

Abstract
Atherosclerosis, a chronic inflammatory disorder, involves both the innate and adaptive arms of the immune response that mediate the initiation, progression, and ultimate thrombotic complications of atherosclerosis. Most fatal thromboses, which may manifest as acute myocardial infarction or ischemic stroke, result from frank rupture or superficial erosion of the fibrous cap overlying the atheroma, processes that occur in inflammatorily active, rupture-prone plaques.
Appreciation of the inflammatory character of atherosclerosis has led to the application of C-reactive protein as a biomarker of cardiovascular risk, and the characterization of the anti-inflammatory and immunomodulatory actions of the statin class of drugs. An improved understanding of the pathobiology of atherosclerosis and further studies of its immune mechanisms provide avenues for the development of future strategies directed toward better risk stratification of patients as well as the identification of novel anti-inflammatory therapies. This review retraces leukocyte subsets involved in innate and adaptive immunity and their contributions to atherogenesis.
Introduction
Atherosclerosis, a chronic inflammatory disease, involves both innate and adaptive arms of immunity which modulate lesion initiation, progression, and potentially devastating thrombotic complications 1 2. Thrombosis often complicates physical disruption of the protective collagen-rich fibrous cap overlying the atheroma, exposing circulating clotting factors to procoagulants expressed within lesions as a result of inflammatory activation and initiation of the coagulation cascade 3. Importantly, inflammation also decisively influences the propensity of a given plaque disruption to lead to a sustained and occlusive thrombus that may manifest clinically as an acute coronary syndrome or ischemic stroke by controlling the balance between fibrinolytic enzymes and their endogenous inhibitors 4 5.
Appreciation of the inflammatory character of atherosclerosis has spawned new avenues in basic, translational, and clinical research. CRP (C-reactive protein), an acute-phase reactant released during inflammatory processes, adds to the predictive power of traditional markers of cardiovascular risk 6. Basic research suggests that treatment with statins (3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors) — initially developed to decrease low density lipoprotein (LDL) cholesterol levels — also reduces leukocyte adhesion, accumulation of macrophages, protease production, procoagulant and pro-inflammatory mediator expression, antigen presentation, and T-cell activation 7. Additional support for the anti-inflammatory and immunomodulatory actions of statins came from clinical research. The magnitude of risk reduction associated with statin therapy may exceed that expected on the basis of LDL-cholesterol lowering alone. The CARE (Cholesterol And Recurrent Events) trial first demonstrated that statin therapy lowers plasma levels of CRP in addition to LDL-cholesterol 8. Retrospective evidence supported the utility of targeting the inflammatory marker CRP with statins in normocholesterolemic patients in both primary 9 and secondary prevention 10 of adverse cardiovascular events. Prospective evidence provided by the JUPITER trial (Justification for the Use of Statins in Primary Prevention: an Intervention Trial Evaluating Rosuvastatin) demonstrated that patients with LDL-cholesterol levels considered near optimal but elevated CRP levels benefit significantly from statin treatment in the prevention of adverse cardiovascular events. This direct result of the clinical application of the science of inflammation in atherosclerosis has potentially far reaching implications for everyday medical practice and public health.
To portray the chronic inflammation in atherothrombosis, we review here the leukocytes involved in innate and adaptive immunity with established or emerging roles in the disease process, and detail their cellular and molecular contributions. Beyond macrophages and CD4+ T-cells, new research highlights the important role of regulatory T-cells, dendritic cells, and mast cells in the disease process. Most of the cited experimental work relies on genetically altered atherosclerosis-prone mice, namely apolipoprotein E (ApoE)-deficient mice, which develop hypercholesterolemia and atherosclerotic disease spontaneously 11, and low-density lipoprotein receptor-deficient mice, which require a high-fat diet to develop hypercholesterolemia and atherogenesis 12.
The innate immune response in atherosclerosis
Monocytes and macrophages — the most numerous leukocytes at all stages of atherosclerosis — comprise the central cellular effectors of disease progression
Accumulation of lipid-laden macrophage-derived foam cells characterizes fatty streaks, the initial asymptomatic lesion of atherosclerosis 1. The precursor lesion of atherosclerotic plaques, fatty streaks, have focal increases in the content of lipoproteins within regions of the intima where they associate with constituents of the extracellular matrix such as proteoglycans, slowing their egress 13. This retention sequesters lipoproteins within the intima, protecting them from plasma antioxidants, thus favoring their oxidative modification. Laboratory and clinical data suggest that oxidized or glycated LDL evokes an inflammatory response in the artery wall, unleashing many of the biological processes thought to participate in atherosclerosis initiation, progression, and complication 14.
Endothelial cells (ECs) normally resist leukocyte adhesion. Pro-inflammatory stimuli that include hypercholesterolemia, hyperglycemia, hypertension, and smoking trigger the endothelial expression of adhesion molecules such as vascular cell adhesion molecule-1 (VCAM-1) and P-selectin that mediate the attachment of circulating monocytes and other leukocytes 15 16 17 18. Chemoattractant factors, including monocyte chemoattractant protein-1 (MCP-1) produced by vascular wall cells in response to modified lipoproteins, direct the migration and diapedesis of adherent monocytes. MCP-1 binds to CCR2 on the surface of migrating monocytes to exert this effect. Genetic absence of MCP-1 in LDLR-deficient mice dramatically decreases atherosclerotic disease with marked inhibition of monocyte recruitment 19. Similar results hold true upon deletion of CCR2 expression in ApoE-deficient mice 20. During diapedesis, monocytes release the matrix metalloproteinase MMP-9 that can degrade type IV collagen in the intimal basement membrane to help them enter the growing intimal atherosclerotic lesion 21. Experimental evidence and human observations support the involvement of several other chemokines in monocyte recruitment into the nascent atherosclerotic lesion, including IL-8, which binds CXCR2 22 23, and fractalkine (CX3CL1), which binds CX3CR1 24 25. Importantly, low shear stress also induces expression of MCP-1, fractalkine, and other chemokines involved in atherogenesis 26. Monocytes infiltrate the lesion and localize particularly in the shoulder region where the atheroma grows. In addition, neovessels arising from the artery's vasa vasorum provide another entry route for monocytes into established atherosclerotic lesions 27.
Within the intima, monocytes mature into macrophages under the influence of macrophage colony-stimulating factor (M-CSF), overexpressed in the inflamed intima 28 29. Importantly, M-CSF stimulation also leads to increased macrophage expression of scavenger receptors, pattern-recognition receptors involved in innate immunity, which engulf modified lipoproteins and apoptotic bodies through receptor-mediated endocytosis, leading to their lysosomal degradation. Scavenger receptors involved in macrophage foam cell formation include CD36, CD68, CXCL16, LOX1 (lectin-type oxidized low-density lipoprotein receptor 1), SR (scavenger receptor)-A, and SR-B 1. Accumulation of cholesteryl esters in the cytoplasm produces the characteristic change of macrophages into foam cells. Another type of pattern-recognition receptor, Toll-like receptors (TLRs), directly elicit inflammatory responses 1. In particular, monocytes in human atherosclerotic plaques have markedly enhanced expression of TLR1, TLR2, and TLR4 30. A majority of these monocytes show nuclear translocation of the transcription factor NF-κB (nuclear factor-κB), consistent with their inflammatory activation in lesions 30. Within atherosclerotic lesions, IκB kinase 2 (IKK-2, or IKK-β) phosphorylates IκBα, leading to its ubiquitination and degradation. IKK-2 thereby terminates the inhibitory action of IκBα on NF-κB, and allows the transcription of pro-inflammatory cytokines and proteases 31. A large number of pathogen-associated molecular patterns can activate TLRs. Heat shock proteins (hsp60) 32 and oxLDL 33 34 mediate at least part of their effects within plaques through TLR4 binding. In support of the notion that TLR4 and downstream effectors such as MyD88 (myeloid differentiation primary-response gene 88) mediate inflammatory activation in atherosclerosis, their genetic abrogation reduces disease burden 35 36. Apolipoprotein CIII, a constituent of certain atherogenic triglyceride-rich lipoproteins, can activate cells involved in atherogenesis through TLR2 37. TLR2, expressed on cells not derived from bone marrow, also appears to promote atherogenesis in mice 38 39.
Macrophages proliferate and amplify the inflammatory response through the secretion of numerous growth factors and cytokines, including tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β). These 2 key cytokines are central mediators of inflammatory pathways relevant to atherosclerosis (Figure 1). Among a myriad of actions, they induce expression of adhesion molecules such as VCAM-1, chemokines such as MCP-1, growth factors such as M-CSF, and proteases such as MMPs by cellular effectors present within lesions 1. In human lesions, IL-18 colocalizes with mononuclear phagocytes while ECs, SMCs, and macrophages all express the IL-18 receptor 40. Importantly, IL-18 signaling evokes essential effectors involved in atherogenesis, e.g., adhesion molecules (VCAM-1), chemokines (IL-8), cytokines (IL-6), and matrix metalloproteinases (MMP-1/-9/-13) 40. Recent evidence supports selective mobilization during hypercholesterolemia in mice of a pro-inflammatory subset of monocytes that bear high levels of a surface marker denoted Ly6C. These Ly-6Chi monocytes increase dramatically in the blood of fat-fed apoE-deficient mice, and preferentially adhere to activated endothelium, infiltrate lesions, and become lesional macrophages 41. Importantly, pro-inflammatory Ly-6Chi monocytes are CCR2+ and CX3CR1+ and rely on these chemokine receptors to enter lesions 42.
Figure 1. Leukocytes and platelets release mediators that control inflammation in atherosclerotic plaques and determine lesion fate.
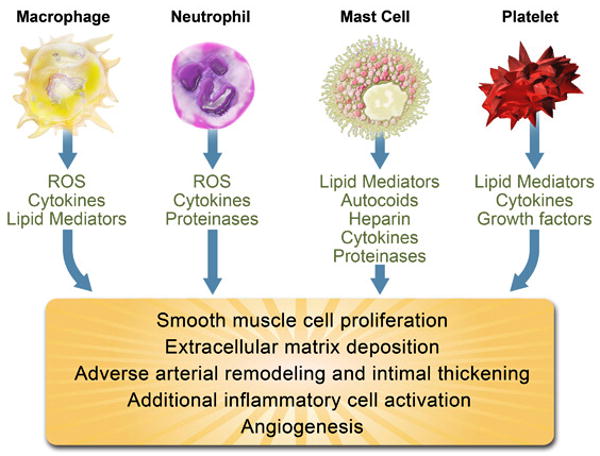
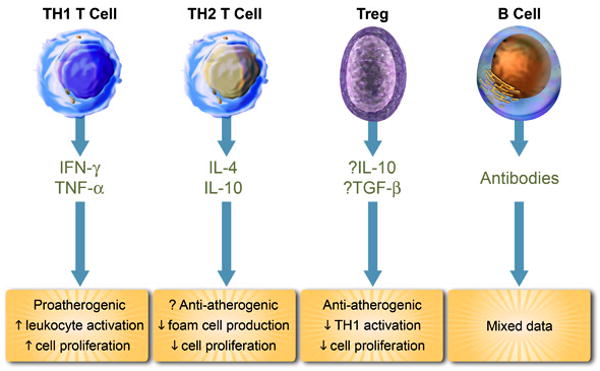
Abbreviations: TH1 T Cell; T helper 1 T cell, Treg; Regulatory T Cell, ROS; reactive oxygen species, IFN-γ; interferon-γ, TNF-α; tumor necrosis factor-α, IL; interleukin, TGF-β; transforming growth factor-β.
Advanced atheromata may exhibit a paucity of SMCs and abundant macrophages, key histological characteristics of plaques that have ruptured and caused fatal thrombosis. The fibrous cap covering the atherosclerotic plaque owes its biomechanical strength to interstitial collagens (types I and III). Inflammation interferes with the integrity of the fibrous cap by limiting the creation of new collagen by SMCs 43 and by stimulating the destruction of existing collagen fibers (Figure 2). Indeed, CD40L as well as IL-1 produced by T-cells induce macrophages to release interstitial collagenases, including MMP-1 44, MMP-8 45, and MMP-13 44 46, which mediate the initial attack on interstitial collagen. MMP-14, a membrane-associated MMP, activates MMP-13 and also appears to contribute to collagenolysis 47. Macrophage-derived foam cells contain MMP-9, a member of the gelatinase class of the metalloproteinase family, in human plaques 48. The catalytically active MMP-9 may contribute to the dysregulation of extracellular matrix that leads to plaque rupture. MMP activity overwhelms regulation mediated by TIMPs (tissue inhibitors of metalloproteinases) produced by macrophages and other cells in atheromata, hence influencing plaque stability 49 50. In addition, macrophages in lesions constitutively produce the serine protease neutrophil elastase, and release this enzyme upon CD40 ligation 51. By inactivating TIMP-1, neutrophil elastase favors MMP activity and collagen breakdown 51. Direct in vivo evidence in collagenase-resistant atherosclerosis-prone mice confirms the role of MMPs in plaque collagen turnover 52.
Figure 2. TH1 cells and macrophages promote atherosclerosis progression.
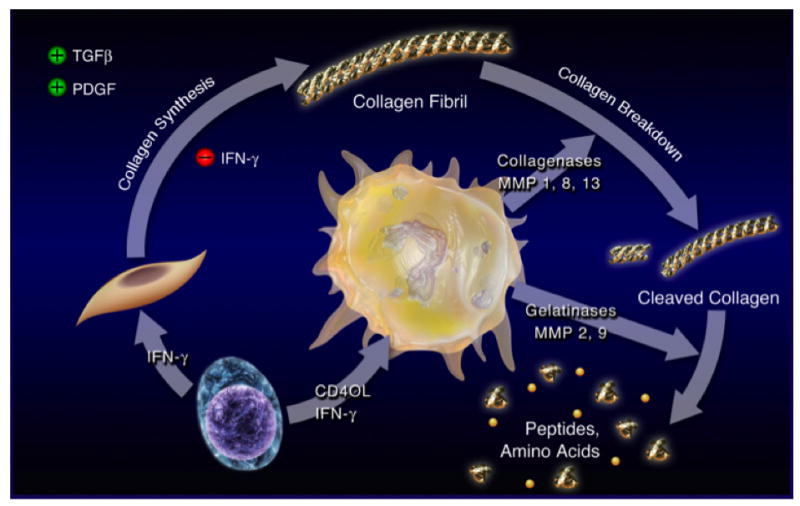
TH1-biased T lymphocytes (lower left) express IFN-γ and CD40L within plaques. IFN-γ inhibits the de novo production of collagen in response to TGF-β and PDGF by vascular smooth muscle cells (middle left). In parallel, CD40L induces the synthesis and release of proteases by macrophages (center), including the interstitial collagenases MMP-1, MMP-8, and MMP-13, which mediate the initial proteolytic attack of intact collagen fibrils. In addition, CD40L also simulates the production of the gelatinases MMP-2 and MMP-9 as well as other proteases that continue the proteolytic degradation of collagen fibrils. These combined effects orchestrated by TH1 cells and macrophages weaken the fibrous cap covering the atherosclerotic plaque — which owes its biomechanical strength to interstitial collagen (types I and III) fibrils — and render the lesion rupture-prone.
