

Abstract
Peroxisome proliferator‐activated receptor δ (PPARδ) agonism increases HDL cholesterol and has therefore the potential to stimulate macrophage‐to‐feces reverse cholesterol transport (RCT). To test whether PPAR™ activation promotes RCT in mice, in vivo macrophage RCT was assessed using cholesterol‐loaded/3H‐cholesterol‐labeled macrophages injected intraperitoneally. PPAR™ agonist GW0742 (10 mg/kg per day) did not change 3H‐tracer plasma appearance, but increased fecal 3H‐free sterols excretion by 103% (p < 0.005) over 48 hours. Total free cholesterol efflux from macrophages to serum (collected from both control and GW0742 groups) was not different, although ABCA1‐mediated efflux was significantly higher with GW0742. The metabolic fate of HDL labeled with 3H‐cholesteryl ether or 3H‐cholesteryl oleate was also measured. While 3H‐cholesteryl ether tissue uptake was unchanged, the 3H‐tracer recovered in fecal free sterol fraction after 3H‐cholesteryl oleate injection increased by 88% with GW0742 (p < 0.0005). This was associated with a lower Niemann‐Pick C1 like 1 (NPC1L1) mRNA expression in the small intestine (p < 0.05). The same experiments in mice treated with ezetimibe, which blocks NPC1L1, showed a similar 2‐fold increase in fecal free sterol excretion after labeled macrophages or HDL injection. In conclusion, PPAR™ activation enhances excretion of macrophage or HDL‐derived cholesterol in feces through reduced NPC1L1 expression in mice, comparable to the effect of ezetimibe.
Keywords: intestine, cholesterol, lipoproteins, reverse cholesterol transport
Introduction
Peroxisome proliferator‐activated receptors (PPARs) represent a subgroup of the nuclear receptor superfamily, including PPARα (NR1C1), PPARδ (NR1C2), and PPARγ (NR1C3). All of these heterodimerize with the 9‐cis‐retinoic acid receptor (RXR) and, upon ligand binding, become transcriptionally active and control a series of genes involved in lipid and energy metabolism. 1 The roles of PPARα and PPARγ have been extensively studied with the use of their respective agonists, fibrates and thiazolidinediones, which are already used clinically. 2 However, less is known about the role of the ubiquitously expressed PPARδ due to the lack of selective ligands.
The recent development of PPARδ agonists has suggested a beneficial effect on lipid and lipoprotein metabolism. PPARδ agonists have been shown to reduce triglycerides and small dense LDL levels and increase HDL cholesterol in insulinresistant animals. 3 , 4 The first in vivo experiment in healthy humans also showed improved triglycerides clearance post‐fat feeding and modestly increased HDL cholesterol levels. 5 These changes have been linked to enhanced fatty acid oxidation in skeletal muscle following PPARδ activation, as observed in obese insulin‐resistant mice 6 and human skeletal muscle cells. 5 Although some other preclinical studies showed that PPARδ agonism failed to inhibit macrophage foam cell formation, 7 , 8 the associated HDL increase may have atheroprotective effects. 3 , 4 , 5 , 9
Higher HDL levels in response to PPARδ activation could upregulate cholesterol transport from peripheral cells to the liver for further excretion into bile and feces. We therefore performed in vitro and in vivo experiments to test the hypothesis that PPARδ activation promotes macrophage‐to‐feces reverse cholesterol transport (RCT).
Methods
Animals and diets
Wild‐type C57BL/6 female mice (8–12 weeks old) were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). Mice were fed ad libitum for 2 weeks with either a control or PPARδ agonist GW0742 (10 mg/kg per day) or ezetimibe (5 mg/kg per day) supplemented chow diet (Purina #5002). For plasma lipid analysis, animals were fasted for 4 hours and then bled from the retro‐orbital plexus. All animals were housed according to guidelines of the Institutional Animal Care and Use Committee of the University of Pennsylvania. All protocols were approved by the Institutional Animal Care and Use Committee.
Plasma lipid analysis
Plasma total cholesterol, free cholesterol, HDL cholesterol, and triglycerides were measured on a Cobas Fara with the use of Sigma Diagnostic reagents. Cholesteryl ester concentration was calculated as the difference between total cholesterol and free cholesterol. Pooled plasma samples (140 μL) were used for lipoprotein separation by fast protein liquid chromatography (FPLC). The cholesterol concentration in the FPLC fractions was then determined with an enzymatic assay (Wako).
Preparation of bonemarrow‐derived macrophages
C57BL6/J mice were euthanized and dissected to remove the femur of each hind leg. Bone marrow was flushed from femur and tibia of each leg using PBS‐heparin (100 μg/mL). Cells were washed with PBS and resuspended in bone marrow growth medium (DMEM containing 30% L‐929 cell‐conditioned medium and 10% FBS). Bone marrow‐derived cells were seeded in 12‐well plates (for in vitro experiments) and cultured at 37°C and 5% CO2. Four days After plating, nonadherent cells were removed by washing. Adherent cells were fed with fresh bone marrow growth medium and cultured for an additional 3 days. Aliquots of bone marrow‐derived cells were analyzed for expression of markers specific for macrophages (CD11b, CD18), T‐lymphocytes (TCRb), and B‐lymphocytes CD19. After 7 days in culture under our experimental conditions, more than 99% of cells subcloned from bone marrow using L‐929 conditioned medium (collected from plates) were positive for CD11b and CD18, while less than 1% of cells were positive for TCRβ or CD19, markers of T or B cells, respectively.
