

Abstract
Non-clinical QT-related assays aligned to the pharmaceutical drug discovery and development phases are used in several ways. During the early discovery phases, assays are used for hazard identification and wherever possible for hazard elimination. The data generated enable us to: (i) establish structure–activity relationships and thereby; (ii) influence the medicinal chemistry design and provide tools for effective decision making; and provide structure–activity data for in silico predictive databases; (iii) solve problems earlier; (iv) provide reassurance for compound or project to progress; and (v) refine strategies as scientific and technical knowledge grows. For compounds progressing into pre-clinical development, the ‘core battery’ QT-related data enable an integrated risk assessment to: (i) fulfil regulatory requirements; (ii) assess the safety and risk–benefit for compound progression to man; (iii) contribute to defining the starting dose during the phase I clinical trials; (iv) influence the design of the phase I clinical trials; (v) identify clinically relevant safety biomarkers; and (vi) contribute to the patient risk management plan. Once a compound progresses into clinical development, QT-related data can be applied in the context of risk management and risk mitigation. The data from ‘follow-up’ studies can be used to: (i) support regulatory approval; (ii) investigate discrepancies that may have emerged within and/or between non-clinical and clinical data; (iii) understand the mechanism of an undesirable pharmacodynamic effect; (iv) provide reassurance for progression into multiple dosing in humans and/or large-scale clinical trials; and (v) assess drug–drug interactions. Based on emerging data, the integrated risk assessment is then reviewed in this article, and the benefit–risk for compound progression was re-assessed. Project examples are provided to illustrate the impact of non-clinical data to support compound progression throughout the drug discovery and development phases, and regulatory approval.
This article is part of a themed section on QT safety. To view this issue visit http://www3.interscience.wiley.com/journal/121548564/issueyear?year=2010
Keywords: QT interval prolongation, hazard identification, risk assessment, risk management and mitigation
Introduction
Since the release a decade ago of the draft points to consider document providing guidance on the assessment for the potential for QT prolongation by non-cardiovascular medicinal products, there have been growing regulatory concerns regarding drug-induced sudden deaths associated with the development of a cardiac arrhythmia known as Torsades de Pointes (for review see Redfern et al., 2003; Pugsley et al., 2008). Although it should be acknowledged that drug-induced QT interval prolongation was recognized as a clinical issue well before it became recognized as a regulatory concern (for review, see Redfern et al., 2003; Pugsley et al., 2008). The increased regulatory concern led to the withdrawal from the market of several products and the release of several guidance documents (Anon., 2000; 2005a,b;). Over the last decade, there have been significant scientific and technological advancements that have enabled the pharmaceutical industry to develop and apply strategies at all stages of the pharmaceutical discovery and development process in order to reduce the likelihood of developing novel and innovative medicines carrying such safety liabilities. The objective of this review in the context of the 2008 EPHAR symposium on ‘reducing QT liability and proarrhythmic risk in drug discovery and development’ is to illustrate, via project examples, the impact of non-clinical data to support compound progression throughout the drug discovery and development phases, and regulatory approval.
Evaluation of hERG/QT liabilities in the pharmaceutical discovery and development process
To achieve an effective evaluation of hERG/QT liability in the drug discovery and development process, studies can be aligned and applied to the different phases of pharmaceutical drug discovery and development (Anon., 2000; 2005a; Valentin and Hammond, 2006; 2008; Pugsley et al., 2008). More specifically, the non-clinical safety studies should aim to: (i) identify undesirable pharmacodynamic properties of a substance that may have relevance to its human safety; (ii) evaluate adverse pharmacodynamic and/or pathophysiological effects of a substance observed in toxicology and/or clinical studies; and (iii) investigate the mechanism of the adverse pharmacodynamic effects observed and/or suspected (Anon., 2000). The impact of the studies aimed at assessing hERG/QT liability in relation to these objectives refer primarily to: (i) hazard identification and elimination; (ii) risk assessment; and (iii) risk management and mitigation, respectively (Figure 1).
Figure 1.
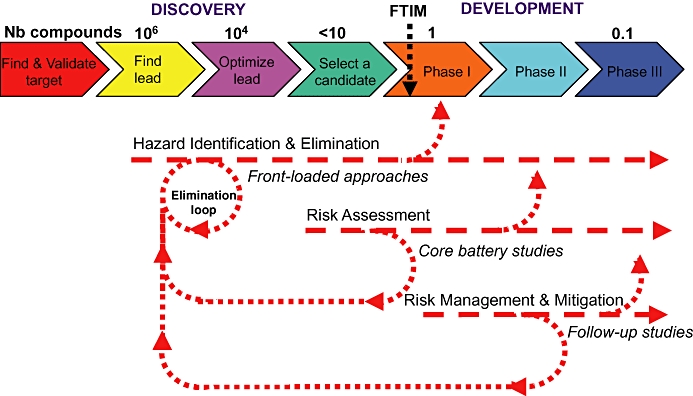
Main objectives of the studies intended at assessing hERG/QT liability and their alignment to the drug discovery and development process. Nb compounds, approximate number of compounds synthesized per project; FTIM, first time in man.
Hazard identification and elimination: front-loaded approaches
Although hazard can be identified at any stage of the pharmaceutical drug discovery and development process, it is most cost-effective to identify and wherever possible to eliminate hazards during the early drug discovery phases. Approaches that are being used should be amenable to the chemistry make-test cycle, by having rapid turn-around time and low compound requirements. Consequently, the approaches used involve primarily in silico paradigms and in vitro assays. The impact of the assays is primarily to: (i) establish structure–activity relationships and thereby; (ii) influence the medicinal chemistry design and provide tools for effective decision making, and provide structure–activity data for in silico predictive databases; (iii) solve problems earlier; (iv) provide reassurance for compound or project to progress; and (v) refine strategies as scientific and technical knowledge grows.
