

Abstract
Long-term synaptic plasticity is a putative mechanism for learning in adults. However, there is little understanding of how synaptic plasticity mechanisms develop or whether their maturation depends on experience. Since inhibitory synapses are particularly malleable to sensory stimulation, long-lasting potentiation of inhibitory synapses was characterized in auditory thalamocortical slices. Intracortical high-frequency electrical stimulation led to a 67% increase in inhibitory synaptic currents. In the absence of stimulation, inhibitory potentiation was induced by a brief exposure to exogenous brain-derived neurotrophic factor (BDNF). BDNF exposure occluded any additional potentiation by high-frequency afferent stimulation, suggesting that BDNF signaling is sufficient to account for inhibitory potentiation. Moreover, inhibitory potentiation was reduced significantly by extracellular application of a BDNF scavenger or by intracellular blockade of BDNF receptor [tropomyosin-related kinase B (TrkB)] signaling. In contrast, glutamatergic or GABAergic antagonists did not prevent the induction of inhibitory potentiation. Since BDNF and TrkB expression are influenced strongly by activity, we predicted that inhibitory potentiation would be diminished by manipulations that decrease central auditory activity, such as hearing loss. Two forms of hearing loss were examined: conductive hearing loss in which the cochleae are not damaged or sensorineural hearing loss in which both cochleae are removed. Both forms of hearing loss were found to reduce significantly the magnitude of inhibitory potentiation. These data indicate that early experience is necessary for the normal development of BDNF-mediated long-lasting inhibitory potentiation, which may be associated with perceptual deficits at later ages.
Introduction
Neurotrophins are implicated in the development and plasticity of inhibitory connections. In particular, brain-derived neurotrophic factor (BDNF) signaling through its cognate receptor, tropomyosin-related kinase B (TrkB), increases the strength and number of GABAergic terminals (Rutherford et al., 1997; Huang et al., 1999; Marty et al., 2000; Kohara et al., 2007; Abidin et al., 2008). Endogenous BDNF–TrkB signaling is also required for inhibitory synaptic long-term potentiation (iLTP) in the hippocampus, visual cortex, and optic tectum (Gubellini et al., 2005; Liu et al., 2007; Inagaki et al., 2008; Kuczewski et al., 2008b). Since BDNF and TrkB expression are influenced strongly by activity (Lu, 2003; Nagappan and Lu, 2005), we predicted that inhibitory potentiation would not only depend on BDNF signaling, but be diminished by manipulations that decrease central activity, such as hearing loss. Therefore, we characterized a BDNF-dependent form of long-lasting enhancement of inhibitory transmission in the developing auditory cortex (ACx) and asked whether moderate or severe hearing loss would disrupt long-lasting inhibitory potentiation.
The dependence of BDNF expression on activity was discovered in epileptic foci but has since been demonstrated to occur during sensory stimulation, environmental enrichment, learning, and long-term synaptic plasticity (Zafra et al., 1990; Ernfors et al., 1991; Isackson et al., 1991; Castrén et al., 1992; Falkenberg et al., 1992; Patterson et al., 1992; Rocamora et al., 1996; Kesslak et al., 1998; Hall et al., 2000; Ickes et al., 2000). Recent reports exploring this concept in animal models of hearing loss revealed a consistent decrease in BDNF expression (Oh et al., 2007; Rüttiger et al., 2007; Tan et al., 2008). For example, when cochleae are damaged with antibiotics, ACx displays a significant reduction in BDNF, and normal levels are restored by electrically stimulating peripheral auditory neurons (Tan et al., 2008). Therefore, if normal BDNF expression is necessary for synaptic plasticity, we would expect hearing loss to impair plasticity.
There are both correlative and causal links between ACx plasticity and acoustically based learning (Bao et al., 2004; Fritz et al., 2005; Rutkowski and Weinberger, 2005; Sakai and Kudoh, 2005; Witte and Kipke, 2005; Ono et al., 2006; Rybalko et al., 2006; Froemke et al., 2007). Several forms of excitatory synaptic plasticity that could contribute to such learning have been described in ACx, and bilateral blockade of NMDA receptors prevents passive sound exposure from enhancing sound discrimination learning in rats (Kudoh and Shibuki, 1996, 1997; Wakatsuki et al., 1998; Seki et al., 1999, 2001, 2003; Sakai and Kudoh, 2005). In contrast, the plasticity of ACx inhibitory synapses has not been well explored.
We recently found that excitatory LTP failed to emerge when cochlear function was eliminated during development (Kotak et al., 2007). To better understand the relationship between developmental experience and the maturation of synaptic plasticity, we characterized inhibitory potentiation in auditory cortex and demonstrated its dependence on BDNF. We then asked whether even a moderate reduction in auditory experience, conductive hearing loss (CHL), would prevent the maturation of inhibitory potentiation. Our results illustrate that developmental hearing loss interfered with the emergence of BDNF-dependent long-lasting inhibitory potentiation.
Materials and Methods
Animals.
Gerbil (Meriones unguiculatus) pups at postnatal day 18 (P18) to P21 were used. All animals were housed under a 12 h light/dark cycle with food and water provided ad libitum. All protocols were reviewed and approved by New York University Institutional Animal Care and Use Committee.
Hearing loss surgery.
Gerbil pups at P10 were anesthetized with the halogenated ethyl methyl ether methoxyflurane (Metofane). Anesthetic induction occurred within 10 min and produced complete elimination of responses to nociceptive stimuli. Conductive hearing loss was induced by tympanic membrane puncture and malleus extirpation (Tucci et al., 1999; Xu et al., 2007). A postauricular skin incision was made, and the tympanic membrane was visualized and punctured with a forceps. The malleus was then removed through this opening. When brain slices were prepared, the stapes was visualized to ensure stability within the oval window; malleus removal did not disrupt the stapes in any of the animals used for this study. The postauricular wound was closed with cyanoacrylate glue, and the procedure was repeated on the other side. Sensorineural hearing loss (SNHL) was induced using procedures similar to those described previously (Sanes et al., 1992; Vale and Sanes, 2002). In brief, a small hole was made in the cochlear wall, and the contents were rapidly removed with a forceps. A piece of Gelfoam was then placed in the cavity, and the wound was closed. The procedure was then repeated on the other side. After surgery, animals were warmed on a heating pad and returned to the litter when respiration and motor activity had recovered. Animals were reared for 8–11 d after surgery, with their parents, under conditions identical with those for control pups. Before each brain slice experiment, successful induction of CHL was confirmed by opening the middle ear under a dissecting microscope and observing the absence of malleus and presence of intact stapes. Successful SNHL was confirmed by opening the inner wall of the cochlea and observing the absence of cochlear tissue and the presence of a Gelfoam insert. The age of surgery was chosen based on the finding that anteroventral cochlear nucleus cell number is unaffected by cochlear ablation after P9 in gerbils (Tierney et al., 1997).
Brain slice preparation.
Brain slices (500 μm) retaining much of the connectivity between the ventral medial geniculate nucleus (MGv) and the core ACx, were generated from P18–P21 animals as previously reported (Cruikshank et al., 2002; Kotak et al., 2005). Slices were transferred to a submerged recovery chamber containing oxygenated artificial CSF (ACSF) at room temperature for at least 1 h before physiological recordings. The ACSF contained the following (in mm): 125 NaCl, 4 KCl, 1.2 KH2PO4, 1.3 MgSO4, 26 NaHCO3, 15 glucose, 2.4 CaCl2, and 0.4 l-ascorbic acid, pH 7.4 (when bubbled with 95% O2/5% CO2).
Whole-cell recordings.
Recording electrodes were fabricated from borosilicate glass microcapillaries (outer diameter, 1.5 mm) with a micropipette puller (model P-97; Sutter Instrument). The internal patch solution contained the following (in mm): 127.5 potassium gluconate, 0.6 EGTA, 10 HEPES, 2 MgCl2, 5 KCl, 2 ATP, 0.3 GTP, and 5 phosphocreatine, pH 7.2 with KOH. To block sodium-dependent action potentials, 5 mm lidocaine N-ethyl bromide (QX-314) was included in the pipette solution to block voltage-gated sodium channels. The tip resistance of the patch electrode filled with the internal solution was 4–8 MΩ. Experiments were performed in a recording chamber mounted on the stage of an upright microscope (BX51WI; Olympus) equipped with infrared differential interference contrast optics. ACSF was superfused in the recording chamber at 2 ml/min at 32°C. Before each experiment, extracellular responses were evoked with stimulating MGv to confirm that the recording site is thalamorecipient ACx. The whole-cell patch-clamp recordings were then obtained from pyramidal neurons in supragranular layers using a PC-501A amplifier (Warner Instruments). To prevent ionotropic glutamatergic transmission, 50 μm AP-5 and 20 μm DNQX were added to ACSF to block NMDA receptor and AMPA receptor, respectively. To evoke inhibitory postsynaptic responses, a bipolar insulated platinum electrode was placed in layer IV, and electrical stimuli were delivered via a stimulus isolator (model BSI-950; Dagan Corporation). Because IPSP amplitudes are small at the resting membrane potential, and sensitive to membrane potential fluctuation, we recorded IPSCs under voltage clamp at a holding potential of −45 mV. The access resistance varied between 15 and 30 MΩ and was determined manually at the beginning of each experiment with a brief voltage pulse. At the end of each experiment, the access resistance was checked again, and data were discarded if access resistance changed by >15% during an experiment. In addition, we monitored whether there was a change in holding current during the recording and found that it did not change significantly for either control or conditioned neurons (final − initial holding current for the neurons shown in Fig. 1B,C; conditioned neurons, −10.5 ± 3.4 pA; control neurons, −12.5 ± 5.0 pA; t = 0.31, df = 14, p = 0.76). All chemicals were from Sigma-Aldrich unless otherwise indicated.
Figure 1.