Abbreviations: CD40L; CD40 Ligand, IFN-γ; interferon-γ, MMP; matrix metalloproteinase, PDGF; platelet-derived growth factor, TGF-β; transforming growth factor-β. (Figure reproduced with kind permission of Springer Science and Business Media 215.)
Cathepsins, members of the cysteine protease family, also participate in plaque evolution and destabilization 53. Macrophages in human atheromata express most of the proteolytically active cathepsins S and cathepsins K, which display elevated elastolytic activities 54. Experimental atherosclerotic lesions also express cathepsins L, and B 55. Cathepsin S 56, cathepsin L 57, or cathepsin K 58 deficiency in atherosclerosis-prone mice reduces collagen and elastin degradation as well as CD4+ T-cell, macrophage, and smooth muscle cell accumulation and overall plaque burden. Importantly, arteries under physiologic conditions abundantly express cystatin C, an endogenous inhibitor of cathepsins, whereas human atheromata exhibit very low cystatin C levels 59.
Recently, proteases of the ADAMTS (a disintegrin and metalloprotease with thrombospondin motif) family have also garnered attention for their role in matrix protein turnover and progression of atherosclerosis 60 61. The combined effects of all these classes of proteases, released mostly by macrophage foam cells within lesions, favors matrix and fibrous cap remodeling that may lead to plaque rupture with ensuing thrombosis and clinical manifestations 62.
Mast cells
Mast cells although numerically minor constituents of the leukocytic population in the atherosclerotic intima can also populate the adventita 63 64. In human coronary artery specimens, mast cell numbers rise in parallel to the severity of clinical presentation 65, and these leukocytes accumulate in the shoulder region of plaques, where they degranulate and release proteases and cytokines 66. Recent studies suggest they can contribute to lesion progression in mice (Figure 1). These data require cautious interpretation in regard to the human disease, as rodents rely more on innate immunity and possess a more complex variety of mast cell functions and proteases 67.
The chemokine eotaxin, found in atherosclerotic plaques, can mediate lesional mast cell recruitment by binding to cellular CCR3 68. Utilization of a mast cell-deficient mouse demonstrates decreased lesion size, lipid deposition, T-cell and macrophage numbers, and cell proliferation and apoptosis, but increased collagen content and fibrous cap development in a mast cell-derived IFN-γ and IL-6-dependent manner 69. Periadventitial injury leads to lesion progression and activation of mast cells with ensuing intraplaque hemorrhage, macrophage apoptosis, vascular permeabilization, and recruitment of further leukocytes 70. Importantly, treatment with the mast cell stabilizer chromoglycate prevents all these adverse events elicited by mast cell degranulation 70.
Mast cells produce certain MMPs, including MMP-1 71 and MMP-9 72. In addition, the mast cell serine proteases tryptase and chymase can activate MMPs in human carotid endarterectomy samples, and MMP-1 and -3 colocalize with degranulated mast cells in the shoulder regions of atherosclerotic plaques 73. Mast cell chymase also processes pro-MMP-2 and -9 into their active forms 74. These results suggest that the direct release of certain MMPs and the activation of MMPs by mast cell-derived proteases may promote atherosclerotic plaque rupture.
The proportion of intimal mast cells expressing basic FGF (fibroblast growth factor) — a potent angiogenic mediator — rises with increasing severity of atherosclerosis, suggesting a role for these leukocytes in angiogenesis, plaque neo-vascularization, and disease progression 75. Mast cell chymase also functions as an angiotensin-converting enzyme (particularly in rodents) and may thus contribute to the local regulation of vascular tone 76.
Activated mast cells induce endothelial death by chymase-mediated inactivation of focal adhesion kinase (FAK) and Akt-dependent cell survival signaling, as well as TNF-α-mediated apoptosis, functions that contribute to plaque erosion 77. Mast cell chymase can also inhibit smooth muscle cell proliferation and collagen expression 78 and induce SMC apoptosis 79 80. Histamine — abundantly present in mast cells — reportedly induces tissue factor expression in human aortic endothelial and vascular smooth muscle cells 81, promoting the plaque's thrombotic potential.
Degranulation of mast cells may facilitate SMC and macrophage uptake of LDL and their development into foam cells 82 83, mediated in part by chymase-dependent degradation of the ApoB moiety of LDL 84. In addition, mast cells inhibit cellular cholesterol efflux and reverse cholesterol transport, in part by chymase-mediated degradation of certain apolipoproteins such as ApoE 85 and tryptase-mediated degradation of HDL 86.
Natural Killer cells
Nature killer (NK) cells, cellular effectors of innate immunity, play a critical role in the defense against infectious organisms, particularly viruses 87. Contrary to T-cells and B-cells, these bone marrow-derived lymphocytes do not require antigen receptor gene rearrangement during cellular development and do not express T-cell receptors or surface immunoglobulins. NK cells receive dual signals from inhibition and activation surface receptors 87. NK cell inhibitory receptors such as Ly49A are specific for MHC class I molecules on target cells and prevent NK cell activation, cytotoxicity, and cytokine secretion. Cells that have lost their expression of MHC-I molecules — typically virus-infected cells — are susceptible to NK cell attack. NK cells also express activation receptors such as Ly49D and Ly49H, structurally related to the inhibitory receptors, which recognize target cell ligands and can trigger perforin-dependent natural killing. In addition, NK cells express a receptor that binds the Fc portion of antibodies known as FcγRIII (CD16). Cross-linking of Fc receptors by IgG antibody-coated target cells may constitute a second form of activation that signals the NK cell to kill the target.
Limited direct evidence supports NK cell involvement in atherogenesis. The shoulder region of human plaques contains modest numbers of CD56+NK cells 88. Given their presence within lesions, and the abundance of cytokines known to activate NK cells such as IFN-α/β, IL-12, IL-15, and IL-18, these cells plausibly contribute to lesion progression. Upon activation, NK cells in turn secrete numerous cytokines and growth factors including IFN-γ, TNF-α, GM-CSF, and trigger perforin-mediated natural killing 87. Indeed, atherosclerosis-prone mice with genetically impaired NK inhibitory signaling have decreased plaque burden 89.
The possible role of neutrophils in atherothrombosis
Neutrophils comprise a minority of the inflammatory cell composition of atherosclerotic lesions. However, neutrophil numbers rise in ruptured human coronary plaques 90, consistent with their role of endocytosing and clearing damaged tissue, but also raising the possibility of their presence before clinical events. In parallel, high circulating neutrophil counts predict myocardial infarction better than any other leukocyte subset, including total white blood cell, lymphocyte, or monocyte count 91. Intraplaque hemorrhage contributes importantly to the progression of atherothrombosis 92, and analyses of human carotid endarterectomy samples suggest intraplaque hemorrhage as an entry route for neutrophils — which constitute 60% of circulating leukocytes — into lesions 93. Atheromatous lesions demonstrate markers of neutrophil degranulation (α1-antitrypsin/elastase complexes, myeloperoxidase, and α-defensins) and the presence of proteases preferentially released by these leukocytes [NGAL (neutrophil gelatinase-associated lipocalin)/MMP-9 heterodimers, and HLE (human leukocyte elastase)], suggesting the presence of active neutrophils within lesions 93 (Figure 1). Atherosclerotic plaques contain NGAL that inhibits MMP-9 inactivation and thereby promotes its proteolytic and matrix-degrading capabilities 94. Another report confirms the colocalization of neutrophils with myeloperoxidase in lesions, suggesting a source for this enzyme beyond a macrophage subset 95. Myeloperoxidase generates the reactive oxygen species hypochlorous acid, which contributes to endothelial apoptosis and tissue factor expression and lesion advancement 96. In addition to mediating chlorination of tyrosyl residues, myeloperoxidase also leads to LDL protein nitration and lipid peroxidation, facilitating the uptake of these modified LDL particles by macrophages and contribution to foam cell formation 97.
Under physiologic circumstances, neutrophils arise exclusively in the bone marrow, and the chemokine ligand CXCL12 (SDF-1) expressed on bone marrow stromal cells and its receptor CXCR4 expressed on neutrophils allow not only their retention but also the homing of senescent neutrophils back to the bone marrow 98. Experimental antagonism of CXCR4 leads to increased circulating neutrophil levels and their enrichment within atherosclerotic lesions in response to CXCL1 expressed within lesions interacting with CXCR2 on neutrophils 99. Neutrophil recruitment also depends on the expression of neutrophilic Mac-1 and endothelial P-selectin for leukocyte rolling 100. These recruited neutrophils can secrete numerous enzymes including proteases. In addition, recruited neutrophils increase lesion size and enhance intraplaque IFN-γ, tissue factor, and CXCL1 levels, further amplifying their recruitment 99. These results suggest intraplaque hemorrhage and the more classic transendothelial route as sources of neutrophil recruitment, enriching the oxidative and proteolytic content and overall inflammatory activation of lesions.
Dendritic cells link the innate and adaptive arms of the immune response
Dendritic cells (DCs), an innate immune cell type, populate atherosclerotic plaques, particularly in the rupture-prone shoulder region of lesions 101, in part under the control of CX3CR1 102. Granulocyte-macrophage colony-stimulating factor (GM-CSF), produced locally in response to oxidized low-density lipoprotein cholesterol (oxLDL), also regulates DC numbers within lesions 103, probably by controlling their differentiation from monocyte precursors. Importantly, stimuli known to accelerate atherogenesis, such as oxLDL or TNF-α, increase DC adhesion to the endothelium and their subsequent transmigration 104. Nicotine increases DC expression of MHC-II, costimulatory molecules, and adhesion molecules, and promotes the production of IL-12 by DCs, thereby promoting a Th1 response 105. In addition, CD11c+ leukocytes with dendritic processes inhabit regions of the normal arterial intima predisposed to atherosclerosis 106. In human lesions, DC numbers increase in parallel to lesion complexity 107, as does the expression of CD83, a marker of DC activation 108.
Hypercholesterolemia may impede the emigration of a proportion of DCs, retaining them in peripheral tissues 109 110 where they locally promote immune responses, for example by re-stimulating effector CD4+ T-cells 111. Contrary to macrophages 112, DCs retain antigen presenting function under conditions typical of atherosclerotic plaques 111. This property may result from superior DC defenses against oxidative stress, displayed by elevated levels of superoxide dismutase and peroxiredoxin-1 113, and an apparent resistance to cholesterol-induced cytotoxicity, displayed by the absence of expression of the transcription factor CHOP (C/EBP-homologous protein), a marker of unfolded protein response (UPR) induction, after loading of DCs with unesterified cholesterol 111. Indeed, DCs embedded in ‘artificial arteries’ made of ECs, VSMCs, and type I collagen stimulate CD4+ T-cells upon exposure to lipopolysaccharide robustly and superiorly to embedded monocytes and macrophages 114.
DCs constitute a heterogeneous family, with different subsets characterized by varying tissue distributions, surface markers, cytokine profiles, and ensuing functions in the orchestration of immune responses 115. Plasmacytoid dendritic cells (pDCs) specialize in sensing bacterial and viral products and produce IFN-α abundantly 115. Through the release of key cytokines, PDCs contribute to the regulation of VSMC numbers within lesions. Upon microbial stimulation, pDCs release IFN-α, inducing tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) on the surface of CD4+ T-cells 116. TRAIL binds to death receptor 5 (DR-5) and thereby mediates VSMC death in an alternate pathway to the canonical CD8+ T-cell mediated cytotoxicity 117. Plasmacytoid-derived IFN-α also amplifies the inflammatory response by enhancing the production of TNF-α and IL-12 by ‘classic’ CD11c+ DCs 118. PDCs may thus provide a potential link between infections and disease progression, an association often evoked in atherosclerosis 119 120.
DCs prime T-cells in secondary lymphoid organs, enabling T-cell antigen-specific differentiation into effectors and the targeting of select tissues in the periphery such as atherosclerotic vessels. To emigrate toward regional lymph nodes, where they regulate adaptive immune responses, DCs must induce expression of CCR7 121 and CCR8 122 in tissues such as plaques.
Co-stimulatory and co-inhibitory molecule expression patterns by DCs drive antigen-dependent activation of naïve T-cells and the initiation of adaptive immunity (Figure 3) 123. Importantly, overlapping co-stimulatory and co-inhibitory molecules control both effector T-cell and regulatory T-cell responses, a complexity discussed elsewhere 123. Two major families comprise the costimulatory molecules: the B7 124 and TNF 125 families that bind to the CD28 and TNF receptor families, respectively 123.
Figure 3. Dendritic cells express co-stimulatory and co-inhibitory molecules involved in T cell activation.

Dendritic cells (DCs) deliver co-stimulatory (denoted by a (+) sign) and co-inhibitory (denoted by a (-) sign) signals to T cells through molecules belonging to the B-7 or TNF family. In the B-7 family, co-stimulatory molecules include CD80 (B7-1), CD86 (B7-2), and ICOS-L, and co-inhibitory molecules include PD-L1, PD-L2. T cells can render a co-stimulatory signal delivered by CD80 or CD86 co-inhibitory by replacing the CD28 receptor with CTLA-4. In the TNF family, co-stimulatory molecules include OX40-L and CD137-L. T cells also activate DCs by CD40-L, which increases DC expression of CD80 and CD86.
