activation of NF-κB by IKK. Expression of the IκBα-SR had no significant effect on transactivation by GAL4-REL in the absence of IKKβ-SS/EE (Fig. 2A). However, cotransfection of the IκBα-SR blocked the ability of IKKβ-SS/EE to enhance transactivation by GAL4-REL (Fig. 2A), suggesting that the effect of IKKβ-SS/EE on GAL4-REL transactivation requires increased NF-κB activity.
Figure 2.
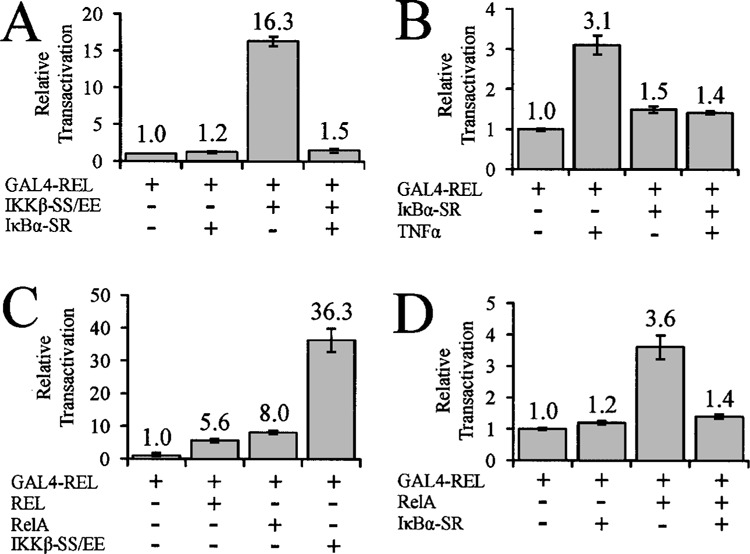
The IκBα super-repressor blocks IKKβ- and NF-κB-mediated enhancement of GAL4-REL transactivation. (A) Cells were transfected with 100 ng of pSG-REL expression vector, 0.5 μg pcDNA, IκBα-SR, or IKKβ-SS/EE, and 0.5 μg of GAL4-luciferase reporter plasmid. (B) Cells were transfected with 100 ng of pSG-REL expression vector, 0.5 μg pcDNA or IκBα-SR, and 0.5 μg of GAL4-luciferase plasmid. Cells were incubated for 24 h, serum starved for 16 h, and then treated with 20 ng/ml TNF-α for 6 h or left untreated. (C) Cells were transfected with 100 ng of pSG-REL expression vector; 0.5 μg pcDNA, REL, RelA, or IKKβ-SS/EE; and 0.5 μg of GAL4-luciferase plasmid. (D) Cells were transfected with 100 ng of pSG-REL GAL4-fusion expression vector; 0.5 μg pcDNA, IκBα-SR, or RelA; and 0.5 μg of GAL4-luciferase plasmid. In all reporter assays, cells were incubated for 48 h, then luciferase and β-galactosidase activities were determined, and values were normalized as indicated for each panel (1.0). In all cases, values are the averages of three experiments performed with triplicate samples.
The cytokine TNF-α can activate the canonical NF-κB pathway, which proceeds through activation of the IKK complex (5). TNF-α has also been shown to enhance GAL4-REL transactivation in cell-based GAL4-site reporter gene assays (13,14,22). To determine whether the effect of TNF-α on GAL4-REL is due to IKK activation of NF-κB signaling, A293 cells were transfected with GAL4-REL and either vector alone or IκBα-SR. These cells were then either treated with TNF-α or left untreated. As previously observed (13), TNF-α treatment increased GAL4-REL transactivation by approximately threefold compared to untreated cells (Fig. 2B). However, expression of the IKBα-SR blocked the TNF-α-induced increase in GAL4-REL transactivation (Fig. 2B). These data show that the IKBα-SR blocks the ability of both overexpressed IKKβ- and TNF-α-activated endogenous IKK to enhance GAL4-REL transactivation, suggesting that downstream activation of NF-κB is required for the effects of IKKβ on GAL4-REL.