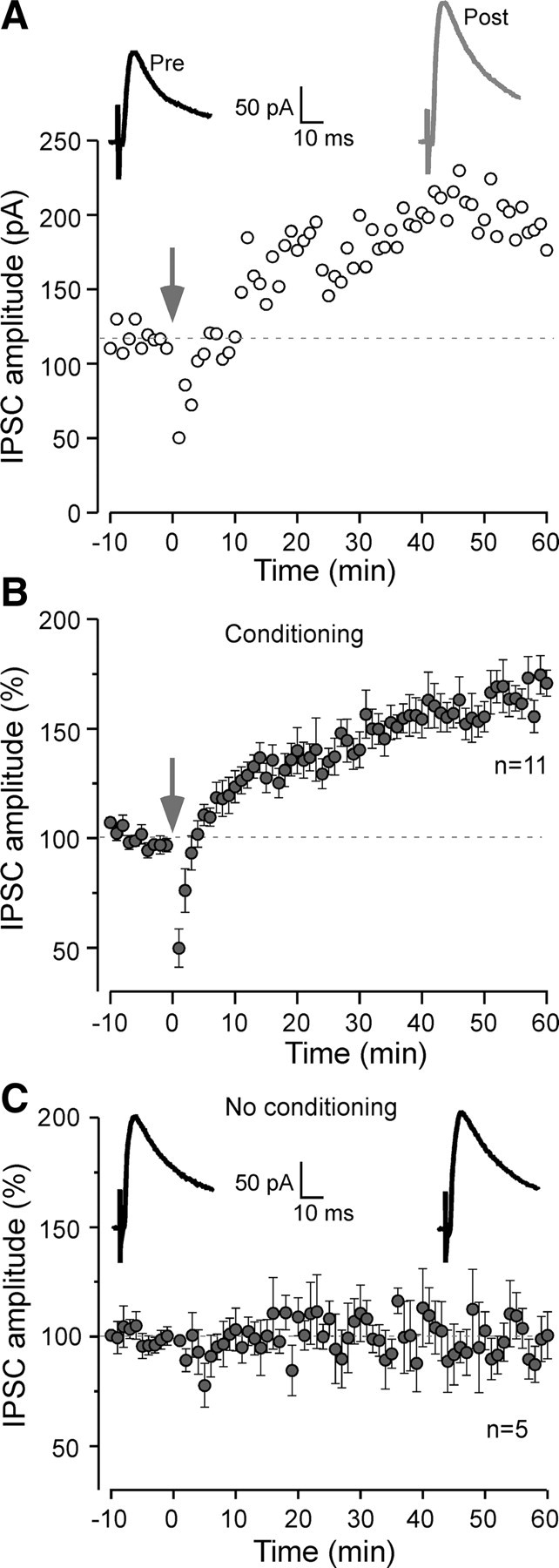
Long-lasting inhibitory potentiation in the auditory cortex. A, Representative L4-evoked IPSCs of a L2/3 pyramidal neuron before and after conditioning (arrow) at a holding potential of −45 mV. Data points represent the peak IPSC amplitude. The dashed line represents preconditioning baseline IPSC amplitude. Averaged IPSC traces before (black; pre) and 50–60 min after (gray; post) conditioning are shown. In this and subsequent figures, 10 consecutively acquired IPSCs were averaged. B, Time course of mean IPSC amplitude for 11 neurons. The IPSC amplitude was normalized to preconditioning baseline (acquired for 10 min) for each recorded neuron. The dashed line represents the mean normalized IPSC value before conditioning (arrow). C, Note that there is no change in averaged IPSCs (top) and normalized IPSC amplitude in the absence of any conditioning stimulation (n = 5). Inset, Representative averaged IPSCs during the first 10 min (left) and last 10 min (right) of the recording session. Error bars indicate SEM.
Induction of synaptic plasticity.
To establish a preconditioning baseline of synaptic response, stimulus intensity was adjusted to obtain IPSC amplitudes of ∼150 pA. The stimulus intensities used were typically in the range of 100–200 μA and were well below the level required to evoke maximum amplitude IPSCs. This relatively low stimulus level, and the brief duration of the pulses (100 μs), was chosen to minimize damage to afferents by the charge buildup during the conditioning protocol. In addition, to make unlikely that use-dependent plasticity would happen during baseline recording period, IPSCs were induced at a low repetition rate of once per minute.
After obtaining stable IPSCs for at least 10 min, conditioning stimuli to induce inhibitory potentiation were delivered through stimulation electrode while recording in voltage-clamp mode. Recordings were obtained from L2/3 pyramidal cells, which were ≈150 μm from the stimulation electrode in L4. Action potentials were not observed in the recorded neurons because of the inclusion of QX-314 in the pipette solution as an effort to avoid contamination of IPSCs by action potentials. However, pilot recordings conducted without QX-314 in the pipette indicated that action potentials were readily evoked in recorded pyramidal cells during conditioning stimulation. Therefore, it is likely that conditioning stimuli elicited action potentials in nearby pyramidal neurons, and the attendant influx of calcium led to the release of BDNF.
The conditioning protocol was comprised of 30 trains (five pulses, 50 Hz) delivered at 2 s intervals, and it was designed to emulate the temporal discharge pattern of the ACx neurons, which are well suited for representing low-frequency modulations (Creutzfeldt et al., 1980; Ter-Mikaelian et al., 2007). For example, we previously reported that the average sound-evoked discharge rate for gerbil cortical neurons recorded in awake animals is ∼40 spikes per second at the onset of sound, and adapts with a time constant of 73 ms [Ter-Mikaelian et al. (2007), their Fig. 6C]. Since our conditioning stimulus consisted of five pulses at 50 Hz, delivered at 2 s interval, it is likely that we did not drive the system into an adapted state that would have resulted in extremely low firing. The conditioning stimulus intensity was set two times as large as that used to acquire preconditioning IPSCs. Immediately after the conditioning stimuli, IPSCs were acquired once per minute for an additional hour with the same stimulus intensity used during the preconditioning period.
Figure 6.

GABAA, GABAB, or mGlu receptors do not mediate long-lasting inhibitory potentiation. The IPSC amplitude, normalized to the initial baseline, was examined before and after conditioning (arrows) when 10 μm bicuculline was present during conditioning stimuli (A), 20 μm SCH-50911 was present during conditioning stimuli (B), or when 0.5 mm MCPG was present during conditioning stimuli (C). The thick gray lines in A–C show the time when each drug was present in the bath. The dashed lines represent the mean normalized IPSCs amplitude before conditioning stimuli. D, The bar graph shows that inhibitory potentiation magnitude did not differ significantly from control (NS) when GABAA, GABAB or mGlu receptors were blocked. Error bars indicate SEM.
A paired-pulse stimulation protocol was used to measure paired-pulse ratio (PPR) of the IPSC amplitude to probe transmitter release probability (Zucker and Regehr, 2002), which was calculated as the ratio between the second IPSC (IPSC2) and the first IPSC (IPSC1). The IPSC reversal potential (EIPSC) was determined by varying the holding potential of the recorded cell in 10 mV increments and measuring IPSC amplitude. Linear regression was used to calculate a best-fit line for the voltage dependence of IPSCs, and the interpolated intercept of this line with the abscissa was taken as the EIPSC value. The slope of this line was used as the corresponding synaptic conductance.
Electrophysiological data collection and analysis.
Data were collected using a Macintosh G4 computer running a custom-designed IGOR (version 3.14; Wavemetrics) macro called SLICE and analyzed off-line using a second IGOR macro called SLICE ANALYSIS. Both the data acquisition and analysis macros are available with complete documentation online at http://www.cns.nyu.edu/∼sanes/slice_software/. The peak amplitude of IPSCs was measured from baseline just before the response onset. Statistical tests (ANOVA, paired t test, Wilcoxon's test) were performed using statistical software (JMP 5; SAS Institute). Data are presented as mean ± SEM unless otherwise indicated. In all cases, statistical significance was defined as p < 0.05.
Pharmacological manipulations.
Several manipulations were performed to assess whether glutamatergic, GABAergic, and neurotropin signaling participated. To examine involvement of metabotropic glutamatergic signaling, a broad-spectrum antagonist (S)-α-methyl-4-carboxyphenylglycine (MCPG) (500 μm) was bath applied during conditioning. To examine GABAA and GABAB receptor participation, bicuculline (10 μm) or (+)-(2S)-5,5-dimethyl-2-morpholineacetic acid (SCH-50911) (20 μm) were separately bath applied during conditioning. To test whether BDNF–Trk signaling participated, three manipulations were performed. First, to assess whether BDNF (100 ng/ml) could alter synaptic strength, it was bath applied for 3 min in the absence of conditioning stimulation. Second, to test whether IPSC potentiation was mediated by endogenous BDNF, slices were incubated in a specific scavenger for TrkB receptor ligands (TrkB-Fc; 2 μg/ml) for at least 1 h before and during physiological recordings. Third, to test whether TrkB signaling was required for synaptic plasticity, a TrkB receptor inhibitor (200 nm k252a) was added to the internal pipette solution. All the agents were obtained from Sigma-Aldrich unless otherwise noted.
Results
High-frequency stimuli modified inhibitory transmission
Before each experiment, thalamorecipient ACx was verified by recording a robust MGv-evoked extracellular response. Whole-cell patch-clamp recordings were then obtained from visually identified pyramidal neurons within layer 2/3. In a subset of experiments, pyramidal neuron location and morphology were confirmed by biocytin labeling.
We first examined whether intracortical high-frequency stimulation altered the strength of evoked IPSCs in ACx (see Materials and Methods). Under our experimental conditions, IPSCs were outward currents and were abolished by GABAA receptor (GABAAR) antagonist bicuculline methiodide (10 μm). After stable IPSCs were recorded for at least 10 min, conditioning stimuli were delivered (see Materials and Methods). During the conditioning protocol in the presence of ionotropic glutamate receptor (iGluR) antagonists (AP-5 and DNQX), a train of outward synaptic currents was observed. A suppression of IPSC amplitude was observed for 2–3 min immediately after conditioning (Fig. 1A,B). Subsequently, a potentiation of IPSCs emerged over 40–50 min and persisted for as long as stable recording was maintained (at least 60 min). At 50–60 min after conditioning, the amplitude was potentiated to 167 ± 10% (mean ± SEM; n = 11) of the average baseline value before conditioning (Fig. 1B). In control experiments in which neurons were not subjected to conditioning stimuli, no change in IPSC amplitude was observed over the same duration (98 ± 8%; n = 5; paired t test, p = 0.81) (Fig. 1C).
Mechanistic basis for long-lasting inhibitory potentiation in auditory cortex
The IPSC amplitude increase could result from a rise in either presynaptic transmitter release or postsynaptic GABA-induced response. To provide a preliminary assessment of these mechanisms, we measured the paired-pulse ratio (PPR = IPSC2/IPSC1) in a subset of neurons that displayed inhibitory potentiation. The PPR measured at an interpulse interval of 50 ms was not altered after inhibitory potentiation was induced (0.87 ± 0.06 before induction and 0.86 ± 0.05 at 50–60 min after induction; n = 6; paired t test, p = 0.75) (Fig. 2A). Similarly, analysis of the coefficient of variation (CV) of IPSC amplitude displayed no significant change after induction (0.12 ± 0.02 before induction and 0.13 ± 0.03 at 50–60 min after induction; n = 11; paired t test, p = 0.69) (Fig. 2B). These results suggest that presynaptic GABA release probability is not affected by the conditioning stimulus.
Figure 2.
