

Abstract
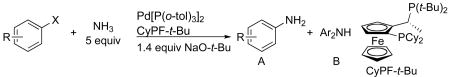
We report that the complex generated from Pd[P(o-tol)3]2 and the alkylbisphosphine CyPF-t-Bu is a highly active and selective catalyst for the coupling of ammonia with aryl chlorides, bromides, iodides, and sulfonates. The couplings of ammonia with this catalyst conducted with a solution of ammonia in dioxane form primary arylamines from a variety of aryl electrophiles in high yields. Catalyst loadings as low as 0.1 mol % were sufficient for reactions of many aryl chlorides and bromides. In the presence of this catalyst, aryl sulfonates also coupled with ammonia for the first time in high yields. A comparison of reactions in the presence of this catalyst versus those in the presence of existing copper and palladium systems revealed a complementary, if not broader substrate scope. The utility of this method to generate amides, imides and carbamates is illustrated by a one-pot synthesis of a small library of these carbonyl compounds from aryl bromides and chlorides. Mechanistic studies show that Pd[P(o-tol)3]2 and CyPF-t-Bu generate a more active and general catalyst than that generated from CyPF-t-Bu and palladiun(II) precursors because of the low concentration of active catalyst that is generated from the combination of palladium(II), ammonia and base.
Introduction
Although ammonia is among the least expensive bulk chemicals, its application in transition-metal catalysis is rare. Because of ammonia's high basicity, small size and strong N-H bond (107 kcal/mol),1 most of its reactions with transition metal complexes yield Lewis acid-base adducts that are often highly stable.2 Thus, catalyst turnover in the presence of ammonia has been difficult to achieve. There are only a few transition-metal-catalyzed processes that incorporate ammonia into more complex molecules, and most of these reactions were published recently. These reactions include rhodium-catalyzed reductive amination of aldehydes,3 iridium-catalyzed reductive amination of α-keto acids,4 rhodium- and iridium-catalyzed hydroaminomethylation of olefins,5 ruthenium-catalyzed synthesis of primary amines from primary alcohols and ammonia,6 gold-catalyzed hydroamination of alkynes and allenes,7 palladium- and iridium-catalyzed allylic amination,8,9 and copper-10-12 and palladium-catalyzed13-15 coupling of ammonia with aryl halides.
The copper- and palladium-catalyzed coupling of ammonia with aryl halides has attracted attention recently because of the value of primary arylamines in the preparation of agrochemicals, dyes and pharmaceuticals.16,17 The copper-catalyzed method has been known since the 1960s,18 but significant progress has only been made recently.10-12 In spite of the recent advances, copper-catalyzed coupling with ammonia is limited in scope and requires high loadings of copper and ligand. The copper-catalyzed coupling does not occur in high yield with electron-rich aryl bromides or ortho-substituted aryl bromides or iodides. The copper-catalyzed reactions also do not occur in substantial yields with aryl chlorides or sulfonates, which are less expensive and more accessible than aryl iodides and bromides or aryl triflates, respectively.11 Finally, catalyst loadings of greater than 10 mol % are usually required. This high loading can complicate isolation of basic products from the copper catalyst.
The palladium-catalyzed coupling of ammonia with aryl halides was first achieved in our laboratory in 2006 (eq 1).13 This process occurred with complexes generated from a Pd(II) precursor ligated by the electron-rich and sterically bulky Josiphos ligand (CyPF-t-Bu). The reactivity of the catalyst containing this ligand for the arylation of ammonia has been attributed to two factors.19 First, the rigid backbone that arises from the orientation of the methyl and ferrocenyl group directs the phosphorus atoms and their electron pairs toward the metal center, creating tight chelation and resulting stability toward displacement by strong Lewis bases, such as ammonia. Second, the strong electron-donation and steric bulk of the ligand make palladium(0) complexes of it highly reactive toward oxidative addition of aryl chlorides and tosylates.20 This work and subsequent reports by Buchwald14 in 2007 and Beller21 in 2009 constitute the only published palladium-catalyzed couplings of ammonia with aryl halides.
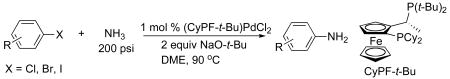 |
(1) |
Although palladium-catalysts have the potential to create a process that occurs with broader scope than the copper-catalyzed reactions, a detailed evaluation of the reaction scope was not included in the first reports of palladium-catalyzed coupling of ammonia. Only one example of the reaction of an aryl chloride was reported in our preliminary communication,13 and Buchwald's report did not include any examples of the reactions of aryl chlorides. The reactions of aryl bromides and aryl chlorides reported by Beller required temperatures of 120 °C, and were conducted under 10 bar of N2 to obtain good yields of primary arylmines.15 Finally, the reactions of our studies in 2006 were conducted with a high pressure of ammonia to achieve good selectivity for formation of the monoarylamine over the corresponding diarylamine.
Moreover, the coupling of aryl sulfonates with ammonia has not been reported. Cleavage of the S-O bond of the sulfonyl group was reported to occur in the presence of ammonia and the strong base NaO-t-Bu faster than C-N coupling.13 For similar reasons, aryl halides containing base-sensitive functional groups such as esters, nitriles, and ketones containing enolizable protons underwent reactions at the functional group faster than they underwent C-N coupling.
To address these limitations, we studied reactions in the presence of a catalyst that would react under milder conditions than the one we used for our initial studies. We recently reported a combination of Pd(0) precursor and the Josiphos ligand that circumvents the difficulties with generation of the active catalyst from the typical Pd(0) source Pd2(dba)3 or the typical Pd(II) source Pd(OAc)2.22 With the strongly electron-donating Josiphos ligand, back-donation into the remaining dba ligand of the (CyPF-t-Bu)Pd(dba) leads to slow dissociation of dba to generate the catalytically active (CyPF-t-Bu)Pd(0).23 With a reagent lacking β-hydrogens, a clear pathway to generate the Pd(0) species from Pd(OAc)2 and the Josiphos ligand does not exist. A catalyst precursor, in combination with CyPF-t-Bu, that generates a higher concentration of active catalyst could allow the coupling of ammonia to occur with less reactive aryl chlorides and tosylates. Identification of such a catalyst could also allow the reactions to be conducted under conditions that are mild enough to be compatible with base-sensitive functional groups and under conditions involving a lower pressure of ammonia.
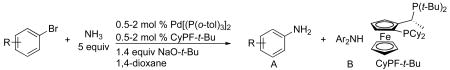
Herein, we report a general method for the coupling of ammonia with aryl chlorides, bromides, iodides and tosylates catalyzed by a combination of Pd[P(o-tol)3]2 and CyPF-t-Bu. The coupling occurs in high yield and with high selectivity for formation of the primary arylamines in the presence of catalyst loadings as low as 0.1 mol %. The substrate scope now includes a wide range of aryl bromides and chlorides, including those containing base-sensitive functional groups. For the first time, the coupling of aryl tosylates and ammonia is also shown to occur. The utility of this catalyst for sequential, one-pot arylations of ammonia to prepare unsymmetrical diarylamines and for the arylation of ammonia to form N-aryl imides, amides, and carbamates by a one-pot, two-step sequence is demonstrated. Preliminary mechanistic studies reveal the origins of the greater reactivity of this catalyst system than that of the CyPF-t-Bu systems reported previously.
2. Results and Discussion
2.1. Identification of Conditions for the Amination of 1-Bromo-4-tert-butylbenzene in a 0.5 M Solution of Ammonia
To identify the conditions for coupling of aryl halides with ammonia at ambient pressure, we examined reactions of the electronically neutral and sterically unhindered 1-bromo-4-tert-butylbenzene in 0.5 M solutions of ammonia in either 1,2-dimethoxyethane (DME) or 1,4-dioxane. These two solvents were chosen because previous studies of the coupling of aryl halides with ammonia in our laboratory showed that reactions in ethereal solvents occurred to better conversions than those in hydrocarbon solvents, such as toluene. Catalysts generated from either one-component precatalysts or combinations of a precatalyst and the CyPF-t-Bu ligand were tested. NaO-t-Bu was used as the base. The conversion of the aryl halide and the selectivity for formation of monoarylamines versus diarylamines were measured by GC and 1H NMR spectroscopy respectively.
We compared reactions initiated with CyPF-t-Bu as ligand and the commercially available Pd(OAc)2 or Pd2(dba)3 as palladium source with reactions initiated with the one-component catalyst (CyPF-t-Bu)PdCl2 that we used in our initial work on the coupling of ammonia.13 Catalysts generated from Pd(OAc)2 exhibited much lower reactivity than those generated from Pd(dba)2 or (CyPF-t-Bu)PdCl2. The test reaction catalyzed by the complex generated from equimolar amounts of Pd(OAc)2 and CyPF-t-Bu occurred to only 50% conversion after 12 h (Table 1, entry 1), whereas reactions catalyzed by complexes generated from Pd(dba)2, (CyPF-t-Bu)PdCl2 or Pd[P(o-tol)3]2 as palladium sources occurred to full conversion after 12 h (Table 1, entries 2-6). This difference in reactivity contrasts results recently reported by our group on the coupling of heteroaryl chloride with primary alkylamines in which complexes generated from Pd(OAc)2 were much more reactive than those generated from Pd(dba)2.19
Table 1.