Measurement of cholesterol efflux in vitro
For in vitro efflux studies, bone marrow macrophages were isolated and grown in 12‐well plates as described above followed by labeling with 3H‐cholesterol (2 μCi/mL; Amersham) in the presence of acetylated LDL (acLDL) (25 μg/mL) for 24 hours. Then cells were washed and equilibrated overnight in serum‐free medium. For the cholesterol efflux, medium containing 2.5% mouse serum was added to cells. After 4 hours, aliquots of the medium were removed, and the 3H‐cholesterol released into the medium was measured by liquid scintillation counting. The 3H‐cholesterol present in the cells was determined by extracting the cell lipids with isopropanol and measured by liquid scintillation counting. To assess the relative contribution of ABCA1‐dependent pathway, cells were pretreated with probucol (20 μM), an ABCA1 inhibitor, for 2 hours before measuring efflux of free cholesterol. 10
Preparation of J774 cells for in vivo reverse cholesterol transport study
J774 cells, obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA), were grown in suspension in RPMI/HEPES supplemented with 10% FBS and 0.5% gentamicin. J774 cells were cultured in suspension in Nalgene Teflon flasks and radiolabeled with 5 μCi/mL 3H‐cholesterol and cholesterol loaded with 25 μg/mL acLDL, as previously described. 11 After 48 hours, radiolabeled cells were washed with RPMI/HEPES and equilibrated for 4 hours in fresh RPMI/HEPES supplemented with 0.2% BSA and gentamicin. Cells were pelletted by lowspeed centrifugation and resuspended in RPMI/HEPES prior to injection into mice.
In vivo reverse cholesterol transport study
3H‐cholesterol‐labeled and acLDL‐loaded J774 cells (typically 4.5 × 106 cells containing 4 × 106 cpm in 0.5 mL minimum essential medium) were injected intraperitoneally into individually caged mice as described previously. 11 Mice had free access to food and water. Blood was collected at 6, 24, and 48 hours to measure radioactivity released into plasma. Feces were collected continuously from 0 to 48 hours and stored at 4°C before extraction of cholesterol and bile acid.
In vivo HDL turn over studies
HDL was prepared from pooled human plasma by sequential ultracentrifugation (density 1.063 < d < 1.21 g/mL). After extensive dialysis against buffer (0.15 M NaCl, EDTA 1 mM, pH 7.4), HDL was exchange labeled with cholesteryl hexadecyl ether (cholesteryl‐1,2‐3H, Perkin Elmer) or cholesteryl oleate (cholesteryl‐1,2,6,7‐3H, Perkin Elmer), as previously described. 12 After labeling, the HDL was reisolated by ultracentrifugation (density 1.063 < d < 1.21 g/mL), extensively dialyzed, fi lter sterilized, and stored at 4°C until injection.
To measure the HDL fractional catabolic rate (FRC) and organ uptake of the HDL cholesteryl esters, 3H‐cholesteryl oleate or 3Hcholesteryl ether‐labeled HDL (1–2 million cpm per animal) was injected intravenously via tail veins into mice (n= 6 per group). Blood samples were drawn by retro‐orbital bleeding at 2 minutes, 1 hour, 3 hours, 6 hours, 9 hours, 24 hours, and 48 hours (≈25 μL at each time point). Feces were collected continuously from 0 to 48 hours and stored at 4°C before extraction of cholesterol and bile acids. At study termination (48 hours After injection), mice were exsanguinated, perfused with ice‐cold PBS, and liver samples were collected for analysis. The small intestine was also removed, flushed with ice‐cold PBS, and sectioned into three segments with lengths ratio of 1:3:2 (duodenum–jejunum–ileum), as described by Duan et al. 13 Each segment was fl ash‐frozen before lipid extraction. A portion of liver and jejunum samples were placed in RNAlater (Ambion) and stored at −80°C for mRNA expression analysis.
Plasma decay curves for both tracers were normalized to radioactivity at the initial 2‐minute time point After tracer injection. Fractional catabolic rates (FCRs) were calculated from the area under the plasma disappearance curves fitted to a bicompartmental model, using the SAAM II soft ware. The HDL cholesteryl esters pool size, calculated by multiplying the HDL cholesteryl esters concentration by the plasma volume (3.5% of the body weight), was multiplied by the FCR to obtain the absolute production rate (APR). To measure the organ uptake of HDL 3H‐cholesteryl ether, lipid extraction was performed according to the procedure of Bligh and Dyer. 11 Briefly, a 50 mg piece of tissue was homogenized in water, and then lipids were extracted with a mixture of chloroform–methanol 2:1 (vol/vol). The lipid layer was collected, evaporated, resuspended in toluene, and counted in a liquid scintillation counter (LSC).
Organ uptake for each tracer was expressed as a percentage of the injected dose. The injected dose was calculated by multiplying the initial plasma counts (2‐minute time point) by the estimated plasma volume.
Fecal cholesterol and bile acid extraction
Fecal cholesterol and bile acid extraction was performed as previously described, 11 with minor modifications. The total feces collected from 0 to 48 hours were weighed and soaked in Millipore water (1 mL water per 100 mg feces) overnight at 4°C. The following day, an equal volume of absolute ethanol was added and the mixtures were homogenized. To extract the 3H‐cholesterol and 3H‐bile acid fractions, 1 mL of the homogenized samples was combined with 1 mL ethanol, a known amount of 14C‐cholic acid as an internal standard, and 200 μL NaOH. The samples were saponified at 95°C for 1 hour and cooled to room temperature, and then 3H‐cholesterol was extracted two times with 3 mL hexane. The extracts were pooled, evaporated, resuspended in toluene, and then counted in an LSC. To extract 3H‐bile acids, the remaining aqueous portion of the feces was acidified with concentrated HCl and then extracted two times with 3 mL ethyl acetate. The extracts were pooled together, evaporated, resuspended in ethyl acetate, and counted in an LSC.
