

Abstract
Introduction:
Doxorubicin, an anthracycline widely used in the treatment of a broad range of tumours, causes acute QT prolongation. Dexrazoxane has been shown to prevent the QT prolongation induced by another anthracycline, epirubicin, but has not yet been reported to prevent that induced by doxorubicin. Thus, the present study was designed to test whether the acute QT effects induced by doxorubicin could be blocked by dexrazoxane and to explore the mechanism. Results were compared with those obtained with a reference human ether-a-go-go (hERG) channel blocker, moxifloxacin.
Methods:
The effects of moxifloxacin (100 µM) and doxorubicin (30 µM), with or without dexrazoxane (from 3 to 30 µM), have been evaluated on the QTc interval in guinea-pig isolated hearts and on IKr (rapid component of the delayed rectifier current) and IKs (slow component of the delayed rectifier current) currents stably expressed in human embryonic kidney 293 cells.
Results:
Moxifloxacin (100 µM), a potent hERG blocker, prolonged QTc by 22%, and this effect was not prevented by dexrazoxane. Doxorubicin (30 µM) also prolonged QTc by 13%, did not significantly block hERG channels and specifically inhibited IKs (IC50: 4.78 µM). Dexrazoxane significantly reduced the doxorubicin-induced QTc prolongation and prevented doxorubicin-induced inhibition of IKs.
Conclusion and implications:
Doxorubicin acutely prolonged the QT interval in guinea-pig heart by selective IKs blockade. This effect was prevented by dexrazoxane. This result is important because it illustrates the danger of neglecting IKs in favour of hERG screening alone, for early preclinical testing for possible induction of torsade de pointes.
This article is part of a themed section on QT safety. To view this issue visit http://www3.interscience.wiley.com/journal/121548564/issueyear?year=2010
Keywords: doxorubicin, dexrazoxane, moxifloxacin, isolated guinea-pig heart, QT interval, hERG, IKr, KvLQT1/MinK, IKs, ICHS7B guideline
Introduction
Doxorubicin, an anthracycline drug, is widely used for chemotherapy of a broad spectrum of solid tumours and haematological malignancies (Carter and Blum, 1974). Apart from its well-known long-term cardiotoxicity (Saltiel and McGuire, 1983; Yeh et al., 2004), doxorubicin has also been shown to induce ECG abnormalities such as QT interval prolongation and QT dispersion, during chemotherapy (Nousiainen et al., 1999). These doxorubicin-induced electrophysiological abnormalities occur during the initiation of the treatment. Indeed, tachyarrhythmias, ventricular premature beats and QT prolongations are often observed during the 24 h after doxorubicin infusion (Steinberg et al., 1987). Doxorubicin has also been involved in one sudden death in a leukaemic patient presenting with severe hypokalaemia (Lacasse and Bolduc, 1992). Acute effects of doxorubicin on ventricular repolarization have been confirmed in animal studies: doxorubicin prolongs action potential duration in guinea-pig isolated myocytes and papillary muscles and blocks the delayed rectifier potassium current IK in guinea-pig myocytes (Wang and Korth, 1995; Wang et al., 2001). Moreover, doxorubicin lengthens monophasic action potential, QT interval and reduces repolarization reserve in rabbits (Milberg et al., 2007).
Dexrazoxane, clinically used to prevent the delayed doxorubicin-induced cardiotoxicity linked with oxygen radical formation (Imondi et al., 1996; Hasinoff et al., 2003) interestingly prevents the acute QT dispersion induced by another anthracycline, epirubicin, in humans (Galetta et al., 2005).
However, a possible prevention by dexrazoxane of the acute QT prolongation induced by doxorubicin has not been reported in the literature. The present study was designed to test whether the acute QT effects induced by doxorubicin could be blocked by dexrazoxane and to explore the associated mechanism. As doxorubicin-induced prolongation of the cardiac action potential has been linked with a reduction of IK in guinea-pig (Wang and Korth, 1995), we investigated the effects of doxorubicin, associated or not with dexrazoxane on QT prolongation in guinea-pig isolated perfused hearts and on IKs (slow component of the delayed rectifier current) and human ether-a-go-go (hERG) currents, the two components of IK, in transfected human embryonic kidney 293 (HEK-293) cells. The present results showed that dexrazoxane efficiently prevented the doxorubicin-induced QT prolongation and that this effect was associated with a prevention of doxorubicin-induced inhibition of IKs. These results have been confirmed by the lack of effects of dexrazoxane on the QT prolongation induced by a reference hERG channel blocker, moxifloxacin (Chen et al., 2005; Alexandrou et al., 2006).
The present study also highlighted a gap in the ICHS7B guidelines (http://www.ich.org/cache/compo/502-272-1.html#S7B). The recommendations of these guidelines are excessively focused on the hERG channel inhibition whereas the potential proarrhythmic effects of effects on other currents such as IKs are not taken into account.
Methods
All animal care and experimental procedures were performed in accordance with the provisions of the European Convention on the protection of vertebrate animals, which are used for experimental and other scientific purposes and with the Appendices A and B, made at Strasbourg on March 18, 1986 (Belgian Act of October 18, 1991).
Effects of drugs in experiments on isolated Langendorff-perfused hearts from guinea-pigs
The isolated heart preparation was set up as follows. Guinea-pigs weighing around 350 g were anaesthetized with i.p. injection of sodium pentobarbital (90 mg·kg−1, 54.7 mg·mL−1; Ceva Santé Animale, Libourne, France). In addition, heparin (250 IU, Choay; Sanofi-Aventis, Paris, France) was also injected i.p.
A sternotomy followed by a pericardiotomy allowed the heart to be quickly removed. It was rinsed afterwards in physiological saline (NaCl 0.9%) at 4°C. A perfusion cannula was inserted in the aorta for retrograde perfusion. Hearts were mounted in a thermostatic chamber and perfused at a pressure of 60 mmHg with a Krebs–Henseleit solution containing (in mM): 118.1 NaCl, 4.7 KCl, 11.1 glucose, 25 NaHCO3, 1.2 MgSO4, 1.8 CaCl2 and 1.2 KH2PO4 (continuously warmed, 36.5–37.0°C), buffered (pH 7.4) and gassed with 95% O2/5% CO2. The perfusion pump (Ismatec) was regulated by a pressure transducer coupled with a PID (proportional integrator differentiator, Gefran 600). A cannula with a fluid filled balloon was inserted in the left ventricle through the mitral orifice, and the balloon was connected to a pressure transducer for monitoring left ventricular pressure and cardiac frequency. Hearts were allowed to stabilize for at least 45 min before recordings.