Effects of Precatalysts and Solvents on the Coupling of 4-Bromo-tert-butylbenzene with Ammonia in 0.5 M Solutionsa
 | ||||||
|---|---|---|---|---|---|---|
| entry | catalyst | loading (%) | solvent | conc. [M]b | conversion (%)c | A:Bd |
| 1 | Pd(OAc)2/CyPF-t-Bu | 0.5 | 1,4-dioxane | 0.038 | 50 | -- |
| 2 | Pd2(dba)3 / CyPF-t-Bu | 0.25e | 1,4-dioxane | 0.038 | 100 | 17:1 |
| 3 | (CyPF-t-Bu)PdCl2 | 0.5 | 1,4-dioxane | 0.038 | 100 | 7:1 |
| 4 | (CyPF-t-Bu)PdCl2 | 2.0 | DME | 0.038 | 100 | 0.7:1.0 |
| 5 | Pd[P(o-tol)3]2/CyPF-t-Bu | 0.5 | 1,4-dioxane | 0.038 | 100 | >30:1 |
| 6 | Pd[P(o-tol)3]2/CyPF-t-Bu | 0.5 | DME | 0.038 | 100 | 0.7:1.0 |
Reactions conducted with 0.5 mmol 4-bromo-tert-butylbenzene, 5 mL of 0.5 M ammonia solutions and 1.4 equiv NaOtBu.
Concentration of 4-bromo-tert-butylbenzene.
Determined by GC with dodecane as an internal standard.
Determined by 1H NMR spectroscopy.
Reaction conducted with 0.25 mol % Pd2(dba)3 and 0.5 mol % CyPF-t-Bu.
Of the reactions that occurred to full conversion, those conducted in 1,4-dioxane occurred with much greater selectivity for formation of monoarylamines versus diarylamines than did reactions conducted in DME (Table 1, entries 3 and 5 vs. 4 and 6). In 1,4-dioxane, high selectivity for formation of the primary arylamine was observed from reactions catalyzed by complexes generated from Pd2(dba)3 or Pd[P(o-tol)3]2 (Table 1, entries 2 and 5). To determine the method that generates the most active catalyst, we studied reactions conducted with Pd2(dba)3 or Pd[P(o-tol)3]2 as precatalyst under the conditions of entries 2 and 5 of Table 1 for the coupling of 4-chlorotoluene with ammonia. Although both systems led to reactions of aryl bromides to high conversions, the system comprising the combination of Pd[P(o-tol)3]2 and CyPF-t-Bu was substantially more active than that comprising Pd2(dba)3 and CyPF-t-Bu for the coupling of ammonia with aryl chlorides.23 More specifically, the reaction of 4-bromo-tert-butylbenzene with ammonia catalyzed by complexes generated from Pd[P(o-tol)3]2 and CyPF-t-Bu occurred with conversion and selectivity that was similar to that of the reaction catalyzed by complexes generated from Pd2(dba)3 and CyPF-t-Bu. However, the reaction of 4-chlorotoluene with ammonia in the presence of 0.5 mol % of Pd[P(o-tol)3]2 and CyPF-t-Bu occurred to full conversion after 12 h, whereas the reaction catalyzed by 0.5 mol % of the combination of Pd2(dba)3 and CyPF-t-Bu occurred to less than 10% conversion of 4-chlorotoluene after 24 h.
Having established that the combination of Pd[P(o-tol)3]2 and CyPF-t-Bu provides the more active catalyst system and that a 0.5 M solution of ammonia in dioxane is a suitable reaction medium for the coupling of ammonia with aryl chlorides and bromides, we sought reaction conditions for a general coupling of aryl halides with ammonia at low catalyst loadings. We identified two general sets of conditions. The first set of reaction conditions was identified for reactions of meta- and para-substituted aryl halides. A substrate concentration of 0.038-0.05 M was used to ensure an excess of ammonia is present and a catalyst loading of 0.5 mol % was used to ensure suitable rates. At this concentration and catalyst loading, the reaction of 4-bromo-tert-butylbenzene occurred to full conversion and high selectivity (Table 2, entry 1). Reactions of these classes of aryl bromides conducted with catalyst loadings below 0.5 mol % occurred to low conversion (Table 2, entry 2). Reactions conducted with higher concentrations of the bromoarene (0.1 M) occurred to completion with only 0.25 mol % of catalyst, but the selectivity for the monoarylation product was lower (Table 2, entry 3). Thus, we found the optimal concentration and catalyst loading for reactions of meta- and para-substituted aryl halides to be 0.038-0.05 M and 0.5 mol % respectively.
Table 2.
Determination of the Catalyst Loading Required for the Coupling of Ammonia with ortho-Substituted Aryl Halidesa
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | aryl halide | loading (%) | concentration [M] | T [°C]b | t [h] | conv. (%)c | A:Bd |
| 1 | 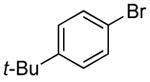 |
0.5 | 0.04 | 80 | 24 | 100 | >30:1 |
| 2 | 0.25 | 0.04 | 80 | 24 | <10 | -- | |
| 3 | 0.1 | 0.1 | 100 | 24 | 100 | 2:1 | |
| 4 | 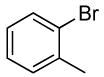 |
0.1 | 0.1 | 100 | 24 | 100 | 17:1 |
| 5 | 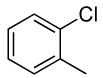 |
0.1 | 0.1 | 100 | 24 | 100 | 17:1 |
Reactions conducted with 1:1 ratio of metal to ligand, 0.5 mmol aryl bromide, 5 mL of 0.5 M ammonia solution, 1.4 equiv NaOtBu in 1,4-dioxane.
Temperature of bath.
Determined by GC analysis.
Determined by 1H NMR spectroscopy.
For reactions of ortho-substituted aryl halides, a second set of conditions was identified. Reactions of 2-bromotoluene and 2-chlorotoluene with ammonia at 0.1 M of the haloarene occurred with selectivities that are similar to those of reactions at 0.05 M concentration (Table 2, entries 4 and 5). At the 0.1 M concentration, these reactions occurred to full conversion of the aryl halides in the presence of only 0.1 mol % catalyst.
2.2. Coupling of Ammonia with Aryl Bromides
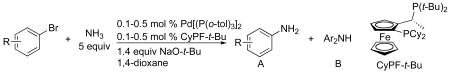
Using the reaction conditions identified in section 2.1, we evaluated the scope of the coupling of aryl bromides with ammonia as a 0.5 M solution in dioxane. The results of this study are summarized in Tables 3 and 4. The scope of these reactions encompasses those of aryl bromides that are electron-rich, electron-neutral and electron-poor and those of aryl bromides that have a range of substitution patterns.
Table 3.
Coupling of ortho-Substituted Aryl Bromides with Ammoniaa
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | aryl bromide | loading (%) | concentration [M] | T [°C]b | t [h] | product | yield (%)c | A:Bd | |
| 1 |  |
0.5 | 0.038 | 80 | 4 |  |
83 | >50:1 | |
| 2 | 0.1 | 0.1 | 100 | 12 | 71 | 17:1 | |||
| 3 | 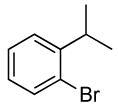 |
0.5 | 0.07 | 80 | 4 | 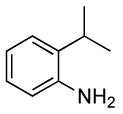 |
93 | >50:1 | |
| 4 | 0.1 | 0.1 | 100 | 12 | 84 | >50:1 | |||
| 5 | 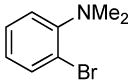 |
0.5 | 0.07 | 80 | 4 | 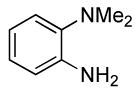 |
89 | >50:1 | |
| 6 | 0.1 | 0.1 | 100 | 12 | 82 | >50:1 | |||
| 7 | 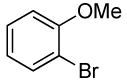 |
0.5 | 0.038 | 80 | 4 | 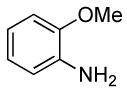 |
85 | >50:1 | |
| 8 | 0.1 | 0.1 | 100 | 12 | 95 | >50:1 | |||
| 9 | 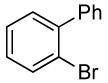 |
0.5 | 0.038 | 80 | 12 | 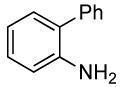 |
96 | >50:1 | |
| 10 | 0.1 | 0.1 | 100 | 24 | 99 | >50:1 | |||
| 11 | 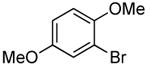 |
0.1 | 0.1 | 100 | 12 |  |
95 | >50:1 | |
| 12 | 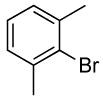 |
0.5 | 0.1 | 80 | 15 | 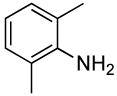 |
88 | >50:1 | |
| 13 | 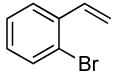 |
0.5 | 0.038 | 80 | 5 | 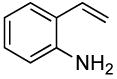 |
66 | -- | |
Reactions conducted with 1:1 ratio of metal to ligand, 0.5 mmol aryl bromide, 5 mL of 0.5 M ammonia solution, 1.4 equiv NaOtBu in 1,4-dioxane.
Temperature of bath.
Isolated yield after purification by flash column chromatography.
Determined by 1H NMR spectroscopy.
Table 4.