RNA extraction and gene expression analysis
Tissue for mRNA analysis was homogenized, and RNA was isolated using Trizol reagent according to the manufacturer's instructions. Real‐time quantitative polymerase chain reaction (PCR) assays were performed with an Applied Biosystems 7300 sequence detector. Briefly, 5 μg of total RNA was reverse transcribed with the use of an Applied Biosystems High Capacity cDNA archive kit according to the manufacturer's instructions. Each 25 μL amplification reaction contained 100 ng cDNA, 900 nmol/L forward primer, 900 nmol/L reverse primer, 200 nmol/L fl uorescent probe, and 2 × universal PCR master mix. Primer and probe sequences are available on request. Data were expressed as fold change ± SD versus control and normalized to β‐actin mRNA.
Statistical analysis
Values are presented as mean ± SD. Comparisons between groups were conducted using the Student t test (two tailed). A two‐tailed p‐value of <0.05 was considered statistically significant.
Results
PPARδ agonist GW0742 promotes reverse cholesterol transport in vivo
Treatment with GW0742 (10 mg/kg per day) for 2 weeks in mice increased plasma total cholesterol (70.0 ± 8.8 mg/dL vs. 102.3 ± 9.8 mg/dL, p < 0.0005), HDL cholesterol (55.3 ± 4.0 mg/dL vs. 80.3 ± 7.4 mg/dL, p < 0.0005) as well as non‐HDL cholesterol (14.7 ± 8.1 mg/dL vs. 21.9 ± 3.4 mg/dL, p < 0.01). Triglycerides were unchanged (20.3 ± 8.5 mg/dL vs. 22.8 ± 4.7 mg/dL). As shown in the FPLC lipoprotein profi les in Figure 1 , cholesterol levels were higher not only in HDL but also in VLDL and LDL.
Figure 1.
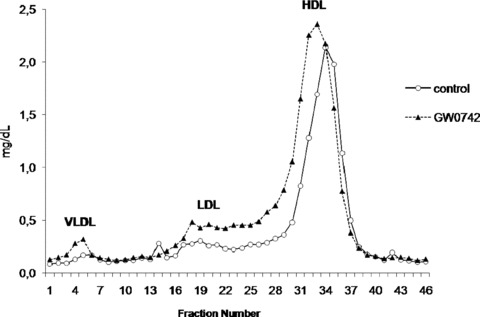
Fast protein liquid chromatography (FPLC) separation of plasma lipoproteins from pooled individual plasma samples from control (full line) and GW0742‐treated (dashed lines) C57BL6/J mice. Cholesterol content in each fraction was measured.
To test the hypothesis that PPARδ activation promotes RCT in vivo, cholesterol‐loaded/3H‐cholesterol‐labeled J774 macrophages were injected intraperitoneally in wild‐type C57BL6/J mice fed with GW0742 or control diet. As shown in Figure 2A , 3H‐tracer present in plasma was unchanged in GW0742‐treated mice. At the same time, we saw a 103% increase (p < 0.005) in the radioactivity recovered After free sterol extraction from the feces ( Figure 2B ). No change was seen in excretion of 3H‐bile acids. These data indicate that PPARδ activation by agonist GW0742 increased fecal free sterol excretion without any major change in 3H‐tracer plasma levels.
Figure 2.
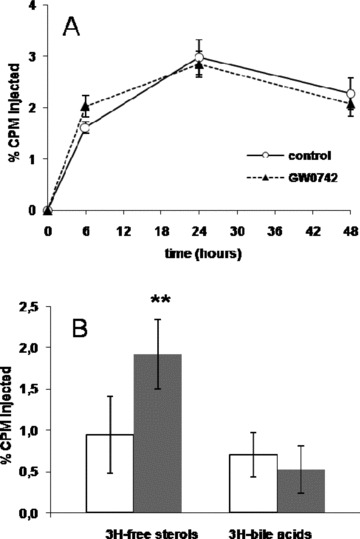
(A) Time course of plasma 3H‐cholesterol distribution over 48 hours in control (full line) and GW0742‐treated (dashed line) C57BL6/J mice. (B) Fecal 3H‐tracer distribution 48 hours after injection of 3H‐cholesterol‐labeled macrophages in control (white bars) and GW0742‐treated (gray bars) C57BL6/J mice (**p < 0.005).
PPARδ agonist GW0742 has no effect on total free cholesterol efflux in vitro
To investigate whether PPARδ activation infl uences the ability of HDL to accept cholesterol effluxed from macrophages, we incubated serum collected from mice fed control diet or diet containing PPARδ agonist GW0742 with labeled bone marrow macrophages. In this experiment ( Figure 3 ), we cholesterol loaded the bone marrow macrophages with acLDL, which is known to stimulate ABCA1 and ABCG1 expression and inhibit SR‐BI expression. Efflux studies were performed in presence/absence of probucol, an ABCA1 inhibitor, to differentiate ABCA1 dependent from ABCA1‐independent efflux. There was a significant 48% increase in ABCA1‐mediated efflux to serum from PPARδ‐treated mice (p < 0.05). There was no change in the non‐ABCA1‐mediated efflux with a resultant nonsignificant trend toward an increase total cholesterol efflux. These data suggest that promotion of macrophage cholesterol efflux by PPARδ is modest relative to the large increase in macrophage RCT seen in vivo.
Figure 3.