The hearts were not electrically stimulated and followed their spontaneous rhythm. Two ECG electrodes were held lightly against the epicardium, one on the apex and the other on the right atria, to generate a bipolar electrocardiogram with well-defined P waves, QRS complex and T waves from which measurements of QT interval were done. Corrected QT intervals in milliseconds (Fridericia, 1920; QTc = QT/RR0.33 and Bazett, 1920; QTc = QT/RR0.5) were calculated to express the QT prolongation independently of the heart rate. ECG signals were digitized at 2 kHz and recorded on a Biopac MP 150 physiological data-acquisition system (Biopac Systems, CA, USA). After the stabilization period, in each group, hearts were first perfused with Krebs–Henseleit solution with distilled water for 5 min (baseline). Drugs were added afterwards in the Krebs–Henseleit solution and perfused during 45 min in all groups. The different groups were: dexrazoxane (30 µM, n= 6), doxorubicin (30 µM, n= 6), doxorubicin (30 µM) + dexrazoxane (3, 10 or 30 µM) perfused simultaneously (n= 6 for each group), moxifloxacin (100 µM, n= 4), moxifloxacin (100 µM) + dexrazoxane (30 µM, n= 4), control group perfused with Krebs–Henseleit solution (n= 10). Measurements were performed on the last 30 s of baseline and of all the following periods on five successive ECG complexes.
Effects of drugs on IKr (rapid component of the delayed rectifier current) stably expressed in HEK-293 cells
The current carried by the hERG channel will be called IKr even if the regulatory subunit of IKr is lacking.
Complementary deoxyribonucleic acid (cDNA) encoding for the human ether-a-go-go related gene product (HERG; GenBank Acc. No. U04270) was cloned into pcDNA3.1-Zeo (Invitrogen, Carlsbad, CA, USA) vector. The plasmid was transfected into HEK-293 cells using Lipofectamin® method in order to establish a clonal cell line using 400 µg·mL−1 Zeocin® (Invitrogen, Carlsbad, CA, USA). The HEK-hERG cells were maintained with 200 µg·mL−1 Zeocin® (Invitrogen, Carlsbad, CA, USA). Protein expression was analysed by means of immunoblotting using anti-HERG antibodies, and functional expression was validated by performing electrophysiological measurements.
Cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) heat inactivated foetal bovine serum in an atmosphere of 95% air/5% CO2. This medium also contained 200 µg·mL−1 Zeocin® and 1% (v/v) penicillin/streptomycin.
Cells used for current recordings were seeded on glass coverslips with the same medium described above without Zeocin® for 1–5 days before use. IKr was recorded using the whole-cell configuration of the patch-clamp technique (Hamill et al., 1981). Pipettes (4–8 MΩ resistance) were drawn from GC150F-15 borosilicate glass capillary tubes (Phymep, Paris, France) and were filled with the following solution (in mM): 130 KCl, 1 MgCl2, 5 EGTA, 10 HEPES and 5 MgATP. The final pH of this intracellular solution was adjusted to 7.20 ± 0.02 with KOH. The composition of the extracellular solution was (in mM): 137 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCL2, 10 HEPES and 10 glucose. The final pH of the extracellular solution was adjusted to 7.40 ± 0.02 with NaOH.
Currents were amplified using a RK300 amplifier (Biologic, Saclay, France) before digitalization to 32 kHz. Stimulation protocols and data acquisition were carried out using a microcomputer (Pentium III), which used commercial software and hardware (Acquis1/Digidata 1200, Biologic, Saclay, France).
The cells were maintained at a holding potential of −80 mV. The following stimulation protocol was successively applied, and the corresponding currents were recorded: IKr was activated by cell depolarization to +30 mV and tail currents occurred at the following repolarization to −40 mV. The amplitude of IKr was calculated as the difference between the peak of the tail current and the initial current recorded before the depolarizing pulse in order to eliminate the leak current. Duration of both depolarization and repolarization pulses were 1 s, and the pulse cycling rate was 20 s. Cells were successively superfused with extracellular solution for stabilization, vehicle during 10 min, three increasing cumulative concentrations of dexrazoxane (1, 10 and 30 µM) or doxorubicin (1, 10 and 30 µM) until steady state, followed by a positive control inhibitor (E-4031, 0.1 µM) to test the viability and the responsiveness of the cells. In other groups, doxorubicin (30 µM) and dexrazoxane (30 µM) were simultaneously superfused, after a 10 min dexrazoxane (30 µM) pretreatment, or immediately after vehicle superfusion. Finally, the spontaneous run-down observed during time-matched experiments was measured in another control group. Each group consisted of five experiments.
Effects of drugs on IKs stably expressed in HEK-293 cells
The human cardiac KCNE1 cDNA (encoding for the subunit minK) that was generously provided by Dr Jacques Barhanin (Institut of Cellular and Molecular Pharmacological, Sophia Antipolis, France) was subcloned into ECO R1/ BamH1 site of pIRES-1-CD8 vector. The human KCNQ1 cDNA (encoding for the subunit KvLQT1) was subcloned into ECO R1/BamH1 site of pIRES-2-eGFP vector (Bioscience Clontech). The vectors of hKCNQ1 (10 µg) and hKCNE1 (1 µg) were cotransfected into HEK-293 cells using calcium/phosphate method in order to establish a clonal cell line. After selection in 1.5 mg·mL−1 geneticin (Sigma Aldrich) for 2 weeks, colonies were picked using cloning cylinders (Sigma Aldrich) and examined for channel expression by whole-cell configuration of patch-clamp technique.
Cells stably transfected with KCNQ1/KCNE1 genes in HEK-293 cells were used for the slow component of the cardiac delayed rectifier potassium current IKs patch-clamp recordings at room temperature. To minimize the spontaneous run-down process, the amphotericin B (Sigma Aldrich) perforated patch-clamp configuration was used to record potassium currents. Pipette (3–5 MΩ resistance) were drawn from GC150T-7.5 borosilicate glass capillary (Phymep, Paris, France) and were filled with the following solution (in mM): 20 KCl, 110 K-aspartate, 1 MgCl2, 5 Na2-phosphocreatine, 10 HEPES and 600 µg·mL−1 amphotericin B (pH adjusted to 7.2 ± 0.02 with KOH). The composition of the extracellular solution was (in mM): 140 NaCl, 5.4 KCl, 1 MgCl2, 1.8 CaCl2, 0.3 NaH2PO4, 5 HEPES and 10 glucose (pH adjusted to 7.3 ± 0.02 with NaOH). Currents were amplified using an Axopatch 200 A amplifier (Axon Instruments). Stimulation protocols and data acquisition were carried out using a microcomputer (Dell Pentium), which used commercial software and hardware (pClamp 8.1/Digidata 1322 A, Axon Instruments). The following stimulation protocol was successively applied, and the corresponding currents were recorded: IKs currents were activated by voltage-clamp steps to membrane potentials of −70 to +100 mV in 10 mV steps applied from a holding potential of −80 mV, and tail currents were generated by the following repolarization to −40 mV. The amplitude of the IKs current was calculated as the peak of the tail current. Duration of both depolarization and repolarization pulses were 2.5 s, and the pulse cycling rate was 15 s. Cells were successively superfused with vehicle until stabilization, then doxorubicin (1 µM, n= 4; 3 µM, n= 6; 10 µM, n= 4; or 30 µM, n= 5) until steady state, followed by a washout period with vehicle. In another group doxorubicin (30 µM) and dexrazoxane (30 µM) were simultaneously superfused, after a 15 min control period and 15 min dexrazoxane (30 µM) pretreatment (n= 4).