To determine whether overexpression of NF-κB subunits could also enhance GAL4-REL transactivation, we cotransfected A293 cells with GAL4-REL, and with vector alone or expression plasmids for REL, RelA, or, as a positive control, IKKβ-SS/EE. As previously observed, IKKβ-SS/EE increased GAL4-REL by approximately 30-fold. REL and RelA increased GAL4-REL transactivation by approximately sixfold and eightfold, respectively (Fig. 2C). Furthermore, the IκBα-SR blocked the ability of RelA to enhance GAL4-REL transactivation (Fig. 2D).
Taken together, these results show that NF-κB activity increases GAL4-REL transactivation and suggest that induction of NF-κB signaling accounts for the effect of IKKβ on GAL4-REL transactivation in these types of reporter gene assays.
The pSG424 GAL4-Fusion Protein Expression Vector Contains κB-Responsive Sites in Its SV40 Promoter Sequences
Upon examination of the Simian virus 40 (SV40) early promoter sequences in the pSG424 vector, which is used for expression of GAL4 and GAL4-REL, we identified two κB sites (5′-GGAAAGTC-CCC-3′) that have been previously identified by Kanno et al. (8). To determine whether the reason for enhancement of GAL4-REL transactivation by NF-κB and IKKβ was due to NF-κB-mediated activation of the promoter in the pSG424 vector, we first sub-cloned the pSG424 SV40 promoter sequences into the luciferase reporter vector pGL3 Promoter Vector. Cotransfection of RelA with pSV40-luciferase resulted in approximately 16-fold higher luciferase activity than the vector control. Additionally, cotransfection of IKKβ-SS/EE with pSV40-luciferase resulted in approximately 55-fold higher luciferase activity than the vector control (Fig. 3).
Figure 3.
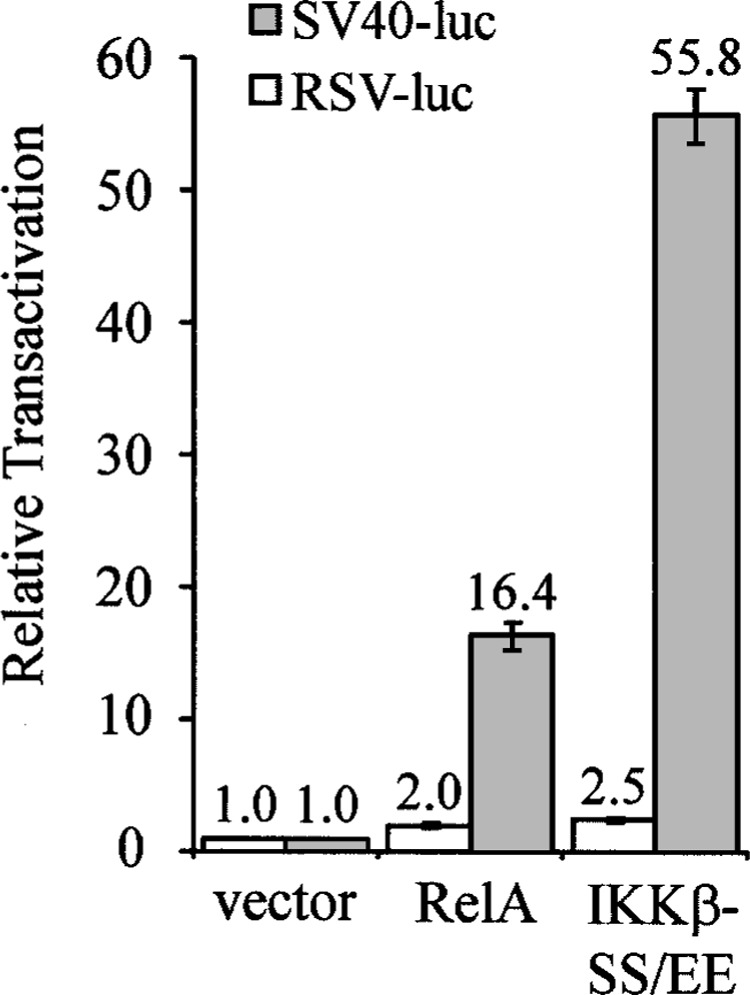
The pSG424 vector contains κB-responsive sites in its SV40 promoter sequences. Cells were transfected with 1 μg pcDNA, RelA, or IKKβ-SS/EE, and either 0.5 μg of SV40-luciferase plasmid (SV40-luc) or 0.5 μg of RSV-luciferase plasmid (RSV-luc). Reporter assays were then conducted as described for Figure 2, and values were normalized to the value obtained with pcDNA alone (vector) (1.0). In all cases, values are the averages of three experiments performed with triplicate samples.