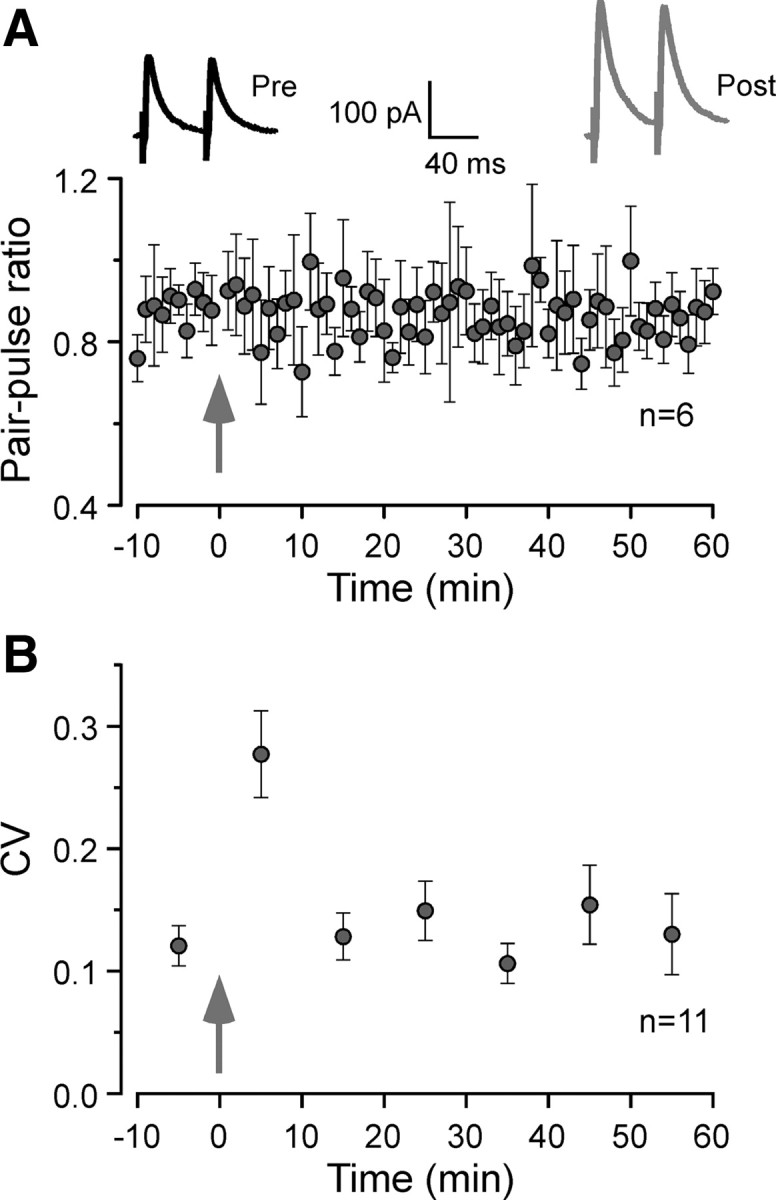
Long-lasting inhibitory potentiation is not associated with changes in PPR or CV. A, Example responses to paired-pulse stimuli are shown before conditioning (Pre; black trace) and 50–60 min after the conditioning stimuli (Post; gray trace). The PPR was computed as the ratio of IPSC2/IPSC1 using a 50 ms interstimulus interval (n = 6). There was no significant difference in PPR before and 50–60 min after conditioning (p = 0.75, paired t test). The arrow indicates the time when conditioning stimuli were applied. B, The CV of IPSCs amplitude throughout the recording session. Ten consecutive IPSCs were used to calculate each CV (SD of IPSCs amplitude divided by their mean amplitude). There was no significant difference in CV before and 50–60 min after the conditioning stimuli (p = 0.69, paired t test). The arrow indicates the time when conditioning stimuli were applied. Error bars indicate SEM.
Postsynaptic mechanisms that could account for inhibitory potentiation include an alteration in IPSC reversal potential (ElPSC) or postsynaptic GABAAR conductance. To explore this issue, we measured IPSCs at different holding potentials (VH) and calculated both EIPSC and IPSC conductance before induction and during IPSC potentiation. As shown in Figure 3A, the IPSC conductance increased substantially after conditioning compared with the value found during the baseline period (16.1 ± 2.8 nS before induction and 22.9 ± 3.2 nS at 50–60 min after induction; n = 11; paired t test, p = 0.0002). However, there was not a significant change in ElPSC before and after induction (−59.7 ± 0.9 mV before conditioning and −59.5 ± 0.9 mV at 50 min after conditioning; n = 11; paired t test, p = 0.64) (Fig. 3B). Thus, the potentiation in IPSC amplitude appeared to be attributable, in part, to a change in synaptic GABAAR conductance.
Figure 3.
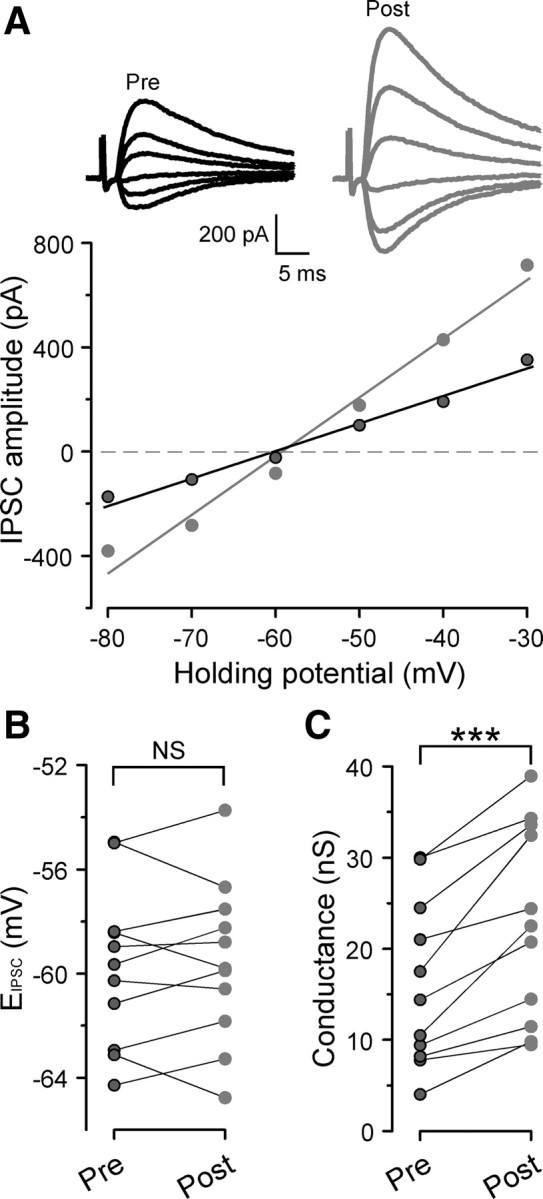
Long-lasting inhibitory potentiation is associated with an increase in synaptic conductance without a change in EIPSC. A, Representative I–V curves for IPSCs before (black circles) and 50–60 min after (gray circles) conditioning. Insets, Example traces of IPSCs at various holding potentials before (black) and 50–60 min after (gray) conditioning. B, Summary plot of EIPSC values measured before (black) and 50–60 min after (gray) conditioning (n = 11). Data from the same cell are connected by a line. NS indicates no significant difference. C, Summary plot of synaptic conductance measured before (black) and 50–60 min after (gray) conditioning (n = 11). Data from the same cell are connected by a line. The difference in synaptic conductance before and 50–60 min after conditioning was highly significant (p = 0.0002, paired t test). ***p < 0.001.
Long-lasting inhibitory potentiation was mediated by BDNF–TrkB signaling
Growing evidence shows that neurotrophins are involved in regulating synaptic transmission and plasticity, including inhibitory synapses (Lu and Figurov, 1997; Schinder and Poo, 2000; Gubellini et al., 2005; Arancio and Chao, 2007; Liu et al., 2007; Inagaki et al., 2008; Kuczewski et al., 2008b). To determine the participation of BDNF signaling in inhibitory potentiation, we first examined whether exogenous BDNF could mimic conditioning-induced potentiation. As shown in Figure 4A, bath application of BDNF (100 ng/ml) for 3 min progressively potentiated IPSCs in a manner similar to that produced by conditioning stimuli. It also took ∼40–50 min for the full expression of inhibitory potentiation (158 ± 5% of initial baseline at 50–60 min; n = 6; paired t test, p < 0.0001) (Fig. 4A), which is comparable with conditioning-induced potentiation, both in its magnitude and time course. The effect of BDNF was mediated by BDNF–TrkB signaling pathway because BDNF (100 ng/ml for 3 min) failed to significantly potentiate IPSC (107 ± 7% of baseline at 50–60 min; n = 5; paired t test, p = 0.11 compared with baseline) when a TrkB receptor inhibitor (200 nm k252a) was included in pipette solution. Together, these results indicate that BDNF is sufficient to induce inhibitory potentiation in the ACx via TrkB receptor activation.
Figure 4.
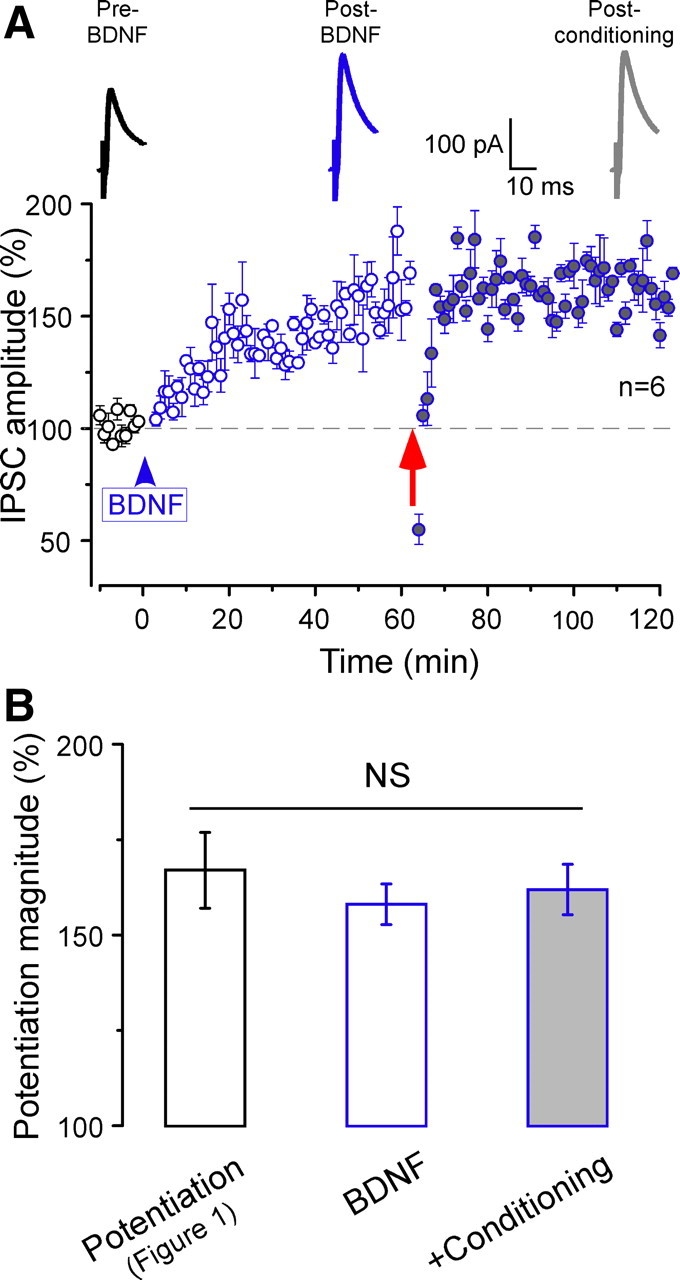
Exogenous BDNF potentiates IPSCs and occludes additional inhibitory potentiation. A, BDNF exposure (100 ng/ml for 3 min; blue arrowhead) progressively increased IPSC amplitude in the absence of conditioning stimulation. Note IPSC potentiation is maximally expressed 50 min after BDNF application. Such BDNF-induced potentiation occluded additional inhibitory potentiation by subsequent conditioning (red arrow). The dashed line represents the mean normalized IPSCs amplitude before BDNF exposure (n = 6). Inset, Average IPSCs before (black; Pre BDNF) and 50–60 min after (blue; Post BDNF) BDNF application and 50–60 min after subsequent conditioning stimuli (gray; Post conditioning). B, The magnitude of long-lasting inhibitory potentiation compared among three experimental conditions: conditioning-induced inhibitory potentiation (control Potentiation), BDNF-induced potentiation (BDNF) and subsequent conditioning-induced potentiation (+Conditioning). NS indicates no significant difference. Error bars indicate SEM.