Examples of impact include the utilization of: (i) pathway mapping analysis to interrogate unstructured data and identify potential liabilities; (ii) in silico models to estimate the potency at the hERG channel; (iii) in vitro assays to increase the separation between activity at the primary target for efficacy and the potency at the hERG channel; and (iv) in silico and in vitro hERG data to refine the strategies regarding utilization and alignment of these approaches to the pharmaceutical drug discovery phases. These four examples are detailed below.
Over the recent years, pathway mapping tools have become available to better understand, deconvolute and optimally utilize unstructured data, either proprietary or from the public domain. For example, one can ask the question as to whether there is a potential link between the primary molecular target and the hERG-encoded K channel. If the answer is yes, the project team may consider assessing the potential QT liability of new chemical entities (NCEs) early in the discovery process. In silico models have been developed and applied to estimate the potency at the hERG channel, enabling millions of virtual compounds to be screened, thus streamlining chemistry to avoid synthesizing compounds likely to be potent hERG blockers (Gavaghan et al., 2007; Lesson and Springthorpe, 2007). Correlation between predicted and measured hERG potencies is usually good, although it may vary from one chemical series to another. Medicinal chemists can now modulate physico-chemical properties known to be associated with increased (dipole moment, positive charges, lipophilicity, size, tertiary amine, aromatic amine) or decreased (polar surface area, negative charges/anion, hydrogen bond donors) hERG potency (Gavaghan et al., 2007; Lesson and Springthorpe, 2007). Historically, one of the limitations of developing in silico tools has been the lack of large and consistent databases. Several companies have expended considerable resources creating proprietary databases focusing on different areas of chemical/pharmacological space so that in silico tools can now be applied more effectively (Armstrong et al., 2008). In vitro assays to assess drug effects on the hERG channel (Bridgland-Taylor et al., 2006) are used during the lead identification and optimization phases in order to establish SAR and therefore influence the medicinal chemistry design and provide tools for effective decision making. Examples of SAR showing increased separation over time between activities at the primary target and hERG resulting in increased safety index are illustrated in Figure 2. Separation was obtained by reducing hERG potency while maintaining the activity at the primary target, or by increasing the potency at the primary target while maintaining low hERG potency. As our scientific understanding and technical capabilities developed, the positioning and utilization of in silico and in vitro models to assess drug effects on the hERG-encoded channel have evolved as illustrated in Figure 3. For example, the manual patch clamp technique used in the late 1990s to screen NCEs against hERG has been superseded by medium-throughput, semi-automated assays (Bridgland-Taylor et al., 2006). Nowadays, the manual patch clamp is only used for good laboratory practice regulatory studies. As the confidence in in silico models increases, one can reasonably ask the question whether they will eventually supersede wet biology testing.
Figure 2.
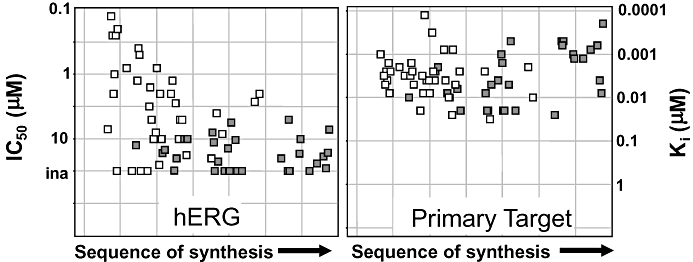
Hazard identification and elimination: Examples of structure activity relationship showing increased separation between activities at the primary target and hERG resulting in increased safety index. Separation was achieved by either reducing hERG potency while maintaining the potency at the primary target (open squares), or by increasing the potency at the primary target while maintaining low hERG potency (filled squares). Each square represents a different compound.
Figure 3.
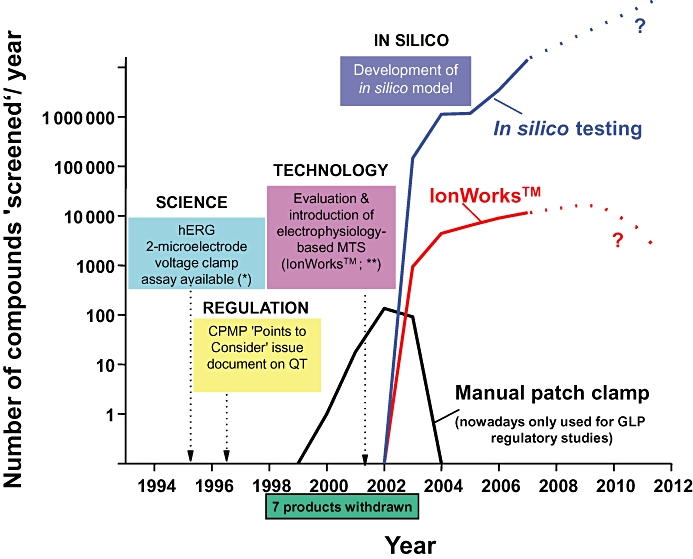
A company perspective illustrating the evolution of the life cycle of IKr/hERG-related assays and associated screening strategies. The manual patch clamp technique used in the late 1990s to screen NCEs against hERG has been superseded by medium-throughput, semi-automated assays (i.e. IonWorks). Nowadays, the manual patch clamp is only used for good laboratory practice (GLP) regulatory studies. As the confidence in in silico models increases, one can reasonably interrogate whether they will eventually supersede wet biology testing. The * refers to Sanguinetti et al. (1995), while ** refers to Schroeder et al. (2003).