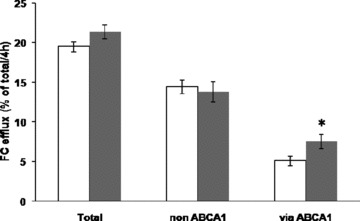
Free cholesterol efflux from C57BL6/J bone marrow macrophages loaded with acLDL was determined in the presence of serum from control mouse (white bars) or GW0742‐treated mouse (gray bars). Probucol (20 μM) was added to obtain non‐ABCA1‐mediated efflux, and ABCA1‐mediated efflux was obtained by subtraction.
PPARδ agonist GW0742 increases the fecal excretion of HDLderived cholesterol despite no effect on HDL cholesterol catabolism
To further characterize the effects of PPARδ agonist GW0742 on RCT, human HDL labeled with 3H‐cholesteryl oleate or 3H‐cholesteryl ether was injected intravenously in wild‐type C57BL6/J mice after 2 weeks of GW0742 treatment (10 mg/kg per day). As shown in Table 1 , there was no effect of PPARδ agonism on the 3H‐cholesteryl oleate HDL FCR. Calculated pool size increased by 28% (p < 0.05) and APR increased by 22% but not significantly. However, in drug‐treated animals, radioactivity recovered in the free sterol fraction was increased by 88% (p < 0.0005), without any change in the bile acids fraction ( Figure 4A ). To further confi rm that PPARδ activation did not affect hepatic and intestinal uptake of HDL cholesteryl ester, a separate group of mice were injected with 3H‐cholesteryl ether‐labeled HDL. Cholesteryl ether, unlike cholesteryl ester, is not metabolized and is trapped in lysosomes once taken up by tissues. As expected, 3H‐cholesteryl ether plasma catabolism was not different between groups ( Table 1 ). Accordingly, hepatic and intestinal uptake was not affected by drug treatment ( Figure 4B ). Intestinal 3H‐tracer uptake was about 15 times lower than hepatic tracer uptake in both groups. Overall, these data indicate that PPARδ activation with the agonist GW0742 increases fecal excretion of HDL‐derived cholesterol without affecting the rate of HDL tissue uptake.
Table 1.
Kinetic parameters in C57BL/6 mice fed a control chow diet or GW0742 diet (10 mg/kg per day) for 2 weeks (*p < 0.05).
| 3H‐cholesteryl oleate FCR (per hour) | 0.136 ± 0.021 | 0.128 ± 0.011 |
| HDL cholesteryl esters pool size (mg/dL) | 34.2 ± 4.8 | 44.0 ± 6.8* |
| 3H‐cholesteryl oleate APR (μg/kg per hour) | 0.046 ± 0.009 | 0.056 ± 0.009 |
| 3H‐cholesteryl ether FCR (per hour) | 0.179 ± 0.026 | 0.168 ± 0.010 |
Figure 4.
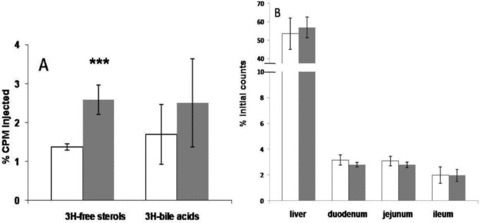
(A) Fecal 3H‐tracer distribution 48 hours after injection of 3H‐cholesteryl oleate‐labeled human HDL in control (white bars) and GW0742‐treated (gray bars) C57BL6/J mice (***p < 0.0005). (B) Uptake of 3H‐cholesteryl ether‐labeled HDL in liver, duodenum, jejunum, and ileum 48 hours after administration of tracer in control (white bars) and GW0742‐treated (gray bars) C57BL6/J mice.
PPARδ agonist GW0742 decreases NPC1L1 mRNA expression in the small intestine
We next analyzed genes involved in hepatic/intestinal secretion and intestinal absorption of cholesterol. Gene expression of ABCA1, ABCG1, and ABCG5 was measured in liver and jejunum by real‐time PCR. As shown in Figure 5A , GW0742 treatment has no effect on ABCA1 and ABCG1 mRNA hepatic expression, but there was a significant increase in ABCG5 mRNA expression (1.7‐fold, p < 0.05) that could result in increased cholesterol secretion into the bile. No significant change was seen for those genes in the small intestine ( Figure 5B ), but expression of NPC1L1 was decreased by 40% (p < 0.05) After GW0742 treatment. The lower expression of NPC1L1 in the intestine suggests that the fecal accumulation of 3H‐free sterols (in vivo RCT and 3H‐cholesteryl oleate kinetic study) may be related to a lower intestinal reabsorption of macrophage/HDL‐derived cholesterol.
Figure 5.
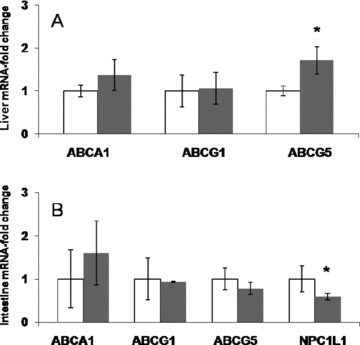
mRNA expression in liver (A) and small intestine (B) from control (white bars) and GW0742‐treated (gray bars) C57BL6/J mice (*p < 0.05).