The Rous sarcoma virus (RSV) promoter is not known to contain κB sites. To determine whether IKKβ and NF-κB could affect expression from the RSV promoter, we subcloned the RSV promoter into the pGL3 Promoter Vector. We cotransfected RelA and IKKβ-SS/EE with pRSV-luciferase and observed only minor increases (approximately twofold) in luciferase activity when compared with the vector control (Fig. 3).
These results suggest that pSG424 contains functional κB sites and that NF-κB increases expression from the promoter in pSG424, which accounts for the NF-κB- and IKKβ-mediated increases in GAL4-REL transactivation.
Replacement of the SV40 Promoter With the RSV Promoter in the pSG424 Vector Abolishes IKKβ- and NF-κB-Mediated Enhancement of GAL4-REL Transactivation
To show more directly that the κB site-containing promoter sequences in the pSG424 vector account for IKKβ-mediated induction of GAL4-REL, we replaced the SV40 promoter sequences in the pSG424 vector with the RSV promoter sequences to create the pRG424 vector, and then assessed whether IKKβ-SS/EE could affect GAL4-REL transactivation (Fig. 4A). As a control, cotransfection of pSG-REL with IKKβ-SS/EE resulted in an approximately 15-fold increase in transactivation by GAL4-REL compared to cotransfection with the vector control. However, cotransfection of the IKKβ-SS/EE expression vector with pRG-REL or pRG-REL-S484,494A did not increase GAL4-REL transactivation above the level seen with the vector control (Fig. 4A); in fact, we even noted a decrease in GAL4-REL transactivation in the presence of IKKβ-SS/EE. In addition, we observed only a minor difference in the basal transactivation by pRG-REL and pRG-REL-S484,494A.
Figure 4.
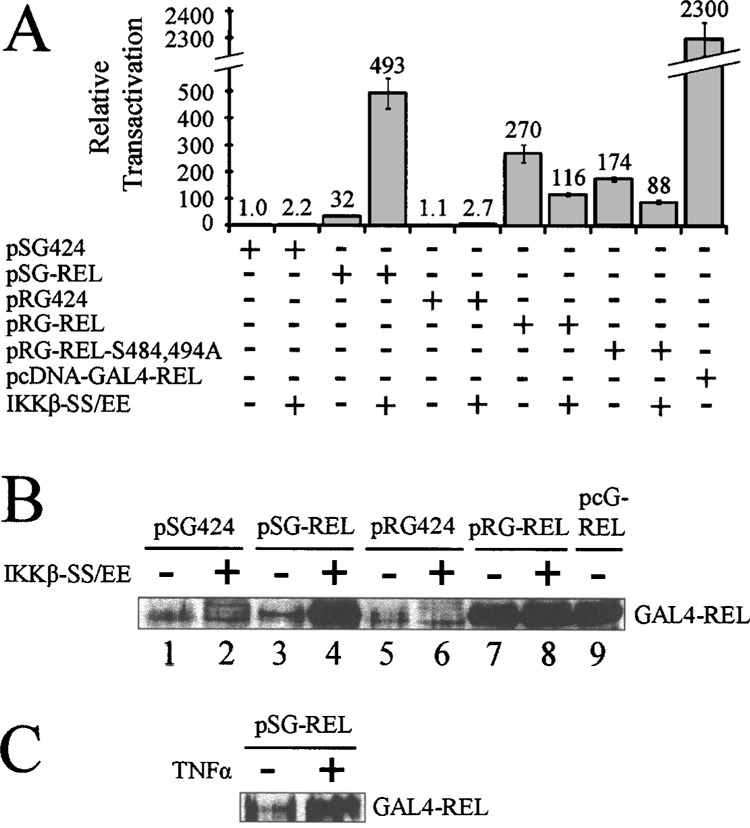
Replacement of the SV40 promoter with the RSV promoter in the pSG424 vector abolishes IKKβ-mediated enhancement of GAL4-REL transactivation. (A) Cells were transfected with 100 ng of pSG424 GAL4-fusion expression vectors, 10 ng of pRG424 GAL4-fusion expression vectors, or 100 ng of pcDNA-GAL4-REL; 0.5 μg pcDNA or IKKβ-SS/EE; and 0.5 μg of GAL4-luciferase plasmid. Cells were incubated 48 h, then luciferase activities and β-galactosidase activities were determined, and values normalized (1.0). Values are the averages of three experiments performed with triplicate samples. (B) Cells were transfected with 1.5 μg of the indicated pSG424 GAL4-fusion expression vector, 1.0 μg of the indicated pRG424 GAL4-fusion expression vector, or 0.5 μg pcDNA-GAL4-REL (pcG-REL); and with 0.5 μg pcDNA or IKKβ-SS/EE. (C) Cells were transfected with 2.0 μg of pSG-REL and were treated (or untreated) with TNF-α for 6 h prior to lysis. In (B) and (C), 40 μg of nuclear extract protein was separated by SDS-PAGE and subjected to anti-REL Western blotting. The positions of the GAL4-REL protein are indicated.