To probe whether exogenous BDNF-induced potentiation and conditioning-induced potentiation shared the similar cellular mechanism, we delivered conditioning stimuli after full manifestation of BDNF-induced potentiation. Interestingly, the BDNF-induced potentiation mostly occluded additional potentiation by subsequent conditioning (50–60 min after subsequent conditioning, 162 ± 7%; n = 6; paired t test, p = 0.16 vs BDNF-induced potentiation) (Fig. 4A,B). This occlusion, together with the similar expression profile between conditioning-induced potentiation and exogenous BDNF-induced potentiation, suggests that conditioning-induced inhibitory potentiation was likely attributable to TrkB activation by endogenously secreted BDNF.
To test the above idea, a TrkB receptor inhibitor (200 nm k252a) was added to the internal pipette solution. As shown in Figure 5A, the magnitude of inhibitory potentiation induced by conditioning stimuli was significantly reduced in the presence of k252a, when compared with the value in control neurons (with k252a, 123 ± 4%; without k252a, 167 ± 10%; χ2 = 9.01; p < 0.003). Because k252a also inhibits other subtypes of Trk receptors, we next used TrkB-Fc, which is a specific scavenger for TrkB receptor ligands and hence prevents BDNF binding to the TrkB receptor. In the presence of TrkB-Fc (2 μg/ml), conditioning stimuli induced a much smaller potentiation (with TrkB-Fc, 116 ± 7%; without TrkB-Fc, 167 ± 10%; ANOVA, df = 14, t = 3.26, p < 0.006) (Fig. 5B). Together, these observations indicate that an endogenous ligand for TrkB receptors was required for the full expression of inhibitory potentiation in the ACx (Fig. 5C).
Figure 5.
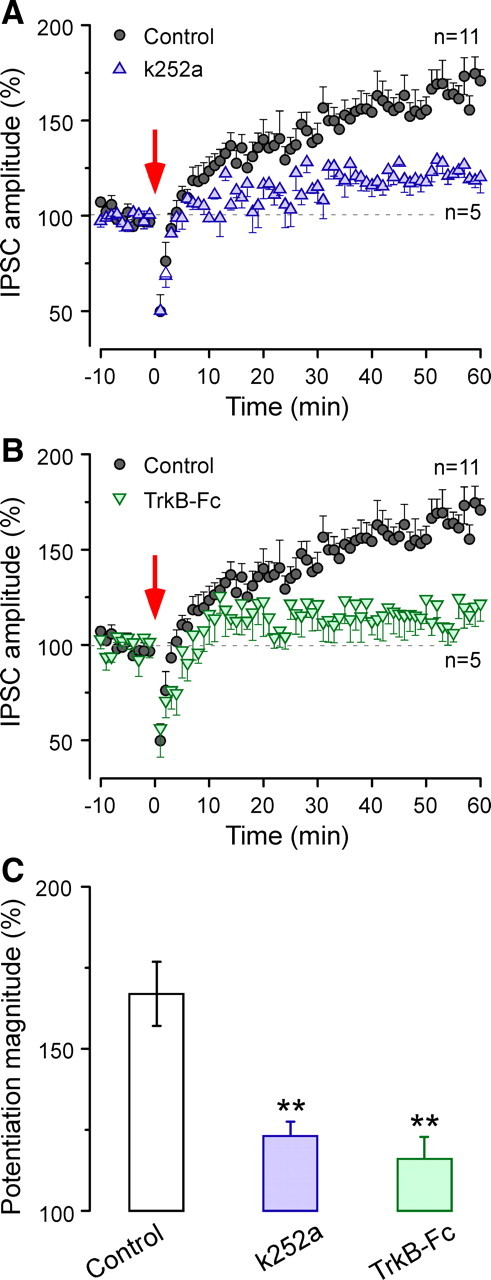
BDNF–TrkB signaling blockade prevents long-lasting inhibitory potentiation. A, Time course of mean IPSC amplitude before and after conditioning (red arrow) in the presence of intracellular k252a, a TrK receptor blocker (n = 5; blue triangles). The control record is the same as in Figure 1B (n = 11; black circles). The dashed line represents the mean normalized IPSCs amplitude before conditioning. B, Time course of mean IPSC amplitude before and after conditioning (red arrow) in the presence of extracellular TrkB-Fc (n = 5; green inverted triangles). The control record is the same as in Figure 1B (n = 11; black circles). The dashed line represents the mean normalized IPSCs amplitude before conditioning. C, A bar graph representation of the magnitude of potentiation shows a significantly reduced inhibitory potentiation in the presence of intracellular k252a or extracellular TrkB-Fc, when compared with controls. **p < 0.01. Error bars indicate SEM.
It has been reported that activation of postsynaptic GABAARs, GABAB receptors (GABABRs), or metabotropic glutamate receptors (mGluRs) is involved in iLTP in hippocampus or visual cortex (Komatsu, 1996; McLean et al., 1996; Patenaude et al., 2003). To determine the dependency of the present inhibitory potentiation on these receptors, we separately blocked each of these receptors during conditioning stimuli pharmacologically (Fig. 6). During the conditioning protocol in the presence of iGluR antagonists (AP-5 and DNQX) plus a GABAAR antagonist (10 μm bicuculline), the outward synaptic currents were eliminated and small inward currents (≈10 pA) were observed only during the 50 Hz train of stimulus pulses. The inhibitory potentiation magnitude was not reduced significantly by blockade of GABAARs. Similarly, blockade of GABABRs (20 μm SCH-50911) or mGluRs (0.5 mm MCPG) did not prevent the induction of inhibitory potentiation (control, 167 ± 10%; bicuculline, 151 ± 16%; SCH-50911, 148 ± 8%; MCPG, 152 ± 6%; ANOVA, df = 3, F = 0.73, p = 0.55) (Fig. 6). These negative findings suggest that these receptors were not required for the inhibitory potentiation observed in the present study.
Conductive or sensorineural hearing loss reduced long-lasting inhibitory potentiation
Developmental hearing loss prevents the maturation of inhibitory transmission in the ACx, indicating that early auditory experience is necessary for the normal development of inhibitory synapses (Kotak et al., 2008; Sarro et al., 2008). To determine whether auditory experience is also required for the proper expression of inhibitory potentiation, we first assessed the consequence of SNHL on the magnitude of long-lasting inhibitory potentiation. To do so, SNHL was induced in animals at P10 just before hearing onset by surgically removing cochleae on both sides (see Materials and Methods). Subsequently, recordings were made at P18–P21. Compared with control neurons, the conditioning stimulus induced a significantly smaller inhibitory potentiation in SNHL neurons (control, 167 ± 10%; SNHL, 127 ± 2%; χ2 = 14.09; p = 0.0002) (Fig. 7A).
Figure 7.
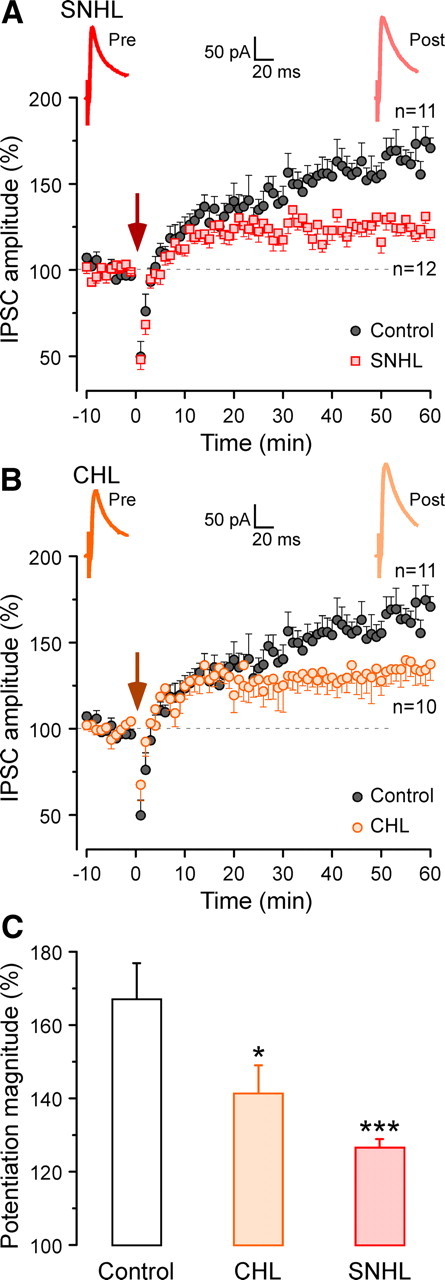
Either sensorineural or conductive hearing loss reduced long-lasting inhibitory potentiation. A, An average IPSC is shown before (red) and 50–60 min after (light red) conditioning for a SNHL neuron. Time course of mean IPSC amplitude before and after conditioning (dark red arrow) for SNHL neurons (n = 12; red squares). The control record is the same as in Figure 1B (n = 11; black circles). The dashed line represents the mean normalized IPSCs amplitude before conditioning. B, An average IPSC is shown before (orange) and 50–60 min after (light orange) conditioning for a CHL neuron. Time course of mean IPSC amplitude before and after conditioning (dark orange arrow) for CHL neurons (n = 10; orange circles). The control record is same as in Figure 1B (n = 11; black circles). The dashed line represents the mean normalized IPSCs amplitude before conditioning. C, A bar graph representation of the magnitude of inhibitory potentiation shows a significantly reduced potentiation in sensorineural or conductive hearing loss animals (SNHL, CHL). Inhibitory potentiation expression in SNHL neurons was the least of the three groups. *p < 0.05 and ***p < 0.001. Error bars indicate SEM.
Our recent study revealed that even a moderate form of CHL modifies intrinsic and synaptic properties within the ACx (Xu et al., 2007). Thus, in a separate set of experiments, we also examined the consequence of CHL on the magnitude of inhibitory potentiation. CHL was surgically induced by malleus extirpation, and this manipulation does not compromise the cochleae but attenuates sound transmission by 40–50 dB (Xu et al., 2007). As shown in Figure 7B, the conditioning stimuli induced a smaller magnitude inhibitory potentiation in CHL neurons, also significantly smaller than control neurons (control, 167 ± 10%; CHL, 141 ± 8%; χ2 = 4.77; p < 0.03). Together, these results demonstrated that developmental hearing loss, even at a moderate degree, significantly compromised inhibitory potentiation in the ACx.
Since inhibitory potentiation was dependent on BDNF–TrkB signaling, its impairment after hearing loss could have been attributable to loss of TrkB receptor function. To test this possibility, exogenous BDNF-induced inhibitory potentiation was examined in neurons from SNHL animals. As shown in Figure 8, BDNF exposure (100 ng/ml for 3 min) progressively potentiated IPSCs of SNHL neurons in a similar manner to that observed in control neurons (control from Fig. 4, 158 ± 5%; SNHL, 149 ± 11%; t test, df = 10, p = 0.61). This observation suggests that hearing loss did not disrupt TrkB receptor function that supports inhibitory potentiation and supports the hypothesis that BDNF expression or release was reduced.
Figure 8.

Exogenous BDNF potentiated IPSCs in SNHL neurons. Time course of mean normalized IPSC amplitude after BDNF exposure (100 ng/ml for 3 min; arrowhead) shows a progressive increase in SNHL neurons (n = 6). The dashed line represents the mean normalized IPSCs amplitude before BDNF exposure. The inhibitory potentiation magnitude observed in these SNHL neurons was equivalent to that observed in control neurons (Fig. 4). Error bars indicate SEM.