Blocking NPC1L1 with ezetimibe also promotes macrophageto‐feces reverse cholesterol transport
To directly demonstrate that a reduction of NPC1L1 activity promotes RCT in vivo, we performed experiments in wild‐type mice treated with the cholesterol absorption inhibitor ezetimibe at 5 mg/kg per day for 2 weeks. Ezetimibe treatment slightly decreased total plasma cholesterol (77 ± 4 mg/dL vs. 83 ± 6 mg/ dL, p < 0.05), but triglycerides and HDL cholesterol levels were unchanged (data not shown). Cholesterol‐loaded/3H‐cholesterollabeled J774 macrophages were injected intraperitoneally in wildtype C57BL6/J mice fed a control diet with or without ezetimibe. Ezetimibe had no significant effect on plasma 3H‐tracer over 48 hours ( Figure 6A ). However, the radioactivity recovered in the feces after free sterol extraction was significantly increased by 87% (p < 0.05), while no change occurred in the bile acids fraction ( Figure 6B ) similar to what was observed for PPARδ.
Figure 6.
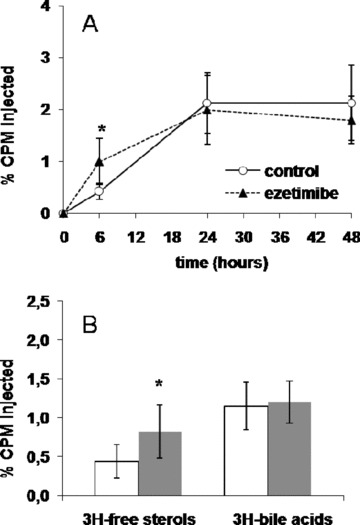
(A) Time course of plasma 3H‐cholesterol distribution over 48 hours in control (full line) and ezetimibe‐treated (dashed line) C57BL6/J mice. (B) Fecal 3H‐tracer distribution 48 hours after injection of 3H‐cholesterol‐labeled macrophages in control (white bars) and ezetimibe‐treated (gray bars) C57BL6/J mice (*p < 0.05).
To investigate whether ezetimibe treatment would affect the excretion of HDL‐derived cholesterol, we also performed HDL cholesteryl ester kinetic studies. Ezetimibe or control‐treated mice were injected with human HDL labeled with 3H‐cholesteryl oleate. Ezetimibe did not change the FCR of HDL labeled with 3H‐cholesteryl oleate (data not shown). However, as shown in Figure 7A , fecal 3H‐free sterols were increased by 109% (p < 0.001), while no significant change was seen in fecal 3H‐bile acids.
Figure 7.
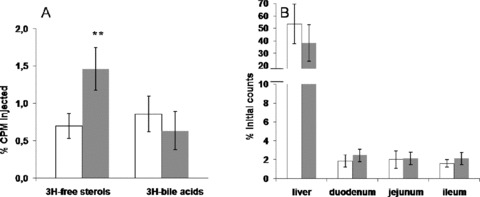
(A) Fecal 3H‐tracer distribution 48 hours after injection of 3H‐cholesteryl oleate‐labeled human HDL in control (white bars) and ezetimibe‐treated (gray bars) C57BL6/J mice (**p < 0.005). (B) Uptake of 3H‐cholesteryl ether‐labeled HDL in liver, duodenum, jejunum, and ileum 48 hours after administration of tracer in control (white bars) and ezetimibe‐treated (gray bars) C57BL6/J mice.
The effect of ezetimibe treatment on HDL cholesteryl ester tissue uptake was also measured by injecting 3H‐cholesteryl etherlabeled human HDL in wild‐type mice. 3H‐cholesteryl ether FCR did not change (data not shown) with ezetimibe treatment, nor was the uptake of 3H‐cholesteryl ether‐labeled HDL in liver ( Figure 7B ). As observed with PPARδ agonism, the direct uptake of 3H‐cholesteryl ether in the small intestine was low (around 2% of the dose injected) and no change was seen in the duodenum, jejunum, and ileum parts ( Figure 7B ). Overall, these data emphasize that blocking NPC1L1 with ezetimibe promotes fecal excretion of macrophage and HDL‐derived cholesterol without any direct effect on HDL cholesterol catabolism or tissue uptake.
Discussion
The present study shows that activation of PPARδ with the selective agonist GW0742 promotes macrophage‐to‐feces RCT by promoting the fecal excretion of HDL‐derived cholesterol. Although we cannot exclude possible contributions of increased ABCA1‐mediated macrophage efflux and increased ABCG5‐mediated biliary secretion of cholesterol, reduced expression of NPC1L1 appeared to be the main driving mechanism. This is supported by studies with ezetimibe, a specific inhibitor of NPC1L1, showing a similar effect on macrophage RCT and fecal excretion of HDL‐derived cholesterol. These studies suggest that reduced NPC1L1 activity through direct inhibition with ezetimibe or downregulation with a PPARδ agonist promotes the fecal excretion of HDL‐derived cholesterol and thus the overall pathway of macrophage RCT.
We first directly demonstrated that PPARδ activation promotes macrophage to feces RCT in vivo. Accordingly, the radioactivity recovered into the fecal free sterols fraction after 3H‐cholesterollabeled macrophage injection was significantly higher in PPARδ agonist GW0742‐treated mice. Because this increase was not associated with a major change in 3H‐tracer plasma level, we then investigated whether serum from GW0742‐treated mice would promote macrophage free cholesterol efflux by using mouse bone marrow macrophages. We saw a significant 48% increase in ABCA1‐mediated efflux when cells were incubated with acLDL ( Figure 3 ). In the present study, macrophages were not pretreated with the PPARδ agonist GW0742. However, some others also showed that GW0742‐treated DBA/1 mice have higher expression of ABCA1 at the peritoneal macrophage level. 9 As well, PPARδ promotes ABCA1 and apolipoprotein AI‐specific efflux in THP1 macrophages. 4 Overall, these data suggest that PPARδ activation may increase macrophage to serum efflux capacity as well as ABCA1 expression. In the present study however, efflux capacities of serum from GW0742‐treated mice were not statistically different from control‐treated mice despite the 48% increase in ABCA1‐mediated efflux. It is also interesting to note that PPARδ activation in human macrophage increases ABCA1 gene expression, but other crucial genes involved in the efflux process, such as apolipoprotein E and cholesterol 27‐hydroxylase, have dramatically lower expression. 8 These changes actually result in macrophages lipid accumulation. 8 This harmful effect has also been suggested in LDL receptor knockout mice where the same agonist GW0742 was unable to prevent atherosclerosis in vivo. 7 Overall, these results indicate that PPARδ activation modulates some aspects of macrophage lipid homeostasis (including ABCA1‐mediated cholesterol efflux) but may not be effective in promoting overall macrophage cholesterol efflux.