To achieve high constitutive expression of GAL4-REL, we subcloned the GAL4-REL sequences into pcDNA 3.1. pcDNA 3.1 contains the cytomegalovirus (CMV) promoter, which is a much stronger promoter than the SV40 promoter (26), presumably directing greater expression of GAL4-REL. Transfection of 100 ng of pcDNA-GAL4-REL resulted in an approximately 70-fold increase in reporter gene expression compared to 100 ng of pSG-REL (Fig. 4A; compare 32 to 2300 relative luciferase units). Of note, the RSV promoter is also about 80-fold stronger than the SV40 promoter: that is, transfection of 10 ng of pRG-REL yields approximately eight times more GAL4-site promoter luciferase activity than transfection of 100 ng of pSG-REL (Fig. 4A; compre 32 to 270 relative luciferase units). Taken together, these results show that increased levels of promoter activity controlling GAL4-REL protein expression are correlated with increased expression of luciferase from the GAL4-site reporter plasmid.
To show that increased GAL4-REL protein expression also correlates with increased GAL4-site luciferase activity, we cotransfected A293 cells with pSG424, pSG-REL, pRG424, or pRG-REL and with pcDNA or pcDNA-IKKβ-SS/EE. Anti-REL Western blot analysis of nuclear extracts showed that cotransfec-tion of IKKβ-SS/EE with pSG-REL results in increased expression of GAL4-REL protein (Fig. 4B, lanes 3 and 4); in contrast, cotransfection of IKKβ-SS/EE did not appreciably affect the levels of GAL4-REL expressed from pRG-REL (Fig. 4B, lanes 7 and 8). Additionally, pcDNA-GAL4-REL directs high levels of expression of GAL4-REL, similar to what is seen with pRG-REL alone and pSG-REL when cotransfected with IKKβ-SS/EE (Fig. 4B, compare lanes 4, 7, and 9). We also show that TNF-α treatment of cells transfected with pSG-REL results in increased expression of GAL4-REL protein (Fig. 4C).
These results show that cotransfection of the IKKβ-SS/EE expression vector and activation of endogenous IKK by TNF-α correlate with both increased GAL4-site luciferase reporter gene activity and increased levels of GAL4-REL protein when using the pSG424 vector, which contains functional κB sites. Conversely, cotransfection of the IKKβ-SS/EE expression plasmid does not result in increased GAL4-site luciferase reporter activity or GAL4-REL protein levels when using the pRG424 vector, which lacks known functional κB sites.