Discussion
Activity-dependent regulation of synaptic plasticity mechanisms, termed metaplasticity, has received increasing attention for excitatory connections (Abraham and Bear, 1996; Thiagarajan et al., 2007; Mockett and Hulme, 2008). For example, activity blockade in vitro can diminish their ability to induce excitatory LTP (Ehlers, 2003; Slutsky et al., 2004). The major finding of this study is that long-lasting inhibitory potentiation in auditory cortex depends on early auditory experience, suggesting a novel form of metaplasticity. This form of plasticity depends on the BDNF–TrkB signaling, which plays a fundamental role in the maturation of inhibitory synapses, indicating that deprivation may affect function and plasticity via the same pathway. Together with a previous report showing that excitatory LTP is disrupted by hearing loss (Kotak et al., 2007), our results demonstrate a significant reduction in the mechanisms that support normal modification of cortical function.
BDNF expression and inhibitory synapse development
The expression of both BDNF and TrkB are affected by excitatory neural activity (Castrén et al., 1992; Bozzi et al., 1995; Singh et al., 1997; Rossi et al., 1999; McLean et al., 2001; Ichisaka et al., 2003; Lu, 2003; Nagappan and Lu, 2005; Oh et al., 2007; Rüttiger et al., 2007; Tan et al., 2008), and BDNF–TrkB signaling enhances strength and number of GABAergic terminals (Rutherford et al., 1997; Huang et al., 1999; Marty et al., 2000; Kohara et al., 2007; Abidin et al., 2008). For example, endogenous BDNF–TrkB signaling facilitates iLTP in the hippocampus, visual cortex, and optic tectum (Gubellini et al., 2005; Liu et al., 2007; Inagaki et al., 2008; Kuczewski et al., 2008b). These findings motivated the hypothesis that long-lasting inhibitory potentiation in the thalamorecipient ACx depends on BDNF–TrkB signaling and would be compromised by hearing loss. Consistent with this, we found that long-lasting inhibitory potentiation was significantly lower after even moderate hearing loss (Fig. 7).
Two lines of evidence suggested that this form of metaplasticity was attributable to decreased BDNF–TrkB signaling. First, blockade of BDNF–TrkB signaling in control neurons duplicated the effect of hearing loss. Treatment with either a TrkB receptor blocker or a BDNF scavenger reduced inhibitory potentiation compared with hearing loss neurons (Fig. 5). The residual inhibitory potentiation could be attributable either to incomplete blockade of BDNF–TrkB signaling or to the presence of another neuromodulatory mechanism (Metherate and Ashe, 1995; Fitzgerald and Sanes, 1999; Nugent et al., 2007). The second line of evidence derives from the observation that a comparable magnitude inhibitory potentiation was induced by application of a low concentration of BDNF, in the absence of afferent conditioning, and this treatment occluded additional potentiation by afferent stimulation (Fig. 4).
Neither glutamatergic nor GABAergic transmission was necessary for the induction of inhibitory potentiation (Fig. 6). Although inhibitory long-term plasticity often depends on these transmitters (Nugent et al., 2007), our data indicate that neurotrophin signaling is sufficient for inhibitory potentiation. Our results are consistent with a demonstration that BDNF exposure alone is sufficient to cause long-term reduction of inhibitory strength (Cheng and Yeh, 2005), and a study showing that estradiol-17β is necessary and sufficient for the decrease of miniature IPSC rate (Tremere et al., 2009).
BDNF is known to be expressed in temporal cortex pyramidal neurons (Conner et al., 1997), and cortical expression increases substantially between P8 and P15 in mice (Baquet et al., 2004). However, BDNF expression is compromised in the auditory CNS after hearing loss (Oh et al., 2007; Rüttiger et al., 2007; Tan et al., 2008). In ACx pyramidal neuron dendrites, cochlear injury leads to a reduction of BDNF levels, and expression can be restored by peripheral stimulation (Tan et al., 2008). Therefore, our data are consistent with the model that conditioning stimuli induce BDNF release from pyramidal cell dendrites, which then induces inhibitory potentiation. We propose that either normal expression or release of BDNF is vulnerable to even a moderate reduction of hearing.
BDNF release requires an elevation of intracellular calcium (Androutsellis-Theotokis et al., 1996; Goggi et al., 2003). Furthermore, several signals have been identified that trigger secretion from the dendrite (Kuczewski et al., 2009). In the present study, inhibitory synaptic responses were isolated by introducing QX-314 in the pipette and glutamate or GABA receptor antagonists in the bath (Fig. 6). Therefore, BDNF could only have been released from the dendrite of the recorded neuron if the stimulating electrode activated another signaling system to that dendrite, which led to calcium influx or release from internal stores. Alternatively, it is possible that the stimulating electrode activated pyramidal neurons nearby the recorded neuron, and propagation of the action potentials into the dendrites of these cells led to the release of BDNF (Kuczewski et al., 2008a). Neither of these putative mechanisms implies how BDNF is released in vivo, but it is plausible that glutamatergic activity would induce release in ACx as it does in other systems (Hartmann et al., 2001).
BDNF signaling can adjust GABAergic strength through either presynaptic or postsynaptic mechanisms (Brünig et al., 2001; Mizoguchi et al., 2003; Wardle and Poo, 2003; Jovanovic et al., 2004; Baldelli et al., 2005; Cheng and Yeh, 2005; Gubellini et al., 2005; Liu et al., 2007; Inagaki et al., 2008). The activity-dependent control of BDNF expression, and inhibitory synapse development, is mediated by at least two transcription factors, cAMP response element-binding protein (CREB) and Npas4 (Tao et al., 1998; Lin et al., 2008). Furthermore, when the CREB promoter is mutated on the mouse BDNF gene, sensory experience does not induce BDNF expression and fewer inhibitory synapses are formed (Hong et al., 2008).
We observed an increased postsynaptic GABAAR conductance (Fig. 3C) that could occur by phosphorylation of existing GABAARs or their de novo insertion. For example, BDNF induces a rapid increase in both the number of GABAARs and GABAAR-mediated currents in rat visual cortex (Mizoguchi et al., 2003), and similar observations have been made in hippocampus (Jovanovic et al., 2004). In contrast, the EIPSC did not shift after induction of inhibitory potentiation, implying that cation-chloride cotransporters are not key participants. Furthermore, cortical inhibitory potentiation was not associated with a change in the PPR or CV (Fig. 2); however, this does not rule out presynaptic mechanisms. It is possible that a change in chloride concentration could contribute to inhibitory potentiation, although this was not tested. Finally, it is possible that inhibitory potentiation is coregulated by NMDAergic inputs in vivo, although this was not assessed in the present paradigm (Grunze et al., 1996; Lin et al., 2008).
Possible role for inhibitory potentiation during inhibitory synapse development
In several auditory regions, inhibitory connections undergo an experience-dependent improvement of specificity that may permit for proper auditory computations (Sanes and Siverls, 1991; Sanes and Takács, 1993; Gabriele et al., 2000; Kapfer et al., 2002; Kim and Kandler, 2003; Franklin et al., 2008; Werthat et al., 2008). Furthermore, activity-dependent adjustment of in synaptic gain is common during the maturation of inhibitory connections (Zheng and Knudsen, 1999; Vale and Sanes, 2002; Yoshimura et al., 2003; Chattopadhyaya et al., 2004, 2007; Carrasco et al., 2005; Tao and Poo, 2005; Jiao et al., 2006; Lien et al., 2006; Maffei et al., 2006; Pallas et al., 2006; Katagiri et al., 2007; Balena and Woodin, 2008; Razak et al., 2008; Wilhelm and Wenner, 2008). We have previously suggested that inhibitory long-term depression could support the elimination of inhibitory synapses (Kotak and Sanes, 2000; Chang et al., 2003). Conversely, synapse strengthening through an iLTP mechanism could create long-term stability of inhibitory synapses (Kim and Kandler, 2003). In cortex, BDNF signaling is a plausible link between inhibitory synapse maturation and plasticity because it regulates both processes (Castrén et al., 1992; Bozzi et al., 1995; Huang et al., 1999; Morales et al., 2002; Gianfranceschi et al., 2003; Chattopadhyaya et al., 2004; Gubellini et al., 2005; Liu et al., 2007; Abidin et al., 2008; Inagaki et al., 2008; Kuczewski et al., 2008b).
Functional consequences
In adult animals, inhibitory plasticity is implicated in normal processing and pathological behaviors (Shumyatsky et al., 2002; Bauer and LeDoux, 2004; Nugent et al., 2007; Szinyei et al., 2007; Kurotani et al., 2008). For example, the sound-evoked escape response in teleost fish mediated by Mauthner neurons can be suppressed by iLTP (Oda et al., 1998; Korn and Faber, 2005). Since inhibition participates in most ACx computations (Wang et al., 2000, 2002; Wehr and Zador, 2003; Wu et al., 2006, 2008; Tan et al., 2007; de la Rocha et al., 2008), it is plausible that enhanced intracortical inhibition via long-lasting potentiation would sharpen neuronal receptive fields by limiting the lateral propagation of recurrent excitation. Indeed, auditory cortical receptive field sharpening is associated with a persistent potentiation of GABAergic inhibition (Froemke et al., 2007).
The ACx can undergo progressive map reorganization throughout life (Bao et al., 2004; Fritz et al., 2005; Rutkowski and Weinberger, 2005; Sakai and Kudoh, 2005; Witte and Kipke, 2005; Ono et al., 2006; Rybalko et al., 2006; Froemke et al., 2007). Although there is no direct evidence linking our findings to behavioral performance, the results may provide a partial explanation for the perceptual difficulties in children with hearing loss. Even transient bouts of conductive hearing loss can disrupt auditory processing, and months or years may be required for normal perception to resolve (Wilmington et al., 1994; Hogan et al., 1996). There are also suggestions from the clinical literature that auditory-based learning and memory mechanisms could be compromised by hearing loss (Davis et al., 1986; Gravel et al., 1996; Cowan et al., 1997; Mody et al., 1999; Burkholder and Pisoni, 2003; Pisoni and Cleary, 2003). For example, children with moderate SNHL performed significantly poorer on tests of phonological discrimination and short-term memory (nonword repetition), even though many subjects had received speech and language therapy (Briscoe et al., 2001). In a study that examined rapid nonsense word learning, normal hearing subjects performed much better than those with hearing loss across a broad age range (Pittman et al., 2005). We propose that the diminished excitatory (Kotak et al., 2007) and inhibitory LTP that attend early hearing loss may be associated with deficits in perception or auditory learning.
Footnotes
This work was supported by National Institutes of Health Grant DC006864 (D.H.S., V.C.K.).