Data analysis
In each isolated heart experiment, values were expressed as mean and standard error of the mean (SEM), absolute variations from baseline or percentage variations from baseline. The first analysis was performed on the baseline values to check that, in all groups, the parameters were homogeneous at the beginning of the experiment, followed by an inter-group comparison, performed on absolute variations from baseline using a two-way anova followed by a Bonferroni test if the P interaction (time × treatment) was significant. In this analysis, doxorubicin (30 µM), dexrazoxane (30 µM) and moxifloxacin (100 µM) values were compared with time-matched values observed in the control group. Moreover, values observed in the groups in which dexrazoxane (3, 10 and 30 µM) was superfused with doxorubicin (30 µM) were compared with these observed in the doxorubicin (30 µM) group to evaluate the potent cardioprotective effects of dexrazoxane. In the same way the moxifloxacin (100 µM) + dexrazoxane (30 µM) group was compared with the moxifloxacin (100 µM) group.
In hERG patch-clamp experiments, results were expressed as percentage inhibition of the peak current ± SEM. Absolute variations from baseline observed in the presence of doxorubicin and dexrazoxane were compared with those of time-matched vehicle superfusion using a two-way anova followed by a Bonferroni test if the P interaction (time × treatment) was significant.
In IKs patch-clamp experiments, results were expressed as tail current amplitude (pA) ± SEM, and the mean percentage inhibition of the peak current ± SEM in the presence of compound were also expressed referred to the baseline. In each doxorubicin group, values of tail current amplitude in the presence drug were compared with baseline values, using a paired Student's t-test.
Materials
Dexrazoxane salt and moxifloxacin salt were gratefully received from APT Pharmaceuticals. Dexrazoxane and doxorubicin (Sigma Chemicals, Saint Louis, MO, USA) were dissolved in distilled water as stock solutions (at 1000-fold their respective highest final concentration) and added to the perfusion medium (Krebs–Henseleit solution for isolated heart experiments or extracellular solutions for patch-clamp experiments). In the same way, moxifloxacin was first dissolved in NaOH (0.1 M) as stock solution (at 1000-fold its highest final concentration) and added to the perfusion medium.
Results
Effects on electrophysiological parameters in isolated hearts
ECG parameters are summarized in Table 1. The baseline means of heart rate and QTc (Fredericia's correction) for the 48 hearts used in the present study were respectively 204 ± 3 beats·min−1 and 248 ± 7 ms.
Table 1.
ECG parameters obtained from experiments with of isolated perfused guinea-pig hearts
| Heart rate (beats·min−1) | PR interval (ms) | QRS interval (ms) | RR interval (ms) | QT interval (ms) | QTc interval Fredericia (ms) | QTc interval Bazett (ms) | |
|---|---|---|---|---|---|---|---|
| Vehicle baseline | 209.0 ± 6.7 | 62.6 ± 1.0 | 18.6 ± 0.7 | 285.6 ± 10.1 | 157.9 ± 3.8 | 239.8 ± 3.2 | 295.6 ± 2.6 |
| Vehicle 45 min | 202.0 ± 6.4 | 64.5 ± 1.2 | 19.1 ± 0.9 | 299.9 ± 11.1 | 162.7 ± 4.2 | 243.1 ± 3.6 | 297.3 ± 3.3 |
| Dox 30 µM baseline | 210.9 ± 8.4 | 64.2 ± 1.8 | 21.3 ± 1.4 | 288.0 ± 11.5 | 166.2 ± 3.0 | 251.8 ± 2.4 | 310.1 ± 3.0 |
| Dox 30 µM 45 min | 183.9 ± 5.1** | 67.3 ± 1.6 | 22.0 ± 1.4 | 328.7 ± 9.1 | 195.3 ± 3.0** | 283.1 ± 2.9** | 340.9 ± 3.5** |
| Dex 30 µM baseline | 214.2 ± 7.4 | 65.7 ± 2.2 | 19.5 ± 0.4 | 281.8 ± 9.2 | 155.8 ± 5.4 | 237.6 ± 6.3 | 293.5 ± 7.0 |
| Dex 30 µM 45 min | 207.6 ± 6.9 | 66.3 ± 2.3 | 19.8 ± 0.6 | 290.0 ± 9.6 | 152.7 ± 5.4 | 230.6 ± 6.5** | 283.5 ± 7.4** |
| Dox 30 µM + Dex 3 µM baseline | 204.3 ± 9.2 | 64.0 ± 2.2 | 18.3 ± 0.8 | 301.5 ± 15.0 | 165.0 ± 4.1 | 246.3 ± 2.7 | 301.1 ± 3.0 |
| Dox 30 µM + Dex 3 µM 45 min | 178.3 ± 7.6 | 66.2 ± 2.5 | 18.8 ± 0.9 | 345.0 ± 13.8 | 182.5 ± 3.6† | 260.4 ± 2.7† | 311.1 ± 3.1† |
| Dox 30 µM + Dex 10 µM baseline | 198.1 ± 8.8 | 66.0 ± 1.3 | 22.3 ± 3.7 | 308.2 ± 13.7 | 171.0 ± 2.8 | 253.5 ± 2.5 | 308.7 ± 4.1 |
| Dox 30 µM + Dex 10 µM 45 min | 170.9 ± 2.9 | 68.5 ± 1.8 | 22.5 ± 3.6 | 353.3 ± 3.5 | 191.5 ± 2.6 | 270.9 ± 3.7† | 322.2 ± 4.5 |
| Dox 30 µM + Dex 30 µM baseline | 192.1 ± 9.3 | 67.0 ± 2.0 | 18.5 ± 0.8 | 316.8 ± 17.0 | 173.7 ± 4.0 | 255.1 ± 2.2 | 309.3 ± 2.9 |
| Dox 30 µM + Dex 30 µM 45 min | 174.7 ± 8.0 | 71.0 ± 1.3 | 19.0 ± 0.7 | 347.8 ± 15.0 | 187.0 ± 4.2†† | 266.0 ± 2.5†† | 317.5 ± 2.1†† |
| Mox 100 µM baseline | 204.3 ± 4.3 | 58.0 ± 1.8 | 18.3 ± 1.8 | 295.5 ± 6.7 | 169.5 ± 4.5 | 254.4 ± 5.0 | 311.7 ± 5.2 |
| Mox 100 µM 45 min | 168.0 ± 5.2** | 70.3 ± 3.1 | 20.3 ± 1.8 | 363.0 ± 12.7 | 220.8 ± 5.8** | 309.4 ± 5.1** | 366.4 ± 4.5** |
| Mox100 µM + Dex 30 µM baseline | 191.5 ± 8.3 | 63.5 ± 1.0 | 18.3 ± 0.9 | 301.0 ± 8.1 | 168.8 ± 4.1 | 251.8 ± 4.4 | 307.6 ± 4.7 |
| Mox 100 µM + Dex 30 µM 45 min | 158.3 ± 3.3 | 73.5 ± 2.5 | 20.0 ± 0.7 | 368.5 ± 7.1 | 218.0 ± 4.1 | 304.1 ± 4.4 | 359.1 ± 4.6 |
Values in the Table are means ± SEM from from 10 hearts in the vehicle group, from six hearts in the groups Dox 30 µM, Dex 30 µM, Dox 30 µM + DEX 3 µM, Dox 30 µM + Dex 10 µM and Dox 30 µM + Dex 30 µM, and from four hearts in the Mox 100 µM and Mox 100 µM + Dex 30 µM groups.