DISCUSSION
Several groups previously demonstrated phosphorylation of REL transactivation domain sequences by IKK in vitro (4,9,23), leading them to suggest a direct IKK → REL cross-talk. We are the first to identify Ser484 and Ser494 as the major sites of in vitro phosphorylation of REL by IKKα and IKKβ. However, we have not been able to establish an in vivo role for phosphorylation of these sites in REL-dependent transactivation. Moreover, we have not detected enhanced phosphorylation of REL by IKK at Ser484 and Ser494 (or any other residues) in vivo (Fig. 1G), nor have we been able to detect a physical interaction between REL and IKK through coimmunoprecipitation (data not shown). Therefore, we conclude that the in vivo effects of IKK on REL that these groups reported (4,9,23) are unlikely to be due to C-terminal phosphorylation of REL by IKKα or IKKβ. As such, it is unclear what role, if any, phosphorylation of these in vitro IKK recognition sites plays in the regulation of REL activity in vivo.
Why can IKK phosphorylate C-terminal sequences of REL in vitro but not in vivo? First, IKK might be in a different subcellular compartment than REL and, therefore, IKK may never be able to access the REL phosphorylation sites in vivo. Second, IKK might have a weak affinity (high K M ) for Ser484 and Ser494, and, therefore, phosphorylation of these residues may occur in vitro where the two proteins are at high concentrations, but not in vivo where the effective concentrations are much lower. Third, we and others have looked at the ability of IKK to phosphorylate in vitro the C-terminal sequences of REL in the context of GST-fusion proteins, whereas our in vivo experiments have been done with full-length REL, wherein the sites of phosphorylation may not be accessible.
It has been reported previously that overexpression of IKKβ and activation of IKK by TNF-α can enhance the transactivation ability of GAL4-REL fusion proteins (13,14,22,23). As we now show, mutation of Ser484 and Ser494 to Ala does not affect the ability of IKK to increase GAL4-REL transactivation in assays similar to those reported previously. Moreover, we show that the effects of IKK on GAL4-REL transactivation are due to IKK-mediated activation of the endogenous NF-κB pathway, which activates transcription from κB sites in the GAL4 expression vector pSG424. NF-κB-mediated activation of the SV40 promoter in these pSG424-based vectors then leads to increased expression of GAL4-REL protein and, consequently, increased expression of the GAL4-site luciferase reporter. Strikingly, we have shown that coexpression of the IκBα super-repressor can block the effects of either overexpressed IKK or TNF-activated IKK on GAL4-REL transactivation.
pSG424 (17) is a commonly used expression vector for GAL4 fusion protein-based reporter gene assays in tissue culture cells; indeed, the short paper describing this vector has now been cited over 500 times (www.isiknowledge.com). Because the pSG424 vector contains κB sites in its promoter (8), several other reports are likely to have misinterpreted effects of NF-κB inducers (e.g., UV, phorbol ester, interleukin-1) on transactivation by GAL4 fusion proteins. For example, GAL4-Jun has been has been reported to be enhanced by UV (1), GAL4-GRIP1 by phorbol ester (16), GAL4-Elk1 and GAL4-MEF2A by interleukin-1 (25), and GAL4-VP16 by TNF-α (2). Similarly, we have found that cotransfection of IKKβ-SS/ EE can also increase transactivation by a GAL4-VP16 fusion protein when expressed from pSG424 (data not shown). The use of our RSV promoter-driven GAL4 system (pRG424) should enable one to avoid problems associated with induction of the endogenous NF-κB pathway in an experimental treatment group, a common problem given the large number of NF-κB inducers (see www.nf-kb.org, under Inducers).
What is the biological function, if any, of the Ser484/Ser494 in vitro IKK phosphorylation sites in REL? Although RelA’s transactivation activity can be increased by IKK phosphorylation on Ser536 (24), we find no evidence that REL transactivation is similarly affected by IKK. That is, although we have shown that the C-terminal half of REL is strongly phosphorylated at Ser484 and Ser494 in vitro by IKKα and IKKβ, IKK does not appear to phosphorylate these sites in vivo, and mutation of these sites to either Ala residues or phosphorylation mimetic Asp residues (data not shown) does not strongly affect GAL4-REL transactivation. Nevertheless, the Ser484 and Ser494 residues are conserved in c-Rel among several mammals (22), suggesting that they have a role in REL activity. Therefore, there may be a kinase(s) (other than IKK) that can phosphorylate Ser484 and Ser494 in vivo, or these two Ser residues may play a role in REL activity that is not modulated by phosphorylation.