Coupling of meta- and para-Substituted Aryl Bromides with Ammoniaa
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | aryl bromide | loading (%) | concentration [M] | T [°C]b | t [h] | product | yield (%)c | A:Bd |
| 1 | 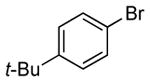 |
0.5 | 0.038 | 80 | 6 | 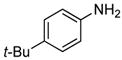 |
88 | 19:1 |
| 2 | 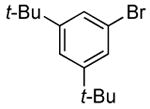 |
0.5 | 0.038 | 80 | 5 | 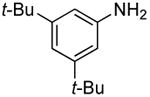 |
78 | -- |
| 3 | 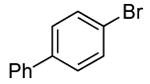 |
0.5 | 0.038 | 80 | 5 | 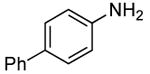 |
98 | -- |
| 4 | 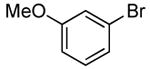 |
0.5 | 0.038 | 80 | 5 | 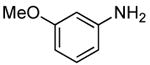 |
99 | >50:1 |
| 5 | 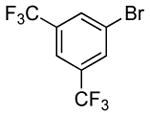 |
0.5 | 0.038 | 80 | 5 |  |
61 | 12:1 |
| 6 | 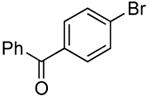 |
0.5 | 0.038 | 80 | 12 | 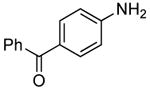 |
77 | -- |
| 7 |  |
1.0 | 0.05 | 100 | 15 | 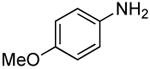 |
53 | 9:1 |
| 8 | 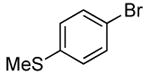 |
1.0 | 0.05 | 90 | 15 | 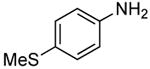 |
91 | >50:1 |
| 9 | 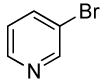 |
0.5 | 0.038 | 80 | 12 | 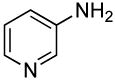 |
60 | -- |
| 10 | 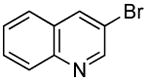 |
0.5 | 0.05 | 80 | 12 | 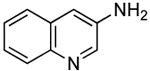 |
64 | -- |
| 11 | 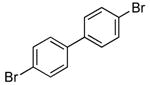 |
2.0 | 0.05 | 100 | 12 | 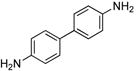 |
79 | >50:1 |
Reactions conducted with 1:1 ratio of metal to ligand, 0.5 mmol aryl bromide, 5 mL of 0.5 M ammonia solution, 1.4 equiv NaOtBu in 1,4-dioxane.
Temperature of bath.
Isolated yield after purification by flash column chromatography.
Determined by 1H NMR spectroscopy.
2.2.1. Reactions of Ortho-Substituted Aryl Bromides
The reactions of ortho-substituted aryl bromides with ammonia encompass substrates containing a wide range of functional groups. For example, ammonia coupled with electron-neutral and sterically hindered bromoarenes (Table 3, entries 1 and 3), as well as ortho-substituted aryl bromides containing aromatic and vinylic substituents (Table 3, entries 9 and 13) to form the primary arylamine products in excellent yields in the presence of 0.5 mol % of the catalyst. It is notable that the reaction of 2-bromostyrene occurred without substantial polymerization. Electron-rich, ortho-substituted aryl halides also coupled with ammonia in high yields to form the primary arylamines. For example, the reactions of ammonia with 2-bromo-N,N-dimethylaniline and 2-bromoanisole occurred with 0.5 mol % of the catalyst at 80 °C in only 4 h to afford excellent yields of the coupled products (Table 3, entries 5 and 7). 2-Bromo-m-xylene is a challenging substrate for both palladium- and copper-catalyzed amination chemistry because of its steric bulk.12 Nevertheless, the reaction of this substrate with ammonia occurred with 0.5 mol % of the catalyst at 100 °C in 15 h to afford excellent yield of the primary arylamine product (Table 3, entry 12).
Reactions of ammonia with a 0.1 M concentration of ortho-substituted aryl halides even occurred with catalyst loadings down to 0.1 mol %. Reactions conducted with 0.1 mol % catalyst required a longer reaction time (12 h) and a higher temperature (100 °C) for complete conversion of ortho-substituted aryl bromides than those conducted with 0.5 mol % of catalyst. However, yields and selectivities of the primary arylamines remained high for substrates containing alkyl, aryl, amino, and methoxy substituents (Table 3, entries 2, 4, 6, 8, 10 and 11). These reactions were conducted with the lowest loadings of any copper or palladium catalyst for the coupling of aryl halides with ammonia.
2.2.2. Reactions of Meta- and para-Substituted Aryl Bromides
In our prior work, the reactions of sterically unhindered aryl bromides occurred with lower selectivity for formation of monarylamine versus diarylamines, especially when a low pressure of ammonia was used.13 Under our new conditions, however, high selectivities were observed for the formation of primary arylamines from reactions of para-, as well as meta-substituted aryl bromides, with ammonia as a 0.5 M solution in dioxane. These results are summarized in Table 4. Electron-neutral 1-bromo-4-tert-butylbenzene and 1-bromo-3,5-di-t-butylbenzene coupled with ammonia to afford primary arylamine products in 88% and 78% yields respectively (Table 4, entries 1 and 2). 4-Bromobiphenyl also coupled with ammonia in excellent yield (Table 4, entry 3).
The scope of this process encompasses aryl bromides possessing a wide range of electronic properties. For example, reactions of electron-deficient 3-bromoanisole and 3,5-bis(trifluoromethyl)bromobenzene with ammonia occurred with 0.5 mol % catalyst loading at substrate concentration of 0.038 M to give the primary arylamine products in excellent yields and selectivities (Table 4, entries 4 and 5). An aryl bromide containing keto functionality also coupled with ammonia in high yield (Table 4, entry 6). Reactions of electron-rich aryl bromides with ammonia occurred to give good yields of primary arylamine products, but required somewhat higher temperatures and catalyst loadings. However, 4-bromoanisole and 4-bromothioanisole still coupled with ammonia in the presence of 1 mol % of the catalyst at 90 – 100 °C to give 53% and 91% yields respectively (Table 4, entries 7 and 8).
Heteroaryl bromides also reacted with ammonia in the presence of Pd[P(o-tol)3]2 and CyPF-t-Bu to give valuable heteroaryl amines in good yields. For example, reactions of 3-bromopyridine and 3-bromoquinoline with ammonia in the presence of 0.5 mol % of the catalyst occurred to full conversion after 12 h at 80°C to give the primary arylamine products in good yields (entries 9 and 10). 4,4′-Benzidine is the core motif of N,N'-bis(3-methylphenyl)-N,N'-diphenylbenzidine (TPD), which is used in hole-transport layer in electroluminescent devices.24 The coupling of 4,4′-dibromobiphenyl with ammonia to form 4,4′-benzidine by formation of two C-N bonds occurred with 2.0 mol % of the catalyst at 100 °C in good yield. Overall, the results presented in Table 4 demonstrate the broadest substrate scope under the mildest conditions for the coupling of ammonia with aryl bromides with any current catalyst.
2.2.3. Comparison to the Reactions Catalyzed by Copper Catalysts
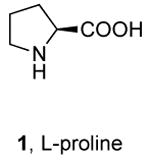
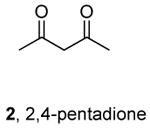
Chang,10 Taillefer11 and Thadani12 recently reported copper-catalyzed coupling of aryl halides with ammonia.25 Chang showed that complexes generated from 20 mol % CuI and 40 mol % of L-proline catalyze the coupling of aryl iodides and electron-poor aryl bromides with NH4Cl or NH4OH. Taillefer reported the coupling of aryl iodides and aryl bromides with NH4OH in the presence of 10 mol % Cu(acac)2 and 40 mol % 2,4-pentadione. Although the reactions conducted with 2,4-pentadione as the ligand precursor occur with broader scope than those with proline, both papers reported that aryl iodides and bromides containing ortho-substituents coupled with ammonia in low yields. In contrast to these two reports, Thadani and coworkers recently reported that the copper-carbene complex 3 catalyzes the coupling of ortho-substituted as well as electron-rich aryl bromides.
Table 5 provides a comparison of the activity of the palladium catalyst generated from Pd[P(o-tol)3]2 and CyPF-t-Bu (condition D) to the copper catalysts generated in situ from CuI and L-proline 1 (condition A) or 2,4-pentadione 2 (condition B) or the discrete complex 3 containing an N-heterocyclic carbene ligand (condition C) for the reactions of ammonia with ortho-substituted 2-iodotoluene and 2-bromoanisole and electron-neutral 4-bromo-tert-butylbenzene. The data in this table were obtained from reactions conducted under the reported optimal conditions for each of the catalyst systems.10-12 The reaction of 2-iodotoluene with ammonia in dioxane in the presence of 0.5 mol % of an equimolar amount of Pd[P(o-tol)3]2 and CyPF-t-Bu gave 73% of ortho-toluidine. The reaction of 2-iodotoluene with ammonia in water catalyzed by copper complexes containing L-proline 1 and 2,4-pentadione 2 gave substantially lower yields. The reaction conducted with complex 3 did not occur to form any coupled product that could be observed by GC/MS. Likewise, the coupling of the ortho-substituted 2-bromoanisole with ammonia in the presence of the copper catalysts occurred with yields that are substantially lower than those of the reaction catalyzed by 0.1 mol% of an equimolar amount of Pd[P(o-tol)3]2 and CyPF-t-Bu (Table 5, entry 2).26
Table 5.