Risk assessment: ‘core battery’ studies
Once an NCE is identified to progress into pre-clinical development, ‘core battery’ studies (Anon, 2000; 2005a;) are conducted and data are used to build an integrated risk assessment for the cardiovascular system with a focus on detecting adverse effects, such as QT-interval prolongation. These studies are primarily, but not exclusively, conducted in relevant animal models. The data generated are used in several ways to: (i) fulfil regulatory requirements; (ii) assess the safety and risk–benefit for compound progression to man; (iii) contribute to defining the starting dose during the phase I clinical trials; (iv) influence the design of the phase I clinical trials; (v) identify clinically relevant safety biomarkers; and (vi) contribute to the patient risk management plan.
Examples of impact of data supporting the safe progression of NCEs through the non-clinical development phases include: (i) establishing an integrated risk assessment for an NCE prior to first administration to humans; and (ii) assessing the potential for delayed effect on QT interval of an NCE following chronic treatment as part of the 1-month dog toxicology study. These examples are further developed below.
Figure 4 summarizes the data set that forms the basis of an integrated risk assessment to assess the liability of an NCE to prolong the duration of the QT interval in relation to the expected therapeutic plasma exposure in man (see section on integrated risk assessment below for further details). In that example, the core battery studies were supplemented by an assessment of the drug's effect on the duration of the cardiac action potential. The recommendation was to thoroughly monitor the duration of the QT interval during the phase I clinical trials, which resulted in the human volunteers data included in Figure 4. The clinical QTc data showed a slight erosion of the safety margin, as assessed by the reduction of the NCE concentration yielding 10% QTc prolongation in humans compared with dogs. Consequently, it was felt that for the intended therapeutic indication and the targeted patient population, the benefit–risk did not warrant progression to patients.
Figure 4.
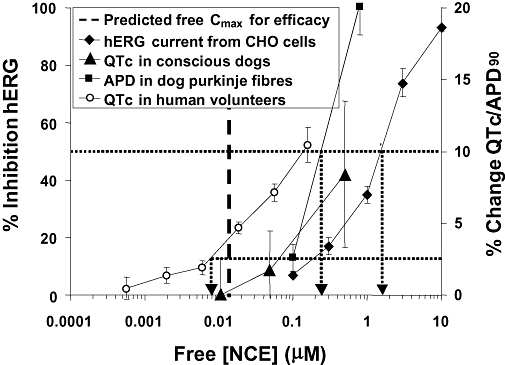
Example of integrated risk assessment to assess the potential of a NCE to prolong the duration of the QT interval in man. APD, action potential duration; QTc, rate corrected QT interval duration in dogs and humans. Black arrows represent unbound (free) plasma concentrations at which 50% hERG block or 10% change in QTc or APD90 occurred. The other arrow shows the free plasma concentration associated with 2.5% change in QTc (i.e. ∼10 ms) in human volunteers. The thick dotted line represents the anticipated free Cmax for efficacy. The recommendation was to thoroughly monitor the duration of the QT interval during the phase I clinical trials, which resulted in the human volunteer data shown. Consequently, it was felt that for the intended therapeutic indication and the targeted patient population, the benefit–risk did not warrant progression to patients.
In another example, the potential for a delayed prolongation of the QT interval duration of an NCE was assessed as part of the 1-month dog toxicology study using a non-invasive jacketed telemetry system (Figure 5). The ECG was recorded once a week for 24 h continuously, and the ECG interval durations were analysed. The NCE dose and time dependently prolonged the duration of the QTc interval. The magnitude of increases in QTc interval duration, almost negligible after 2 days of dosing, became significant after several days of treatment. Consequently, appropriate ECG monitoring was incorporated into the design of the single and most importantly the multiple ascending dose Phase I human clinical trials.
Figure 5.
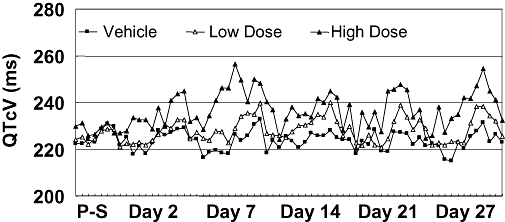
Example of risk assessment to evaluate the liability of an NCE to prolong the QT-interval in a 1-month dog toxicology study using a non-invasive jacketed telemetry system. The ECG was recorded once a week for 24 h continuously on pre-study (P-S) day, and days 2, 7, 14, 21 and 27. The NCE dose and time dependence prolonged the duration of the QTc interval. The magnitude of increases in QTc interval duration, almost negligible at day 2 of dosing, became significant after several days of treatment. Consequently, appropriate ECG monitoring was incorporated into the design of the single and most importantly the multiple ascending dose phase I human studies.