On the basis of these data, we next investigated plasma removal of HDL cholesterol using 3H‐cholesteryl oleate or 3H‐cholesteryl ether‐labeled HDL kinetics. Kinetic parameters show that PPARδ activation has no effect on either production or catabolism of HDL cholesteryl esters ( Table 1 ). Moreover, HDL cholesteryl ester uptake, assessed using 3H‐cholesteryl ether‐labeled HDL, was practically unchanged in both liver and small intestine. Hence, these data indicate that plasma removal of HDL cholesterol is not modified under PPARδ activation. After 3H‐cholesteryl oleatelabeled HDL however, there was still an 88% increase in the radioactivity recovered in the fecal free sterols fraction, indicating that some changes could occur at the biliary or intestinal levels. In the liver, there was an unexpected significant increase of the ABCG5 mRNA expression, which suggests that biliary cholesterol excretion could increase under PPARδ activation but this was not measured in our study. Biliary cholesterol secretion rate has previously been suggested to be unaffected by the agonist GW0742 in mice. 9 At the intestinal level, we measured a lower expression of NPC1L1, the target of cholesterol absorption inhibitor ezetimibe. 14 Activation of PPARδ has previously been shown to decrease NPC1L1 expression and intestinal cholesterol absorption in mice. 9 Consequently, cholesterol derived from macrophage and HDL, once secreted into the intestinal lumen, may be less reabsorbed in the small intestine, thereby increasing the loss of free sterols into the feces.
To confirm this, we then performed in vivo experiments in mice treated with the cholesterol absorption inhibitor ezetimibe. Similar to PPARδ activation, ezetimibe had no effect on HDL metabolism and tissue uptake but cholesterol absorption inhibition resulted in a similar increase (80–100%) in fecal free sterols excretion without affecting bile acid excretion. While this manuscript was in preparation, Sehayek and Hazen briefly reported that inhibition of cholesterol absorption with ezetimibe increased radiolabeled cholesterol fecal excretion from peripheral tissue macrophage. 15 Similar to our study, the authors observed an increased fecal excretion of cholesterol despite no change in plasma tracer appearance. However, the extent of the increase (6‐fold) in fecal total steroid excretion (cholesterol plus bile acids) was much larger. A complete block of absorption would be expected to result in about a 2.5‐fold increase (assuming 60% cholesterol absorption). The reason for the much larger increase is not immediately apparent but may be methodological. The authors used radiolabeled sitostanol as an internal standard and calculated the percentage of fecal excretion as the ratio of cholesterol to sitostanol in the feces divided by the same ratio in the macrophage. 15 Macrophages loaded with cholesterol acLDL for 24 hours contain about 50% of their cholesterol as ester. The sitostanol was incubated briefly with cholesterol—loaded macrophages immediately prior to injection and would be unesterified. Since only free sterol is available for efflux from the macrophage, this might account for the diff erence in the extent of observed increase seen in their study. Also, the plasma turnover of sitostanol is more rapid. 16 Finally, in their studies the macrophages were delivered by subcutaneous injection as opposed to intraperitoneal injection. Overall, the effects of ezetimibe and PPARδ activation thus support that targeting NPC1L1 significantly promotes RCT. Activation of LXR 17 and PPARα 18 also reduces intestinal cholesterol absorption as well as NPC1L1 expression, and both of these may promote RCT at least in part through this mechanism.
Some other studies recently demonstrated an alternative route for RCT, which involves the direct excretion of plasma‐derived cholesterol via the intestine. 19 , 20 , 21 , 22 This mechanism has been suggested to be stimulated in mice by PPARδ activation, without any accelerated intestinal cell turnover or cholesterol synthesis. 9 Our present results indicate that this mechanism is not involving HDL cholesteryl ester uptake, which was not different in the intestine. Moreover, intestinal 3H‐cholesteryl ether uptake was about 15 times lower than the hepatic route ( Figure 4B ). Because intestinal cells are replaced every 3 days in mice, 23 we also checked the 3H‐cholesteryl ether recovery in fecal homogenate (data not shown) but this represented only about 0.4% of the dose injected and no significant change was observed in any case. Our data therefore indicate that this alternative pathway for RCT does not directly involve HDL cholesteryl esters. Recent studies actually suggest that unesterified cholesterol secreted by the liver could follow this nonbiliary route. 21 Whether this occurs upon PPARδ activation requires further studies, but the fact that unesterified cholesterol is rapidly exchanged among different types of lipoproteins would make the investigation more difficult. 22 However, the present study strongly suggests that the reduced intestinal absorption of macrophage or HDL‐derived cholesterol may be responsible for the higher fecal free sterols excretion induced by PPARδ agonism.