References
- Abidin I, Eysel UT, Lessmann V, Mittmann T. Impaired GABAergic inhibition in the visual cortex of brain-derived neurotrophic factor heterozygous knockout mice. J Physiol. 2008;586:1885–1901. doi: 10.1113/jphysiol.2007.148627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham WC, Bear MF. Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 1996;19:126–130. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- Androutsellis-Theotokis A, McCormack WJ, Bradford HF, Stern GM, Pliego-Rivero FB. The depolarisation-induced release of [125I]BDNF from brain tissue. Brain Res. 1996;743:40–48. doi: 10.1016/s0006-8993(96)00981-x. [DOI] [PubMed] [Google Scholar]
- Arancio O, Chao MV. Neurotrophins, synaptic plasticity and dementia. Curr Opin Neurobiol. 2007;17:325–330. doi: 10.1016/j.conb.2007.03.013. [DOI] [PubMed] [Google Scholar]
- Baldelli P, Hernandez-Guijo JM, Carabelli V, Carbone E. Brain-derived neurotrophic factor enhances GABA release probability and nonuniform distribution of N- and P/Q-type channels on release sites of hippocampal inhibitory synapses. J Neurosci. 2005;25:3358–3368. doi: 10.1523/JNEUROSCI.4227-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balena T, Woodin MA. Coincident pre- and postsynaptic activity downregulates NKCC1 to hyperpolarize ECl during development. Eur J Neurosci. 2008;27:2402–2412. doi: 10.1111/j.1460-9568.2008.06194.x. [DOI] [PubMed] [Google Scholar]
- Bao S, Chang EF, Woods J, Merzenich MM. Temporal plasticity in the primary auditory cortex induced by operant perceptual learning. Nat Neurosci. 2004;7:974–981. doi: 10.1038/nn1293. [DOI] [PubMed] [Google Scholar]
- Baquet ZC, Gorski JA, Jones KR. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J Neurosci. 2004;24:4250–4258. doi: 10.1523/JNEUROSCI.3920-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer EP, LeDoux JE. Heterosynaptic long-term potentiation of inhibitory interneurons in the lateral amygdala. J Neurosci. 2004;24:9507–9512. doi: 10.1523/JNEUROSCI.3567-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozzi Y, Pizzorusso T, Cremisi F, Rossi FM, Barsacchi G, Maffei L. Monocular deprivation decreases the expression of messenger RNA for brain-derived neurotrophic factor in the rat visual cortex. Neuroscience. 1995;69:1133–1144. doi: 10.1016/0306-4522(95)00321-9. [DOI] [PubMed] [Google Scholar]
- Briscoe J, Bishop DV, Norbury CF. Phonological processing, language, and literacy: a comparison of children with mild-to-moderate sensorineural hearing loss and those with specific language impairment. J Child Psychol Psychiatry. 2001;42:329–340. [PubMed] [Google Scholar]
- Brünig I, Penschuck S, Berninger B, Benson J, Fritschy JM. BDNF reduces miniature inhibitory postsynaptic currents by rapid downregulation of GABAA receptor surface expression. Eur J Neurosci. 2001;13:1320–1328. doi: 10.1046/j.0953-816x.2001.01506.x. [DOI] [PubMed] [Google Scholar]
- Burkholder RA, Pisoni DB. Speech timing and working memory in profoundly deaf children after cochlear implantation. J Exp Child Psychol. 2003;85:63–88. doi: 10.1016/s0022-0965(03)00033-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco MM, Razak KA, Pallas SL. Visual experience is necessary for maintenance but not development of receptive fields in superior colliculus. J Neurophysiol. 2005;94:1962–1970. doi: 10.1152/jn.00166.2005. [DOI] [PubMed] [Google Scholar]
- Castrén E, Zafra F, Thoenen H, Lindholm D. Light regulates expression of brain-derived neurotrophic factor mRNA in rat visual cortex. Proc Natl Acad Sci U S A. 1992;89:9444–9448. doi: 10.1073/pnas.89.20.9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EH, Kotak VC, Sanes DH. Long-term depression of synaptic inhibition is expressed postsynaptically in the developing auditory system. J Neurophysiol. 2003;90:1479–1488. doi: 10.1152/jn.00386.2003. [DOI] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di Cristo G, Higashiyama H, Knott GW, Kuhlman SJ, Welker E, Huang ZJ. Experience and activity-dependent maturation of perisomatic GABAergic innervation in primary visual cortex during a postnatal critical period. J Neurosci. 2004;24:9598–9611. doi: 10.1523/JNEUROSCI.1851-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di Cristo G, Wu CZ, Knott G, Kuhlman S, Fu Y, Palmiter RD, Huang ZJ. GAD67-mediated GABA synthesis and signaling regulate inhibitory synaptic innervation in the visual cortex. Neuron. 2007;54:889–903. doi: 10.1016/j.neuron.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q, Yeh HH. PLCgamma signaling underlies BDNF potentiation of Purkinje cell responses to GABA. J Neurosci Res. 2005;79:616–627. doi: 10.1002/jnr.20397. [DOI] [PubMed] [Google Scholar]
- Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan RS, DelDot J, Barker EJ, Sarant JZ, Pegg P, Dettman S, Galvin KL, Rance G, Hollow R, Dowell RC, Pyman B, Gibson WP, Clark GM. Speech perception results for children with implants with different levels of preoperative residual hearing. Am J Otol. 1997;18:S125–S126. [PubMed] [Google Scholar]
- Creutzfeldt O, Hellweg FC, Schreiner C. Thalamocortical transformation of responses to complex auditory stimuli. Exp Brain Res. 1980;39:87–104. doi: 10.1007/BF00237072. [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Rose HJ, Metherate R. Auditory thalamocortical synaptic transmission in vitro. J Neurophysiol. 2002;87:361–384. doi: 10.1152/jn.00549.2001. [DOI] [PubMed] [Google Scholar]
- Davis JM, Elfenbein J, Schum R, Bentler RA. Effects of mild and moderate hearing impairments on language, educational, and psychosocial behavior of children. J Speech Hear Disord. 1986;51:53–62. doi: 10.1044/jshd.5101.53. [DOI] [PubMed] [Google Scholar]
- de la Rocha J, Marchetti C, Schiff M, Reyes AD. Linking the response properties of cells in auditory cortex with network architecture: cotuning versus lateral inhibition. J Neurosci. 2008;28:9151–9163. doi: 10.1523/JNEUROSCI.1789-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6:231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Bengzon J, Kokaia Z, Persson H, Lindvall O. Increased levels of messenger RNAs for neurotrophic factors in the brain during kindling epileptogenesis. Neuron. 1991;7:165–176. doi: 10.1016/0896-6273(91)90084-d. [DOI] [PubMed] [Google Scholar]
- Falkenberg T, Mohammed AK, Henriksson B, Persson H, Winblad B, Lindefors N. Increased expression of brain-derived neurotrophic factor mRNA in rat hippocampus is associated with improved spatial memory and enriched environment. Neurosci Lett. 1992;138:153–156. doi: 10.1016/0304-3940(92)90494-r. [DOI] [PubMed] [Google Scholar]
- Fitzgerald KK, Sanes DH. Serotonergic modulation of synapses in the developing gerbil lateral superior olive. J Neurophysiol. 1999;81:2743–2752. doi: 10.1152/jn.1999.81.6.2743. [DOI] [PubMed] [Google Scholar]
- Franklin SR, Brunso-Bechtold JK, Henkel CK. Bilateral cochlear ablation in postnatal rat disrupts development of banded pattern of projections from the dorsal nucleus of the lateral lemniscus to the inferior colliculus. Neuroscience. 2008;154:346–354. doi: 10.1016/j.neuroscience.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz JB, Elhilali M, Shamma SA. Differential dynamic plasticity of A1 receptive fields during multiple spectral tasks. J Neurosci. 2005;25:7623–7635. doi: 10.1523/JNEUROSCI.1318-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froemke RC, Merzenich MM, Schreiner CE. A synaptic memory trace for cortical receptive field plasticity. Nature. 2007;450:425–429. doi: 10.1038/nature06289. [DOI] [PubMed] [Google Scholar]
- Gabriele ML, Brunso-Bechtold JK, Henkel CK. Plasticity in the development of afferent patterns in the inferior colliculus of the rat after unilateral cochlear ablation. J Neurosci. 2000;20:6939–6949. doi: 10.1523/JNEUROSCI.20-18-06939.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianfranceschi L, Siciliano R, Walls J, Morales B, Kirkwood A, Huang ZJ, Tonegawa S, Maffei L. Visual cortex is rescued from the effects of dark rearing by overexpression of BDNF. Proc Natl Acad Sci U S A. 2003;100:12486–12491. doi: 10.1073/pnas.1934836100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goggi J, Pullar IA, Carney SL, Bradford HF. The control of [125I]BDNF release from striatal rat brain slices. Brain Res. 2003;967:201–209. doi: 10.1016/s0006-8993(03)02225-x. [DOI] [PubMed] [Google Scholar]
- Gravel JS, Wallace IF, Ruben RJ. Auditory consequences of early mild hearing loss associated with otitis media. Acta Otolaryngol. 1996;116:219–221. doi: 10.3109/00016489609137827. [DOI] [PubMed] [Google Scholar]
- Grunze HC, Rainnie DG, Hasselmo ME, Barkai E, Hearn EF, McCarley RW, Greene RW. NMDA-dependent modulation of CA1 local circuit inhibition. J Neurosci. 1996;16:2034–2043. doi: 10.1523/JNEUROSCI.16-06-02034.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubellini P, Ben-Ari Y, Gaïarsa JL. Endogenous neurotrophins are required for the induction of GABAergic long-term potentiation in the neonatal rat hippocampus. J Neurosci. 2005;25:5796–5802. doi: 10.1523/JNEUROSCI.0824-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J, Thomas KL, Everitt BJ. Rapid and selective induction of BDNF expression in the hippocampus during contextual learning. Nat Neurosci. 2000;3:533–535. doi: 10.1038/75698. [DOI] [PubMed] [Google Scholar]
- Hartmann M, Heumann R, Lessmann V. Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J. 2001;20:5887–5897. doi: 10.1093/emboj/20.21.5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan SC, Meyer SE, Moore DR. Binaural unmasking returns to normal in teenagers who had otitis media in infancy. Audiol Neurootol. 1996;1:104–111. doi: 10.1159/000259189. [DOI] [PubMed] [Google Scholar]
- Hong EJ, McCord AE, Greenberg ME. A biological function for the neuronal activity-dependent component of BDNF transcription in the development of cortical inhibition. Neuron. 2008;60:610–624. doi: 10.1016/j.neuron.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, Tonegawa S. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999;98:739–755. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- Ichisaka S, Katoh-Semba R, Hata Y, Ohshima M, Kameyama K, Tsumoto T. Activity-dependent change in the protein level of brain-derived neurotrophic factor but no change in other neurotrophins in the visual cortex of young and adult ferrets. Neuroscience. 2003;117:361–371. doi: 10.1016/s0306-4522(02)00771-6. [DOI] [PubMed] [Google Scholar]
- Ickes BR, Pham TM, Sanders LA, Albeck DS, Mohammed AH, Granholm AC. Long-term environmental enrichment leads to regional increases in neurotrophin levels in rat brain. Exp Neurol. 2000;164:45–52. doi: 10.1006/exnr.2000.7415. [DOI] [PubMed] [Google Scholar]
- Inagaki T, Begum T, Reza F, Horibe S, Inaba M, Yoshimura Y, Komatsu Y. Brain-derived neurotrophic factor-mediated retrograde signaling required for the induction of long-term potentiation at inhibitory synapses of visual cortical pyramidal neurons. Neurosci Res. 2008;61:192–200. doi: 10.1016/j.neures.2008.02.006. [DOI] [PubMed] [Google Scholar]