Abbreviations: CTLA-4; cytotoxic T-lymphocyte antigen 4, ICOS; inducible co-stimulatory molecule, ICOS-L; inducible co-stimulatory molecule-ligand, PD-L1; programmed death-ligand 1, PD-1; programmed death-1, TNF; tumor necrosis factor.
CD80 and CD86 (B7-1 and B7-2), the prototypical and best described co-stimulatory molecules on DCs/APCs, initially deliver their signals by binding to CD28 on T-cells. CD80/CD86 deficiency in atherosclerosis-prone mice reduces lesion development and decreases IFN-γ production by CD4+ T-cells upon presentation of the atherosclerosis-associated antigen hsp60 126, suggesting inefficient priming by DCs. Following their activation, T-cells express CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4). CTLA-4 then binds CD80 and CD86 on DCs, and turns the co-stimulatory signal initially delivered by CD28 into a co-inhibitory one, dampening the T-cell response 124.
Contrary to resting naïve T-cells, effector and memory T-cells express ICOS (inducible co-stimulatory molecule), another CD28 family member 127. ICOS-ligand activates ICOS — which figures critically in Treg function — on DCs, with decreased Treg suppressive function and increased lesional CD4+ T-cell content in ICOS-deficient mice 128.
Programmed death-ligand 1 (PD-L1) and PD-L2, B7 family members expressed on several cell types including DCs, inhibit T-cell activation by binding to programmed death-1 (PD-1) on T-cells 129. PD-L1/PD-L2-deficient mice have increased atherosclerotic burden in conjunction with exaggerated systemic immune responses with lymphadenopathy and elevated numbers of activated T-cells due to enhanced stimulation by DCs 130.
DCs and APCs express OX40Ligand (OX40L), a member of the TNF receptor family that provides co-stimulatory signals to T-cells through the TNF family member OX40, enabling long-lasting T-cell responses 125. Genetic studies identified OX40L as an atherosclerosis-susceptibility locus in mice 131. OX40L-deficient mice fed an atherogenic diet have decreased lesion size, whereas transgenic over-expression of OX40L demonstrates opposite effects 131. Antagonist anti-OX40L antibody treatment of atherosclerosis-prone mice also decreases plaque burden 132. Moreover, polymorphisms in the OX40L gene increase the risk of myocardial infarction 131, further highlighting the possible contribution of OX40L/OX40-mediated co-stimulation in promoting atherosclerosis.
CD137Ligand (CD137L), a TNF family member, also belongs to the TNF receptor family 125. The wide cellular distribution of these molecules complicates the elucidation of their contribution to atherosclerotic disease, but antigen recognition induces CD137 expression on T-cells that receive co-stimulatory signals from CD137L expressed on DCs 125. CD137L/CD137 participates importantly in CD8+ T-cell responses 133. Indeed, agonistic anti-CD137 antibody treatment increases CD8+ T-cell infiltration, pro-inflammatory cytokine expression, and overall lesion size 134.
DCs maintain their antigen-processing and -presentation functions and co-stimulatory capabilities under hypercholesterolemic conditions present in experimental atherosclerosis 111. DCs thereby activate the adaptive immune response and efficiently generate monoclonal and polyclonal effector CD4+ T-cells that may subsequently leave secondary lymphoid organs and reach atherosclerotic vessels 111.
The adaptive immune response in atherosclerosis
CD4+ TH1 T-cells promote atherothrombosis
IFN-γ-producing TH1 CD4+ T-cells with αβ T-cell receptors constitute the majority of T lymphocytes present in human 135 and experimental 136 atherosclerotic lesions 1 2 (Figure 1). T-cells enter lesions in response to the chemokines inducible protein-10 (IP-10), monokine induced by IFN-γ (MIG), and IFN-inducible T-cell α-chemoattractant (I-TAC), which bind CXCR3, highly expressed by T lymphocytes in the plaque 137. CD4+ T-cells undergo oligoclonal expansion within lesions, suggesting the occurrence of antigen-driven T-cell proliferation 138. Indeed, CD4+ T-cell clones in plaques recognize oxLDL 139 and hsp60 140.
Experimental evidence supports an important role for CD4+ T-cells in atherosclerosis. Immunodeficient RAG (recombinase activating gene)-deficient 141 or DNA-PK (DNA-dependent protein kinase)-knockout 142 atherosclerosis-prone mice have no lymphocytes and reduced lesions compared to immunocompetent atherosclerosis-prone mice. Adoptive transfer of CD4+ T-cells into immunodeficient animals, however, greatly increases lesion size in parallel to increased T-cell recruitment and MHC-II expression in plaques 142.
In addition to the histopathologic features of macrophage and T-cell accumulation within lesions, several lines of evidence support TH1 predominance in atherosclerosis. Different strains of mice have varying susceptibilities to atherosclerosis 143. TH1-biased C57BL/6 mice develop significantly more atherosclerosis in association with increased serum levels of IL-6 and the acute-phase protein serum amyloid A (SAA) than TH2-biased BALB/c mice 144. Genetic T-bet deficiency (a transcription factor required for TH1 differentiation) 145 or treatment with pentoxifylline (an inhibitor of TH1 differentiation) 146 protects against atherosclerosis. While administration of IL-12 147, the central cytokine driving TH1-cell differentiation, enhances atherosclerosis, genetic deficiency of IL-12 148 or IL-12 blockade through vaccination 149 attenuates the disease. In addition, the genetic deficiency 150 or inhibition 151 of IL-18, which drives TH1-cell differentiation synergistically with IL-12, also decreases disease progression. Deficiency of IFN-γ 152, the prototypical TH1-cytokine, or of the IFN-γ receptor 153, greatly decreases plaque burden, macrophage content, and MHC-II expression within lesions. Administration of recombinant IFN-γ has opposite effects 154. IFN-γ inhibits the proliferation and differentiation of vascular smooth muscle cells 155 and also decreases collagen production by these cells 43, functions that could contribute to the thinning of the collagen-rich fibrous cap (Figure 2). IFN-γ also inhibits endothelial cell proliferation 156 and potentiates the production of pro-inflammatory cytokines by macrophages as well as MHC-II expression 1. Further supporting these observations, human plaques contain an abundance of cells producing the TH1-type cytokines IFN-γ, IL-12, IL-15, IL-18, and TNF-α, but few cells producing the TH2 cytokine IL-4 157. In vivo, IFN-γ appears to augment arteriosclerosis, based on observations on the effects of this cytokine on small human arteries dwelling in immunodeficient mice 158.
Expression of CD40Ligand (CD40L) by activated CD4+ T-cells induces the expression of the procoagulant tissue factor in endothelial cells 159, VSMCs 160, and macrophages 161. CD40L also stimulates production of the interstitial collagenases MMP-1 44, MMP-8 45, and MMP-13 44 by macrophages, which interfere with the integrity of the protective fibrous cap (Figure 2). Recent results suggest that CD40L mediates certain effects in atherosclerosis through Mac-1 binding 162. CD40L on T-cells also activates APCs/DCs 123. Indeed, CD40 ligation increases co-stimulatory molecule expression by APCs, mainly CD80 and CD86, which then bind CD28 on the T-cell and transmit co-stimulatory signaling 163. In addition to T-cells, endothelial cells, VSMCs, macrophages, and platelets all express CD40L and CD40 164 165. As CD40 ligation promotes the inflammatory activation of all the major cell types participating in atherosclerosis, genetic 166 or antibody-mediated 167 disruption of CD40L signaling significantly decreases lesion size, macrophage and T-cell content, as well as VCAM-1 expression, and promotes the formation of a fibrous collagen-rich plaque.
CD8+ T-cells also populate lesions 168, but their relevance and contribution is less well understood 2. Conditions that enhance CD8+ T-cell function and increase their numbers by modulating co-stimulation 134 or co-inhibition 130 promote atherosclerosis in experimental settings. Though CD8+ T-cells kill VSMCs in aortic aneurysms 169, their role in atherosclerosis remains uncertain.
Natural Killer T-cells and γδ T-cells
A subpopulation of T-cells that include Natural Killer T (NKT) cells and γδ T-cells express semi-invariant TCRs. NKT cells constitute a heterogeneous family of cells that has characteristics of both NK cells and conventional T cells 170. NKT cells express TCRs that recognize glycolipid antigens presented on CD1d, an MHC class I–like molecule expressed by antigen-presenting cells 170. The normal development of NKT cells also requires CD1d, found in human lesions 171. CD1d deficiency results in reduction of plaque burden in atherosclerosis-prone mice 172 173 174. Administration of a synthetic glycolipid that activates NKT cells via CD1d induces IFN-γ, MCP-1, TNF-α, IL-2, IL-4, IL-5, and IL-6 production within lesions 172. Lipid antigens present within plaques may activate NKT cells, which appear to participate in the early phases of atherogenesis 175.
Although the majority of T-cells express the classic αβ TCR, a subset expresses γδ TCRs with limited diversity. γδ T-cells represent fewer than 1-5% of circulating T-cells, but are enriched in sites of chronic inflammation 176. Importantly, γδ T-cells may recognize a wide array of antigens in the absence of MHC presentation by APCs 176. Some γδ T-cells recognize lipid antigens presented by CD1. These cells reside in lesions 177 and secrete IFN-γ and other cytokines, but the extent of their contribution to the pathobiology of atherosclerosis remains unclear 178.
Regulatory T-cells and immunosuppression
Regulatory T-cells (Tregs) control other T-cell types and suppress their activation in secondary lymphoid organs, or their effector functions in peripheral tissues such as plaques, either directly or indirectly via actions on APCs (Figure 1). Tregs contribute to the maintenance of tolerance to self-antigens and the regulation of immunity. Of the different Treg subsets, CD4+CD25+FoxP3+ Tregs are the best characterized. 179 FoxP3 serves as a lineage-specific transcription factor involved in Treg suppressive function. These well-characterized ‘natural’ Tregs, generated during thymic development, comprise 5-10% of peripheral CD4+ T-cells. However, naïve CD4+T-cells induce additional Tregs during antigen-specific immune responses in the presence of IL-2 and TGF-β 180. Other surface molecules expressed by Tregs include CTLA-4, GITR (Glucocorticoid-Induced Tumor necrosis factor Receptor), and CD127. These antigen-specific Tregs inhibit effector T-cell activation either by direct contact inhibition or through the release of the anti-inflammatory cytokines transforming growth factor (TGF)-β and IL-10. These cytokines made by Tregs can mitigate atherogenesis in mice.
IL-10-deficient mice fed an atherogenic diet have increased lesion area, effector T-cell infiltration, and IFN-γ expression 181. IL-10-transfected mice demonstrate opposite results, with IL-10 also impeding the modified LDL-mediated endothelial recruitment of monocytes 182. Bone marrow transplantation of transgenic IL-10 overexpressing T-cells also decreases lesion size and inflammation 183.
Inhibition of TGF-β signaling using neutralizing antibodies 184 or recombinant soluble TGF-β receptors 185 accelerates the development of atherosclerotic lesions in ApoE-deficient mice and favors the development of lesions with increased monocyte and lymphocyte accumulation and decreased collagen content. TGF-β exerts its atheroprotective effect by dampening CD4+ T-cell effector function. Indeed, mice carrying dominant-negative TGF-β receptors on CD4+ T-cells (CD4dnTβRII) exhibit increased inflammation and a paucity of mature interstitial collagen fibers within vascular lesions 186 187. CD4dnTβRII effector T-cells inhibit the production of lysyl oxidase, the extracellular enzyme needed for collagen cross-linking, limiting collagen maturation in the atherosclerotic plaque while having little effect on collagen degradation 188. Although numerous cell types produce TGF-β and IL-10, including endothelial cells, smooth muscle cells, macrophages, and platelets, regulatory T-cells may constitute an antigen-specific source of these anti-inflammatory cytokines 189.
In mice, decreased Treg numbers 189 or Treg function secondary to absence of ICOS 128 leads to increased lesional CD4+ T cells and macrophage numbers as well as amplified expression of the pro-inflammatory cytokines IFN-γ and TNF-α. Moreover, patients with acute coronary syndromes have reduced circulating Treg numbers and suppressive function 190. Conversely, the adoptive transfer of Tregs 191 or Treg-induction secondary to measles virus nucleoprotein vaccination (known to induce immunosuppression) 192 inhibits macrophage and T-cell accumulation within lesions. In addition, leptin deficiency in atherosclerosis-prone mice reduces atherosclerotic lesion formation, in association with diminished Th1 responses in addition to a marked increase in the number and suppressive function of Tregs 193. Given that the adipokine leptin increases in obesity, the possibility that leptin limits Treg function presents an additional link between obesity and inflammation. Interestingly, deficiency of the chemokine CXCL10 (IP-10) decreases atherogenesis not only by diminishing the recruitment of CD4+ T-cells, but also by increasing intra-plaque Treg numbers 194.
Therapeutic modulation of Tregs has undergone scrutiny in experimental settings. Anti-CD3 antibody treatment decreases CD3/T-cell receptor complex expression and has immunosuppressive functions by increasing Treg numbers and TGF-β levels, thus reducing lesion initiation and progression 195. Induction of tolerance through the oral administration of oxLDL 196 or hsp60/65 197 to atherosclerosis-prone mice increases Treg numbers in secondary lymphoid organs as well as TGF-β and IL-10 levels, again attenuating experimental atherogenesis. These reports highlight the central role of Tregs in tolerance induction, which may constitute the mechanism of atheroprotection reported in earlier similar studies to hsp65 198 199 and β2-glycoprotein I 200.
B-cells and humoral immunity
B-cells infiltrate the adventitial layer of human coronary lesions 201 and atherosclerosis-prone mice 202 where they may form lymphoid follicles. The plaques of ApoE-deficient mice contain B-cells at all stages of the disease 203 (Figure 1). Splenectomized mice have increased susceptibility to atherosclerosis, a situation reversed by the transfer of B-cells and the production of anti-oxLDL antibodies 204.
Both humans and atherosclerosis-prone animals have antibodies against oxLDL particles 205 206. Germ-line encoded natural anti-oxLDL IgM antibodies produced by B1-cells bind the oxidized phospholipids on oxLDL and also recognize phosphorylcholine in the cell wall of Streptococcus pneumoniae 207. Taking advantage of this molecular mimicry, pneumococcal vaccination of atherosclerosis-prone mice decreases the extent of experimental atherosclerosis 208.