Comparison of the Activity of Copper Catalysts versus Pd[P(o-tol)3]2 and CyPF-t-Bu in the Coupling of Ammonia with Aryl Halides.
| Entry | Aryl Halide | Condition A (CuI/1)a |
Condition B (Cu(acac)2/2)b |
Condition C (3) |
Condition D (Pd/CyPF-t-Bu) |
||
|---|---|---|---|---|---|---|---|
| 1 | X = I, R = 2-Me | 17% | 30% | --e | 73% | ||
| 2 | X = Br, R = 2-OMe | trace | --e | --e | 95% | ||
| 3 | X = Br, R = 4-tBu | 84% | 55% | --e | 88% | ||
 |
 |
 |
|||||
Reaction conditions A: 0.5 mmol aryl halide, 0.65 mmol NH4OH, 0.1 mmol CuI, 0.2 mmol L-proline, 1.5 mmol K2CO3, 1 mL DMSO, 0.05 mL H2O, 80 °C, 24 h.
Reaction conditions B: 1 mmol aryl halide, 300 mL NH4OH, 0.1 mmol Cu(acac)2, 0.4 mmol 2,4-pentadione, 2 mmol Cs2CO3, 2 mL DMF, 90 °C, 24 h.
Reaction conditions C: 1 mmol aryl halide, 0.05 mmol 3, 2 mmol K2CO3, 5 mL (1:1, MeOH/NMP), 90 °C, 24 h.
Reaction conditions D: 0.5 mmol aryl halide, 5 mL of 0.5 M ammonia in dioxane, 0.7 mmol NaOtBu; entry 1: 0.0025 mmol Pd[P(o-tol)3]2/CyPF-t-Bu, 5 ml of 0.5 M ammonia in dioxane, 80 °C; entry 2: 0.0005 mmol Pd[P(o-tol)3]2/CyPF-t-Bu, 5 ml of 0.5 M ammonia in dioxane, 100 °C; entry 3: 0.0025 mmol Pd[P(o-tol)3]2/CyPF-t-Bu, 5 ml of 0.5 M ammonia in dioxane, 5 mL dioxane, 80 °C.
No product observed by GC/MS.
The differences in yields from the reactions of sterically unhindered substrates with the different catalysts were less dramatic, but the reaction catalyzed by 0.5 mol% of Pd[P(o-tol)3]2 and CyPF-t-Bu still formed the primary arylamine in the highest yield. The coupling of 4-bromo-tert-butylbenzene with ammonia in the presence of copper complexes containing 40 mol % 1 and 40 mol % 2 gave 84% and 55% yield of 4-tert-butylaniline, respectively. Again, the reaction catalyzed by complex 3 afforded no coupled product that could be observed by GC/MS. In contrast, the reaction catalyzed by 0.5 mol% of Pd[P(o-tol)3]2 and CyPF-t-Bu formed the primary arylamine in 88% yield.
2.3. Coupling of Ammonia with Aryl Chlorides and Aryl Iodides
Because aryl chlorides are less expensive than aryl bromides and iodides and more derivatives are commercially available, the synthesis of primary arylamines from aryl chlorides would be particularly valuable. However, the carbon-chlorine bonds in these reagents are less reactive than their bromide and iodide counterparts.27,28 Prior to our current work, only one example of the coupling of ammonia with an aryl chloride had been reported to give high yield of the primary arylamine product, and this example was conducted with a catalyst generated from CyPF-t-Bu.13 We show here that the reactions of a series of aryl chlorides with ammonia catalyzed by the combination of Pd[P(o-tol)3]2 and CyPF-t-Bu lead to high yields of primary arylamines with relatively low catalyst loadings.
The scope of the coupling of ammonia with aryl chlorides and iodides is summarized in Table 6. The catalyst generated from Pd[P(o-tol)3]2 and CyPF-t-Bu catalyzed the coupling of ammonia with aryl chlorides in high yields under the same reaction conditions described for the coupling of aryl bromides. The reactions of ortho-subsituted aryl chlorides occurred in particularly high yields and selectivities for the primary arylamine. For example, reactions of 2-chlorotoluene and 2,5-dimethylchlorobenzene with ammonia occurred with 0.5 mol % of the catalyst at 80 °C for 24 h to afford the primary arylamines in 64% and 85% yields respectively (Table 6, entries 1 and 3). Like the couplings of ortho-substituted aryl bromides, the coupling of aryl chlorides with ammonia occurred in the presence of only 0.1 mol % of catalyst at substrate concentration of 0.1 M without significant reduction in yield (Table 6, entries 2 and 4). The electron-rich 2-chloroanisole also reacted under this low catalyst loading to give high yield of ortho-anisidine (Table 6, entry 5). Even the coupling of the 2,6-disubstituted 2-chloro-m-xylene with ammonia in the presence of 1 mol % catalyst occurred to full conversion of the aryl chloride to afford high yield of the primary arylamine product (Table 6, entry 6). The reaction of ammonia with the pseudo ortho-substituted, fused polyarene 1-chloronaphthalene also occurred in high yield (Table 6, entry 7).
Table 6.
Coupling of Aryl Chlorides and Iodides with Ammoniaa
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | aryl bromide | loading (%) | concentration [M] | T [°C]b | t [h] | product | yield (%)c | A:Bd |
| 1 | 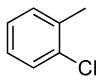 |
0.5 | 0.07 | 100 | 24 | 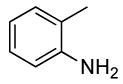 |
64 | 17:1 |
| 2 | 0.1 | 0.1 | 100 | 24 | 63 | -- | ||
| 3 | 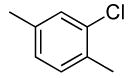 |
0.07 | 0.07 | 100 | 24 |  |
85 | >50:1 |
| 4 | 0.1 | 0.1 | 100 | 24 | 63 | -- | ||
| 5 | 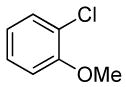 |
0.1 | 0.1 | 100 | 24 | 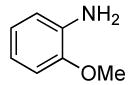 |
84 | -- |
| 6 | 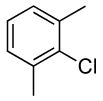 |
1 | 0.1 | 100 | 24 | 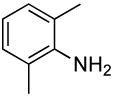 |
89 | >50:1 |
| 7 | 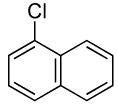 |
0.5 | 0.038 | 100 | 12 | 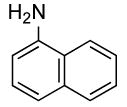 |
89 | -- |
| 8 | 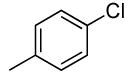 |
1 | 0.038 | 100 | 10 | 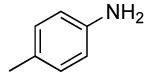 |
55 | 12:1 |
| 9 | 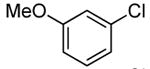 |
0.5 | 0.038 | 100 | 12 | 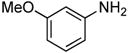 |
99 | -- |
| 10 | 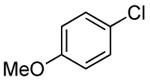 |
1 | 0.05 | 100 | 12 | 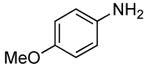 |
69 | -- |
| 11 | 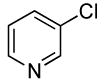 |
1 | 0.038 | 100 | 12 | 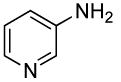 |
64 | -- |
| 12 | 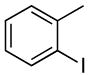 |
0.5 | 0.1 | 80 | 12 | 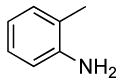 |
73 | -- |
Reactions conducted with 1:1 ratio of metal to ligand, 0.5 mmol aryl chloride, 5 mL of 0.5 M ammonia solution, 1.4 equiv NaOtBu in 1,4-dioxane.
Temperature of bath.
Isolated yield after purification by flash column chromatography.
Determined by 1H NMR spectroscopy.
Sterically unhindered aryl chlorides and heteroaryl chlorides ranging from electron-deficient to electron-rich also coupled with ammonia in high yields and selectivities. For example, reactions of 4-chlorotoluene and 3-chloroanisole with ammonia occurred at 0.038 M concentration of the substrates to afford good yields of coupled products (Table 6, entries 8 and 9). The coupling of electron-rich substrates with ammonia is challenging for reasons that are discussed later in this paper. Nevertheless, the reaction of electron-rich 4-chloroanisole with ammonia occurred with 1 mol % of the catalyst at 100 °C for 12 h to give 69% yield of para-anisidine. The conditions for reactions of aryl chlorides proved to be suitable for the reactions of heteroaryl chlorides. The reaction of 3-chloropyridine with ammonia occurred in 64% isolated yield (Table 6, entry 11).
The generality of this method is further demonstrated by the high yields of primary arylamines derived from aryl iodides. The reactions of aryl iodides with nitrogen nucleophiles are known to occur in yields and selectivities that are often lower than those of reactions of aryl bromides.29-42 Nevertheless, the reaction of 2-iodotoluene with ammonia in the presence of 0.5 mol % of catalyst occurred to full conversion and formed a good yield of the coupled product (Table 6, entry 12).
2.4. Coupling of Aryl Sulfonates with Ammonia
Prior to this work, the coupling of ammonia with aryl sulfonates formed the primary arylamines in low yields.13 The yields were low because the C-N coupling process competes with uncatalyzed cleavage of the S-O bond of the sulfonyl group by ammonia in the presence of the strong base NaO-t-Bu to form aryl alcohols as the major product. In 2003, we showed that the complex generated from Pd[P(o-tol)3]2 and CyPF-t-Bu undergoes the oxidative addition of aryl tosylates rapidly at room temperature.20 More recently, the authors' laboratory showed that the coupling of aryl tosylates with alkyl- and arylamines occurs at room temperature when catalyzed by the species generated from Pd[P(o-tol)3]2 and CyPF-t-Bu.22 The low temperatures of the coupling of primary amines suggested that the coupling of ammonia could occur faster than cleavage of an aryl sulfonates. Indeed, we found that the coupling of ammonia with aryl sulfonates conducted with 2 mol % catalyst, at a temperature of 50 °C, and with a tosylate as the sulfonate group occurred faster than S-O bond cleavage. Reactions of aryl triflates led to the phenol as the major product from S-O bond cleavage.