ACKNOWLEDGMENTS
This work was supported by a grant from the National Institutes of Health (CA47763). We thank Alexander Hoffmann (UCSD) for the IKK knockout mouse fibroblasts, and Dan Starczynowski, Josh Lee-man, and other members of our lab for helpful discussions.
REFERENCES
- 1. Caelles C.; González-Sancho J. M.; Muñoz A. Nuclear hormone receptor antagonism with AP-1 by inhibition of the JNK pathway. Genes Dev. 11:3351–3364; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. De Fabiani E.; Mitro N.; Anzulovich A. C.; Pinelli A.; Galli G.; Crestani M. The negative effects of bile acids and tumor necrosis factor-α on the transcription of cholesterol 7α-hydroxylase gene (CYP7A1) converge to hepatic nuclear factor-4. J. Biol. Chem. 276:30708–30716; 2001. [DOI] [PubMed] [Google Scholar]
- 3. Gilmore T. D.; Kalaitzidis D.; Liang M.-C; Starczynowski D. T. The c-Rel transcription factor and B-cell proliferation: A deal with the devil. Oncogene 23:2275–2286; 2004. [DOI] [PubMed] [Google Scholar]
- 4. Harris J.; Olière S.; Sharma S.; Sun Q.; Lin R.; Hiscott J.; Grandvaux N. Nuclear accumulation of cRel following C-terminal phosphorylation by TBK1/IKKε . J. Immunol. 177:2527–2535; 2006. [DOI] [PubMed] [Google Scholar]
- 5. Hayden M. S.; Ghosh S. Shared principles in NF-κB signaling. Cell 132:344–362; 2008. [DOI] [PubMed] [Google Scholar]
- 6. Herscovitch M.; Comb W.; Ennis T.; Coleman K.; Yong S.; Armstead B.; Kalaitzidis D.; Chandani S.; Gilmore T. D. Intermolecular disulfide bond formation in the NEMO dimer requires Cys54 and Cys347. Biochem. Biophys. Res. Commun. 367:103–108; 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kalaitzidis D.; Davis R. E.; Rosenwald A.; Staudt L. M.; Gilmore T. D. The human B-cell lymphoma cell line RC-K8 has multiple genetic alterations that dysregulate the Rel/NF-κB signal transduction pathway. Oncogene 21:8759–8768; 2002. [DOI] [PubMed] [Google Scholar]
- 8. Kanno M.; Fromental, C; Staub A.; Ruffenach F.; Davison I.; Chambon P. The SV40 TC-II(κB) and the related H-2KB enhansons exhibit different cell type specific and inducible proto-enhancer activities, but the SV40 core sequence and the AP-2 binding sites have no enhanson properties. EMBO J. 8:4205–4214; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lawrence T.; Bebien M.; Liu G. Y.; Nizet V.; Karin M. IKKα limits macrophage NF-κB activation and contributes to the resolution of inflammation. Nature 434:1138–1143; 2005. [DOI] [PubMed] [Google Scholar]
- 10. Leeman J. R.; Weniger M. A.; Barth T. F.; Gilmore T. D. Deletion analysis and alternative splicing define a transactivation inhibitory domain in human oncoprotein REL. Oncogene; in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liang M.-C.; Bardhan S.; Li C.; Pace E. A.; Porco J. A. Jr.; Gilmore T. D. Jesterone dimer, a synthetic derivative of the fungal metabolite jesterone, blocks activation of nuclear factor κB by inhibiting the inhibitor of κB kinase. Mol. Pharm. 64:123–131; 2003. [DOI] [PubMed] [Google Scholar]
- 12. Liang M.-C.; Bardhan S.; Pace E. A.; Rosman D.; Beutler J. A.; Porco J. A. Jr.; Gilmore T. D. Inhibition of transcription factor NF-κB signaling proteins IKKβ and p65 through specific cysteine residues by epoxyquinone A monomer: Correlation with its anti-cancer cell growth activity. Biochem. Pharmacol. 71:634–645; 2006. [DOI] [PubMed] [Google Scholar]