Anti-hsp60 antibodies cross-react between microbial and eukaryotic hsp60/65, a consequence of high sequence conservation. As such, infections, for example by Chlamydia pneumoniae, might result in breaking tolerance to self-hsp60 and promoting auto-immunity and atherogenesis 209. Indeed, experimental results using hsp65 as an immunogen for vaccination 210 211 mediate endothelial cytotoxicity 212 and promote atherogenesis. Over all, B-cells are considered to mediate protective immunity during the development of atherosclerosis, possibly by preventing antigens from reaching lesions.
Conclusion
The evidence reviewed here highlights the extensive role of innate and adaptive immunity in atherosclerosis, from its initiation to its final thrombotic complications. Our improved pathobiologic understanding of this disease allows the detection of patients at high risk and the design and development of novel treatment modalities targeting cellular and molecular mediators. These have already led to the identification of certain inflammatory indicators, such as CRP, as biomarkers of adverse cardiovascular events, allowing a better risk stratification of patients and targeting of therapy 213. In addition, statins have emerged as powerful anti-inflammatory and immunomodulatory agents, with extensive clinical use in primary and secondary prevention. Despite these strides, death from cardiovascular disease continues to increase worldwide, with many patients experiencing cardiovascular events despite statin, anti-platelet and anti-hypertensive treatment 214.
As a systemic and non-selective modulation of immune responses could lead to adverse effects including acquired immunodeficiency with ensuing infectious and oncologic complications, a more subtle and targeted approach of the immune response would probably prove advantageous. Based on the myriad immune mechanisms involved in atherogenesis, a wide range of potential therapies appear on the horizon, with the goal of diminishing the global cost paid by humanity to this scourge.
Acknowledgments
This work was supported by the Fondation Leducq, Paris, France (Dr. Libby) and by NIH HL087282 (Dr. Lichtman) & HL36436 (Dr. Libby).
Contributor Information
René R. S. Packard, Leducq Center for Cardiovascular Research, Brigham and Women's Hospital, Harvard Medical School, Boston, Massachusetts, U.S.A
Andrew H. Lichtman, Division of Cardiovascular Medicine, Department of Medicine, and Immunology and Vascular Research Divisions, Department of Patholog, Brigham and Women's Hospital, Harvard Medical School, Boston, Massachusetts, U.S.A
Peter Libby, Leducq Center for Cardiovascular Research, Brigham and Women's Hospital, Harvard Medical School, Boston, Massachusetts, U.S.A
References
- 1.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–19. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 2.Weber C, Zernecke A, Libby P. The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat Rev Immunol. 2008;8:802–15. doi: 10.1038/nri2415. [DOI] [PubMed] [Google Scholar]
- 3.Libby P. Current concepts of the pathogenesis of the acute coronary syndromes. Circulation. 2001;104:365–72. doi: 10.1161/01.cir.104.3.365. [DOI] [PubMed] [Google Scholar]
- 4.Naghavi M, Libby P, Falk E, Casscells SW, Litovsky S, Rumberger J, Badimon JJ, Stefanadis C, Moreno P, Pasterkamp G, Fayad Z, Stone PH, Waxman S, Raggi P, Madjid M, Zarrabi A, Burke A, Yuan C, Fitzgerald PJ, Siscovick DS, de Korte CL, Aikawa M, Juhani Airaksinen KE, Assmann G, Becker CR, Chesebro JH, Farb A, Galis ZS, Jackson C, Jang IK, Koenig W, Lodder RA, March K, Demirovic J, Navab M, Priori SG, Rekhter MD, Bahr R, Grundy SM, Mehran R, Colombo A, Boerwinkle E, Ballantyne C, Insull W, Jr, Schwartz RS, Vogel R, Serruys PW, Hansson GK, Faxon DP, Kaul S, Drexler H, Greenland P, Muller JE, Virmani R, Ridker PM, Zipes DP, Shah PK, Willerson JT. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: Part I. Circulation. 2003;108:1664–72. doi: 10.1161/01.CIR.0000087480.94275.97. [DOI] [PubMed] [Google Scholar]
- 5.Naghavi M, Libby P, Falk E, Casscells SW, Litovsky S, Rumberger J, Badimon JJ, Stefanadis C, Moreno P, Pasterkamp G, Fayad Z, Stone PH, Waxman S, Raggi P, Madjid M, Zarrabi A, Burke A, Yuan C, Fitzgerald PJ, Siscovick DS, de Korte CL, Aikawa M, Airaksinen KE, Assmann G, Becker CR, Chesebro JH, Farb A, Galis ZS, Jackson C, Jang IK, Koenig W, Lodder RA, March K, Demirovic J, Navab M, Priori SG, Rekhter MD, Bahr R, Grundy SM, Mehran R, Colombo A, Boerwinkle E, Ballantyne C, Insull W, Jr, Schwartz RS, Vogel R, Serruys PW, Hansson GK, Faxon DP, Kaul S, Drexler H, Greenland P, Muller JE, Virmani R, Ridker PM, Zipes DP, Shah PK, Willerson JT. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: Part II. Circulation. 2003;108:1772–8. doi: 10.1161/01.CIR.0000087481.55887.C9. [DOI] [PubMed] [Google Scholar]
- 6.Ridker PM, Rifai N, Rose L, Buring JE, Cook NR. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med. 2002;347:1557–65. doi: 10.1056/NEJMoa021993. [DOI] [PubMed] [Google Scholar]
- 7.Liao JK, Laufs U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol. 2005;45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ridker PM, Rifai N, Pfeffer MA, Sacks F, Braunwald E. Long-term effects of pravastatin on plasma concentration of C-reactive protein. The Cholesterol and Recurrent Events (CARE) Investigators. Circulation. 1999;100:230–5. doi: 10.1161/01.cir.100.3.230. [DOI] [PubMed] [Google Scholar]
- 9.Ridker PM, Rifai N, Clearfield M, Downs JR, Weis SE, Miles JS, Gotto AM., Jr Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med. 2001;344:1959–65. doi: 10.1056/NEJM200106283442601. [DOI] [PubMed] [Google Scholar]
- 10.Ridker PM, Cannon CP, Morrow D, Rifai N, Rose LM, McCabe CH, Pfeffer MA, Braunwald E. C-reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352:20–8. doi: 10.1056/NEJMoa042378. [DOI] [PubMed] [Google Scholar]
- 11.Plump AS, Smith JD, Hayek T, Aalto-Setala K, Walsh A, Verstuyft JG, Rubin EM, Breslow JL. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–53. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- 12.Ishibashi S, Goldstein JL, Brown MS, Herz J, Burns DK. Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. J Clin Invest. 1994;93:1885–93. doi: 10.1172/JCI117179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skalen K, Gustafsson M, Rydberg EK, Hulten LM, Wiklund O, Innerarity TL, Boren J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002;417:750–4. doi: 10.1038/nature00804. [DOI] [PubMed] [Google Scholar]
- 14.Glass CK, Witztum JL. Atherosclerosis. the road ahead. Cell. 2001;104:503–16. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 15.Cybulsky MI, Gimbrone MA., Jr Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science. 1991;251:788–91. doi: 10.1126/science.1990440. [DOI] [PubMed] [Google Scholar]
- 16.Li H, Cybulsky MI, Gimbrone MA, Jr, Libby P. An atherogenic diet rapidly induces VCAM-1, a cytokine-regulatable mononuclear leukocyte adhesion molecule, in rabbit aortic endothelium. Arterioscler Thromb. 1993;13:197–204. doi: 10.1161/01.atv.13.2.197. [DOI] [PubMed] [Google Scholar]
- 17.Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, Davis V, Gutierrez-Ramos JC, Connelly PW, Milstone DS. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. 2001;107:1255–62. doi: 10.1172/JCI11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson RC, Chapman SM, Dong ZM, Ordovas JM, Mayadas TN, Herz J, Hynes RO, Schaefer EJ, Wagner DD. Absence of P-selectin delays fatty streak formation in mice. J Clin Invest. 1997;99:1037–43. doi: 10.1172/JCI119231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu L, Okada Y, Clinton SK, Gerard C, Sukhova GK, Libby P, Rollins BJ. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell. 1998;2:275–81. doi: 10.1016/s1097-2765(00)80139-2. [DOI] [PubMed] [Google Scholar]
- 20.Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2-/-mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–7. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 21.Amorino GP, Hoover RL. Interactions of monocytic cells with human endothelial cells stimulate monocytic metalloproteinase production. Am J Pathol. 1998;152:199–207. [PMC free article] [PubMed] [Google Scholar]
- 22.Boisvert WA, Santiago R, Curtiss LK, Terkeltaub RA. A leukocyte homologue of the IL-8 receptor CXCR-2 mediates the accumulation of macrophages in atherosclerotic lesions of LDL receptor-deficient mice. J Clin Invest. 1998;101:353–63. doi: 10.1172/JCI1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerszten RE, Garcia-Zepeda EA, Lim YC, Yoshida M, Ding HA, Gimbrone MA, Jr, Luster AD, Luscinskas FW, Rosenzweig A. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature. 1999;398:718–23. doi: 10.1038/19546. [DOI] [PubMed] [Google Scholar]
- 24.Lesnik P, Haskell CA, Charo IF. Decreased atherosclerosis in CX3CR1-/- mice reveals a role for fractalkine in atherogenesis. J Clin Invest. 2003;111:333–40. doi: 10.1172/JCI15555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Combadiere C, Potteaux S, Gao JL, Esposito B, Casanova S, Lee EJ, Debre P, Tedgui A, Murphy PM, Mallat Z. Decreased atherosclerotic lesion formation in CX3CR1/apolipoprotein E double knockout mice. Circulation. 2003;107:1009–16. doi: 10.1161/01.cir.0000057548.68243.42. [DOI] [PubMed] [Google Scholar]
- 26.Cheng C, Tempel D, van Haperen R, de Boer HC, Segers D, Huisman M, van Zonneveld AJ, Leenen PJ, van der Steen A, Serruys PW, de Crom R, Krams R. Shear stress-induced changes in atherosclerotic plaque composition are modulated by chemokines. J Clin Invest. 2007;117:616–26. doi: 10.1172/JCI28180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moulton KS, Vakili K, Zurakowski D, Soliman M, Butterfield C, Sylvin E, Lo KM, Gillies S, Javaherian K, Folkman J. Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc Natl Acad Sci U S A. 2003;100:4736–41. doi: 10.1073/pnas.0730843100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rajavashisth TB, Andalibi A, Territo MC, Berliner JA, Navab M, Fogelman AM, Lusis AJ. Induction of endothelial cell expression of granulocyte and macrophage colony-stimulating factors by modified low-density lipoproteins. Nature. 1990;344:254–7. doi: 10.1038/344254a0. [DOI] [PubMed] [Google Scholar]
- 29.Clinton SK, Underwood R, Hayes L, Sherman ML, Kufe DW, Libby P. Macrophage colony-stimulating factor gene expression in vascular cells and in experimental and human atherosclerosis. Am J Pathol. 1992;140:301–16. [PMC free article] [PubMed] [Google Scholar]
- 30.Edfeldt K, Swedenborg J, Hansson GK, Yan ZQ. Expression of toll-like receptors in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation. 2002;105:1158–61. [PubMed] [Google Scholar]
- 31.Monaco C, Andreakos E, Kiriakidis S, Mauri C, Bicknell C, Foxwell B, Cheshire N, Paleolog E, Feldmann M. Canonical pathway of nuclear factor kappa B activation selectively regulates proinflammatory and prothrombotic responses in human atherosclerosis. Proc Natl Acad Sci U S A. 2004;101:5634–9. doi: 10.1073/pnas.0401060101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kol A, Lichtman AH, Finberg RW, Libby P, Kurt-Jones EA. Cutting edge: heat shock protein (HSP) 60 activates the innate immune response: CD14 is an essential receptor for HSP60 activation of mononuclear cells. J Immunol. 2000;164:13–7. doi: 10.4049/jimmunol.164.1.13. [DOI] [PubMed] [Google Scholar]
- 33.Xu XH, Shah PK, Faure E, Equils O, Thomas L, Fishbein MC, Luthringer D, Xu XP, Rajavashisth TB, Yano J, Kaul S, Arditi M. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation. 2001;104:3103–8. doi: 10.1161/hc5001.100631. [DOI] [PubMed] [Google Scholar]
- 34.Miller YI, Viriyakosol S, Binder CJ, Feramisco JR, Kirkland TN, Witztum JL. Minimally modified LDL binds to CD14, induces macrophage spreading via TLR4/MD-2, and inhibits phagocytosis of apoptotic cells. J Biol Chem. 2003;278:1561–8. doi: 10.1074/jbc.M209634200. [DOI] [PubMed] [Google Scholar]
- 35.Bjorkbacka H, Kunjathoor VV, Moore KJ, Koehn S, Ordija CM, Lee MA, Means T, Halmen K, Luster AD, Golenbock DT, Freeman MW. Reduced atherosclerosis in MyD88-null mice links elevated serum cholesterol levels to activation of innate immunity signaling pathways. Nat Med. 2004;10:416–21. doi: 10.1038/nm1008. [DOI] [PubMed] [Google Scholar]