The results of our study on the scope of the coupling of aryl tosylates with ammonia under these conditions are shown in Table 7. Reactions of sterically hindered aryl tosylates with ammonia produced primary arylamines in good to excellent yields. For example, 2-methylphenyl p-toluenesulfonate and 2,4,6-trimethylphenyl p-toluenesulfonate coupled with ammonia in the presence of 2 mol % of the catalyst at 50 °C for 24 h to give primary arylamines in 65% and 86% yield respectively (Table 7, entries 1 and 2). Less sterically hindered aryl tosylates reacted with ammonia to give slightly lower yields of the arylamine than did ortho-substituted aryl tosylates. We attributed this result to the faster rates of formation of aryl alcohol from less sterically hindered tosylates. For example, the reactions of 1-naphthyl p-toluenesulfonate and 2-naphthyl p-toluenesulfonate with ammonia occurred with 2 mol % of the catalyst to afford the coupled products in 67% yield in both cases (Table 7, entries 3 and 4). Heteroaryl tosylates also coupled with ammonia to form synthetically useful, albeit lower, yields of the primary heteroarylamines. For example, the reaction of 6-quinolinyl p-toluenesulfonate with ammonia afforded 55% yield of the coupled product (Table 7, entry 5). Improvements in the reactions of sterically unhindered aryl tosylates will be the focus of future work, but these results represent the first coupling of aryl sulfonates with ammonia and demonstrate that the synthesis of primary arylamines from aryl alcohols by this approach is feasible.
Table 7.
Coupling of Ammonia with Aryl Tosylatesa
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | aryl bromide | loading (%) | concentration [M] | T [°C]b | t [h] | product | yield (%)c | A:Bd |
| 1 | 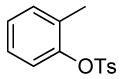 |
2 | 0.1 | 50 | 24 | 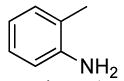 |
65 | 17:1 |
| 2 | 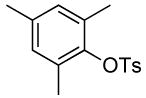 |
2 | 0.1 | 50 | 24 | 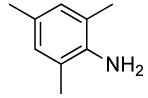 |
86 | >50:1 |
| 3 | 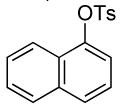 |
2 | 0.05 | 50 | 24 | 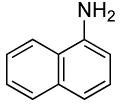 |
67 | >50:1 |
| 4 | 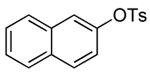 |
2 | 0.05 | 50 | 24 | 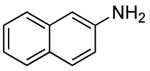 |
67 | --e |
| 5 | 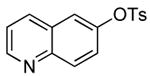 |
2 | 0.05 | 50 | 24 | 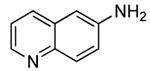 |
55 | --e |
Reactions conducted with 1:1 ratio of metal to ligand, 0.5 mmol aryl chloride, 5 mL of 0.5 M ammonia solution, 1.4 equiv NaO-t-Bu in 1,4-dioxane.
Temperature of bath.
Isolated yield after purification by flash column chromatography.
Determined by 1H NMR spectroscopy.
Selectivity was not determined due to overlapping resonances in the aromatic region of the 1H NMR spectra of the crude reaction mixtures.
2.5. Coupling of Ammonia with Aryl Halides Containing Base-sensitive Functional Groups
2.5.1. Identification of Reaction Conditions
The scope of the palladium-catalyzed coupling of ammonia with aryl halides containing base-sensitive functional groups, such as ketones with enolizable protons, esters and nitriles, has been limited. We presumed that this limitation in scope results from the use of the strong base NaO-t-Bu.
Thus, our initial attempts to expand the scope of the coupling of ammonia to encompass base-sensitive functional groups focused on conducting reactions with weak bases, such as Cs2CO3 and K3PO4 that have helped to address issues of functional group compatibility in the coupling of amines.43,44 We studied reactions of ammonia with ethyl 4-bromobenzoate to evaluate conditions required to obtain high yield and selectivity of the primary arylamine from base-sensitive haloarenes.
The reaction of ammonia with ethyl 4-bromobenzoate conducted in 0.5 M solutions of dioxane in the presence of 0.5 mol % of equimolar amounts of Pd[P(o-tol)3]2 and CyPF-t-Bu and 5 equiv of K3PO4 occurred to full conversion, but only the diarylamine product was observed. Apparently the more acidic arylamine product reacts faster than ammonia in reactions conducted with the weaker base. However, reactions conducted with higher concentration of ammonia occurred with higher selectivity for formation of the primary arylamine. For example, the reaction of ethyl 4-bromobenzoate with 200 psi of ammonia in the presence of 0.5 mol % of equimolar amounts of Pd[P(o-tol)3]2 and CyPF-t-Bu and 5 equiv of K3PO4 occurred to full conversion and gave a 30:1 ratio of the primary arylamine to the diarylamine.
2.5.2. Scope of the Coupling of Ammonia with Aryl Halides and Sulfonates Containing Base-Sensitive Functional Groups
Using the conditions described in section 2.5.1, we studied the scope of the coupling of ammonia with aryl halides that contain base-sensitive functional groups. The results of this study are summarized in Table 8. Aryl halides and tosylates containing base-sensitive, electron-withdrawing functional groups at the para-position coupled with ammonia in high yields. For example, reactions of ethyl 4-bromobenzoate and methyl 4-bromobenzoate with ammonia occurred with only 0.5 mol % of the catalyst to give 94% and 83% yields of primary arylamines respectively (Table 8, entries 1 and 3). Aryl halides containing ketones with enolizable protons such as 4′-bromoacetophenone and 4′-bromopropiophenone coupled with ammonia to give high yields of coupled products (Table 8, entries 5 and 7).
Table 8.
Coupling of Ammonia with Aryl Halides and Sulfonates Containing Base-sensitive Functional Groupsa
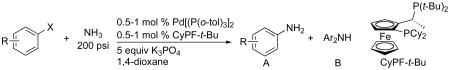 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | aryl halide | loading (%) | con. [M] | T [°C]b | t [h] | product | yield (%)c | A:Bd |
| 1 | 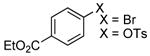 |
0.5 | 0.1 | 70 | 12 | 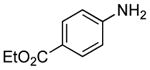 |
94 | 30:1 |
| 2 | 0.5 | 0.1 | 70 | 12 | 73 | 6:1 | ||
| 3 | 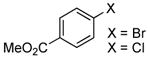 |
0.5 | 0.1 | 70 | 12 | 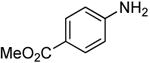 |
83 | >30:1 |
| 4 | 0.5 | 0.1 | 70 | 12 | 79 | >30:1 | ||
| 5 | 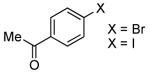 |
0.5 | 0.1 | 70 | 12 | 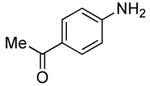 |
76 | -- |
| 6 | 1 | 0.1 | 70 | 12 | 78 | -- | ||
| 7 | 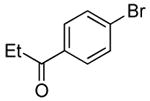 |
0.5 | 0.1 | 70 | 12 | 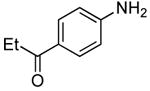 |
85 | -- |
| 8 |  |
0.5 | 0.1 | 70 | 12 |  |
71 | -- |
| 9 | 0.5 | 0.1 | 70 | 12 | 44 | |||
| 10 | 0.5 | 0.1 | 70 | 12 | 77 | |||
Reactions conducted with 1:1 ratio of metal to ligand, 0.5 mmol aryl chloride, and 200 psi of ammonia in a 50 mL Parr bomb, 1.4 equiv NaOtBu in 1,4-dioxane.
Temperature of bath.
Isolated yield after purification by flash column chromatography.
Determined by 1H NMR spectroscopy.
Cyano functionality is typically stable toward reactions in the presence of NaO-t-Bu and either alkyl-or arylamine nucleophiles,29,45 but 4-halobenzonitriles reacted with ammonia in the presence of NaO-t-Bu with and without the palladium catalyst to form multiple products. In contrast to the reactions of ammonia in the presence of the strong alkoxide base, the reaction of 4-bromobenzonitrile with ammonia and K3PO4 as base catalyzed by Pd[P(o-tol)3]2 and CyPF-t-Bu occurred to give 71% yield of the coupled product (Table 8, entry 8).
The ability of the catalyst generated from Pd[P(o-tol)3]2 and CyPF-t-Bu to add aryl halides under mild conditions and the apparent stability of this catalyst toward iodide byproduct also led to increases in reaction scope. For example, reactions of ammonia with methyl 4-chlorobenzoate and 4-chlorobenzonitrile occurred to give high yields of primary arylamines without formation of amide or amidine side products (Table 8, entries 4 and 9). Moreover, the reaction of the electron-poor 4′-iodoacetophenone with ammonia in the presence of 1 mol % of Pd[P(o-tol)37]2 and CyPF-t-Bu afforded good yield of the aniline product (Table 8, entry 6).