An important question that remains unanswered is the mechanism by which PPARδ agonism increases HDL cholesterol levels. Expression of ABCA1 in the liver and the small intestine significantly aff ects HDL cholesterol in mice. 24 , 25 In our study however, PPARδ activation did not upregulate ABCA1 expression in either liver or small intestine. Higher ABCA1 expression could occur in the macrophage 9 and muscle, 5 but their subsequent contribution to HDL cholesterol levels is likely to be limited for macrophage ABCA1 26 and remains unknown for muscle ABCA1. We also observed that PPARδ agonism did not change the HDL cholesteryl esters kinetics (neither production nor catabolism) and HDL cholesterol–total cholesterol ratio, which would explain the lack of effect on HDL metabolism. Moreover, GW0742 treatment in mice increases plasma cholesterol levels in all lipoprotein fractions and not only through HDL particles, as shown in the FPLC profi les ( Figure 1 ). The mechanism involved in plasma HDL cholesterol and total cholesterol increase thus remains to be established.
Conclusion
We demonstrated that PPARδ agonist GW0742 promotes macrophage‐to‐feces RCT by reducing NPC1L1 expression rather than by stimulating macrophage cholesterol efflux. The effects of PPARδ activation on macrophage RCT are very similar to those of ezetimibe. Whether this mechanism of RCT promotion is itself antiatherosclerotic remains to be determined.
Disclosure
Colin Macphee and Max Walker are full‐time employees of GlaxoSmithKline.
Rader has served as a consultant too and has research funding from GlaxoSmithKline, Merck, Merck/Schering‐Plough, and Schering‐Plough.
Acknowledgments
We are indebted to Dawn Marchadier, Aisha Wilson, Edwige Edouard, David Chang, Michelle Joshi, Anna DiFlorio, and Linda Morrell for their excellent technical assistance. This work was supported by P01‐HL22633 and P01‐HL59407 from the National Heart, Lung, and Blood Institute, and an Alternative Drug Discovery Initiative award to the University of Pennsylvania from GlaxoSmithKline.
References
- 1. Duval C, Chinetti G, Trottein F, Fruchart JC, Staels B. The role of PPARs in atherosclerosis. Trends Mol Med. 2002; 8: 422–430. [DOI] [PubMed] [Google Scholar]
- 2. Staels B, Fruchart JC. Therapeutic roles of peroxisome proliferator‐activated receptor agonists. Diabetes. 2005; 54: 2460–2470. [DOI] [PubMed] [Google Scholar]
- 3. Leibowitz MD, Fiévet C, Hennuyer N, Peinado‐Onsurbe J, Duez H, Bergera J, Cullinan CA, Sparrow CP, Baffic J, Berger GD, Santini C, Marquis RW, Tolman RL, Smith RG, Moller DE, Auwerx J. Activation of PPARδ alters lipid metabolism in db/db mice. FEBS Lett. 2000; 473: 333–336. [DOI] [PubMed] [Google Scholar]
- 4. Oliver WR Jr, Shenk JL, Snaith MR, Russell CS, Plunket KD, Bodkin NL, Lewis MC, Winegar DA, Sznaidman ML, Lambert MH, Xu HE, Sternbach DD, Kliewer SA, Hansen BC, Willson TM. A selective peroxisome proliferator‐activated receptor delta agonist promotes reverse cholesterol transport. Proc Natl Acad Sci USA. 2001; 98: 5306–5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sprecher DL, Massien C, Pearce G, Billin AN, Perlstein I, Willson TM, Hassall DG, Ancellin N, Patterson SD, Lobe DC, Johnson TG. Triglyceride: high‐density lipoprotein cholesterol effects in healthy subjects administered a peroxisome proliferator activated receptor delta agonist. Arterioscler Thromb Vasc Biol. 2007; 27: 359–365. [DOI] [PubMed] [Google Scholar]
- 6. Tanaka T, Yamamoto J, Iwasaki S, Asaba H, Hamura H, Ikeda Y, Watanabe M, Magoori K, Ioka RX, Tachibana K, Watanabe Y, Uchiyama Y, Sumi K, Iguchi H, Ito S, Doi T, Hamakubo T, Naito M, Auwerx J, Yanagisawa M, Kodama T, Sakai J. Activation of peroxisome proliferator‐activated receptor delta induces fatty acid beta‐oxidation in skeletal muscle and attenuates metabolic syndrome. Proc Natl Acad Sci USA. 2003; 100: 15924–15929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li AC, Binder CJ, Gutierrez A, Brown KK, Plotkin CR, Pattison JW, Valledor AF, Davis RA, Willson TM, Witztum JL, Palinski W, Glass CK. Differential inhibition of macrophage foam‐cell formation and atherosclerosis in mice by PPARalpha, beta/delta, and gamma. J Clin Invest. 2004; 114: 1564–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vosper H, Patel L, Graham TL, Khoudoli GA, Hill A, Macphee CH, Pinto I, Smith SA, Suckling KE, Wolf CR, Palmer CN. The peroxisome proliferator‐activated receptor delta promotes lipid accumulation in human macrophages. J Biol Chem. 2001; 276: 44258–44265. [DOI] [PubMed] [Google Scholar]
- 9. Van Der Veen JN, Kruit JK, Havinga R, Baller JF, Chimini G, Lestavel S, Staels B, Groot PH, Groen AK, Kuipers F. Reduced cholesterol absorption upon PPARdelta activation coincides with decreased intestinal expression of NPC1L1. J Lipid Res. 2005; 46: 526–534. [DOI] [PubMed] [Google Scholar]