P < 0.01 different from the control group.
P < 0.05 different from Dox 30 µM group.
P < 0.01 different from Dox 30 µM group.
Dex, Dexrazoxane; Dox, Doxorubicin; Mox, Moxifloxacin.
The effects of drugs on heart rate are presented in the Figure 1. In the control group, heart rate remained constant during the entire experiment (fall of 3.2 ± 1.5% after 45 min of vehicle superfusion). Doxorubicin (30 µM), perfused during 45 min, induced a significant decrease in heart rate compared with time-matched vehicle perfusion in the control group (P < 0.01). In the same way, moxifloxacin (100 µM), a potent hERG channel inhibitor decreased the heart rate (P < 0.01). Finally, dexrazoxane (30 µM) did not affect the heart rate and did not prevent the bradycardia induced by doxorubicin or by moxifloxacin.
Figure 1.
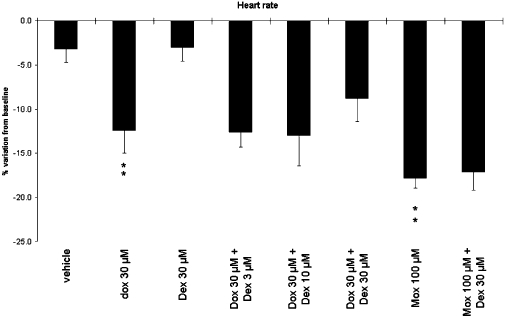
Effects of compounds on heart rate in guinea-pig isolated hearts. Preparations were superfused with Krebs–Henseleit solution (baseline) followed by Krebs–Henseleit solution (control group, n= 10), Dox (30 µM, n= 6), Dex (30 µM, n= 6), Dox (30 µM) + Dex (3, 10 or 30 µM, n= 6 per concentration), Mox (100 µM, n= 4) or Dex (30 µM) + Mox (100 µM, n= 4). Mean values over the 45 min treatment period are shown. Data are expressed as percentage variations from baseline + SEM. **P < 0.01 versus time-matched control group (anova followed by Bonferroni's test performed on absolute variations from baseline). Dex, dexrazoxane; Dox, doxorubicin; Mox, moxifloxacin.
The effects of dexrazoxane, doxorubicin and moxifloxacin have also been evaluated on the QTc interval (Fredericia's correction; Figure 2). In the control group, this parameter remained constant during the entire experiment (45 min of vehicle superfusion). Dexrazoxane (30 µM) superfused alone induced a slight but significant decrease in QTc compared with the control group (P < 0.01). As expected, moxifloxacin (100 µM) significantly lengthened QT interval (P < 0.01). Doxorubicin (30 µM) also induced significant increases in QT interval duration compared with time-matched control group (P < 0.01). Our results also showed that dexrazoxane (30 µM) did not significantly prevent the QTc prolongation induced by moxifloxacin (100 µM).
Figure 2.
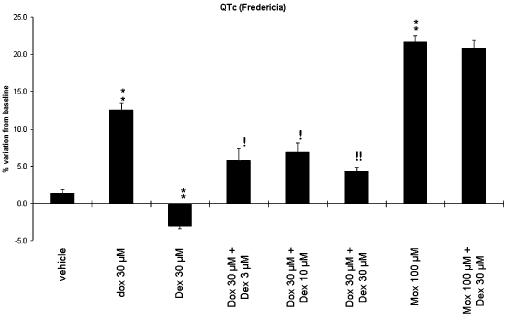
Effects of compounds on QTc (Fredericia's correction) in guinea-pig isolated hearts. Preparations were superfused with Krebs–Henseleit solution (baseline) followed by Krebs–Henseleit solution (control group, n= 10), Dox (30 µM, n= 6), Dex (30 µM, n= 6), Dox (30 µM) + Dex (3, 10 or 30 µM, n= 6 per concentration), Mox (100 µM, n= 4) or Dex (30 µM) + Mox (100 µM, n= 4). Mean values over the 45 min treatment period are shown. Data are expressed as percentage variations from baseline + SEM. **P < 0.01 versus time-matched control group (anova followed by Bonferroni's test performed on absolute variations from baseline). !P < 0.05 and !!P < 0.01 versus time-matched Dox 30 µM group (anova followed by Bonferroni's test performed on absolute variations from baseline). Dex, dexrazoxane; Dox, doxorubicin; Mox, moxifloxacin.
Dexrazoxane from 3 µM, perfused with doxorubicin (30 µM), significantly prevented the doxorubicin-induced QT prolongation [P < 0.05 for dexrazoxane (3 and 10 µM) + doxorubicin (30 µM) compared with doxorubicin (30 µM) perfused alone, P < 0.01 for dexrazoxane (30 µM) + doxorubicin (30 µM) compared with doxorubicin (30 µM) perfused alone].
Effects on IKr
In order to investigate why dexrazoxane prevented doxorubicin- but not moxifloxacin-induced QT lengthening, both dexrazoxane and doxorubicin were tested on IKr and IKs, both expressed in HEK-293 cell lines. Moxifloxacin was not tested on these currents, as its electrophysiological profile is already well defined (Kang et al., 2001; Alexandrou et al., 2006).