Risk management and mitigation: ‘follow-up’ studies
Once an NCE progresses into clinical development, data can be applied in the context of risk management and risk mitigation. In this context, the studies can be conducted at any stage during the pharmaceutical development process, based on cause for concern that emerges from clinical and/or toxicological studies, literature information and knowledge about the class and/or pharmacology of that specific or similar drugs (Anon., 2000; 2005a;). The data are used in several ways which include, but are not limited to: (i) supporting regulatory approval; (ii) investigating discrepancies that may have emerged within and/or between non-clinical and clinical data; (iii) understanding the mechanism of an undesirable pharmacodynamic effect; (iv) providing reassurance for progression into multiple dosing in humans and/or large-scale clinical trials; and (v) assessing drug–drug interactions. Based on emerging data, the integrated risk assessment is reviewed, and the benefit–risk for NCE progression re-assessed.
Examples of impact of data in support to the safe progression of NCEs through the clinical development phases include: (i) assessing the potential for an additive or synergistic effect on QT interval prolongation of known hERG blocking and QT-prolonging drugs; (ii) identifying and validating potential novel biomarker of drug-induced Torsades de Pointes; (iii) investigating discrepancies between non-clinical and clinical effect on cardiac repolarization to support progression throughout clinical development; and (iv) investigating the pro-arrhythmic potential of an NCE to support registration. These examples are presented below.
A set of experiments was conducted to test the hypothesis that two known hERG blocking and QT-prolonging drugs [ondansetron (Benedict et al., 1996) and a proprietary NCE] could have an additive or synergistic effect on QT interval duration in vivo. In an anaesthetized dog model, two successive dose–response curves to ondansetron were established in the presence of the proprietary NCE or its vehicle. The results (Figure 6) demonstrated an absence of additive or synergistic effect of the two compounds on QT interval duration. Rather, an apparent interference between ondansetron and compound Z, compared with ondansetron and vehicle alone, was noted. Although the mechanistic basis for this outcome is unclear, these empirical findings supported, with careful monitoring in place, the proposed clinical combination.
Figure 6.
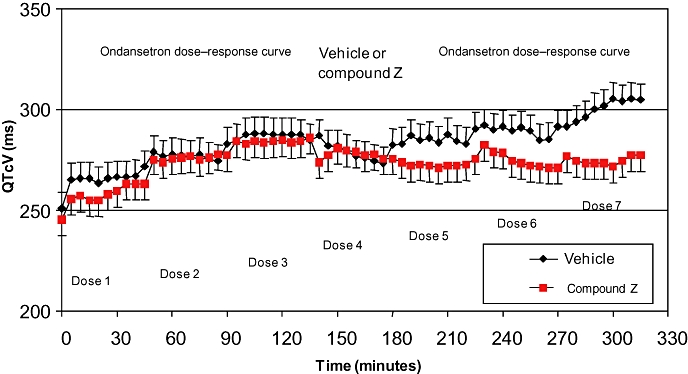
Example of risk management and mitigation to evaluate the in vivo combination of two QT-prolonging drugs. The combination did not lead to an additive or synergistic effect on QT interval duration. Rather, an apparent interference between ondansetron and compound Z, compared with ondansetron and vehicle alone, was noted. Although the mechanistic basis for this outcome is unclear, these empirical findings supported, with careful monitoring in place, the proposed clinical combination. In that study, eight dogs anaesthetized with propofol and alfentanil were divided into two groups of four. Group 1 (red symbols) received three i.v. doses of ondansetron (1, 1.5 and 3 mg·kg−1; doses 1–3) followed by 20.75 mg·kg−1 of compound Z (dose 4) and a repeat of the previous ondansetron doses (doses 5–7). [Note that in a previous study, this dose of compound Z alone had been shown to prolong the QT interval in this model by around 18%]. In group 2 (black symbols), the same sequence of doses was given except dose 4 was the vehicle for compound Z (15% HP-b-cyclodextrin in 1.6% mannitol in water). Parameters were monitored for the 45 min during each intravenous infusion; there was no recovery period between doses. The first ondansetron dose–response curve (doses 1–3) caused a dose-related increase in QTcV in both groups (17% for group 1 and 14% for group 2) relative to the group baseline. A recovery in the increased QTcV was apparent in both groups during vehicle or compound Z infusion (dose 4). Administration of the second ondansetron dose–response curve (doses 5–7) caused further increases in QTcV in group 2; however, no additional increase in QTcV was apparent in group 1.
The duration of the QT interval is used as a surrogate marker of drug-induced pro-arrhythmia, although it is widely recognized that it might be an imperfect marker. Additional electrophysiological indices of pro-arrhythmic risk are being evaluated as potential biomarkers of drug-induced TdP; such markers include, but are not limited to: transmural and spatial dispertion of repolarization, Tpeak−Tend, triangulation, reverse use dependence, instability, early after depolarization and T wave morphology (Lawrence et al., 2008). Furthermore, short-term variability of ventricular repolarization could be a useful biomarker of torsadogenic risk, although additional validation is required. In mid-myocardial canine ventricular myocytes, the short-term variability in APD90 was greater in cells that developed early after depolarizations compared with cells that did not, despite similar degree of APD90 lengthening (Abi-Gerges et al., 2010). The human equivalent of such findings have been described by Hinterseer et al. (2008) showing that patients with history of drug-induced long QT Syndrome had significantly greater short-term variability compared to their respective age and sex-matched controls. Thus, short-term variability of ventricular repolarization could prove to be a useful biomarker of torsadogenic risk, although further validation is required.