- 10. Favari E, Zanotti I, Zimetti F, Ronda N, Bernini F, Rothblat GH. Probucol inhibits ABCA1‐mediated cellular lipid efflux. Arterioscler Thromb Vasc Biol. 2004; 24: 2345–2350. [DOI] [PubMed] [Google Scholar]
- 11. Naik SU, Wang X, Da Silva JS, Jaye M, Macphee CH, Reilly MP, Billheimer JT, Rothblat GH, Rader DJ. Pharmacological activation of liver X receptors promotes reverse cholesterol transport in vivo. Circulation. 2006; 113: 90–97. [DOI] [PubMed] [Google Scholar]
- 12. Tietge UJ, Maugeais C, Cain W, Grass D, Glick JM, De Beer FC, Rader DJ. Overexpression of secretory phospholipase A(2) causes rapid catabolism and altered tissue uptake of high density lipoprotein cholesteryl ester and apolipoprotein A‐I. J Biol Chem. 2000; 275: 10077–10084. [DOI] [PubMed] [Google Scholar]
- 13. Duan LP, Wang HH, Ohashi A, Wang DQ. Role of intestinal sterol transporters Abcg5, Abcg8, and Npc1l1 in cholesterol absorption in mice: gender and age effects. Am J Physiol Gastrointest Liver Physiol. 2006; 290: G269–276. [DOI] [PubMed] [Google Scholar]
- 14. Garcia‐Calvo M, Lisnock J, Bull HG, Hawes BE, Burnett DA, Braun MP, Crona JH, Davis HR Jr, Dean DC, Detmers PA, Graziano MP, Hughes M, Macintyre DE, Ogawa A, O’neill KA, Iyer SP, Shevell DE, Smith MM, Tang YS, Makarewicz AM, Ujjainwalla F, Altmann SW, Chapman KT, Thornberry NA. The target of ezetimibe is Niemann‐Pick C1‐like 1 (NPC1L1). Proc Natl Acad Sci USA. 2005; 102: 8132–8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sehayek E, Hazen SL. Cholesterol absorption from the intestine is a major determinant of reverse cholesterol transport from peripheral tissue macrophages. Arterioscler Thromb Vasc Biol. 2008; 28: 1296–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ostlund RE, McGill JB, Zeng CM, Covey DF, Stearns J, Stenson WF, Spilburg CA. Gastrointestinal absorption and plasma kinetics of soy delta‐(5)‐phytosterols and phytostanols in humans. Am J Physiol Endocrinol Metab. 2002; 282: E911–916. [DOI] [PubMed] [Google Scholar]
- 17. Duval C, Touche V, Tailleux A, Fruchart JC, Fievet C, Clavey V, Staels B, Lestavel S. Niemann‐Pick C1 like 1 gene expression is down‐regulated by LXR activators in the intestine. Biochem Biophys Res Commun. 2006; 340: 1259–1263. [DOI] [PubMed] [Google Scholar]
- 18. Valasek MA, Clarke SL, Repa JJ. Fenofibrate reduces intestinal cholesterol absorption via PPARalpha‐dependent modulation of NPC1L1 expression in mouse. J Lipid Res. 2007; 48: 2725–2735. [DOI] [PubMed] [Google Scholar]
- 19. Kruit JK, Plösch T, Havinga R, Boverhof R, Groot PH, Groen AK, Kuipers F. Increased fecal neutral sterol loss upon liver X receptor activation is independent of biliary sterol secretion in mice. Gastroenterology. 2005; 128: 147–156. [DOI] [PubMed] [Google Scholar]
- 20. Kruit JK, Groen AK, Van Berkel TJ, Kuipers F. Emerging roles of the intestine in control of cholesterol metabolism. World J Gastroenterol. 2006; 12: 6429–6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brown JM, Bell TA III, Alger HM, Sawyer JK, Smith TL, Kelley K, Shah R, Wilson MD, Davis MA, Lee RG, Graham MJ, Crooke RM, Rudel LL. Targeted depletion of hepatic ACAT2‐driven cholesterol esterification reveals a non‐biliary route for fecal neutral sterol loss. J Biol Chem. 2008; 283: 10522–10534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Van Der Velde AE, Vrins CL, Van Den Oever K, Kunne C, Oude Elferink RP, Kuipers F, Groen AK. Direct intestinal cholesterol secretion contributes significantly to total fecal neutral sterol excretion in mice. Gastroenterology. 2007; 133: 967–975. [DOI] [PubMed] [Google Scholar]
- 23. De Aguilar‐Nascimento JE. Evaluation of intestinal trophism: review of current methods and techniques. Curr Opin Clin Nutr Metab Care. 2006; 9: 257–262. [DOI] [PubMed] [Google Scholar]
- 24. Brunham LR, Kruit JK, Pape TD, Parks JS, Kuipers F, Hayden MR. Tissue‐specific induction of intestinal ABCA1 expression with a liver X receptor agonist raises plasma HDL cholesterol levels. Circ Res. 2006; 99: 672–674. [DOI] [PubMed] [Google Scholar]
- 25. Singaraja RR, Van Eck M, Bissada N, Zimetti F, Collins HL, Hildebrand RB, Hayden A, Brunham LR, Kang MH, Fruchart JC, Van Berkel TJ, Parks JS, Staels B, Rothblat GH, Fiévet C, Hayden MR. Both hepatic and extrahepatic ABCA1 have discrete and essential functions in the maintenance of plasma high‐density lipoprotein cholesterol levels in vivo . Circulation. 2006; 114: 1301–1309. [DOI] [PubMed] [Google Scholar]
- 26. Haghpassand M, Bourassa PA, Francone OL, Aiello RJ. Monocyte/macrophage expression of ABCA1 has minimal contribution to plasma HDL levels. J Clin Invest. 2001; 108: 1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]