The average value of IKr tail current during baseline was 535.7 ± 34.0 pA. Effects of cumulative concentrations of dexrazoxane and doxorubicin on IKr are illustrated in Figure 3. Dexrazoxane only showed a weak but significant effect on IKr compared with the spontaneous rundown observed in the vehicle group: the mean inhibition values were 9.8 ± 1.3%, 17.3 ± 1.8% and 23.8 ± 3.1% in the presence of dexrazoxane (1, 10 and 30 µM) compared with 3.2 ± 1.0%, 6.2 ± 1.1%, 7.1 ± 1.3% over the time-matched period of vehicle perfusion (P < 0.05 at 10 µM and P < 0.01 at 30 µM). The net inhibition values after subtraction of the rundown were then 6.7 ± 1.3%, 11.1 ± 1.8% and 16.7 ± 3.1% in the presence of 1, 10 and 30 µM dexrazoxane respectively.
Figure 3.
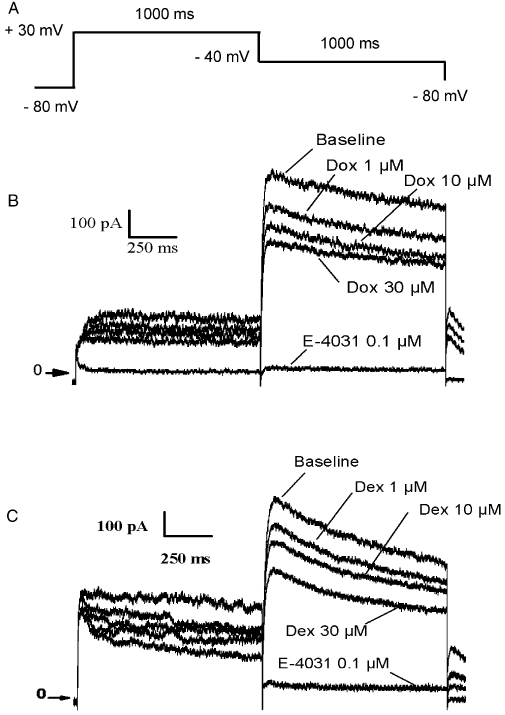
Effects of doxorubicin (Dox) and dexrazoxane (Dex) on the human ether-a-go-go (hERG) current in human embryonic kidney 293 cells. (A) Schematic representation of the pulse protocol (see Methods for details). (B) Whole cell hERG currents recorded from the same cell in the presence of three cumulative concentrations of doxorubicin (1, 10 and 30 µM), followed by E-4031 (0.1 µM) used as a reference control to validate the cell response to hERG inhibition. (C) Whole cell hERG currents recorded from the same cell in the presence of three cumulative concentrations of dexrazoxane (1, 10 and 30 µM), followed by E-4031 (0.1 µM) used as a reference control to validate the cell response to hERG inhibition.
Doxorubicin inhibited the hERG current in the same range as dexrazoxane, by 9.9 ± 1.4%, 18.3 ± 3.3% and 24.4 ± 1.9% at 1, 10 and 30 µM, respectively, compared with the 3.2 ± 1.0, 5.8 ± 1.0 and 7.0 ± 1.3% inhibition observed in the time-matched vehicle groups (P < 0.05 at 1 µM and P < 0.01 at 10 and 30 µM). The net inhibition values after subtraction of the rundown were then 6.8 ± 1.4%, 12.4 ± 3.3% and 17.3 ± 1.9% in the presence of 1, 10 and 30 µM doxorubicin, respectively, suggesting an IC50 value >30 µM.
The simultaneous superfusion of doxorubicin (30 µM) + dexrazoxane (30 µM) induced an inhibition of 20.1 ± 1.4% compared with baseline values. In cells pretreated with dexrazoxane (30 µM), the simultaneous perfusion of the two compounds led to an inhibition of 19.1 ± 1.5% of the hERG current compared with baseline values. These values are in the same range as the others observed with the two compounds superfused alone at 30 µM and clearly indicate no additive blocking effects of the compounds on the hERG channels.
Effects on IKs
The average values of IKs tail current during baseline was 386 ± 33 pA.
We also examined whether IKs could be modulated by doxorubicin. The effects of doxorubicin on the potassium current tracings (IKs) obtained from cell line stably transfected with KCNQ1/KCNE1 channels are illustrated in Figure 4.
Figure 4.
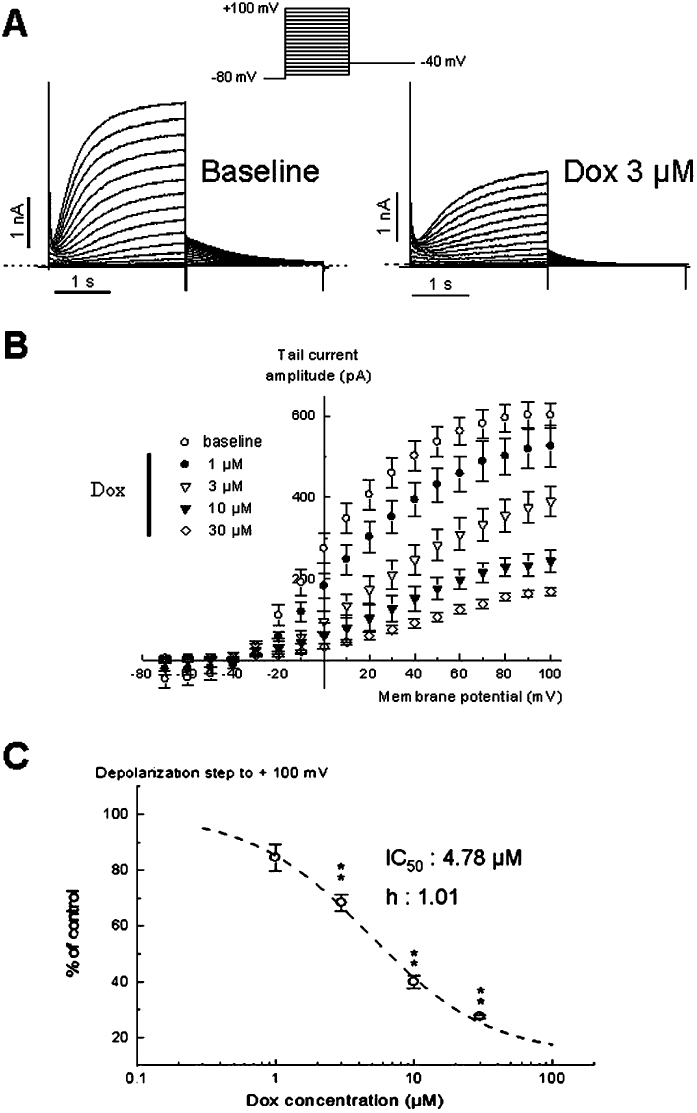
Effect of doxorubicin on IKs current in human embryonic kidney 293 cells. (A) Representative traces illustrating IKs in control and in the presence of 3 µM doxorubicin (Dox). The currents were obtained from the protocol in insert. (B) Tail current amplitude–voltage relationships in control and in the presence of 1, 3, 10 and 30 µM doxorubicin. (C) Concentration–response relationship of IKs inhibition by doxorubicin from 14 cells. The data were fitted with the expression f(x) =[A1 −A2/(1 + (x/IC50)nH)]+ A2, where A1 is the maximal current in control conditions, A2 the current observed at the maximal inhibitory effects of the compound for each concentration and nH (1.01) is the Hill coefficient. From this analysis the IC50 value was 4.78 µM. **P < 0.01 versus baseline (paired Student's t-test).