The ability to conduct these couplings with K3PO4 as base also allowed us to develop the coupling of aryl tosylates containing base-sensitive functionality. The rate of formation of aryl alcohol side products should be slower in the presence of ammonia and K3PO4 than in the presence of ammonia and NaO-t-Bu, while the catalyst should add the electron-poor aryl tosylate with a rate that is faster than it adds electron-neutral and electron-rich aryl tosylates. Consistent with these expectations, reactions of ammonia with 4-cyanophenyl p-toluenesulfonate and ethyl 4-{[(4-methylphenyl)sulfonyl]oxy}benzoate occurred with only 0.5 mol % of the catalyst to afford 73% and 79% yields of coupled products, respectively (Table 8, entries 2 and 10). Because the aryl halides shown in Table 8 are electron-poor, control reactions were conducted without the palladium catalyst to determine if the reactions were truly metal catalyzed. No measurable quantity of primary arylamine product was observed in the absence of palladium for reactions of any of the aryl halides shown in Table 8. Some conversion was observed during reactions of aryl tosylates with ammonia and K3PO4 in the absence of catalyst at 80 °C, but these reactions formed the corresponding aryl alcohols by S-O bond cleavage, not the desired arylamines.
The reactions of ammonia with more electron-rich aryl halides and sulfonates that contain base-sensitive functional groups gave low yields of primary arylamines in the presence of Pd[P(o-tol)3]2 and CyPF-t-Bu as catalyst. A rationalization for this result will be provided later in this paper. Despite this limitation, our method allows for the coupling of ammonia with a set of aryl iodides, bromides, chlorides and sulfonates that contain base-sensitive functional groups. The ability to conduct these reactions with this range of halides and with sulfonates constitutes a significant expansion of the scope of related reactions described previously with other palladium or copper catalysts.10-14
2.6. Sequential Reactions Initiated by the Coupling of Ammonia with Aryl Halides
Although primary arylamines are often the desired end product, in many instances the primary arylamine would be used to generate diarylamines, amides, sulfonamides, imides or other products containing an aromatic C-N bond. In some cases, the reagents one would use for the direct coupling to form these classes of products have not been shown to undergo C-N coupling in a reliable fashion, whereas, in other cases, one might wish to generate the arylamine as a means to generate a library of compounds derived from the initial arylamine product. Having developed an efficient synthesis of arylamines, we sought to demonstrate the utility of the arylation of ammonia as a means to generate arylamine derivatives. Two examples of such sequential reaction chemistry are shown here.
2.6.1. Sequential Arylation of Ammonia by a Single Catalyst in One-Pot
Unsymmetrical diarylamines are intermediates to unsymmetrical triarylamines, which have numerous applications in photonic, organic polymers.46 The work described here has demonstrated that the combination of Pd[P(o-tol)3]2 and CyPF-t-Bu is a highly active catalyst for the coupling of aryl halides and tosylates with ammonia to form primary arylamines, and the authors' group has recently shown that this same combination of metal and ligand catalyzes the coupling of aryl and heteroaryl tosylates with arylamines at room temperature to form mixed diarylamines.22 Thus, we sought to develop a sequential coupling of ammonia with aryl halides and sulfonates in the presence of a single catalyst to form mixed diarylamines.
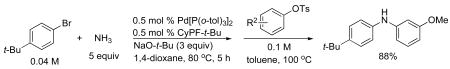
The catalyst generated from Pd[P(o-tol)3]2 and CyPF-t-Bu does allow the sequential coupling of ammonia with two different aryl electrophiles to occur in one pot. As shown in eq 2, the arylation of ammonia with 4-bromo-tert-butylbenzene occurs in the presence of 0.5 mol % of Pd[P(o-tol)3]2 and CyPF-t-Bu and 3 equiv of NaO-t-Bu. After full conversion of the aryl bromide, evaporation of the dioxane, followed by addition of the aryl tosylate and toluene and heating led to the formation of the unsymmetrical diarylamine in high yield (eq 2). Sequential coupling of two different aryl halides with ammonia can also be conducted on one pot and will be reported in due course.
 |
(2) |
2.6.2. One-pot Synthesis of Amides and Imides from Aryl Halides
The coupling of ammonia with aryl halides creates the ability to conduct a one-pot synthesis of aniline derivatives that are challenging to obtain directly by C-N coupling. These derivatives include amides, imides and carbamates. The amide moiety is present in many biologically active compounds, and work has been conducted to develop methods to access libraries of amides for studies on structure-reactivity relationships.47-50 Palladium-51,52 and copper-catalyzed53,54 amidation have limitations in substrate scope, require high catalyst loadings, and are sufficiently challenging in many cases that parallel synthesis would require attention to the development of conditions for individual substrate combinations.
N-Aryl imides, in particular phthalimides, have been used extensively as intermediates in the manufacture of dyes,55 polymers,56,57 and pesticides,58 and this class of compound has also been shown to be biologically active.59,60 Amides and imides are widely prepared by reactions between amines and acid chlorides or anhydrides, but this procedure requires an arylamine reagent. The N-arylation of imides has not been reported with palladium catalysts and has not been reported with modern copper catalysts.
The preparation of N-Boc-arylamines via palladium or copper-catalyzed coupling of aryl halides with tert-butyl carbamate is limited to a few examples conducted with palladium catalysts generated from either xantphos, P(t-Bu)3, or tert-butyl X-Phos as ligands.45,61-63 No reactions of aryl chlorides or sulfonates have been reported, and the reactions of ortho-substituted aryl halides were limited to one electron-poor62 and two electron-neutral examples.45,63
Thus, the palladium-catalyzed coupling of an aryl halide with ammonia, followed by the uncatalyzed reaction of the primary amine product with an acid chloride, cyclic anhydide, or Boc anhydride could provide a convenient route to a family of N-aryl amides, -imides and -carbamates from a single aryl halide. Our demonstration of this sequence focused on reactions of ortho-substituted aryl halides because of the direct coupling of this class of aryl halides with primary arylamides, tert-butyl carbamate and phthalimide has been challenging. Yet, a sequence initiated with meta- or para-substituted aryl halides would lead to analogous products.
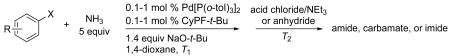
Our data illustrating the two-step, one-pot synthesis of a family of N-aryl amides, imides and carbamates from a single aryl halide are summarized in Table 9. Ortho-substituted aryl halides, such as 2-bromo-N,N-dimethylaniline and 2-chloroanisole, reacted with ammonia in the presence of only 0.1 mol % of the palladium catalysts. After evaporation of excess ammonia under reduced pressure for 5 min, the addition of acid chlorides that range from electron-rich to electron-poor, along with 1 equiv of Et3N to neutralize the released HCl, afforded the N-aryl amides in 70-94% yields (Table 9, entries 1, 3, 5 and 6).
Table 9.
Synthesis of Amides and Imides from Aryl Bromides and Chloridesa
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | aryl halide | electrophile | loading (%) | T1 (°C)b | T2 (°C)b | product | yield (%)c |
| 1d |  |
 |
0.1 | 100 | 25 | 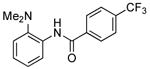 |
89 |
| 2 | 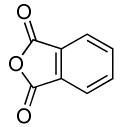 |
0.1 | 100 | 25 |  |
51 | |
| 3d | 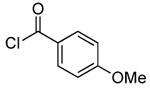 |
0.1 | 100 | 50 | 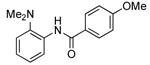 |
74 | |
| 4 | Boc2 | 0.1 | 100 | 80 | 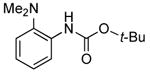 |
75 | |
| 5d | 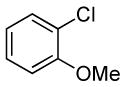 |
 |
0.1 | 100 | 50 | 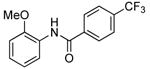 |
82 |
| 6d | 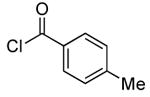 |
0.1 | 100 | 50 | 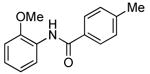 |
94 | |
| 7 | Boc2 | 0.1 | 100 | 80 | 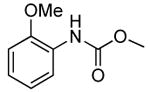 |
68 | |
| 8 | 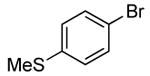 |
 |
0.1 | 90 | 25 | 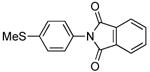 |
75 |
Reactions conducted with 1:1 ratio of metal to ligand.
Temperature of bath.
Isolated yield after purification by flash column chromatography.
1 equiv of Et3N was added.
This one-pot method also formed N-aryl carbamates in good yield. The conversion of 2-bromo-N,N-dimethylaniline and 2-chloroanisole with low loading of the palladium catalyst (0.1 mol %) followed by addition of Boc-anhydride formed the corresponding N-Boc protected anilines in 75% and 68% yields, respectively (Table 9, entries 4 and 7).
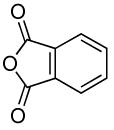
Finally, this sequence conducted with phthalic anhydride in the second step occurred to form N-aryl imides. The reactions of phthalic anhydride with sterically hindered arylamines formed by the C-N coupling of ortho-substituted aryl halides gave lower yields of N-aryl phthalimide than did those of sterically unhindered arylamines. However, the imide derived from 2-bromo-N,N-dimethylaniline was formed by the two-step, one-pot protocol in 51% yield. The coupling of ammonia with the less hindered 4-bromothioanisole, followed by addition of phthalic anhydride, gave 75% yield of the corresponding N-aryl imide.