Viozan (Sibenadet) is a dual D2 dopamine receptor and β2-adrenoceptor agonist that was developed in the 1990s as an inhaled treatment for chronic obstructive pulmonary diseases. In 32 human volunteers, Viozan (250–750 µg inhaled) dose-dependently increased heart rate, and consequently shortened uncorrected QT interval (ranging from ∼10 to 25 beats per min, and ∼10 to 30 ms placebo subtracted at the low and high dose respectively). When corrected for heart rate, using Bazett, Fridericia or a subject-specific correction formula, QTc was dose-dependently prolonged in all cases (Newbold et al., 2007). Although Fridericia and subject-specific QTc correction resulted in a much reduced QTc prolongation compared with Bazett-corrected QTc, the magnitude of QTc changes was still >10 ms at the lowest, clinically relevant, dose tested (Newbold et al., 2007). Nowadays, such a magnitude of change in QTc would be considered as ‘positive’ in the context of a ‘thorough QT/QTc study’ (Anon, 2005b; Table 1). Furthermore, there had been no report of Torsades de Pointes in patients treated with Viozan, although the number of patients that received Viozan was limited. Retrospectively, we sought to determine whether there was evidence from non-clinical studies that Viozan affected cardiac repolarization (Table 1; Valentin et al., 2006). Viozan, at concentrations up to a significant multiple (i.e. >100-fold) of the clinical free plasma Cmax, failed to inhibit the hERG tail current amplitude or to prolong the duration of the action potential in canine Purkinje fibres or prolong the Van de Water-corrected QTc(VdW) in conscious dogs (either telemetered or restrained) treated once daily for up to 1 year. Furthermore, when QTc(VdW) was plotted against heart rate and logCmax, the slope for each curve was not statistically different from zero. Finally, in rabbit isolated Langendorff hearts, Viozan failed to elicit any prolongation of the action potential duration or overt signs of pro-arrhythmia. Despite prolonging the rate-corrected QT interval in human volunteers, Viozan, at clinically relevant concentrations, did not appear to affect cardiac repolarization in non-clinical models and did not appear to be pro-arrhythmic. The retrospective analysis of the non-clinical studies, conducted during clinical development, contributed to demonstrate the safety profile of Viozan with respect to its effect on cardiac repolarization and pro-arrhythmic potential. The non-clinical data provided reassurance to the product team to proceed with the clinical development of Viozan.
Table 1.
Effect of Viozan and Ranolazine on indices of cardiac repolarization and pro-arrhythmia
| Test system | Species | Viozan | Ranolazine |
|---|---|---|---|
| Cardiac repolarization indices | |||
| in vivo QT/QTc interval* | Human | Positive*** | Positive |
| in vitro IKr/hERG | Human | Negative | Positive |
| in vitro APD** | Dog | Negative | Positive |
| in vivo QT/QTc interval | Dog | Negative | Positive |
| Pro-arrhythmia indices | |||
| Isolated heart | Rabbit | Negative | Negative |
| Wedge preparation | Dog | – | Negative |
| Isolated myocyte | Guinea-pig | – | Negative |
| A-V block in vivo | Dog | – | Negative |
| Torsades de Pointes | Human | Negative | Negative |
Negative: no statistically/biologically significant effect at exposures up to 100-fold the clinically relevant free plasma concentration.
Positive clinically according to the criteria defined by the ICHE14, although neither Viozan or Ranolazine were evaluated in a dedicated thorough QT/QTc study.
Purkinje fibre and epicardial cells for Viozan and Ranolazine respectively. –, not tested.
Irrespective of the correction formulae considered, i.e. Bazett, Fridericia or subject specific. Data extracted from: Valentin and Hammond (2006), Valentin et al. (2006, 2007) and Newbold et al. (2007).
Ranolazine (Raxena) is an anti-anginal agent approved for the treatment of chronic stable angina pectoris for use as a combination therapy when angina is not adequately controlled with other anti-anginal agents (for review, see Valentin et al., 2007). Although the exact mechanism of action of Ranolazine remains unclear, a partial inhibition of fatty acid oxidation and inhibition of the late sodium current, and subsequent reduction in intracellular sodium-dependent calcium overload have been postulated. Nevertheless, its anti-ischaemic and anti-anginal effects do not appear to depend upon changes in blood pressure or heart rate. In patients, Ranolazine has been associated with a slight, but dose-dependent, prolongation of QTc. In contrast to Viozan, Ranolazine was shown to affect cardiac repolarization in non-clinical models (Table 1). However, the effect of Ranolazine on ventricular repolarization was shown to be mechanistically different from that of other hERG blockers that possess pro-arrhythmic effects; in effect, Ranolazine did not exhibit pro-arrhythmic properties in several non-clinical pro-arrhythmia models. Because Ranolazine was shown to possess multi-ion channel blocking properties, including IKr, IKs, ICaL, INa within a clinically relevant concentration range, it can be hypothesized that such pharmacological properties could contribute to its overall non-pro-arrhythmic potential. Non-clinical studies, conducted in parallel to clinical development, contributed to demonstrate the safety profile of Ranolazine with respect to a pro-arrhythmic/torsadogenic potential. The non-clinical data were instrumental in supporting the approval of Ranolazine. Moreover, recent non-clinical studies demonstrated that Ranolazine possess anti-arrhythmic properties, thereby opening the door for its potential use in additional therapeutic indications as an anti-arrhythmic agent.
Integrated risk assessment
The integrated risk assessment is the stepwise and holistic evaluation of non-clinical study results in conjunction with any other relevant information, and should be scientifically based and individualized for an NCE. Such an assessment can contribute to the design of clinical investigations and the interpretation of their findings. The ICH S7B guideline recommends combining the in vitro IKr and in vivo QT data to establish a non-clinical integrated risk assessment prior to administration of the NCE to man (Anon., 2005a).