The drug reduced both IKs amplitude and corresponding tail currents recorded at −40 mV (15%, 32%, 60% and 73% of inhibition at respectively 1, 3, 10 and 30 µM with respect of the baseline IKs amplitude). Moreover, the tail current amplitude–voltage relationship shows that the component inhibited IKs at every potential in a concentration-dependent manner. The concentration–response curve for IKs blockade is illustrated in Figure 4C. The effect was evaluated on the deactivation phase (at −40 mV) of the slow potassium current pre-activated at the potential test of +100 mV. The IC50 value and the Hill coefficient were estimated at 4.78 µM and 1.01, respectively, indicating clearly that doxorubicin was more potent on IKs than on IKr.
To evaluate possible interactions between doxorubicin and dexrazoxane, sequential superfusions of 30 µM dexrazoxane, then 30 µM doxorubicin + 30 µM dexrazoxane were applied (Figure 5). Following the control period, subsequent superfusion of dexrazoxane did not significantly change the tail potassium current amplitude. Interestingly, the simultaneous application of both dexrazoxane and doxorubicin did not induce any modification of the current, contrary to the effect obtained without dexrazoxane (see insert in Figure 5). This result suggests that dexrazoxane prevented the inhibitory effect of doxorubicin on IKs.
Figure 5.
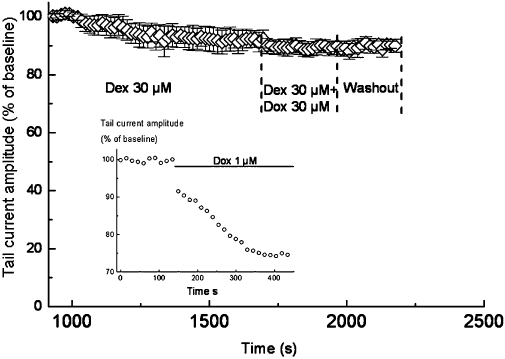
Prevention of doxorubicin (Dox)-induced IKs inhibition by dexrazoxane (Dex). Time course of tail current amplitude (normalized) during the sequential perfusions of vehicle (baseline), 30 µM dexrazoxane, 30 µM doxorubicin + 30 µM dexrazoxane and washout. The tail current amplitudes correspond to the deactivation of the slow potassium current (IKs) during the return to −40 mV after activation at +100 mV. Results are expressed as mean± SEM (n= 4). The insert shows the effects of doxorubicin (1 µM) on the IKs deactivation (pre-activated at +100 mV) in the absence of dexrazoxane, in one of five experiments.
Discussion
The first main finding of the present study was the prevention of doxorubicin-induced QT prolongation by dexrazoxane, in contrast to the lack of effects of dexrazoxane on moxifloxacin-induced QT lengthening.
The effects of moxifloxacin (100 µM) on cardiac repolarization observed in the present study are in full agreement with those already published. Indeed, moxifloxacin is responsible for QT prolongation and for an increase in action potential duration in various animal models (Chen et al., 2005; Wisialowski et al., 2006; Lu et al., 2007). In humans, moxifloxacin has been shown to significantly increase the QT interval duration (Demolis et al., 2000) and has been involved in a case of torsade de pointes arrhythmia (Dale et al., 2007). A major contributor to cardiac repolarization is the delayed rectifier current IK, which can be subdivided into two components, a rapidly activating current, IKr and a slowly activating current, IKs (Li et al., 1996; Cheng and Kodama, 2004). The current carried by the hERG channel corresponds to IKr (Abbott et al., 1999) whereas IKs is expressed by the co-assembly of the subunits KvLQT1 and MinK (Barhanin et al., 1996; Sanguinetti et al., 1996). The adverse effects of moxifloxacin on ventricular repolarization are strongly suspected to be due to this compound inhibiting the hERG potassium current, with IC50 of 65 µM and 129 µM in HEK-293 and Chinese hamster ovary transfected cells respectively (Kang et al., 2001; Alexandrou et al., 2006). Indeed, a good correlation has been observed concerning hERG inhibition and QT prolongation with moxifloxacin when compared with other specific hERG channel blockers such as dofetilide or E-4031 (Chen et al., 2005). Moxifloxacin has also been shown not to interfere with the KvLQT1/minK channel (Kang et al., 2001).
A critical assessment of the literature reveals that the knowledge about the electrophysiological effects of dexrazoxane is still lacking. We showed that dexrazoxane (30 µM) had only slight effects on the hERG current and no effects on IKs. Moreover, we have also demonstrated that dexrazoxane did not prevent moxifloxacin-induced QT prolongation. This result confirmed the lack of significant effects of dexrazoxane on the hERG current. Dexrazoxane was not completely soluble at concentrations higher than 30 µM. Thus, possible effects of higher concentrations of dexrazoxane, on moxifloxacin-induced QT prolongation cannot be excluded
Doxorubicin (30 µM), as moxifloxacin, also prolonged the QTc interval. Even if a reduction of the QT interval has been already observed in guinea-pig isolated hearts (Stark et al., 1990), our results are in agreement with the QT prolongation clinically observed in the 24 h following doxorubicin infusions (Steinberg et al., 1987). Moreover, doxorubicin has been shown to prolong APD90 (action potential duration at 90% of final repolarization) in guinea-pig ventricular myocardium (Wang and Korth, 1995).