- 36.Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, Akira S, Rajavashisth TB, Arditi M. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci U S A. 2004;101:10679–84. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawakami A, Osaka M, Aikawa M, Uematsu S, Akira S, Libby P, Shimokado K, Sacks FM, Yoshida M. Toll-like receptor 2 mediates apolipoprotein CIII-induced monocyte activation. Circ Res. 2008;103:1402–9. doi: 10.1161/CIRCRESAHA.108.178426. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by Toll-like receptor 2. J Clin Invest. 2005;115:3149–56. doi: 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tobias PS, Curtiss LK. TLR2 in murine atherosclerosis. Semin Immunopathol. 2008;30:23–7. doi: 10.1007/s00281-007-0102-3. [DOI] [PubMed] [Google Scholar]
- 40.Gerdes N, Sukhova GK, Libby P, Reynolds RS, Young JL, Schonbeck U. Expression of interleukin (IL)-18 and functional IL-18 receptor on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for atherogenesis. J Exp Med. 2002;195:245–57. doi: 10.1084/jem.20011022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, Lira SA, Habenicht AJ, Randolph GJ. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–94. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Amento EP, Ehsani N, Palmer H, Libby P. Cytokines and growth factors positively and negatively regulate interstitial collagen gene expression in human vascular smooth muscle cells. Arterioscler Thromb. 1991;11:1223–30. doi: 10.1161/01.atv.11.5.1223. [DOI] [PubMed] [Google Scholar]
- 44.Sukhova GK, Schonbeck U, Rabkin E, Schoen FJ, Poole AR, Billinghurst RC, Libby P. Evidence for increased collagenolysis by interstitial collagenases-1 and -3 in vulnerable human atheromatous plaques. Circulation. 1999;99:2503–9. doi: 10.1161/01.cir.99.19.2503. [DOI] [PubMed] [Google Scholar]
- 45.Herman MP, Sukhova GK, Libby P, Gerdes N, Tang N, Horton DB, Kilbride M, Breitbart RE, Chun M, Schonbeck U. Expression of neutrophil collagenase (matrix metalloproteinase-8) in human atheroma: a novel collagenolytic pathway suggested by transcriptional profiling. Circulation. 2001;104:1899–904. doi: 10.1161/hc4101.097419. [DOI] [PubMed] [Google Scholar]
- 46.Deguchi JO, Aikawa E, Libby P, Vachon JR, Inada M, Krane SM, Whittaker P, Aikawa M. Matrix metalloproteinase-13/collagenase-3 deletion promotes collagen accumulation and organization in mouse atherosclerotic plaques. Circulation. 2005;112:2708–15. doi: 10.1161/CIRCULATIONAHA.105.562041. [DOI] [PubMed] [Google Scholar]
- 47.Schneider F, Sukhova GK, Aikawa M, Canner J, Gerdes N, Tang SM, Shi GP, Apte SS, Libby P. Matrix-metalloproteinase-14 deficiency in bone-marrow-derived cells promotes collagen accumulation in mouse atherosclerotic plaques. Circulation. 2008;117:931–9. doi: 10.1161/CIRCULATIONAHA.107.707448. [DOI] [PubMed] [Google Scholar]
- 48.Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94:2493–503. doi: 10.1172/JCI117619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fabunmi RP, Sukhova GK, Sugiyama S, Libby P. Expression of tissue inhibitor of metalloproteinases-3 in human atheroma and regulation in lesion-associated cells: a potential protective mechanism in plaque stability. Circ Res. 1998;83:270–8. doi: 10.1161/01.res.83.3.270. [DOI] [PubMed] [Google Scholar]
- 50.Newby AC. Metalloproteinase expression in monocytes and macrophages and its relationship to atherosclerotic plaque instability. Arterioscler Thromb Vasc Biol. 2008;28:2108–14. doi: 10.1161/ATVBAHA.108.173898. [DOI] [PubMed] [Google Scholar]
- 51.Dollery CM, Owen CA, Sukhova GK, Krettek A, Shapiro SD, Libby P. Neutrophil elastase in human atherosclerotic plaques: production by macrophages. Circulation. 2003;107:2829–36. doi: 10.1161/01.CIR.0000072792.65250.4A. [DOI] [PubMed] [Google Scholar]
- 52.Fukumoto Y, Deguchi JO, Libby P, Rabkin-Aikawa E, Sakata Y, Chin MT, Hill CC, Lawler PR, Varo N, Schoen FJ, Krane SM, Aikawa M. Genetically determined resistance to collagenase action augments interstitial collagen accumulation in atherosclerotic plaques. Circulation. 2004;110:1953–9. doi: 10.1161/01.CIR.0000143174.41810.10. [DOI] [PubMed] [Google Scholar]
- 53.Liu J, Sukhova GK, Sun JS, Xu WH, Libby P, Shi GP. Lysosomal cysteine proteases in atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:1359–66. doi: 10.1161/01.ATV.0000134530.27208.41. [DOI] [PubMed] [Google Scholar]
- 54.Sukhova GK, Shi GP, Simon DI, Chapman HA, Libby P. Expression of the elastolytic cathepsins S and K in human atheroma and regulation of their production in smooth muscle cells. J Clin Invest. 1998;102:576–83. doi: 10.1172/JCI181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jormsjo S, Wuttge DM, Sirsjo A, Whatling C, Hamsten A, Stemme S, Eriksson P. Differential expression of cysteine and aspartic proteases during progression of atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol. 2002;161:939–45. doi: 10.1016/S0002-9440(10)64254-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sukhova GK, Zhang Y, Pan JH, Wada Y, Yamamoto T, Naito M, Kodama T, Tsimikas S, Witztum JL, Lu ML, Sakara Y, Chin MT, Libby P, Shi GP. Deficiency of cathepsin S reduces atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2003;111:897–906. doi: 10.1172/JCI14915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kitamoto S, Sukhova GK, Sun J, Yang M, Libby P, Love V, Duramad P, Sun C, Zhang Y, Yang X, Peters C, Shi GP. Cathepsin L deficiency reduces diet-induced atherosclerosis in low-density lipoprotein receptor-knockout mice. Circulation. 2007;115:2065–75. doi: 10.1161/CIRCULATIONAHA.107.688523. [DOI] [PubMed] [Google Scholar]
- 58.Lutgens E, Lutgens SP, Faber BC, Heeneman S, Gijbels MM, de Winther MP, Frederik P, van der Made I, Daugherty A, Sijbers AM, Fisher A, Long CJ, Saftig P, Black D, Daemen MJ, Cleutjens KB. Disruption of the cathepsin K gene reduces atherosclerosis progression and induces plaque fibrosis but accelerates macrophage foam cell formation. Circulation. 2006;113:98–107. doi: 10.1161/CIRCULATIONAHA.105.561449. [DOI] [PubMed] [Google Scholar]
- 59.Shi GP, Sukhova GK, Grubb A, Ducharme A, Rhode LH, Lee RT, Ridker PM, Libby P, Chapman HA. Cystatin C deficiency in human atherosclerosis and aortic aneurysms. J Clin Invest. 1999;104:1191–7. doi: 10.1172/JCI7709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jonsson-Rylander AC, Nilsson T, Fritsche-Danielson R, Hammarstrom A, Behrendt M, Andersson JO, Lindgren K, Andersson AK, Wallbrandt P, Rosengren B, Brodin P, Thelin A, Westin A, Hurt-Camejo E, Lee-Sogaard CH. Role of ADAMTS-1 in atherosclerosis: remodeling of carotid artery, immunohistochemistry, and proteolysis of versican. Arterioscler Thromb Vasc Biol. 2005;25:180–5. doi: 10.1161/01.ATV.0000150045.27127.37. [DOI] [PubMed] [Google Scholar]
- 61.Wagsater D, Bjork H, Zhu C, Bjorkegren J, Valen G, Hamsten A, Eriksson P. ADAMTS-4 and -8 are inflammatory regulated enzymes expressed in macrophage-rich areas of human atherosclerotic plaques. Atherosclerosis. 2008;196:514–22. doi: 10.1016/j.atherosclerosis.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 62.Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481–8. doi: 10.1161/CIRCULATIONAHA.105.537878. [DOI] [PubMed] [Google Scholar]
- 63.Cairns A, Constantinides P. Mast cells in human atherosclerosis. Science. 1954;120:31–2. doi: 10.1126/science.120.3105.31. [DOI] [PubMed] [Google Scholar]
- 64.Jeziorska M, McCollum C, Woolley DE. Mast cell distribution, activation, and phenotype in atherosclerotic lesions of human carotid arteries. J Pathol. 1997;182:115–22. doi: 10.1002/(SICI)1096-9896(199705)182:1<115::AID-PATH806>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 65.Kaartinen M, van der Wal AC, van der Loos CM, Piek JJ, Koch KT, Becker AE, Kovanen PT. Mast cell infiltration in acute coronary syndromes: implications for plaque rupture. J Am Coll Cardiol. 1998;32:606–12. doi: 10.1016/s0735-1097(98)00283-6. [DOI] [PubMed] [Google Scholar]
- 66.Kovanen PT, Kaartinen M, Paavonen T. Infiltrates of activated mast cells at the site of coronary atheromatous erosion or rupture in myocardial infarction. Circulation. 1995;92:1084–8. doi: 10.1161/01.cir.92.5.1084. [DOI] [PubMed] [Google Scholar]
- 67.Libby P, Shi GP. Mast cells as mediators and modulators of atherogenesis. Circulation. 2007;115:2471–3. doi: 10.1161/CIRCULATIONAHA.107.698480. [DOI] [PubMed] [Google Scholar]
- 68.Haley KJ, Lilly CM, Yang JH, Feng Y, Kennedy SP, Turi TG, Thompson JF, Sukhova GH, Libby P, Lee RT. Overexpression of eotaxin and the CCR3 receptor in human atherosclerosis: using genomic technology to identify a potential novel pathway of vascular inflammation. Circulation. 2000;102:2185–9. doi: 10.1161/01.cir.102.18.2185. [DOI] [PubMed] [Google Scholar]
- 69.Sun J, Sukhova GK, Wolters PJ, Yang M, Kitamoto S, Libby P, MacFarlane LA, Mallen-St Clair J, Shi GP. Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat Med. 2007;13:719–24. doi: 10.1038/nm1601. [DOI] [PubMed] [Google Scholar]
- 70.Bot I, de Jager SC, Zernecke A, Lindstedt KA, van Berkel TJ, Weber C, Biessen EA. Perivascular mast cells promote atherogenesis and induce plaque destabilization in apolipoprotein E-deficient mice. Circulation. 2007;115:2516–25. doi: 10.1161/CIRCULATIONAHA.106.660472. [DOI] [PubMed] [Google Scholar]
- 71.Di Girolamo N, Wakefield D. In vitro and in vivo expression of interstitial collagenase/MMP-1 by human mast cells. Dev Immunol. 2000;7:131–42. doi: 10.1155/2000/82708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Baram D, Vaday GG, Salamon P, Drucker I, Hershkoviz R, Mekori YA. Human mast cells release metalloproteinase-9 on contact with activated T cells: juxtacrine regulation by TNF-alpha. J Immunol. 2001;167:4008–16. doi: 10.4049/jimmunol.167.7.4008. [DOI] [PubMed] [Google Scholar]
- 73.Johnson JL, Jackson CL, Angelini GD, George SJ. Activation of matrix-degrading metalloproteinases by mast cell proteases in atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 1998;18:1707–15. doi: 10.1161/01.atv.18.11.1707. [DOI] [PubMed] [Google Scholar]
- 74.Tchougounova E, Lundequist A, Fajardo I, Winberg JO, Abrink M, Pejler G. A key role for mast cell chymase in the activation of pro-matrix metalloprotease-9 and pro-matrix metalloprotease-2. J Biol Chem. 2005;280:9291–6. doi: 10.1074/jbc.M410396200. [DOI] [PubMed] [Google Scholar]
- 75.Lappalainen H, Laine P, Pentikainen MO, Sajantila A, Kovanen PT. Mast cells in neovascularized human coronary plaques store and secrete basic fibroblast growth factor, a potent angiogenic mediator. Arterioscler Thromb Vasc Biol. 2004;24:1880–5. doi: 10.1161/01.ATV.0000140820.51174.8d. [DOI] [PubMed] [Google Scholar]
- 76.Caughey GH, Raymond WW, Wolters PJ. Angiotensin II generation by mast cell alpha- and beta-chymases. Biochim Biophys Acta. 2000;1480:245–57. doi: 10.1016/s0167-4838(00)00076-5. [DOI] [PubMed] [Google Scholar]
- 77.Heikkila HM, Latti S, Leskinen MJ, Hakala JK, Kovanen PT, Lindstedt KA. Activated mast cells induce endothelial cell apoptosis by a combined action of chymase and tumor necrosis factor-alpha. Arterioscler Thromb Vasc Biol. 2008;28:309–14. doi: 10.1161/ATVBAHA.107.151340. [DOI] [PubMed] [Google Scholar]
- 78.Wang Y, Shiota N, Leskinen MJ, Lindstedt KA, Kovanen PT. Mast cell chymase inhibits smooth muscle cell growth and collagen expression in vitro: transforming growth factor-beta1-dependent and -independent effects. Arterioscler Thromb Vasc Biol. 2001;21:1928–33. doi: 10.1161/hq1201.100227. [DOI] [PubMed] [Google Scholar]
- 79.Leskinen MJ, Lindstedt KA, Wang Y, Kovanen PT. Mast cell chymase induces smooth muscle cell apoptosis by a mechanism involving fibronectin degradation and disruption of focal adhesions. Arterioscler Thromb Vasc Biol. 2003;23:238–43. doi: 10.1161/01.atv.0000051405.68811.4d. [DOI] [PubMed] [Google Scholar]
- 80.Leskinen MJ, Heikkila HM, Speer MY, Hakala JK, Laine M, Kovanen PT, Lindstedt KA. Mast cell chymase induces smooth muscle cell apoptosis by disrupting NF-kappaB-mediated survival signaling. Exp Cell Res. 2006;312:1289–98. doi: 10.1016/j.yexcr.2005.12.033. [DOI] [PubMed] [Google Scholar]
- 81.Steffel J, Akhmedov A, Greutert H, Luscher TF, Tanner FC. Histamine induces tissue factor expression: implications for acute coronary syndromes. Circulation. 2005;112:341–9. doi: 10.1161/CIRCULATIONAHA.105.553735. [DOI] [PubMed] [Google Scholar]