Risk assessment in terms of protecting Phase I clinical trial participants is relatively straightforward, as it does not take into account any consideration of the therapeutic target (unless the Phase I trials include patients) or the degree of unmet medical need. Therefore, the assessment of the safety data (including cardiac repolarization assays) has to take into consideration the ‘severity’ of the outcome in any given test (see below, bearing in mind the sensitivity and specificity of the assays), and the plasma concentration at which it occurred relative to the expected exposure in the clinical trial. Depending on the stage of drug development, the integrated risk assessment should consider contribution of metabolites, as well as metabolic differences between humans and animals.
In terms of early risk assessment of NCE viability (i.e. as a potential product), the situation is more complex. In an attempt to simplify and standardize how safety data can contribute to early risk assessment of project viability, Redfern et al. (2002) proposed a matrix-type approach described below. This requires a grading process: each of the factors in the risk assessment can be graded into, for example, three categories (low, medium and high). Starting with the outcome of the safety pharmacology tests themselves, they can be categorized as follows: (i) minor – predictive of non-serious, reversible side effects (e.g. minor and transient increase in heart rate); (ii) moderate – predictive of impairment of ‘quality of life’ (e.g. syncope); and (iii) major – predictive of potentially life-threatening effects (e.g. prolonged QT interval, pronounced hypotension). The next step consists of grading the therapeutic target according to disease severity: (i) ‘minor/moderate’ disease (e.g. eczema, rhinitis, Raynaud's syndrome); (ii) ‘debilitating’ disease (e.g. asthma, epilepsy, Parkinson's, stroke, angina); and (iii) ‘life-threatening’ disease (e.g. cancer, AIDS, myocardial infarction). The third component that can be considered is the existing therapy; the NCE must be anticipated to be superior to existing therapy. Therefore, the existing therapy cannot be classified as ‘excellent’; instead, it can be rated as: (i) good; (ii) partially effective with side effects; and (iii) poor/non-existent. Once collected, the set of information can be put together in a matrix that also takes into account the plasma concentration level at which the effects were observed in the non-clinical assays, in comparison to the expected clinical exposure (preferably free plasma concentration), as shown in Table 2. Effects to the left of the line of crosses are acceptable without further debate, those to the right of it are unacceptable and those situated on or near to the line of crosses would require further discussion. Hypothetical examples are presented to illustrate the usefulness of such a matrix (Table 2). In the first example, an NCE was targeted at Raynaud's syndrome – a ‘minor/moderate’ disease for which existing therapy is poor. The NCE is found to block the hERG channel and prolong the QT interval at a relatively low multiple (e.g. ∼10-fold) above the expected therapeutic plasma concentration, which are risk factors for Torsade de Pointes (Redfern et al., 2003; De Bruin et al., 2005). According to the matrix analysis, the decision would be either to discontinue the progression of the NCE into development or to accept embarking on an extensive and expensive clinical programme in compliance with the ICH E14 guidance (Anon., 2005b). At the other end of the spectrum, an NCE is targeted at treating leukaemia, where the existing therapy is partially effective and has marked side effects. The NCE was found to cause QT prolongation at a large multiple (e.g. ∼100-fold) of the therapeutic exposure; this would not be seen as a major issue to progress into clinical development. However, this could become a challenge if the NCE eventually reaches the market, and has to compete with a rival NCE of similar efficacy, but lacking this side effect. Other factors should be considered such as: (i) the target population: for example, syncope as a side effect may be more problematic in elderly patients; and (ii) the ultimate project objective: for example, QT interval prolongation as a side effect may be manageable and acceptable for an NCE aimed at demonstrating proof of mechanism or proof of principle. Overall, the evidence of risk, as part of an integrated risk assessment can support the planning and interpretation of subsequent clinical studies.
Table 2.
QT liability integrated risk assessment matrix
|
Severity of QT liability assessment outcome |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Minor | Moderate | Major | ||||||||
| Therapeutic target | Existing therapy | 100× | 10× | 1× | 100× | 10× | 1× | 100× | 10× | 1× |
| Good | × | |||||||||
| Minor | Partially effective | × | ||||||||
| Poor/none | × | ⇒ | ⇒ | ⇒ | ⇒ |  |
||||
| Good | × | |||||||||
| Debilitating | Partially effective | × | ||||||||
| Poor/none | × | |||||||||
| Good | × | |||||||||
| Life threatening | Partially effective |
 ⇐ ⇐ |
× | |||||||
| Poor/none | × | |||||||||
Outcomes situated to the left of the cross line are acceptable, those outcomes situated to the right are unacceptable, whereas outcomes on or near the line of identity would require further discussion and possibly further investigations. Source: Modified fromRedfern et al. (2002). 100×, 10×, 1× represents the exposure levels at which a particular QT liability outcome is observed, and are fixed at 100, 10 or 1-fold the therapeutic dose/concentration respectively.