Contrary to the lack of effect on moxifloxacin, dexrazoxane significantly prevented the QT prolongation induced by doxorubicin. This discrepancy between effects of dexrazoxane on moxifloxacin and doxorubicin-induced QT prolongations implied different mechanisms involved in the proarrhythmic potential between the two drugs. Prolongation of action potential duration and, by extension, of QT interval may involve different mechanisms: reduction of outward potassium currents such as hERG current or IKs and/or increase in inward sodium or calcium currents. Wang and Korth (1995) have demonstrated that the increase in action potential duration observed in guinea-pig papillary muscle in the presence of doxorubicin did not involve increases in sodium or calcium current, but a reduction of IK. Therefore, based on this result, we put forward the following hypothesis. Doxorubicin prolonged the QT interval, not through a hERG inhibition but through a reduction of the second component of the delayed rectifier potassium current, IKs. Our experiments performed with the patch-clamp technique strengthened this hypothesis. Indeed, in contrast to moxifloxacin, doxorubicin showed only slight effects on hERG, but for the first time, we showed that doxorubicin significantly inhibited the current carried by the combination of KvLQT1 and minK subunits. In addition, dexrazoxane prevented the reduction of IKs induced by doxorubicin. Thus, dexrazoxane might protect the heart from acute doxorubicin-induced QT prolongation by preventing the doxorubicin-induced IKs inhibition. However this explanation remains uncertain, as dexrazoxane did not show a direct effect on IKs. Therefore, further studies are required to support this molecular mechanism.
This interaction between dexrazoxane and doxorubicin may have important clinical applications during the initiation of chemotherapy with doxorubicin. Indeed, the efficient dose ratio dexrazoxane/doxorubicin for the prevention of doxorubicin-induced cardiotoxicity is relatively high, from 10:1 in mice and rat (Imondi et al., 1996). A clinical study has shown, in women with advanced breast cancer, a lower incidence of cardiac events when dexrazoxane (ratio 10:1 and 20:1) was given about 30 min prior to doxorubicin infusion compared with placebo recipients (Wiseman and Spencer, 1998). However, at these concentrations, dexrazoxane is not recommended at the beginning of therapy because of possibly of reducing the anticancer effects of the anthracyclines (Yeh et al., 2004). In our study, dexrazoxane (from 3 µM) significantly prevented the acute QT prolongation induced by doxorubicin (30 µM), corresponding to a ratio of 1:10. A new prevention protocol may then be tested in which lower doses of dexrazoxane would be used in the early phase of the treatment to prevent acute QT prolongation induced by doxorubicin, followed by a gradual increase in dexrazoxane concentration when the chemotherapeutic effects are installed. Moreover, it would be interesting to test the potential cardioprotective effects of dexrazoxane on the reduction of IKs induced by other specific inhibitors, in order to prevent iatrogenic LQT1 syndrome.
Beyond the chemotherapeutic interest of the present study findings, the QT prolongation observed in the presence of doxorubicin and its links to IKs inhibition adds a new example opposed to the recommendations of the ICHS7B guidelines. These guidelines deal with the strategy for the detection of possible torsadogenic effects of new chemical entities, linked with delayed ventricular repolarization. A direct correlation between QT prolongation and torsade de pointes lethal arrhythmias does not exist, as a considerable number of compounds induce QT prolongation but are devoid of torsadogenic risk clinically. Other factors such as spatial dispersion of repolarization and instability are implicated in the genesis of ventricular arrhythmias. However, QT prolongation remains a strong marker of torsadogenic potential in preclinical safety studies. As almost all compounds associated with QT prolongation have been shown to reduce the activity of IKr, the ICHS7B recommendations have been focused on the hERG channel assay. However, other ion channels may be involved in the prolongation of QT interval. Indeed, the congenital long QT syndromes LQT1 and LQT3 are respectively defined by mutations in the genes encoding for the proteins responsible for the currents IKs and INa (sodium current).
Moreover, if specific IKs inhibitors such as HMR1556 have not been associated with QT lengthening and torsade de pointes in humans under normal physiological conditions, IKs is essential in the notion of repolarization reserve. Indeed, due to its slow kinetics of activation, the participation of IKs in human action potential repolarization is minor. However, in situations where action potential duration is prolonged, for example by hERG inhibitors, bradycardia or hypokalaemia, there is a possibility for more IKs activation to allow an important mean of limiting excessive action potential duration lengthening by a negative feedback mechanism (Jost et al., 2007). Moreover, pharmacological block of IKs has been shown to significantly enhance the susceptibility of heart to develop torsade de pointes in the presence of an IKr block (Jost et al., 2005; So et al., 2006). A decrease of the repolarization reserve associated with torsade de pointes has been already observed in the presence of combined doxorubicin and a selective hERG blocker, erythromycin, in rabbits (Milberg et al., 2007). This decrease of repolarization reserve consequent to doxorubicin-induced IKs inhibition may explain the origin of torsade de pointes, which led to the death of one hypokalaemic patient treated with doxorubicin (Lacasse and Bolduc, 1992).
In conclusion, our study clearly demonstrated that doxorubicin prolonged the QT interval in guinea-pig isolated hearts, associated with a reduced amplitude of IKs and that dexrazoxane prevented this proarrhythmic mechanism. This is important because it illustrates the danger of neglecting IKs in favour of hERG screening alone, for early preclinical testing for torsadogenic potential. The hERG channel assay should be complemented with assays on IKs current and on action potential/QT measurements, in species in which IKs participation to repolarization is effective (guinea-pig) or under conditions favouring its expression.
Acknowledgments
The authors thank APT Pharmaceuticals for the gift of dexrazoxane and doxorubicin and for the financial support. The authors also thank Dr Jacques Barhanin for the generous gift of the KCNE1 cDNA.
Glossary
Abbreviations:
- APD90
action potential duration at 90% of final repolarization
- cDNA
complementary deoxyribonucleic acid
- h
Hill coefficient
- HEK-293
human embryonic kidney 293
- hERG
human ether-a-go-go
- IK
delayed rectifier current
- IKr
rapid component of the delayed rectifier current
- IKs
slow component of the delayed rectifier current
- INa
sodium current
- LQT1
long QT type 1
- LQT3
long QT type 3
- Mox
moxifloxacin
- PID
proportional integrator differentiator
Conflicts of interest
This study was funded by APT Pharmaceuticals, and one author (SD) is an employee of this company.