- 82.Wang Y, Lindstedt KA, Kovanen PT. Mast cell granule remnants carry LDL into smooth muscle cells of the synthetic phenotype and induce their conversion into foam cells. Arterioscler Thromb Vasc Biol. 1995;15:801–10. doi: 10.1161/01.atv.15.6.801. [DOI] [PubMed] [Google Scholar]
- 83.Ma H, Kovanen PT. IgE-dependent generation of foam cells: an immune mechanism involving degranulation of sensitized mast cells with resultant uptake of LDL by macrophages. Arterioscler Thromb Vasc Biol. 1995;15:811–9. doi: 10.1161/01.atv.15.6.811. [DOI] [PubMed] [Google Scholar]
- 84.Kokkonen JO, Kovanen PT. Proteolytic enzymes of mast cell granules degrade low density lipoproteins and promote their granule-mediated uptake by macrophages in vitro. J Biol Chem. 1989;264:10749–55. [PubMed] [Google Scholar]
- 85.Lee M, Calabresi L, Chiesa G, Franceschini G, Kovanen PT. Mast cell chymase degrades apoE and apoA-II in apoA-I-knockout mouse plasma and reduces its ability to promote cellular cholesterol efflux. Arterioscler Thromb Vasc Biol. 2002;22:1475–81. doi: 10.1161/01.atv.0000029782.84357.68. [DOI] [PubMed] [Google Scholar]
- 86.Lee M, Sommerhoff CP, von Eckardstein A, Zettl F, Fritz H, Kovanen PT. Mast cell tryptase degrades HDL and blocks its function as an acceptor of cellular cholesterol. Arterioscler Thromb Vasc Biol. 2002;22:2086–91. doi: 10.1161/01.atv.0000041405.07367.b5. [DOI] [PubMed] [Google Scholar]
- 87.Yokoyama WM, Kim S, French AR. The dynamic life of natural killer cells. Annu Rev Immunol. 2004;22:405–29. doi: 10.1146/annurev.immunol.22.012703.104711. [DOI] [PubMed] [Google Scholar]
- 88.Millonig G, Malcom GT, Wick G. Early inflammatory-immunological lesions in juvenile atherosclerosis from the Pathobiological Determinants of Atherosclerosis in Youth (PDAY)-study. Atherosclerosis. 2002;160:441–8. doi: 10.1016/s0021-9150(01)00596-2. [DOI] [PubMed] [Google Scholar]
- 89.Whitman SC, Rateri DL, Szilvassy SJ, Yokoyama W, Daugherty A. Depletion of natural killer cell function decreases atherosclerosis in low-density lipoprotein receptor null mice. Arterioscler Thromb Vasc Biol. 2004;24:1049–54. doi: 10.1161/01.ATV.0000124923.95545.2c. [DOI] [PubMed] [Google Scholar]
- 90.Naruko T, Ueda M, Haze K, van der Wal AC, van der Loos CM, Itoh A, Komatsu R, Ikura Y, Ogami M, Shimada Y, Ehara S, Yoshiyama M, Takeuchi K, Yoshikawa J, Becker AE. Neutrophil infiltration of culprit lesions in acute coronary syndromes. Circulation. 2002;106:2894–900. doi: 10.1161/01.cir.0000042674.89762.20. [DOI] [PubMed] [Google Scholar]
- 91.Horne BD, Anderson JL, John JM, Weaver A, Bair TL, Jensen KR, Renlund DG, Muhlestein JB. Which white blood cell subtypes predict increased cardiovascular risk? J Am Coll Cardiol. 2005;45:1638–43. doi: 10.1016/j.jacc.2005.02.054. [DOI] [PubMed] [Google Scholar]
- 92.Kolodgie FD, Gold HK, Burke AP, Fowler DR, Kruth HS, Weber DK, Farb A, Guerrero LJ, Hayase M, Kutys R, Narula J, Finn AV, Virmani R. Intraplaque hemorrhage and progression of coronary atheroma. N Engl J Med. 2003;349:2316–25. doi: 10.1056/NEJMoa035655. [DOI] [PubMed] [Google Scholar]
- 93.Leclercq A, Houard X, Philippe M, Ollivier V, Sebbag U, Meilhac O, Michel JB. Involvement of intraplaque hemorrhage in atherothrombosis evolution via neutrophil protease enrichment. J Leukoc Biol. 2007;82:1420–9. doi: 10.1189/jlb.1106671. [DOI] [PubMed] [Google Scholar]
- 94.Hemdahl AL, Gabrielsen A, Zhu C, Eriksson P, Hedin U, Kastrup J, Thoren P, Hansson GK. Expression of neutrophil gelatinase-associated lipocalin in atherosclerosis and myocardial infarction. Arterioscler Thromb Vasc Biol. 2006;26:136–42. doi: 10.1161/01.ATV.0000193567.88685.f4. [DOI] [PubMed] [Google Scholar]
- 95.van Leeuwen M, Gijbels MJ, Duijvestijn A, Smook M, van de Gaar MJ, Heeringa P, de Winther MP, Tervaert JW. Accumulation of myeloperoxidase-positive neutrophils in atherosclerotic lesions in LDLR-/- mice. Arterioscler Thromb Vasc Biol. 2008;28:84–9. doi: 10.1161/ATVBAHA.107.154807. [DOI] [PubMed] [Google Scholar]
- 96.Sugiyama S, Kugiyama K, Aikawa M, Nakamura S, Ogawa H, Libby P. Hypochlorous acid, a macrophage product, induces endothelial apoptosis and tissue factor expression: involvement of myeloperoxidase-mediated oxidant in plaque erosion and thrombogenesis. Arterioscler Thromb Vasc Biol. 2004;24:1309–14. doi: 10.1161/01.ATV.0000131784.50633.4f. [DOI] [PubMed] [Google Scholar]
- 97.Podrez EA, Schmitt D, Hoff HF, Hazen SL. Myeloperoxidase-generated reactive nitrogen species convert LDL into an atherogenic form in vitro. J Clin Invest. 1999;103:1547–60. doi: 10.1172/JCI5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Martin C, Burdon PC, Bridger G, Gutierrez-Ramos JC, Williams TJ, Rankin SM. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity. 2003;19:583–93. doi: 10.1016/s1074-7613(03)00263-2. [DOI] [PubMed] [Google Scholar]
- 99.Zernecke A, Bot I, Djalali-Talab Y, Shagdarsuren E, Bidzhekov K, Meiler S, Krohn R, Schober A, Sperandio M, Soehnlein O, Bornemann J, Tacke F, Biessen EA, Weber C. Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ Res. 2008;102:209–17. doi: 10.1161/CIRCRESAHA.107.160697. [DOI] [PubMed] [Google Scholar]
- 100.Woollard KJ, Suhartoyo A, Harris EE, Eisenhardt SU, Jackson SP, Peter K, Dart AM, Hickey MJ, Chin-Dusting JP. Pathophysiological levels of soluble P-selectin mediate adhesion of leukocytes to the endothelium through Mac-1 activation. Circ Res. 2008;103:1128–38. doi: 10.1161/CIRCRESAHA.108.180273. [DOI] [PubMed] [Google Scholar]
- 101.Bobryshev YV. Dendritic cells in atherosclerosis: current status of the problem and clinical relevance. Eur Heart J. 2005;26:1700–4. doi: 10.1093/eurheartj/ehi282. [DOI] [PubMed] [Google Scholar]
- 102.Liu P, Yu YR, Spencer JA, Johnson AE, Vallanat CT, Fong AM, Patterson C, Patel DD. CX3CR1 deficiency impairs dendritic cell accumulation in arterial intima and reduces atherosclerotic burden. Arterioscler Thromb Vasc Biol. 2008;28:243–50. doi: 10.1161/ATVBAHA.107.158675. [DOI] [PubMed] [Google Scholar]
- 103.Shaposhnik Z, Wang X, Weinstein M, Bennett BJ, Lusis AJ. Granulocyte macrophage colony-stimulating factor regulates dendritic cell content of atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2007;27:621–7. doi: 10.1161/01.ATV.0000254673.55431.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Weis M, Schlichting CL, Engleman EG, Cooke JP. Endothelial determinants of dendritic cell adhesion and migration: new implications for vascular diseases. Arterioscler Thromb Vasc Biol. 2002;22:1817–23. doi: 10.1161/01.atv.0000036418.04998.d5. [DOI] [PubMed] [Google Scholar]
- 105.Aicher A, Heeschen C, Mohaupt M, Cooke JP, Zeiher AM, Dimmeler S. Nicotine strongly activates dendritic cell-mediated adaptive immunity: potential role for progression of atherosclerotic lesions. Circulation. 2003;107:604–11. doi: 10.1161/01.cir.0000047279.42427.6d. [DOI] [PubMed] [Google Scholar]
- 106.Jongstra-Bilen J, Haidari M, Zhu SN, Chen M, Guha D, Cybulsky MI. Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J Exp Med. 2006;203:2073–83. doi: 10.1084/jem.20060245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yilmaz A, Lochno M, Traeg F, Cicha I, Reiss C, Stumpf C, Raaz D, Anger T, Amann K, Probst T, Ludwig J, Daniel WG, Garlichs CD. Emergence of dendritic cells in rupture-prone regions of vulnerable carotid plaques. Atherosclerosis. 2004;176:101–10. doi: 10.1016/j.atherosclerosis.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 108.Erbel C, Sato K, Meyer FB, Kopecky SL, Frye RL, Goronzy JJ, Weyand CM. Functional profile of activated dendritic cells in unstable atherosclerotic plaque. Basic Res Cardiol. 2007;102:123–32. doi: 10.1007/s00395-006-0636-x. [DOI] [PubMed] [Google Scholar]
- 109.Llodra J, Angeli V, Liu J, Trogan E, Fisher EA, Randolph GJ. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc Natl Acad Sci U S A. 2004;101:11779–84. doi: 10.1073/pnas.0403259101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Angeli V, Llodra J, Rong JX, Satoh K, Ishii S, Shimizu T, Fisher EA, Randolph GJ. Dyslipidemia associated with atherosclerotic disease systemically alters dendritic cell mobilization. Immunity. 2004;21:561–74. doi: 10.1016/j.immuni.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 111.Packard RR, Maganto-Garcia E, Gotsman I, Tabas I, Libby P, Lichtman AH. CD11c(+) dendritic cells maintain antigen processing, presentation capabilities, and CD4(+) T-cell priming efficacy under hypercholesterolemic conditions associated with atherosclerosis. Circ Res. 2008;103:965–73. doi: 10.1161/CIRCRESAHA.108.185793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, Sweeney M, Rong JX, Kuriakose G, Fisher EA, Marks AR, Ron D, Tabas I. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003;5:781–92. doi: 10.1038/ncb1035. [DOI] [PubMed] [Google Scholar]
- 113.Rivollier A, Perrin-Cocon L, Luche S, Diemer H, Strub JM, Hanau D, van Dorsselaer A, Lotteau V, Rabourdin-Combe C, Rabilloud T, Servet-Delprat C. High expression of antioxidant proteins in dendritic cells: possible implications in atherosclerosis. Mol Cell Proteomics. 2006;5:726–36. doi: 10.1074/mcp.M500262-MCP200. [DOI] [PubMed] [Google Scholar]
- 114.Han JW, Shimada K, Ma-Krupa W, Johnson TL, Nerem RM, Goronzy JJ, Weyand CM. Vessel wall-embedded dendritic cells induce T-cell autoreactivity and initiate vascular inflammation. Circ Res. 2008;102:546–53. doi: 10.1161/CIRCRESAHA.107.161653. [DOI] [PubMed] [Google Scholar]
- 115.Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002;2:151–61. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- 116.Niessner A, Sato K, Chaikof EL, Colmegna I, Goronzy JJ, Weyand CM. Pathogen-sensing plasmacytoid dendritic cells stimulate cytotoxic T-cell function in the atherosclerotic plaque through interferon-alpha. Circulation. 2006;114:2482–9. doi: 10.1161/CIRCULATIONAHA.106.642801. [DOI] [PubMed] [Google Scholar]
- 117.Sato K, Niessner A, Kopecky SL, Frye RL, Goronzy JJ, Weyand CM. TRAIL-expressing T cells induce apoptosis of vascular smooth muscle cells in the atherosclerotic plaque. J Exp Med. 2006;203:239–50. doi: 10.1084/jem.20051062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Niessner A, Shin MS, Pryshchep O, Goronzy JJ, Chaikof EL, Weyand CM. Synergistic proinflammatory effects of the antiviral cytokine interferon-alpha and Toll-like receptor 4 ligands in the atherosclerotic plaque. Circulation. 2007;116:2043–52. doi: 10.1161/CIRCULATIONAHA.107.697789. [DOI] [PubMed] [Google Scholar]
- 119.Kalayoglu MV, Libby P, Byrne GI. Chlamydia pneumoniae as an emerging risk factor in cardiovascular disease. Jama. 2002;288:2724–31. doi: 10.1001/jama.288.21.2724. [DOI] [PubMed] [Google Scholar]
- 120.Madjid M, Vela D, Khalili-Tabrizi H, Casscells SW, Litovsky S. Systemic infections cause exaggerated local inflammation in atherosclerotic coronary arteries: clues to the triggering effect of acute infections on acute coronary syndromes. Tex Heart Inst J. 2007;34:11–8. [PMC free article] [PubMed] [Google Scholar]
- 121.Forster R, Schubel A, Breitfeld D, Kremmer E, Renner-Muller I, Wolf E, Lipp M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 122.Qu C, Edwards EW, Tacke F, Angeli V, Llodra J, Sanchez-Schmitz G, Garin A, Haque NS, Peters W, van Rooijen N, Sanchez-Torres C, Bromberg J, Charo IF, Jung S, Lira SA, Randolph GJ. Role of CCR8 and other chemokine pathways in the migration of monocyte-derived dendritic cells to lymph nodes. J Exp Med. 2004;200:1231–41. doi: 10.1084/jem.20032152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gotsman I, Sharpe AH, Lichtman AH. T-cell costimulation and coinhibition in atherosclerosis. Circ Res. 2008;103:1220–31. doi: 10.1161/CIRCRESAHA.108.182428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–48. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- 125.Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 126.Buono C, Pang H, Uchida Y, Libby P, Sharpe AH, Lichtman AH. B7-1/B7-2 costimulation regulates plaque antigen-specific T-cell responses and atherogenesis in low-density lipoprotein receptor-deficient mice. Circulation. 2004;109:2009–15. doi: 10.1161/01.CIR.0000127121.16815.F1. [DOI] [PubMed] [Google Scholar]