2.7. Mechanistic Studies
Although a full evaluation of the mechanism of the coupling of ammonia with aryl halides awaits further study, several initial mechanistic experiments provide insight into the factors that differentiate the current catalyst from those used previously containing the CyPF-t-Bu ligand and that differentiate the reactivity of aryl halides containing electron-withdrawing groups in the para and meta positions. These experiments evaluated the efficiency of the generation of the active palladium(0) catalyst from the palladium(II) precursors and identified the turnover-liminting steps of the reactions of electron-poor aryl halides.
2.7.1. Studies on the Reactions of Palladium(II) Precursors with Ammonia and Base
To address the origin of the higher reactivity of complexes generated from Pd[P(o-tol)3]2 than those generated from Pd(OAc)2 or (CyPF-t-Bu)PdCl2, we studied the catalytic reactions of 4-bromo-tert-butylbenzene with ammonia catalyzed by (CyPF-t-Bu)PdCl2 or the combination of CyPF-t-Bu and Pd(OAc)2. 31P NMR spectra of reactions conducted with (CyPF-t-Bu)PdCl2 or the combination of CyPF-t-Bu and Pd(OAc)2 produced complex mixtures of palladium species, whereas the reaction catalyzed by the combination of Pd[P(o-tol)3]2 and CyPF-t-Bu generated a single major species. From this observation, we concluded that the combination of Pd[P(o-tol)3]2 and CyPF-t-Bu generates a higher concentration of active catalyst than other catalyst precursors.
To address the fate of the Pd(II) species in the presence of ammonia and base in more detail, we determined the amount of (CyPF-t-Bu)Pd(0) species generated from the reaction of (CyPF-t-Bu)PdCl2 with ammonia and base. (CyPF-t-Bu)PdCl2 was treated with NaO-t-Bu and ammonia in the presence of P(o-tol)3 to trap any Pd(0) complex formed. This reaction generated a complex mixture of products that contained less than 10% of the Pd(0) species. Because the alkoxide base and ammonia both lack hydrogens α to the heteroatom, the Pd(II) species cannot be reduced to Pd(0) by the combination of β-hydrogen elimination from an alkoxo or amido intermediate and reductive elimination of amine or alcohol. Because the aryl group serves as the electrophilic component, rather than the nucleophilic component, formation of a bis-aryl complex and reductive elimination of an organic biaryl product also does not lead to formation of Pd(0) in high yield. In contrast, the combination of Pd[P(o-tol)3]2 and CyPF-t-Bu generates Pd[P(o-tol)3]2 and (CyPF-t-Bu)Pd[P(o-tol)3] that adds aryl halides and sulfonates rapidly.20,23
2.7.2. Studies on the Turnover-Limiting Step of Reactions with Weak Base
We also conducted studies to determine whether the use of a weak base would cause transmetalation to be the turnover-limiting step. The reaction of 4′-bromoacetophenone with ammonia at 200 psi in the presence of 5 equiv of K3PO4 was monitored by 31P NMR spectroscopy after approximately 50% conversion of the aryl bromide (Scheme 1). The resting state of the catalyst was identified to be the arylpalladium bromide complex by comparison of the 31P NMR chemical shifts of the species in the catalytic reaction to those of the species formed from the reaction of Pd[P(o-tol)3]2 and CyPF-t-Bu with 4′-bromoacetophenone. This result indicates that transmetalation is, indeed, the turnover-limiting step in the reactions of aryl bromides with ammonia in the presence of the weak base K3PO4.
Scheme 1.

The same experiment was conducted on reactions of aryl halides containing electron-withdrawing groups in the meta-position, such as 3′-bromopropiophenone. Again, the arylpalladium halide complex was observed. Because catalytic reactions of the meta-substituted, electron-poor aryl halides occur in much lower yields than those of the para-substituted analogs, it appears that the rates of the transmetalation and reductive elimination portion of the catalytic cycle are affected strongly by the relative electron-withdrawing ability of the aryl group in the meta- or para-position. Because the pKa values of ammonia and monohydrogen phosphate are so different (33 for ammonia64 and 12 for HPO42-65 respectively), we assume that the formation of the amido complex is facilitated by coordination of ammonia to palladium to increase the acidity of the ammonia, and the aryl group bound to palladium affects the Lewis acidity of the metal center (Scheme 2).
Scheme 2.

3. Conclusions
We have shown that the catalyst generated from the combination of Pd[P(o-tol)3]2 and the sterically hindered alkyl bis-phosphine ligand CyPF-t-Bu is highly active and selective for the coupling of ammonia with aryl halides and sulfonates to form primary arylamine products. For example, reactions of ortho-substituted aryl bromides and chlorides occurred with only 0.1 mol % of catalyst loading. This catalyst leads to a reaction scope that is expanded over that of couplings conducted with previous catalysts. This scope now encompasses aryl chlorides, bromides and iodides that possess or lack an ortho-substituent and for the first time encompasses aryl sulfonates. Moreover, this scope now includes reactions of certain aryl halides containing base-sensitive functional groups such as ketones, esters, and nitriles. The utility of this coupling process to form not only primary arylamines, but mixed secondary arylamines, N-aryl amides, N-aryl imides, and N-aryl carbamates by a one-pot procedure has also been demonstrated.
The efficiency of the catalyst arises from the high yield of a reactive (CyPF-t-Bu)Pd fragment from equimolar amounts of Pd[P(o-tol)3]2 and CyPF-t-Bu. Preliminary studies on the generation of the active catalyst suggest that low yields of the active palladium(0) species are generated from palladium(II) precursors, ammonia and sodium tert-butoxide base. Additional studies imply that the reactions with weak base occur by turnover-limiting transmetalation and that the rate of this step depends on the position of the electron-withdrawing group on the aryl ligand. Detailed mechanistic studies on the catalyst generated from Pd[P(o-tol)3]2 and CyPF-t-Bu will be reported in due course.
Supplementary Material
Acknowledgments
We thank the NIH NIGMS for support of this work (GM-55382), Johnson-Matthey for gifts of palladium complexes, and Solvias AG for gifts of the Josiphos ligand.
Footnotes
Supporting Information Available. Full experimental procedures and characterizations of reaction products. This information is available free of charge via the Internet at www.pubs.acs.org.
References
- 1.Roundhill DM. Chem Rev. 1992;92:1–27. [Google Scholar]
- 2.Zhao J, Goldman AS, Hartwig JF. Science. 2005;307:1080–1082. doi: 10.1126/science.1109389. [DOI] [PubMed] [Google Scholar]
- 3.Gross T, Seayad AM, Ahmad M, Beller M. Org Lett. 2002;4:2055–2058. doi: 10.1021/ol0200605. [DOI] [PubMed] [Google Scholar]
- 4.Ogo S, Uehara K, Abura T, Fukuzumi S. J Am Chem Soc. 2004;126:3020–3021. doi: 10.1021/ja031633r. [DOI] [PubMed] [Google Scholar]
- 5.Zimmermann B, Herwig J, Beller M. Angew Chem Int Ed. 1999;38:2372–2375. doi: 10.1002/(sici)1521-3773(19990816)38:16<2372::aid-anie2372>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 6.Gunanathan C, Milstein D. Angew Chem Int Ed. 2008;47:8661–8664. doi: 10.1002/anie.200803229. [DOI] [PubMed] [Google Scholar]
- 7.Lavallo V, Frey GD, Donnadieu B, Soleilhavoup M, Bertrand G. Angew Chem Int Ed. 2008;47:5224–5228. doi: 10.1002/anie.200801136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagano T, Kobayashi S. J Am Chem Soc. 2009;131:4200–4201. doi: 10.1021/ja900328x. [DOI] [PubMed] [Google Scholar]
- 9.Pouy MJ, Leitner A, Weix DJ, Ueno S, Hartwig JF. Org Lett. 2007;9:3949–3952. doi: 10.1021/ol701562p. [DOI] [PubMed] [Google Scholar]
- 10.Kim J, Chang S. Chem Comm. 2008:3052–3054. doi: 10.1039/b804637a. [DOI] [PubMed] [Google Scholar]
- 11.Xia N, Taillefer M. Angew Chem Int Ed. 2009;48:337–339. doi: 10.1002/anie.200802569. [DOI] [PubMed] [Google Scholar]
- 12.Ntaganda R, Dhudshia B, Macdonald CLB, Thadani AN. Chem Comm. 2008:6200–6202. doi: 10.1039/b815757j. [DOI] [PubMed] [Google Scholar]; After the submission of our manuscript, this paper on the coupling of aryl bromides with ammonia catalyzed by copper complexes of carbene ligands was retracted and deleted from the journal website.