- Isackson PJ, Huntsman MM, Murray KD, Gall CM. BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: temporal patterns of induction distinct from NGF. Neuron. 1991;6:937–948. doi: 10.1016/0896-6273(91)90234-q. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Zhang C, Yanagawa Y, Sun QQ. Major effects of sensory experiences on the neocortical inhibitory circuits. J Neurosci. 2006;26:8691–8701. doi: 10.1523/JNEUROSCI.2478-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Thomas P, Kittler JT, Smart TG, Moss SJ. Brain-derived neurotrophic factor modulates fast synaptic inhibition by regulating GABAA receptor phosphorylation, activity, and cell-surface stability. J Neurosci. 2004;24:522–530. doi: 10.1523/JNEUROSCI.3606-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapfer C, Seidl AH, Schweizer H, Grothe B. Experience-dependent refinement of inhibitory inputs to auditory coincidence-detector neurons. Nat Neurosci. 2002;5:247–253. doi: 10.1038/nn810. [DOI] [PubMed] [Google Scholar]
- Katagiri H, Fagiolini M, Hensch TK. Optimization of somatic inhibition at critical period onset in mouse visual cortex. Neuron. 2007;53:805–812. doi: 10.1016/j.neuron.2007.02.026. [DOI] [PubMed] [Google Scholar]
- Kesslak JP, So V, Choi J, Cotman CW, Gomez-Pinilla F. Learning upregulates brain-derived neurotrophic factor messenger ribonucleic acid: a mechanism to facilitate encoding and circuit maintenance? Behav Neurosci. 1998;112:1012–1019. doi: 10.1037//0735-7044.112.4.1012. [DOI] [PubMed] [Google Scholar]
- Kim G, Kandler K. Elimination and strengthening of glycinergic/GABAergic connections during tonotopic map formation. Nat Neurosci. 2003;6:282–290. doi: 10.1038/nn1015. [DOI] [PubMed] [Google Scholar]
- Kohara K, Yasuda H, Huang Y, Adachi N, Sohya K, Tsumoto T. A local reduction in cortical GABAergic synapses after a loss of endogenous brain-derived neurotrophic factor, as revealed by single-cell gene knock-out method. J Neurosci. 2007;27:7234–7244. doi: 10.1523/JNEUROSCI.1943-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu Y. GABAB receptors, monoamine receptors, and postsynaptic inositol trisphosphate-induced Ca2+ release are involved in the induction of long-term potentiation at visual cortical inhibitory synapses. J Neurosci. 1996;16:6342–6352. doi: 10.1523/JNEUROSCI.16-20-06342.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn H, Faber DS. The Mauthner cell half a century later: a neurobiological model for decision-making? Neuron. 2005;47:13–28. doi: 10.1016/j.neuron.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Kotak VC, Sanes DH. Long-lasting inhibitory synaptic depression is age- and calcium-dependent. J Neurosci. 2000;20:5820–5826. doi: 10.1523/JNEUROSCI.20-15-05820.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotak VC, Fujisawa S, Lee FA, Karthikeyan O, Aoki C, Sanes DH. Hearing loss raises excitability in the auditory cortex. J Neurosci. 2005;25:3908–3918. doi: 10.1523/JNEUROSCI.5169-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotak VC, Breithaupt AD, Sanes DH. Developmental hearing loss eliminates long-term potentiation in the auditory cortex. Proc Natl Acad Sci U S A. 2007;104:3550–3555. doi: 10.1073/pnas.0607177104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotak VC, Takesian AE, Sanes DH. Hearing loss prevents the maturation of GABAergic transmission in the auditory cortex. Cereb Cortex. 2008;18:2098–2108. doi: 10.1093/cercor/bhm233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczewski N, Porcher C, Ferrand N, Fiorentino H, Pellegrino C, Kolarow R, Lessmann V, Medina I, Gaiarsa JL. Backpropagating action potentials trigger dendritic release of BDNF during spontaneous network activity. J Neurosci. 2008a;28:7013–7023. doi: 10.1523/JNEUROSCI.1673-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczewski N, Langlois A, Fiorentino H, Bonnet S, Marissal T, Diabira D, Ferrand N, Porcher C, Gaiarsa JL. Spontaneous glutamatergic activity induces a BDNF-dependent potentiation of GABAergic synapses in the newborn rat hippocampus. J Physiol. 2008b;586:5119–5128. doi: 10.1113/jphysiol.2008.158550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczewski N, Porcher C, Lessmann V, Medina I, Gaiarsa JL. Activity-dependent dendritic release of BDNF and biological consequences. Mol Neurobiol. 2009;39:37–49. doi: 10.1007/s12035-009-8050-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudoh M, Shibuki K. Long-term potentiation of supragranular pyramidal outputs in the rat auditory cortex. Exp Brain Res. 1996;110:21–27. doi: 10.1007/BF00241370. [DOI] [PubMed] [Google Scholar]
- Kudoh M, Shibuki K. Importance of polysynaptic inputs and horizontal connectivity in the generation of tetanus-induced long-term potentiation in the rat auditory cortex. J Neurosci. 1997;17:9458–9465. doi: 10.1523/JNEUROSCI.17-24-09458.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurotani T, Yamada K, Yoshimura Y, Crair MC, Komatsu Y. State-dependent bidirectional modification of somatic inhibition in neocortical pyramidal cells. Neuron. 2008;57:905–916. doi: 10.1016/j.neuron.2008.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien CC, Mu Y, Vargas-Caballero M, Poo MM. Visual stimuli-induced LTD of GABAergic synapses mediated by presynaptic NMDA receptors. Nat Neurosci. 2006;9:372–380. doi: 10.1038/nn1649. [DOI] [PubMed] [Google Scholar]
- Lin Y, Bloodgood BL, Hauser JL, Lapan AD, Koon AC, Kim TK, Hu LS, Malik AN, Greenberg ME. Activity-dependent regulation of inhibitory synapse development by Npas4. Nature. 2008;455:1198–1204. doi: 10.1038/nature07319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhang LI, Tao HW. Heterosynaptic scaling of developing GABAergic synapses: dependence on glutamatergic input and developmental stage. J Neurosci. 2007;27:5301–5312. doi: 10.1523/JNEUROSCI.0376-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem. 2003;10:86–98. doi: 10.1101/lm.54603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Figurov A. Role of neurotrophins in synapse development and plasticity. Rev Neurosci. 1997;8:1–12. doi: 10.1515/revneuro.1997.8.1.1. [DOI] [PubMed] [Google Scholar]
- Maffei A, Nataraj K, Nelson SB, Turrigiano GG. Potentiation of cortical inhibition by visual deprivation. Nature. 2006;443:81–84. doi: 10.1038/nature05079. [DOI] [PubMed] [Google Scholar]
- Marty S, Wehrlé R, Sotelo C. Neuronal activity and brain-derived neurotrophic factor regulate the density of inhibitory synapses in organotypic slice cultures of postnatal hippocampus. J Neurosci. 2000;20:8087–8095. doi: 10.1523/JNEUROSCI.20-21-08087.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean HA, Caillard O, Ben-Ari Y, Gaiarsa JL. Bidirectional plasticity expressed by GABAergic synapses in the neonatal rat hippocampus. J Physiol. 1996;496:471–477. doi: 10.1113/jphysiol.1996.sp021699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean JH, Darby-King A, Bonnell WS. Neonatal olfactory sensory deprivation decreases BDNF in the olfactory bulb of the rat. Brain Res Dev Brain Res. 2001;128:17–24. doi: 10.1016/s0165-3806(01)00144-4. [DOI] [PubMed] [Google Scholar]
- Metherate R, Ashe JH. Synaptic interactions involving acetylcholine, glutamate, and GABA in rat auditory cortex. Exp Brain Res. 1995;107:59–72. doi: 10.1007/BF00228017. [DOI] [PubMed] [Google Scholar]
- Mizoguchi Y, Kanematsu T, Hirata M, Nabekura J. A rapid increase in the total number of cell surface functional GABAA receptors induced by brain-derived neurotrophic factor in rat visual cortex. J Biol Chem. 2003;278:44097–44102. doi: 10.1074/jbc.M305872200. [DOI] [PubMed] [Google Scholar]
- Mockett BG, Hulme SR. Metaplasticity: new insights through electrophysiological investigations. J Integr Neurosci. 2008;7:315–336. doi: 10.1142/s0219635208001782. [DOI] [PubMed] [Google Scholar]
- Mody M, Schwartz RG, Gravel JS, Ruben RJ. Speech perception and verbal memory in children with and without histories of otitis media. J Speech Lang Hear Res. 1999;42:1069–1079. doi: 10.1044/jslhr.4205.1069. [DOI] [PubMed] [Google Scholar]
- Morales B, Choi SY, Kirkwood A. Dark rearing alters the development of GABAergic transmission in visual cortex. J Neurosci. 2002;22:8084–8090. doi: 10.1523/JNEUROSCI.22-18-08084.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagappan G, Lu B. Activity-dependent modulation of the BDNF receptor TrkB: mechanisms and implications. Trends Neurosci. 2005;28:464–471. doi: 10.1016/j.tins.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Nugent FS, Penick EC, Kauer JA. Opioids block long-term potentiation of inhibitory synapses. Nature. 2007;446:1086–1090. doi: 10.1038/nature05726. [DOI] [PubMed] [Google Scholar]
- Oda Y, Kawasaki K, Morita M, Korn H, Matsui H. Inhibitory long-term potentiation underlies auditory conditioning of goldfish escape behaviour. Nature. 1998;394:182–185. doi: 10.1038/28172. [DOI] [PubMed] [Google Scholar]
- Oh SH, Kim CS, Song JJ. Gene expression and plasticity in the rat auditory cortex after bilateral cochlear ablation. Acta Otolaryngol. 2007;127:341–350. doi: 10.1080/00016480701275246. [DOI] [PubMed] [Google Scholar]
- Ono K, Kudoh M, Shibuki K. Roles of the auditory cortex in discrimination learning by rats. Eur J Neurosci. 2006;23:1623–1632. doi: 10.1111/j.1460-9568.2006.04695.x. [DOI] [PubMed] [Google Scholar]
- Pallas SL, Wenner P, Gonzalez-Islas C, Fagiolini M, Razak KA, Kim G, Sanes D, Roerig B. Developmental plasticity of inhibitory circuitry. J Neurosci. 2006;26:10358–10361. doi: 10.1523/JNEUROSCI.3516-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patenaude C, Chapman CA, Bertrand S, Congar P, Lacaille JC. GABAB receptor- and metabotropic glutamate receptor-dependent cooperative long-term potentiation of rat hippocampal GABAA synaptic transmission. J Physiol. 2003;553:155–167. doi: 10.1113/jphysiol.2003.049015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson SL, Grover LM, Schwartzkroin PA, Bothwell M. Neurotrophin expression in rat hippocampal slices: a stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron. 1992;9:1081–1088. doi: 10.1016/0896-6273(92)90067-n. [DOI] [PubMed] [Google Scholar]