- 127.Dong C, Juedes AE, Temann UA, Shresta S, Allison JP, Ruddle NH, Flavell RA. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature. 2001;409:97–101. doi: 10.1038/35051100. [DOI] [PubMed] [Google Scholar]
- 128.Gotsman I, Grabie N, Gupta R, Dacosta R, MacConmara M, Lederer J, Sukhova G, Witztum JL, Sharpe AH, Lichtman AH. Impaired regulatory T-cell response and enhanced atherosclerosis in the absence of inducible costimulatory molecule. Circulation. 2006;114:2047–55. doi: 10.1161/CIRCULATIONAHA.106.633263. [DOI] [PubMed] [Google Scholar]
- 129.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8:239–45. doi: 10.1038/ni1443. [DOI] [PubMed] [Google Scholar]
- 130.Gotsman I, Grabie N, Dacosta R, Sukhova G, Sharpe A, Lichtman AH. Proatherogenic immune responses are regulated by the PD-1/PD-L pathway in mice. J Clin Invest. 2007;117:2974–82. doi: 10.1172/JCI31344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang X, Ria M, Kelmenson PM, Eriksson P, Higgins DC, Samnegard A, Petros C, Rollins J, Bennet AM, Wiman B, de Faire U, Wennberg C, Olsson PG, Ishii N, Sugamura K, Hamsten A, Forsman-Semb K, Lagercrantz J, Paigen B. Positional identification of TNFSF4, encoding OX40 ligand, as a gene that influences atherosclerosis susceptibility. Nat Genet. 2005;37:365–72. doi: 10.1038/ng1524. [DOI] [PubMed] [Google Scholar]
- 132.van Wanrooij EJ, van Puijvelde GH, de Vos P, Yagita H, van Berkel TJ, Kuiper J. Interruption of the Tnfrsf4/Tnfsf4 (OX40/OX40L) pathway attenuates atherogenesis in low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2007;27:204–10. doi: 10.1161/01.ATV.0000251007.07648.81. [DOI] [PubMed] [Google Scholar]
- 133.Dawicki W, Bertram EM, Sharpe AH, Watts TH. 4-1BB and OX40 act independently to facilitate robust CD8 and CD4 recall responses. J Immunol. 2004;173:5944–51. doi: 10.4049/jimmunol.173.10.5944. [DOI] [PubMed] [Google Scholar]
- 134.Olofsson PS, Soderstrom LA, Wagsater D, Sheikine Y, Ocaya P, Lang F, Rabu C, Chen L, Rudling M, Aukrust P, Hedin U, Paulsson-Berne G, Sirsjo A, Hansson GK. CD137 is expressed in human atherosclerosis and promotes development of plaque inflammation in hypercholesterolemic mice. Circulation. 2008;117:1292–301. doi: 10.1161/CIRCULATIONAHA.107.699173. [DOI] [PubMed] [Google Scholar]
- 135.Hosono M, de Boer OJ, van der Wal AC, van der Loos CM, Teeling P, Piek JJ, Ueda M, Becker AE. Increased expression of T cell activation markers (CD25, CD26, CD40L and CD69) in atherectomy specimens of patients with unstable angina and acute myocardial infarction. Atherosclerosis. 2003;168:73–80. doi: 10.1016/s0021-9150(03)00024-8. [DOI] [PubMed] [Google Scholar]
- 136.Roselaar SE, Kakkanathu PX, Daugherty A. Lymphocyte populations in atherosclerotic lesions of apoE -/- and LDL receptor -/- mice. Decreasing density with disease progression. Arterioscler Thromb Vasc Biol. 1996;16:1013–8. doi: 10.1161/01.atv.16.8.1013. [DOI] [PubMed] [Google Scholar]
- 137.Mach F, Sauty A, Iarossi AS, Sukhova GK, Neote K, Libby P, Luster AD. Differential expression of three T lymphocyte-activating CXC chemokines by human atheroma-associated cells. J Clin Invest. 1999;104:1041–50. doi: 10.1172/JCI6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Paulsson G, Zhou X, Tornquist E, Hansson GK. Oligoclonal T cell expansions in atherosclerotic lesions of apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2000;20:10–7. doi: 10.1161/01.atv.20.1.10. [DOI] [PubMed] [Google Scholar]
- 139.Stemme S, Faber B, Holm J, Wiklund O, Witztum JL, Hansson GK. T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc Natl Acad Sci U S A. 1995;92:3893–7. doi: 10.1073/pnas.92.9.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Xu Q, Kleindienst R, Waitz W, Dietrich H, Wick G. Increased expression of heat shock protein 65 coincides with a population of infiltrating T lymphocytes in atherosclerotic lesions of rabbits specifically responding to heat shock protein 65. J Clin Invest. 1993;91:2693–702. doi: 10.1172/JCI116508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Song L, Leung C, Schindler C. Lymphocytes are important in early atherosclerosis. J Clin Invest. 2001;108:251–9. doi: 10.1172/JCI11380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhou X, Nicoletti A, Elhage R, Hansson GK. Transfer of CD4(+) T cells aggravates atherosclerosis in immunodeficient apolipoprotein E knockout mice. Circulation. 2000;102:2919–22. doi: 10.1161/01.cir.102.24.2919. [DOI] [PubMed] [Google Scholar]
- 143.Paigen B, Morrow A, Brandon C, Mitchell D, Holmes P. Variation in susceptibility to atherosclerosis among inbred strains of mice. Atherosclerosis. 1985;57:65–73. doi: 10.1016/0021-9150(85)90138-8. [DOI] [PubMed] [Google Scholar]
- 144.Schulte S, Sukhova GK, Libby P. Genetically programmed biases in Th1 and Th2 immune responses modulate atherogenesis. Am J Pathol. 2008;172:1500–8. doi: 10.2353/ajpath.2008.070776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Buono C, Binder CJ, Stavrakis G, Witztum JL, Glimcher LH, Lichtman AH. T-bet deficiency reduces atherosclerosis and alters plaque antigen-specific immune responses. Proc Natl Acad Sci U S A. 2005;102:1596–601. doi: 10.1073/pnas.0409015102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Laurat E, Poirier B, Tupin E, Caligiuri G, Hansson GK, Bariety J, Nicoletti A. In vivo downregulation of T helper cell 1 immune responses reduces atherogenesis in apolipoprotein E-knockout mice. Circulation. 2001;104:197–202. doi: 10.1161/01.cir.104.2.197. [DOI] [PubMed] [Google Scholar]
- 147.Lee TS, Yen HC, Pan CC, Chau LY. The role of interleukin 12 in the development of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 1999;19:734–42. doi: 10.1161/01.atv.19.3.734. [DOI] [PubMed] [Google Scholar]
- 148.Davenport P, Tipping PG. The role of interleukin-4 and interleukin-12 in the progression of atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol. 2003;163:1117–25. doi: 10.1016/S0002-9440(10)63471-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hauer AD, Uyttenhove C, de Vos P, Stroobant V, Renauld JC, van Berkel TJ, van Snick J, Kuiper J. Blockade of interleukin-12 function by protein vaccination attenuates atherosclerosis. Circulation. 2005;112:1054–62. doi: 10.1161/CIRCULATIONAHA.104.533463. [DOI] [PubMed] [Google Scholar]
- 150.Elhage R, Jawien J, Rudling M, Ljunggren HG, Takeda K, Akira S, Bayard F, Hansson GK. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc Res. 2003;59:234–40. doi: 10.1016/s0008-6363(03)00343-2. [DOI] [PubMed] [Google Scholar]
- 151.Mallat Z, Corbaz A, Scoazec A, Graber P, Alouani S, Esposito B, Humbert Y, Chvatchko Y, Tedgui A. Interleukin-18/interleukin-18 binding protein signaling modulates atherosclerotic lesion development and stability. Circ Res. 2001;89:E41–5. doi: 10.1161/hh1901.098735. [DOI] [PubMed] [Google Scholar]
- 152.Buono C, Come CE, Stavrakis G, Maguire GF, Connelly PW, Lichtman AH. Influence of interferon-gamma on the extent and phenotype of diet-induced atherosclerosis in the LDLR-deficient mouse. Arterioscler Thromb Vasc Biol. 2003;23:454–60. doi: 10.1161/01.ATV.0000059419.11002.6E. [DOI] [PubMed] [Google Scholar]
- 153.Gupta S, Pablo AM, Jiang X, Wang N, Tall AR, Schindler C. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J Clin Invest. 1997;99:2752–61. doi: 10.1172/JCI119465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Whitman SC, Ravisankar P, Elam H, Daugherty A. Exogenous interferon-gamma enhances atherosclerosis in apolipoprotein E-/- mice. Am J Pathol. 2000;157:1819–24. doi: 10.1016/s0002-9440(10)64820-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hansson GK, Hellstrand M, Rymo L, Rubbia L, Gabbiani G. Interferon gamma inhibits both proliferation and expression of differentiation-specific alpha-smooth muscle actin in arterial smooth muscle cells. J Exp Med. 1989;170:1595–608. doi: 10.1084/jem.170.5.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Friesel R, Komoriya A, Maciag T. Inhibition of endothelial cell proliferation by gamma-interferon. J Cell Biol. 1987;104:689–96. doi: 10.1083/jcb.104.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Frostegard J, Ulfgren AK, Nyberg P, Hedin U, Swedenborg J, Andersson U, Hansson GK. Cytokine expression in advanced human atherosclerotic plaques: dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis. 1999;145:33–43. doi: 10.1016/s0021-9150(99)00011-8. [DOI] [PubMed] [Google Scholar]
- 158.Tellides G, Tereb DA, Kirkiles-Smith NC, Kim RW, Wilson JH, Schechner JS, Lorber MI, Pober JS. Interferon-gamma elicits arteriosclerosis in the absence of leukocytes. Nature. 2000;403:207–11. doi: 10.1038/35003221. [DOI] [PubMed] [Google Scholar]
- 159.Bavendiek U, Libby P, Kilbride M, Reynolds R, Mackman N, Schonbeck U. Induction of tissue factor expression in human endothelial cells by CD40 ligand is mediated via activator protein 1, nuclear factor kappa B, and Egr-1. J Biol Chem. 2002;277:25032–9. doi: 10.1074/jbc.M204003200. [DOI] [PubMed] [Google Scholar]
- 160.Schonbeck U, Mach F, Sukhova GK, Herman M, Graber P, Kehry MR, Libby P. CD40 ligation induces tissue factor expression in human vascular smooth muscle cells. Am J Pathol. 2000;156:7–14. doi: 10.1016/S0002-9440(10)64699-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Mach F, Schonbeck U, Bonnefoy JY, Pober JS, Libby P. Activation of monocyte/macrophage functions related to acute atheroma complication by ligation of CD40: induction of collagenase, stromelysin, and tissue factor. Circulation. 1997;96:396–9. doi: 10.1161/01.cir.96.2.396. [DOI] [PubMed] [Google Scholar]
- 162.Zirlik A, Maier C, Gerdes N, MacFarlane L, Soosairajah J, Bavendiek U, Ahrens I, Ernst S, Bassler N, Missiou A, Patko Z, Aikawa M, Schonbeck U, Bode C, Libby P, Peter K. CD40 ligand mediates inflammation independently of CD40 by interaction with Mac-1. Circulation. 2007;115:1571–80. doi: 10.1161/CIRCULATIONAHA.106.683201. [DOI] [PubMed] [Google Scholar]
- 163.Grewal IS, Flavell RA. CD40 and CD154 in cell-mediated immunity. Annu Rev Immunol. 1998;16:111–35. doi: 10.1146/annurev.immunol.16.1.111. [DOI] [PubMed] [Google Scholar]
- 164.Mach F, Schonbeck U, Sukhova GK, Bourcier T, Bonnefoy JY, Pober JS, Libby P. Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for CD40-CD40 ligand signaling in atherosclerosis. Proc Natl Acad Sci U S A. 1997;94:1931–6. doi: 10.1073/pnas.94.5.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, Kroczek RA. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. 1998;391:591–4. doi: 10.1038/35393. [DOI] [PubMed] [Google Scholar]
- 166.Lutgens E, Gorelik L, Daemen MJ, de Muinck ED, Grewal IS, Koteliansky VE, Flavell RA. Requirement for CD154 in the progression of atherosclerosis. Nat Med. 1999;5:1313–6. doi: 10.1038/15271. [DOI] [PubMed] [Google Scholar]
- 167.Mach F, Schonbeck U, Sukhova GK, Atkinson E, Libby P. Reduction of atherosclerosis in mice by inhibition of CD40 signalling. Nature. 1998;394:200–3. doi: 10.1038/28204. [DOI] [PubMed] [Google Scholar]
- 168.Nadareishvili ZG, Koziol DE, Szekely B, Ruetzler C, LaBiche R, McCarron R, DeGraba TJ. Increased CD8(+) T cells associated with Chlamydia pneumoniae in symptomatic carotid plaque. Stroke. 2001;32:1966–72. doi: 10.1161/hs0901.095633. [DOI] [PubMed] [Google Scholar]
- 169.Henderson EL, Geng YJ, Sukhova GK, Whittemore AD, Knox J, Libby P. Death of smooth muscle cells and expression of mediators of apoptosis by T lymphocytes in human abdominal aortic aneurysms. Circulation. 1999;99:96–104. doi: 10.1161/01.cir.99.1.96. [DOI] [PubMed] [Google Scholar]
- 170.Park SH, Bendelac A. CD1-restricted T-cell responses and microbial infection. Nature. 2000;406:788–92. doi: 10.1038/35021233. [DOI] [PubMed] [Google Scholar]
- 171.Melian A, Geng YJ, Sukhova GK, Libby P, Porcelli SA. CD1 expression in human atherosclerosis. A potential mechanism for T cell activation by foam cells. Am J Pathol. 1999;155:775–86. doi: 10.1016/S0002-9440(10)65176-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Tupin E, Nicoletti A, Elhage R, Rudling M, Ljunggren HG, Hansson GK, Berne GP. CD1d-dependent activation of NKT cells aggravates atherosclerosis. J Exp Med. 2004;199:417–22. doi: 10.1084/jem.20030997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Nakai Y, Iwabuchi K, Fujii S, Ishimori N, Dashtsoodol N, Watano K, Mishima T, Iwabuchi C, Tanaka S, Bezbradica JS, Nakayama T, Taniguchi M, Miyake S, Yamamura T, Kitabatake A, Joyce S, Van Kaer L, Onoe K. Natural killer T cells accelerate atherogenesis in mice. Blood. 2004;104:2051–9. doi: 10.1182/blood-2003-10-3485. [DOI] [PubMed] [Google Scholar]