Integrated risk assessment may be conducted for any measured parameter, and at its most simplistic, can involve creation of a series of normalized concentration–response curves for the parameter of interest; these can also be related to the predicted or measured effective human plasma concentration (total or free). This is illustrated in the example provided above (Figure 4). The concentration/exposure effect curves of QTc prolongation in human volunteers and conscious dogs, the prolongation of APD90 in canine Purkinje fibres and the inhibition of hERG current measured in CHO cells are presented in Figure 4. The dotted line is drawn at the 50% inhibition of hERG and at the 10% change level, and where it intersects with the data from conscious dogs and dog Purkinje fibres the effective concentration (free) of NCE can be read from the X-axis. The 10% change is used because it reflects the statistical power of these non-clinical models, whereas the 50% hERG inhibition represents the pharmacological measure of the IC50. The thin dotted line is drawn at the 2.5% change in QTc level to illustrate the sensitivity required in the ‘thorough QT/QTc study’ (i.e. ∼10 ms; Anon, 2005b). In this case, where it intersects with data from human volunteers, the effective concentration (free) can again be read from the X-axis. In that example, the clinical QTc data showed a slight erosion of the safety margin, as assessed by the reduction of the NCE concentration yielding 10% QTc prolongation in humans compared to dogs; the mechanism(s) of which is (are) not fully understood. Consequently, it was felt that for the intended therapeutic indication and the targeted patient population, the benefit–risk did not warrant progression to patients. To further increase our confidence in the predictive value of non-clinical models, it is of primary importance to understand correlations between assays/models including human data, such attempts are on-going within individual organizations (e.g. Pfizer; Wallis, 2010) and via consortia efforts (e.g. ILSI-HESI; Trepakova et al., 2009).
Summary and conclusion
Non-clinical data sets related to QT interval prolongation are being generated at all stages of the pharmaceutical drug discovery and development process. The non-clinical assays are used in the context of: (i) hazard identification and elimination, primarily during the early discovery phases; (ii) risk assessment, primarily before first administration to man; and (iii) risk management and mitigation, primarily during clinical development and life cycle management. The outcomes from these assays are currently added to an integrated risk assessment, and used to aid internal and external decision making to provide a rational basis for drug progression in relation to risk assessment, management and mitigation based on an assessment of the risk versus benefit for the intended patient population. Over the last decade, significant scientific and technological advancements have been made that enable the development of NCEs that have a low likelihood to prolong the duration of the QT interval in humans.
However, some significant challenges remain to be addressed, in particular understanding: (i) the incidence, impact and mechanisms underpinning non-hERG-mediated QT prolongation; (ii) the molecular, pharmacological and physiological mechanisms underpinning discrepancies within non-clinical assays and between non-clinical assays and the clinical outcome; (iii) whether non-hERG-mediated mechanisms of QT interval prolongation do carry similar pro-arrhythmogenic risk; and lastly, (iv) the incidence, impact and mechanisms underpinning drug-induced QT interval shortening (see review from Shah, 2010 in this issue). Although significant scientific and technological progresses have been made over the last few years to address the challenges listed above (for review, see e.g. Hoffmann and Warner, 2006), and some are being addressed via consortia efforts (e.g. ILSI-HESI; Trepakova et al., 2009), they do remain a significant limitation to our ability to develop safer medicines.
Acknowledgments
The work described in this review has been made possible through interactions and collaborations with many colleagues from AstraZeneca. The authors wish to thank colleagues from the Safety Pharmacology Departments, within Global Safety Assessment for their contribution over the years; in particular our thanks go to Najah Abi-Gerges, Duncan Armstrong, Russ Bialecki, Joanne Bowes, Matthew Bridgland-Taylor, Clare Draper, Alison Easter, Lorna Ewart, Alex Harmer, Silvana Lindgren, Jackie Moors, Karen Philp, Helen Prior, Will Redfern, Mike Rolf, Jason Schofield, Matt Skinner, Sharon Storey, Ann Woods; and colleagues within AstraZeneca: Scott Boyer, Leif Carlsson, Stewart Davis, Goran Duker, Chris Lawrence, Karina Meachin, Paul Newbold, Kathryn Owen, Brian Springthorpe, Nick Sanders, Stots Reele and Roger Yates.
Glossary
Abbreviations:
- ECG
electrocardiogram
- EPHAR
The Federation of European Pharmacological Societies
- hERG
human ether-a-go-go-related gene
- ICH
International Conference on Harmonization
- NCE
New Chemical Entity
- QT
duration of the QT interval of the electrocardiogram
- QTc
rate corrected duration of the QT interval
- SAR
structure–activity relationship
References
- Abi-Gerges N, Valentin JP, Pollard CE. Beagle dog left ventricular midmyocardial myocytes for in vitro assessment of drug-induced delayed repolarisation Action potential short-term variability may predict the proarrhythmic potential of a drug. Br J Pharmacol. 2010;159:77–92. doi: 10.1111/j.1476-5381.2009.00338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anonymous. ICHS7A: safety pharmacology studies for human pharmaceuticals. 2000. CPMP/ICH/539/00. London, November 16, 2000. [WWW document]. URL http://www.emea.europa.eu/pdfs/human/ich/042302en.pdf[accessed on 23 October 2009.