- Pisoni DB, Cleary M. Measures of working memory span and verbal rehearsal speed in deaf children after cochlear implantation. Ear Hear. 2003;24:106S–120S. doi: 10.1097/01.AUD.0000051692.05140.8E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittman AL, Lewis DE, Hoover BM, Stelmachowicz PG. Rapid word-learning in normal-hearing and hearing-impaired children: effects of age, receptive vocabulary, and high-frequency amplification. Ear Hear. 2005;26:619–629. doi: 10.1097/01.aud.0000189921.34322.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razak KA, Richardson MD, Fuzessery ZM. Experience is required for the maintenance and refinement of FM sweep selectivity in the developing auditory cortex. Proc Natl Acad Sci U S A. 2008;105:4465–4470. doi: 10.1073/pnas.0709504105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocamora N, Welker E, Pascual M, Soriano E. Upregulation of BDNF mRNA expression in the barrel cortex of adult mice after sensory stimulation. J Neurosci. 1996;16:4411–4419. doi: 10.1523/JNEUROSCI.16-14-04411.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi FM, Bozzi Y, Pizzorusso T, Maffei L. Monocular deprivation decreases brain-derived neurotrophic factor immunoreactivity in the rat visual cortex. Neuroscience. 1999;90:363–368. doi: 10.1016/s0306-4522(98)00463-1. [DOI] [PubMed] [Google Scholar]
- Rutherford LC, DeWan A, Lauer HM, Turrigiano GG. Brain-derived neurotrophic factor mediates the activity-dependent regulation of inhibition in neocortical cultures. J Neurosci. 1997;17:4527–4535. doi: 10.1523/JNEUROSCI.17-12-04527.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski RG, Weinberger NM. Encoding of learned importance of sound by magnitude of representational area in primary auditory cortex. Proc Natl Acad Sci U S A. 2005;102:13664–13669. doi: 10.1073/pnas.0506838102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüttiger L, Panford-Walsh R, Schimmang T, Tan J, Zimmermann U, Rohbock K, Köpschall I, Limberger A, Müller M, Fraenzer JT, Cimerman J, Knipper M. BDNF mRNA expression and protein localization are changed in age-related hearing loss. Neurobiol Aging. 2007;28:586–601. doi: 10.1016/j.neurobiolaging.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Rybalko N, Suta D, Nwabueze-Ogbo F, Syka J. Effect of auditory cortex lesions on the discrimination of frequency-modulated tones in rats. Eur J Neurosci. 2006;23:1614–1622. doi: 10.1111/j.1460-9568.2006.04688.x. [DOI] [PubMed] [Google Scholar]
- Sakai M, Kudoh M. Characteristics of sound discrimination enhancement after sound exposure in adult rats. Behav Neurosci. 2005;119:961–973. doi: 10.1037/0735-7044.119.4.961. [DOI] [PubMed] [Google Scholar]
- Sanes DH, Siverls V. Development and specificity of inhibitory terminal arborizations in the central nervous system. J Neurobiol. 1991;22:837–854. doi: 10.1002/neu.480220805. [DOI] [PubMed] [Google Scholar]
- Sanes DH, Takács C. Activity-dependent refinement of inhibitory connections. Eur J Neurosci. 1993;5:570–574. doi: 10.1111/j.1460-9568.1993.tb00522.x. [DOI] [PubMed] [Google Scholar]
- Sanes DH, Markowitz S, Bernstein J, Wardlow J. The influence of inhibitory afferents on the development of postsynaptic dendritic arbors. J Comp Neurol. 1992;321:637–644. doi: 10.1002/cne.903210410. [DOI] [PubMed] [Google Scholar]
- Sarro EC, Kotak VC, Sanes DH, Aoki C. Hearing loss alters the subcellular distribution of presynaptic GAD and postsynaptic GABAA receptors in the auditory cortex. Cereb Cortex. 2008;18:2855–2867. doi: 10.1093/cercor/bhn044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinder AF, Poo M. The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci. 2000;23:639–645. doi: 10.1016/s0166-2236(00)01672-6. [DOI] [PubMed] [Google Scholar]
- Seki K, Kudoh M, Shibuki K. Long-term potentiation of Ca2+ signal in the rat auditory cortex. Neurosci Res. 1999;34:187–197. doi: 10.1016/s0168-0102(99)00049-8. [DOI] [PubMed] [Google Scholar]
- Seki K, Kudoh M, Shibuki K. Sequence dependence of post-tetanic potentiation after sequential heterosynaptic stimulation in the rat auditory cortex. J Physiol. 2001;533:503–518. doi: 10.1111/j.1469-7793.2001.0503a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki K, Kudoh M, Shibuki K. Polysynaptic slow depolarization and spiking activity elicited after induction of long-term potentiation in rat auditory cortex. Brain Res. 2003;988:114–120. doi: 10.1016/s0006-8993(03)03351-1. [DOI] [PubMed] [Google Scholar]
- Shumyatsky GP, Tsvetkov E, Malleret G, Vronskaya S, Hatton M, Hampton L, Battey JF, Dulac C, Kandel ER, Bolshakov VY. Identification of a signaling network in lateral nucleus of amygdala important for inhibiting memory specifically related to learned fear. Cell. 2002;111:905–918. doi: 10.1016/s0092-8674(02)01116-9. [DOI] [PubMed] [Google Scholar]
- Singh TD, Mizuno K, Kohno T, Nakamura S. BDNF and trkB mRNA expression in neurons of the neonatal mouse barrel field cortex: normal development and plasticity after cauterizing facial vibrissae. Neurochem Res. 1997;22:791–797. doi: 10.1023/a:1022075508176. [DOI] [PubMed] [Google Scholar]
- Slutsky I, Sadeghpour S, Li B, Liu G. Enhancement of synaptic plasticity through chronically reduced Ca2+ flux during uncorrelated activity. Neuron. 2004;44:835–849. doi: 10.1016/j.neuron.2004.11.013. [DOI] [PubMed] [Google Scholar]
- Szinyei C, Narayanan RT, Pape HC. Plasticity of inhibitory synaptic network interactions in the lateral amygdala upon fear conditioning in mice. Eur J Neurosci. 2007;25:1205–1211. doi: 10.1111/j.1460-9568.2007.05349.x. [DOI] [PubMed] [Google Scholar]
- Tan AY, Atencio CA, Polley DB, Merzenich MM, Schreiner CE. Unbalanced synaptic inhibition can create intensity-tuned auditory cortex neurons. Neuroscience. 2007;146:449–462. doi: 10.1016/j.neuroscience.2007.01.019. [DOI] [PubMed] [Google Scholar]
- Tan J, Widjaja S, Xu J, Shepherd RK. Cochlear implants stimulate activity-dependent CREB pathway in the deaf auditory cortex: implications for molecular plasticity induced by neural prosthetic devices. Cereb Cortex. 2008;18:1799–1813. doi: 10.1093/cercor/bhm206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao HW, Poo MM. Activity-dependent matching of excitatory and inhibitory inputs during refinement of visual receptive fields. Neuron. 2005;45:829–836. doi: 10.1016/j.neuron.2005.01.046. [DOI] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- Ter-Mikaelian M, Sanes DH, Semple MN. Transformation of temporal properties between auditory midbrain and cortex in the awake Mongolian gerbil. J Neurosci. 2007;27:6091–6102. doi: 10.1523/JNEUROSCI.4848-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajan TC, Lindskog M, Malgaroli A, Tsien RW. LTP and adaptation to inactivity: overlapping mechanisms and implications for metaplasticity. Neuropharmacology. 2007;52:156–175. doi: 10.1016/j.neuropharm.2006.07.030. [DOI] [PubMed] [Google Scholar]
- Tierney TS, Russell FA, Moore DR. Susceptibility of developing cochlear nucleus neurons to deafferentation-induced death abruptly ends just before the onset of hearing. J Comp Neurol. 1997;378:295–306. doi: 10.1002/(sici)1096-9861(19970210)378:2<295::aid-cne11>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Tremere LA, Jeong JK, Pinaud R. Estradiol shapes auditory processing in the adult brain by regulating inhibitory transmission and plasticity-associated gene expression. J Neurosci. 2009;29:5949–5963. doi: 10.1523/JNEUROSCI.0774-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucci DL, Cant NB, Durham D. Conductive hearing loss results in a decrease in central auditory system activity in the young gerbil. Laryngoscope. 1999;109:1359–1371. doi: 10.1097/00005537-199909000-00001. [DOI] [PubMed] [Google Scholar]
- Vale C, Sanes DH. The effect of bilateral deafness on excitatory and inhibitory synaptic strength in the inferior colliculus. Eur J Neurosci. 2002;16:2394–2404. doi: 10.1046/j.1460-9568.2002.02302.x. [DOI] [PubMed] [Google Scholar]
- Wakatsuki H, Gomi H, Kudoh M, Kimura S, Takahashi K, Takeda M, Shibuki K. Layer-specific NO dependence of long-term potentiation and biased NO release in layer V in the rat auditory cortex. J Physiol. 1998;513:71–81. doi: 10.1111/j.1469-7793.1998.071by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Caspary D, Salvi RJ. GABA-A antagonist causes dramatic expansion of tuning in primary auditory cortex. Neuroreport. 2000;11:1137–1140. doi: 10.1097/00001756-200004070-00045. [DOI] [PubMed] [Google Scholar]
- Wang J, McFadden SL, Caspary D, Salvi R. Gamma-aminobutyric acid circuits shape response properties of auditory cortex neurons. Brain Res. 2002;944:219–231. doi: 10.1016/s0006-8993(02)02926-8. [DOI] [PubMed] [Google Scholar]
- Wardle RA, Poo MM. Brain-derived neurotrophic factor modulation of GABAergic synapses by postsynaptic regulation of chloride transport. J Neurosci. 2003;23:8722–8732. doi: 10.1523/JNEUROSCI.23-25-08722.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehr M, Zador AM. Balanced inhibition underlies tuning and sharpens spike timing in auditory cortex. Nature. 2003;426:442–446. doi: 10.1038/nature02116. [DOI] [PubMed] [Google Scholar]
- Werthat F, Alexandrova O, Grothe B, Koch U. Experience-dependent refinement of the inhibitory axons projecting to the medial superior olive. Dev Neurobiol. 2008;68:1454–1462. doi: 10.1002/dneu.20660. [DOI] [PubMed] [Google Scholar]
- Wilhelm JC, Wenner P. GABAA transmission is a critical step in the process of triggering homeostatic increases in quantal amplitude. Proc Natl Acad Sci U S A. 2008;105:11412–11417. doi: 10.1073/pnas.0806037105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmington D, Gray L, Jahrsdoerfer R. Binaural processing after corrected congenital unilateral conductive hearing loss. Hear Res. 1994;74:99–114. doi: 10.1016/0378-5955(94)90179-1. [DOI] [PubMed] [Google Scholar]
- Witte RS, Kipke DR. Enhanced contrast sensitivity in auditory cortex as cats learn to discriminate sound frequencies. Brain Res Cogn Brain Res. 2005;23:171–184. doi: 10.1016/j.cogbrainres.2004.10.018. [DOI] [PubMed] [Google Scholar]
- Wu GK, Li P, Tao HW, Zhang LI. Nonmonotonic synaptic excitation and imbalanced inhibition underlying cortical intensity tuning. Neuron. 2006;52:705–715. doi: 10.1016/j.neuron.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GK, Arbuckle R, Liu BH, Tao HW, Zhang LI. Lateral sharpening of cortical frequency tuning by approximately balanced inhibition. Neuron. 2008;58:132–143. doi: 10.1016/j.neuron.2008.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Kotak VC, Sanes DH. Conductive hearing loss disrupts synaptic and spike adaptation in developing auditory cortex. J Neurosci. 2007;27:9417–9426. doi: 10.1523/JNEUROSCI.1992-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura Y, Ohmura T, Komatsu Y. Two forms of synaptic plasticity with distinct dependence on age, experience, and NMDA receptor subtype in rat visual cortex. J Neurosci. 2003;23:6557–6566. doi: 10.1523/JNEUROSCI.23-16-06557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafra F, Hengerer B, Leibrock J, Thoenen H, Lindholm D. Activity dependent regulation of BDNF and NGF mRNAs in the rat hippocampus is mediated by non-NMDA glutamate receptors. EMBO J. 1990;9:3545–3550. doi: 10.1002/j.1460-2075.1990.tb07564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Knudsen EI. Functional selection of adaptive auditory space map by GABAA-mediated inhibition. Science. 1999;284:962–965. doi: 10.1126/science.284.5416.962. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]