- 174.Major AS, Wilson MT, McCaleb JL, Ru Su Y, Stanic AK, Joyce S, Van Kaer L, Fazio S, Linton MF. Quantitative and qualitative differences in proatherogenic NKT cells in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2004;24:2351–7. doi: 10.1161/01.ATV.0000147112.84168.87. [DOI] [PubMed] [Google Scholar]
- 175.Aslanian AM, Chapman HA, Charo IF. Transient role for CD1d-restricted natural killer T cells in the formation of atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2005;25:628–32. doi: 10.1161/01.ATV.0000153046.59370.13. [DOI] [PubMed] [Google Scholar]
- 176.Carding SR, Egan PJ. Gammadelta T cells: functional plasticity and heterogeneity. Nat Rev Immunol. 2002;2:336–45. doi: 10.1038/nri797. [DOI] [PubMed] [Google Scholar]
- 177.Kleindienst R, Xu Q, Willeit J, Waldenberger FR, Weimann S, Wick G. Immunology of atherosclerosis. Demonstration of heat shock protein 60 expression and T lymphocytes bearing alpha/beta or gamma/delta receptor in human atherosclerotic lesions. Am J Pathol. 1993;142:1927–37. [PMC free article] [PubMed] [Google Scholar]
- 178.Elhage R, Gourdy P, Brouchet L, Jawien J, Fouque MJ, Fievet C, Huc X, Barreira Y, Couloumiers JC, Arnal JF, Bayard F. Deleting TCR alpha beta+ or CD4+ T lymphocytes leads to opposite effects on site-specific atherosclerosis in female apolipoprotein E-deficient mice. Am J Pathol. 2004;165:2013–8. doi: 10.1016/s0002-9440(10)63252-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–6. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 180.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–87. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 181.Mallat Z, Besnard S, Duriez M, Deleuze V, Emmanuel F, Bureau MF, Soubrier F, Esposito B, Duez H, Fievet C, Staels B, Duverger N, Scherman D, Tedgui A. Protective role of interleukin-10 in atherosclerosis. Circ Res. 1999;85:e17–24. doi: 10.1161/01.res.85.8.e17. [DOI] [PubMed] [Google Scholar]
- 182.Pinderski Oslund LJ, Hedrick CC, Olvera T, Hagenbaugh A, Territo M, Berliner JA, Fyfe AI. Interleukin-10 blocks atherosclerotic events in vitro and in vivo. Arterioscler Thromb Vasc Biol. 1999;19:2847–53. doi: 10.1161/01.atv.19.12.2847. [DOI] [PubMed] [Google Scholar]
- 183.Pinderski LJ, Fischbein MP, Subbanagounder G, Fishbein MC, Kubo N, Cheroutre H, Curtiss LK, Berliner JA, Boisvert WA. Overexpression of interleukin-10 by activated T lymphocytes inhibits atherosclerosis in LDL receptor-deficient Mice by altering lymphocyte and macrophage phenotypes. Circ Res. 2002;90:1064–71. doi: 10.1161/01.res.0000018941.10726.fa. [DOI] [PubMed] [Google Scholar]
- 184.Mallat Z, Gojova A, Marchiol-Fournigault C, Esposito B, Kamate C, Merval R, Fradelizi D, Tedgui A. Inhibition of transforming growth factor-beta signaling accelerates atherosclerosis and induces an unstable plaque phenotype in mice. Circ Res. 2001;89:930–4. doi: 10.1161/hh2201.099415. [DOI] [PubMed] [Google Scholar]
- 185.Lutgens E, Gijbels M, Smook M, Heeringa P, Gotwals P, Koteliansky VE, Daemen MJ. Transforming growth factor-beta mediates balance between inflammation and fibrosis during plaque progression. Arterioscler Thromb Vasc Biol. 2002;22:975–82. doi: 10.1161/01.atv.0000019729.39500.2f. [DOI] [PubMed] [Google Scholar]
- 186.Robertson AK, Rudling M, Zhou X, Gorelik L, Flavell RA, Hansson GK. Disruption of TGF-beta signaling in T cells accelerates atherosclerosis. J Clin Invest. 2003;112:1342–50. doi: 10.1172/JCI18607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Gojova A, Brun V, Esposito B, Cottrez F, Gourdy P, Ardouin P, Tedgui A, Mallat Z, Groux H. Specific abrogation of transforming growth factor-beta signaling in T cells alters atherosclerotic lesion size and composition in mice. Blood. 2003;102:4052–8. doi: 10.1182/blood-2003-05-1729. [DOI] [PubMed] [Google Scholar]
- 188.Ovchinnikova O, Robertson AK, Wagsater D, Folco EJ, Hyry M, Myllyharju J, Eriksson P, Libby P, Hansson GK. T-cell activation leads to reduced collagen maturation in atherosclerotic plaques of Apoe(-/-) mice. Am J Pathol. 2009;174:693–700. doi: 10.2353/ajpath.2009.080561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ait-Oufella H, Salomon BL, Potteaux S, Robertson AK, Gourdy P, Zoll J, Merval R, Esposito B, Cohen JL, Fisson S, Flavell RA, Hansson GK, Klatzmann D, Tedgui A, Mallat Z. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. 2006;12:178–80. doi: 10.1038/nm1343. [DOI] [PubMed] [Google Scholar]
- 190.Mor A, Luboshits G, Planer D, Keren G, George J. Altered status of CD4(+)CD25(+) regulatory T cells in patients with acute coronary syndromes. Eur Heart J. 2006;27:2530–7. doi: 10.1093/eurheartj/ehl222. [DOI] [PubMed] [Google Scholar]
- 191.Mallat Z, Gojova A, Brun V, Esposito B, Fournier N, Cottrez F, Tedgui A, Groux H. Induction of a regulatory T cell type 1 response reduces the development of atherosclerosis in apolipoprotein E-knockout mice. Circulation. 2003;108:1232–7. doi: 10.1161/01.CIR.0000089083.61317.A1. [DOI] [PubMed] [Google Scholar]
- 192.Ait-Oufella H, Horvat B, Kerdiles Y, Herbin O, Gourdy P, Khallou-Laschet J, Merval R, Esposito B, Tedgui A, Mallat Z. Measles virus nucleoprotein induces a regulatory immune response and reduces atherosclerosis in mice. Circulation. 2007;116:1707–13. doi: 10.1161/CIRCULATIONAHA.107.699470. [DOI] [PubMed] [Google Scholar]
- 193.Taleb S, Herbin O, Ait-Oufella H, Verreth W, Gourdy P, Barateau V, Merval R, Esposito B, Clement K, Holvoet P, Tedgui A, Mallat Z. Defective leptin/leptin receptor signaling improves regulatory T cell immune response and protects mice from atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27:2691–8. doi: 10.1161/ATVBAHA.107.149567. [DOI] [PubMed] [Google Scholar]
- 194.Heller EA, Liu E, Tager AM, Yuan Q, Lin AY, Ahluwalia N, Jones K, Koehn SL, Lok VM, Aikawa E, Moore KJ, Luster AD, Gerszten RE. Chemokine CXCL10 promotes atherogenesis by modulating the local balance of effector and regulatory T cells. Circulation. 2006;113:2301–12. doi: 10.1161/CIRCULATIONAHA.105.605121. [DOI] [PubMed] [Google Scholar]
- 195.Steffens S, Burger F, Pelli G, Dean Y, Elson G, Kosco-Vilbois M, Chatenoud L, Mach F. Short-term treatment with anti-CD3 antibody reduces the development and progression of atherosclerosis in mice. Circulation. 2006;114:1977–84. doi: 10.1161/CIRCULATIONAHA.106.627430. [DOI] [PubMed] [Google Scholar]
- 196.van Puijvelde GH, Hauer AD, de Vos P, van den Heuvel R, van Herwijnen MJ, van der Zee R, van Eden W, van Berkel TJ, Kuiper J. Induction of oral tolerance to oxidized low-density lipoprotein ameliorates atherosclerosis. Circulation. 2006;114:1968–76. doi: 10.1161/CIRCULATIONAHA.106.615609. [DOI] [PubMed] [Google Scholar]
- 197.van Puijvelde GH, van Es T, van Wanrooij EJ, Habets KL, de Vos P, van der Zee R, van Eden W, van Berkel TJ, Kuiper J. Induction of oral tolerance to HSP60 or an HSP60-peptide activates T cell regulation and reduces atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27:2677–83. doi: 10.1161/ATVBAHA.107.151274. [DOI] [PubMed] [Google Scholar]
- 198.Maron R, Sukhova G, Faria AM, Hoffmann E, Mach F, Libby P, Weiner HL. Mucosal administration of heat shock protein-65 decreases atherosclerosis and inflammation in aortic arch of low-density lipoprotein receptor-deficient mice. Circulation. 2002;106:1708–15. doi: 10.1161/01.cir.0000029750.99462.30. [DOI] [PubMed] [Google Scholar]
- 199.Harats D, Yacov N, Gilburd B, Shoenfeld Y, George J. Oral tolerance with heat shock protein 65 attenuates Mycobacterium tuberculosis-induced and high-fat-diet-driven atherosclerotic lesions. J Am Coll Cardiol. 2002;40:1333–8. doi: 10.1016/s0735-1097(02)02135-6. [DOI] [PubMed] [Google Scholar]
- 200.George J, Yacov N, Breitbart E, Bangio L, Shaish A, Gilburd B, Shoenfeld Y, Harats D. Suppression of early atherosclerosis in LDL-receptor deficient mice by oral tolerance with beta 2-glycoprotein I. Cardiovasc Res. 2004;62:603–9. doi: 10.1016/j.cardiores.2004.01.028. [DOI] [PubMed] [Google Scholar]
- 201.Watanabe M, Sangawa A, Sasaki Y, Yamashita M, Tanaka-Shintani M, Shintaku M, Ishikawa Y. Distribution of inflammatory cells in adventitia changed with advancing atherosclerosis of human coronary artery. J Atheroscler Thromb. 2007;14:325–31. doi: 10.5551/jat.e489. [DOI] [PubMed] [Google Scholar]
- 202.Moos MP, John N, Grabner R, Nossmann S, Gunther B, Vollandt R, Funk CD, Kaiser B, Habenicht AJ. The lamina adventitia is the major site of immune cell accumulation in standard chow-fed apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:2386–91. doi: 10.1161/01.ATV.0000187470.31662.fe. [DOI] [PubMed] [Google Scholar]
- 203.Zhou X, Hansson GK. Detection of B cells and proinflammatory cytokines in atherosclerotic plaques of hypercholesterolaemic apolipoprotein E knockout mice. Scand J Immunol. 1999;50:25–30. doi: 10.1046/j.1365-3083.1999.00559.x. [DOI] [PubMed] [Google Scholar]
- 204.Caligiuri G, Nicoletti A, Poirier B, Hansson GK. Protective immunity against atherosclerosis carried by B cells of hypercholesterolemic mice. J Clin Invest. 2002;109:745–53. doi: 10.1172/JCI07272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Palinski W, Rosenfeld ME, Yla-Herttuala S, Gurtner GC, Socher SS, Butler SW, Parthasarathy S, Carew TE, Steinberg D, Witztum JL. Low density lipoprotein undergoes oxidative modification in vivo. Proc Natl Acad Sci U S A. 1989;86:1372–6. doi: 10.1073/pnas.86.4.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Palinski W, Horkko S, Miller E, Steinbrecher UP, Powell HC, Curtiss LK, Witztum JL. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Invest. 1996;98:800–14. doi: 10.1172/JCI118853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Shaw PX, Horkko S, Chang MK, Curtiss LK, Palinski W, Silverman GJ, Witztum JL. Natural antibodies with the T15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J Clin Invest. 2000;105:1731–40. doi: 10.1172/JCI8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Binder CJ, Horkko S, Dewan A, Chang MK, Kieu EP, Goodyear CS, Shaw PX, Palinski W, Witztum JL, Silverman GJ. Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat Med. 2003;9:736–43. doi: 10.1038/nm876. [DOI] [PubMed] [Google Scholar]
- 209.Perschinka H, Mayr M, Millonig G, Mayerl C, van der Zee R, Morrison SG, Morrison RP, Xu Q, Wick G. Cross-reactive B-cell epitopes of microbial and human heat shock protein 60/65 in atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:1060–5. doi: 10.1161/01.ATV.0000071701.62486.49. [DOI] [PubMed] [Google Scholar]
- 210.Xu Q, Dietrich H, Steiner HJ, Gown AM, Schoel B, Mikuz G, Kaufmann SH, Wick G. Induction of arteriosclerosis in normocholesterolemic rabbits by immunization with heat shock protein 65. Arterioscler Thromb. 1992;12:789–99. doi: 10.1161/01.atv.12.7.789. [DOI] [PubMed] [Google Scholar]
- 211.Afek A, George J, Gilburd B, Rauova L, Goldberg I, Kopolovic J, Harats D, Shoenfeld Y. Immunization of low-density lipoprotein receptor deficient (LDL-RD) mice with heat shock protein 65 (HSP-65) promotes early atherosclerosis. J Autoimmun. 2000;14:115–21. doi: 10.1006/jaut.1999.0351. [DOI] [PubMed] [Google Scholar]
- 212.Schett G, Xu Q, Amberger A, Van der Zee R, Recheis H, Willeit J, Wick G. Autoantibodies against heat shock protein 60 mediate endothelial cytotoxicity. J Clin Invest. 1995;96:2569–77. doi: 10.1172/JCI118320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Packard RR, Libby P. Inflammation in atherosclerosis: from vascular biology to biomarker discovery and risk prediction. Clin Chem. 2008;54:24–38. doi: 10.1373/clinchem.2007.097360. [DOI] [PubMed] [Google Scholar]
- 214.Libby P. The forgotten majority: unfinished business in cardiovascular risk reduction. J Am Coll Cardiol. 2005;46:1225–8. doi: 10.1016/j.jacc.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 215.Libby P. Inflammation in cardiovascular disease. In: Morrow DA, editor. Contemporary Cardiology. Totowa, NJ: Humana Press Inc.; 2006. pp. 205–19. [Google Scholar]