- 13. Martin A. G.; Fresno M. Tumor necrosis factor-α activation of NF-κB requires the phosphorylation of Ser-471 in the transactivation domain of c-Rel. J. Biol. Chem. 275:24383–24391; 2000. [DOI] [PubMed] [Google Scholar]
- 14. Martin A. G.; San-Antonio B.; Fresno M. Regulation of nuclear factor κB transactivation: Implication of phosphatydylinositol 3-kinase and protein kinase Cζ in c-Rel activation by tumor necrosis factor α. J. Biol. Chem. 276:15840–15849; 2001. [DOI] [PubMed] [Google Scholar]
- 15. Perkins N. D. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene 25:6717–6730; 2006. [DOI] [PubMed] [Google Scholar]
- 16. Rogatsky I.; Luecke H. F.; Leitman D. C.; Yamamoto K. R. Alternate surfaces of transcriptional coregulator GRIP1 function in different glucocorticoid receptor activation and repression contexts. Proc. Natl. Acad. Sci. USA 99:16701–16706; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sadowski I.; Ptashne M. A vector for expressing GAL4 (1-147) fusions in mammalian cells. Nucleic Acids Res. 17:7539; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sakurai H.; Chiba H.; Miyoshi H.; Sugita T.; Toriumi W. IκB kinases phosphorylate NF-κB p65 subunit on serine 536 in the transactivation domain. J. Biol. Chem. 274:30353–30356; 1999. [DOI] [PubMed] [Google Scholar]
- 19. Sambrook J.; Fritsch E. F.; Maniatis T. Molecular cloning: A laboratory manual, 2nd ed Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 20. Scheidereit C. IκB kinase complexes: Gateways to NF-κB activation and transcription. Oncogene 25:6685–6705; 2006. [DOI] [PubMed] [Google Scholar]
- 21. Starczynowski D. T.; Reynolds J. G.; Gilmore T. D. Deletion of either C-terminal transactivation subdomain enhances the in vitro transforming activity of human transcription factor REL in chicken spleen cells. Oncogene 22:6928–6936; 2003. [DOI] [PubMed] [Google Scholar]
- 22. Starczynowski D. T.; Reynolds J. G.; Gilmore T. D. Mutations of tumor necrosis factor α-responsive serine residues within the C-terminal transactivation domain of human transcription factor REL can enhance its in vitro transforming ability. Oncogene 24:7355–7368; 2005. [DOI] [PubMed] [Google Scholar]
- 23. Starczynowski D. T.; Trautman H.; Pott C.; Harder L.; Arnold N.; Africa J. A.; Leeman J. R.; Siebert R.; Gilmore T. D. Mutation of an IKK phosphorylation site within the transactivation domain of REL in two patients with human B-cell lymphoma enhances REL’s in vitro transforming activity. Oncogene 26:2685–2694; 2007. [DOI] [PubMed] [Google Scholar]
- 24. Yang F.; Tang E.; Guan K.; Wang C. Y. IKKβ plays an essential role in the phosphorylation of RelA/p65 on serine 536 induced by lipopolysaccharide. J. Immunol. 170:5630–5635; 2003. [DOI] [PubMed] [Google Scholar]
- 25. Yang S. H.; Galanis A.; Sharrocks A. D. Targeting of p38 mitogen-activated protein kinases to MEF2 transcription factors. Mol. Cell. Biol. 19:4028–4038; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zarrin A. A.; Malkin L.; Fong I.; Luk K. D.; Ghose A.; Berinstein N. L. Comparison of CMV, RSV, SV40 viral and Vλ1 cellular promoters in B and T lymphoid and non-lymphoid cell lines. Biochim. Biophys. Acta 1446:135–139; 1999. [DOI] [PubMed] [Google Scholar]