- 13.Shen Q, Hartwig JF. J Am Chem Soc. 2006;128:10028–10029. doi: 10.1021/ja064005t. [DOI] [PubMed] [Google Scholar]
- 14.Surry DS, Buchwald SL. J Am Chem Soc. 2007;129:10354–10355. doi: 10.1021/ja074681a. [DOI] [PubMed] [Google Scholar]
- 15.Schulz T, Torborg C, Enthaler S, Schäffner B, Dumrath A, Spannenberg A, Neumann H, Börner A, Beller M. Chem Eur J. 2009;15:4528–4533. doi: 10.1002/chem.200802678. [DOI] [PubMed] [Google Scholar]
- 16.Weissermel K, Arpe H. Industrial Organic Chemistry. Wiley-VCH; Weinheim, Germany: 1997. [Google Scholar]
- 17.Lawrence SA. Amines: Synthesis, Properties, and Application. Cambridge University Press; Cambridge: 2004. [Google Scholar]
- 18.Lindley J. Tetrahedron. 1984;40:1433–1456. [Google Scholar]
- 19.Shen Q, Ogata T, Hartwig JF. J Am Chem Soc. 2008;130:6586–6596. doi: 10.1021/ja077074w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roy AH, Hartwig JF. J Am Chem Soc. 2003;125:8704–8705. doi: 10.1021/ja035835z. [DOI] [PubMed] [Google Scholar]
- 21.Schulz T, Torborg C, Enthaler S, Schäffner B, Dumrath A, Spannenberg A, Neumann H, Börner A, Beller M. Chem Eur J. 2009;15:4528–4533. doi: 10.1002/chem.200802678. [DOI] [PubMed] [Google Scholar]
- 22.Ogata T, Hartwig JF. J Am Chem Soc. 2008;130:13848–13849. doi: 10.1021/ja805810p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alvaro E, Hartwig JF. J Am Chem Soc. 2009;131:7858–7868. doi: 10.1021/ja901793w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen Y, Klein MW, Jacobs DB, Scott JC, Malliaras GG. Phys Rev Lett. 2001;86:3867. doi: 10.1103/PhysRevLett.86.3867. [DOI] [PubMed] [Google Scholar]
- 25.Soon after the submission of this manuscript, a report on ligandless copper-catalyzed coupling of aryl bromides and chlorides with ammonia in water was published:; Xu H, Wolf C. Chem Comm. 2009:3035–3037. doi: 10.1039/b904188e. [DOI] [PubMed] [Google Scholar]; In our hands, the reaction of 2-bromotoluene with aqueous ammonia by the published protocols using various sources of copper iodide, open or closed vessels, and reaction vessels with varying headspaces afforded less than 10% of ortho-toluidine. Thus, we abandonded studies to compare our system to this one.
- 26.While this work was under review, Ma and coworkers reported the coupling of 2-bromoanisole and 2-bromotoluene with ammonia in water in the presence of 20% CuI and 40 mol % 4-hydroxy-L-proline to give 81% and 55% yields of the primary arylamines respectively; Jiang L, Lu X, Zhang H, Jiang Y, Ma D. J Org Chem. 2009;74:4542–4546. doi: 10.1021/jo9006738. [DOI] [PubMed] [Google Scholar]; In contrast to the work with ligandless catalysts in reference 25, we have obtained yields (69% and 60% respectively) that are close to those published, but these yields are still lower than those in the current work with palladium catalysts.
- 27.Grushin VV, Alper H. Chem Rev. 1994;94:1047–1062. [Google Scholar]
- 28.Littke A, Fu GC. Angew Chem Int Ed. 2002;41:4176–4211. doi: 10.1002/1521-3773(20021115)41:22<4176::AID-ANIE4176>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 29.Hamann BC, Hartwig JF. J Am Chem Soc. 1998;120:7369–7370. [Google Scholar]
- 30.Urgaonkar S, Xu JH, Verkade JG. J Org Chem. 2003;68:8416–8423. doi: 10.1021/jo034994y. [DOI] [PubMed] [Google Scholar]
- 31.Grasa GA, Viciu MS, Huang J, Nolan SP. J Org Chem. 2001;66:7729–7737. doi: 10.1021/jo010613+. [DOI] [PubMed] [Google Scholar]
- 32.Wolfe JP, Buchwald SL. J Org Chem. 1996;61:1133–1135. [Google Scholar]
- 33.Driver MS, Hartwig JF. J Am Chem Soc. 1996;118:7217–7218. [Google Scholar]
- 34.Wolfe JP, Buchwald SL. J Org Chem. 1997;62:6066–6068. [Google Scholar]
- 35.Huang J, Grasa G, Nolan SP. Org Lett. 1999;1:1307–1309. [Google Scholar]
- 36.Ali MH, Buchwald SL. J Org Chem. 2001;66:2560–2565. doi: 10.1021/jo0008486. [DOI] [PubMed] [Google Scholar]
- 37.Arterburn JB, Pannala M, Gonzalez AM. Tetrahedron Lett. 2001;42:1475–1477. [Google Scholar]
- 38.Urgaonkar S, Nagarajan M, Verkade JG. J Org Chem. 2003;68:452–459. doi: 10.1021/jo0205309. [DOI] [PubMed] [Google Scholar]
- 39.Enguehard C, Allouchi H, Gueiffier A, Buchwald SL. J Org Chem. 2003;68:4367–4370. doi: 10.1021/jo0341463. [DOI] [PubMed] [Google Scholar]
- 40.Meyers C, Maes BUW, Loones KTJ, Bal G, Lemiere GLF, Dommisse RA. J Org Chem. 2004;69:6010–6017. doi: 10.1021/jo049774e. [DOI] [PubMed] [Google Scholar]
- 41.Gerristma D, Brenstrum T, McNulty J, Capretta A. Tetrahedron Lett. 2004;45:8319–8321. [Google Scholar]
- 42.For recent reports on efficient couplings of aryl iodides, see reference 19 and the following reference:; Fors BP, Davis NR, Buchwald SL. J Am Chem Soc. 2009;131:5766–5768. doi: 10.1021/ja901414u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Åhman J, Buchwald SL. Tetrahedron Lett. 1997;38:6363–6366. [Google Scholar]
- 44.Wolfe JP, Buchwald SL. Tetrahedron Lett. 1997;38:6359–6362. [Google Scholar]
- 45.Hartwig JF, Kawatsura M, Hauck SI, Shaughnessy KH, Alcazar-Roman LM. J Org Chem. 1999;64:5575–5580. doi: 10.1021/jo990408i. [DOI] [PubMed] [Google Scholar]
- 46.Thayumanavan S, Barlow S, Marder SR. Chem Mater. 1997;9:3231–3235. [Google Scholar]
- 47.An H, Cook PD. Chem Rev. 2000;100:3311–3340. doi: 10.1021/cr990014r. [DOI] [PubMed] [Google Scholar]
- 48.Boger DL, Tarby CM, Myers PL, Caporale LH. J Am Chem Soc. 1996;118:2109–2110. [Google Scholar]
- 49.Kim S, Ko H, Kim S, Lee T. J Comb Chem. 2002;4:549–551. doi: 10.1021/cc020044z. [DOI] [PubMed] [Google Scholar]
- 50.Cheng S, Comer DD, Williams JP, Myers PL, Boger DL. J Am Chem Soc. 1996;118:2567–2573. [Google Scholar]
- 51.Hartwig JF. In: Modern Amination Methods. Ricci A, editor. Wiley-VCH; Weinheim, Germany: 2000. [Google Scholar]
- 52.Jiang L, Buchwald SL. In: Metal-Catalyzed Cross-Coupling Reactions. De Meijere A, Diederich F, editors. Vol. 2. Wiley-VCH; Weinheim, Germany: 2004. p. 699. [Google Scholar]
- 53.Ley SV, Thomas AW. Angew Chem Int Ed. 2003;42:5400–5449. doi: 10.1002/anie.200300594. [DOI] [PubMed] [Google Scholar]
- 54.Beletskaya IP, Cheprakov AV. Coord Chem Rev. 2004;248:2337–2364. [Google Scholar]
- 55.Steffanut P, Klein C, Graciet JC, Luecke L, Winter MA. Int Appl. PCT. 2007. [Google Scholar]
- 56.Chae KH, Kim YH. Adv Funct Mater. 2007;17:3470. [Google Scholar]
- 57.Chen G, Zhang X, Zhang S, Chen T, Wu Y. J Appl Polym Sci. 2007;106:2808. [Google Scholar]
- 58.Pawar NS, Kapadi UR, Hundiwale DG, Kumbhar PP. Sci Ind Res. 2002;61:454. [Google Scholar]
- 59.Meng XB, Han D, Zhang SN, Guo W, Cui JR, Li ZJ. Carbohydr Res. 2007;342:1169. doi: 10.1016/j.carres.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 60.Balzarini J, Clercq ED, Kaminska B, Orzeszko A. Antiviral Chem Chemother. 2003;14:139. doi: 10.1177/095632020301400303. [DOI] [PubMed] [Google Scholar]
- 61.Wannberg J, Dallinger D, Kappe CO, Larhed M. J Comb Chem. 2005;7:574–583. doi: 10.1021/cc049816c. [DOI] [PubMed] [Google Scholar]
- 62.Trabanco AA, Vega JA, Fernandez MA. J Org Chem. 2007;72:8146–8148. doi: 10.1021/jo701573w. [DOI] [PubMed] [Google Scholar]
- 63.Bhagwanth S, Waterson AG, Adjabeng GM, Hornberger KR. J Org Chem. 2009;74:4634–4637. doi: 10.1021/jo9004537. [DOI] [PubMed] [Google Scholar]
- 64.pKa value obtained from ionization of liquid ammonia:; Coulter LV, Sinclair JR, Cole AG, Roper GC. J Am Chem Soc. 1959;81:2986–2989. [Google Scholar]
- 65.Perrin DD. Ionization Constants for Inorganic Acids and Bases in Aqueous Solutions. Pergamon; Oxford: 1982. p. 84. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.