- Anonymous. ICH S7B: the nonclinical evaluation of the potential for delayed ventricular repolarization (QT interval prolongation) by human pharmaceuticals. 2005a. London, 25 May 2005. CPMP/ICH/423/02. [WWW document]. URL http://www.emea.europa.eu/pdfs/human/ich/053900en.pdf[accessed on 23 October 2009. [PubMed]
- Anonymous. ICH E14: the clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs. 2005b. London, 25 May 2005. CPMP/ICH/2/04. [WWW document]. URL http://www.emea.europa.eu/pdfs/human/ich/000204en.pdf[accessed on 23 October 2009. [PubMed]
- Armstrong D, Migeon J, Rolf MG, Bowes J, Crowford M, Valentin JP. Secondary pharmacodynamic studies and in vitro pharmacological profiling. In: Gad SC, editor. Preclinical Development Handbook: Toxicology. Hoboken, NJ: Wiley Publishers; 2008. pp. 581–610. [Google Scholar]
- Benedict CR, Arbogast R, Martin L, Patton L, Morrill B, Hahne W. Single-blind study of the effects of intravenous dolasetron mesylate versus ondansetron on electrocardiographic parameters in normal volunteers. J Cardiovasc Pharmacol. 1996;28:53–59. doi: 10.1097/00005344-199607000-00009. [DOI] [PubMed] [Google Scholar]
- Bridgland-Taylor MH, Hargreave AC, Orme E, Harmer A, Henthorn A, Ding DC, et al. Optimisation and validation of a medium-throughput electrophysiology-based hERG assay using IonWorks™ HT. J Pharmacol Toxicol Methods. 2006;54:189–199. doi: 10.1016/j.vascn.2006.02.003. [DOI] [PubMed] [Google Scholar]
- De Bruin ML, Pettersson M, Meyboom RH, Hoes AW, Leufkens HG. Anti-HERG activity and the risk of drug-induced arrhythmias and sudden death. Eur Heart J. 2005;26:590–597. doi: 10.1093/eurheartj/ehi092. [DOI] [PubMed] [Google Scholar]
- Gavaghan CL, Arnby CH, Blomberg N, Strandlund G, Boyer S. Development, interpretation and temporal evaluation of a global QSAR of hERG electrophysiology screening data. J Comput Aided Mol Des. 2007;21:189–206. doi: 10.1007/s10822-006-9095-6. [DOI] [PubMed] [Google Scholar]
- Hinterseer M, Thomsen MB, Beckmann BM, Pfeufer A, Schimpf R, Wichmann HE, et al. Beat-to-beat variability of QT intervals is increased in patients with drug-induced long-QT syndrome: a case control pilot study. Eur Heart J. 2008;29:185–190. doi: 10.1093/eurheartj/ehm586. [DOI] [PubMed] [Google Scholar]
- Hoffmann P, Warner B. Are hERG channel inhibition and QT interval prolongation all there is in drug-induced torsadogenesis? A review of emerging trends. J Pharmacol Toxicol Methods. 2006;53:87–105. doi: 10.1016/j.vascn.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Lawrence CL, Pollard CE, Hammond TG, Valentin JP. In vitro models of proarrhythmia. Br J Pharmacol. 2008;154:1516–1522. doi: 10.1038/bjp.2008.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesson PD, Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Discov. 2007;6:881–890. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- Newbold P, Sanders N, Reele SB. Lack of correlation between exercise and sibenadet-induced changes in heart rate corrected measurement of the QT interval. Br J Clin Pharmacol. 2007;63:279–287. doi: 10.1111/j.1365-2125.2006.02763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugsley MK, Authier S, Curtis MJ. Principles in safety pharmacology. Br J Pharmacol. 2008;154:1382–1399. doi: 10.1038/bjp.2008.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redfern WS, Wakefield ID, Prior H, Hammond TG, Valentin JP. Safety pharmacology – a progressive approach. Fundam Clin Pharmacol. 2002;16:161–173. doi: 10.1046/j.1472-8206.2002.00098.x. [DOI] [PubMed] [Google Scholar]
- Redfern WS, Carlsson L, Davis AS, Lynch WG, MacKenzie I, Palethorpe S, et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res. 2003;58:32–45. doi: 10.1016/s0008-6363(02)00846-5. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- Schroeder K, Neagle B, Trezise DJ, Worley J. IonWorks HT: a new high-throughput electrophysiology measurement platform. J Biomol Screen. 2003;8:50–64. doi: 10.1177/1087057102239667. [DOI] [PubMed] [Google Scholar]
- Shah RR. Drug-induced QT interval shortening: potential implications and regulatory perspectives. Br J Pharmacol. 2010;159:58–69. doi: 10.1111/j.1476-5381.2009.00191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trepakova ES, Koerner J, Pettit SD, Valentin JP, HESI Pro-Arrhythmia Committee A HESI consortium approach to assess the human predictive value of non-clinical repolarization assays. J Pharmacol Toxicol Methods. 2009;60:45–50. doi: 10.1016/j.vascn.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Valentin JP, Hammond TG. Safety pharmacology: past, present and future. In: Smith CG, O'Donnell J, editors. The Process of New Drug Discovery and Development. New York, NY, USA: Informa Healthcare; 2006. pp. 243–290. [Google Scholar]
- Valentin JP, Hammond TG. Safety and secondary pharmacology: successes, threats, challenges and opportunities. J Pharmacol Toxicol Methods. 2008;58:77–87. doi: 10.1016/j.vascn.2008.05.007. [DOI] [PubMed] [Google Scholar]
- Valentin JP, Newbold P, Hammond TG. Predictive value of non-clinical models to assess QT liability in man: sibenadet (viozan, AR-C68397AA), a case study. J Pharmacol Toxicol Methods. 2006;54:253. [Google Scholar]
- Valentin JP, Abi-Gerges N, Hammond TG. Ranolazine: non-clinical safety pharmacology studies supporting regulatory approval? In: Sietsema WK, Schwen R, editors. Nonclinical Drug Safety Assessment – Practical Considerations for Successful Registration. Falls Church, VA, USA: FDA News; 2007. pp. 539–555. [Google Scholar]
- Wallis R. Integrated risk assessment and predictive value to humans of non-clinical repolarization assays. Br J Pharmacol. 2010;159:115–121. doi: 10.1111/j.1476-5381.2009.00395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]