References
- Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, et al. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–187. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- Alexandrou AJ, Duncan RS, Sullivan A, Hancox JC, Leishman DJ, Witchel HJ, et al. Mechanism of hERG K+ channel blockade by the fluoroquinolone antibiotic moxifloxacin. Br J Pharmacol. 2006;147:905–916. doi: 10.1038/sj.bjp.0706678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. KvLQT1 and IsK (minK) proteins associate to form the IKs cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- Bazett HC. An analysis of the time-relations of electrocardiograms. Heart. 1920;7:353–370. [Google Scholar]
- Carter SK, Blum RH. New Chemotherapeutic agents … Bleomycin and Adriamycin. CA Cancer J Clin. 1974;24:322–331. doi: 10.3322/canjclin.24.6.322. [DOI] [PubMed] [Google Scholar]
- Chen X, Cass JD, Bradley JA, Dahm CM, Sun Z, Kadyszewski E, et al. QT prolongation and proarrhythmia by moxifloxacin: concordance of preclinical models in relation to clinical outcome. Br J Pharmacol. 2005;146:792–799. doi: 10.1038/sj.bjp.0706389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JH, Kodama I. Two components of delayed rectifier K+ current in heart: molecular basis, functional diversity, and contribution to repolarization. Acta Pharmacol Sin. 2004;25:137–145. [PubMed] [Google Scholar]
- Dale KM, Lertsburapa K, Kluger J, White CM. Moxifloxacin and torsade de pointes. Ann Pharmacother. 2007;41:336–340. doi: 10.1345/aph.1H474. [DOI] [PubMed] [Google Scholar]
- Demolis JL, Kubitza D, Tennezé L, Funck-Brentano C. Effect of a single oral dose of moxifloxacin (400 mg and 800 mg) on ventricular repolarization in healthy subjects. Clin Pharmacol Ther. 2000;68:658–666. doi: 10.1067/mcp.2000.111482. [DOI] [PubMed] [Google Scholar]
- Fridericia LS. Die Systolendauer im Elektrokardiogramm beinormalen Menschen and bei Herzkranken. Acta Medica Scandinavica. 1920;57:469–486. [Google Scholar]
- Galetta F, Franzoni F, Cervetti G, Cecconi N, Carpi A, Petrini M, et al. Effect of epirubicin-based chemotherapy and dexrazoxane supplementation on QT dispersion in non-Hodgkin lymphoma patients. Biomed Pharmacother. 2005;59:541–544. doi: 10.1016/j.biopha.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hasinoff BB, Schroeder PE, Patel D. The metabolites of the cardioprotective drug dexrazoxane do not protect myocytes from doxorubicin-induced cytotoxicity. Mol Pharmacol. 2003;64:670–678. doi: 10.1124/mol.64.3.670. [DOI] [PubMed] [Google Scholar]
- Imondi AR, Torre PD, Mazué G, Sullivan TM, Robbins TL, Hagerman LM, et al. Dose-response relationship of dexrazoxane for prevention of doxorubicin-induced cardiotoxicity in mice, rats, and dogs. Cancer Res. 1996;56:4200–4204. [PubMed] [Google Scholar]
- Jost N, Virág L, Bitay M, Takács J, Lengyel C, Biliczki P, et al. Restricting excessive cardiac action potential and QT prolongation: a vital role for IKs in human ventricular muscle. Circulation. 2005;112:1392–1399. doi: 10.1161/CIRCULATIONAHA.105.550111. [DOI] [PubMed] [Google Scholar]
- Jost N, Papp JG, Varró A. Slow delayed rectifier potassium current (IKs) and the repolarization reserve. Ann Noninvasive Electrocardiol. 2007;12:64–78. doi: 10.1111/j.1542-474X.2007.00140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Wang L, Chen XL, Triggle DJ, Rampe D. Interactions of a series of fluoroquinolone antibacterial drugs with the human cardiac K+ channel HERG. Mol Pharmacol. 2001;59:122–126. doi: 10.1124/mol.59.1.122. [DOI] [PubMed] [Google Scholar]
- Lacasse Y, Bolduc P. Sudden death in leukemic patient treated with doxorubicin. Can J Cardiol. 1992;8:53–56. [PubMed] [Google Scholar]
- Li GR, Feng J, Yue L, Carrier M, Nattel S. Evidence for two components of delayed rectifier K+ current in human ventricular myocytes. Circ Res. 1996;78:689–696. doi: 10.1161/01.res.78.4.689. [DOI] [PubMed] [Google Scholar]
- Lu HR, Vlaminckx E, Van de Water A, Rohrbacher J, Hermans A, Gallacher DJ. Corrigendum to ‘In-vitro experimental models for the risk assessment of antibiotic-induced QT prolongation.’. Eur J Pharmacol. 2007;577:222–232. doi: 10.1016/j.ejphar.2007.07.070. [DOI] [PubMed] [Google Scholar]
- Milberg P, Fleischer D, Stypmann J, Osada N, Mönning G, Engelen MA, et al. Reduced repolarization reserve due to anthracycline therapy facilitates torsade de pointes induced by IKr blockers. Basic Res Cardiol. 2007;102:42–51. doi: 10.1007/s00395-006-0609-0. [DOI] [PubMed] [Google Scholar]
- Nousiainen T, Vanninen E, Rantala A, Jantunen E, Hartikainen J. QT dispersion and late potentials during doxorubicin therapy for non-Hodgkin's lymphoma. J Intern Med. 1999;245:359–364. doi: 10.1046/j.1365-2796.1999.00480.x. [DOI] [PubMed] [Google Scholar]
- Saltiel E, McGuire W. Doxorubicin (Adriamycin) cardiomyopathy. West J Med. 1983;139:332–341. [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, et al. Coassembly of KvLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- So PP, Hu XD, Backx PH, Puglisi JL, Dorian P. Blockade of IKs by HMR 1556 increases the reverse rate-dependence of refractoriness prolongation by dofetilide in isolated rabbit ventricles. Br J Pharmacol. 2006;148:255–263. doi: 10.1038/sj.bjp.0706721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark G, Stark U, Samonigg H, Kasparek K, Lueger A, Nagl S, et al. Comparison of acute effects of anthracyclines on cardiac electrophysiological parameters of isolated guinea-pig hearts. Cancer Chemother Pharmacol. 1990;26:415–418. doi: 10.1007/BF02994091. [DOI] [PubMed] [Google Scholar]
- Steinberg JS, Cohen AJ, Wasserman AG, Cohen P, Ross AM. Acute arrhythmogenicity of doxorubicin administration. Cancer. 1987;60:1213–1218. doi: 10.1002/1097-0142(19870915)60:6<1213::aid-cncr2820600609>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Wang GX, Wang YX, Zhou XB, Korth M. Effects of doxorubicinol on excitation-contraction coupling in guinea pig ventricular myocytes. Eur J Pharmacol. 2001;423:99–107. doi: 10.1016/s0014-2999(01)01096-2. [DOI] [PubMed] [Google Scholar]
- Wang YX, Korth M. Effects of doxorubicin on excitation-concentration coupling in guinea pig ventricular myocardium. Circ Res. 1995;76:645–653. doi: 10.1161/01.res.76.4.645. [DOI] [PubMed] [Google Scholar]
- Wiseman LR, Spencer CM. Dexrazoxane. A review of its use as a cardioprotective agent in patients receiving anthracycline-based chemotherapy. Drugs. 1998;56:385–403. doi: 10.2165/00003495-199856030-00009. [DOI] [PubMed] [Google Scholar]
- Wisialowski T, Crimin K, Engtrakul J, O'Donnell J, Fermini B, Fossa AA. Differentiation of arrhythmia risk of the antibacterials moxifloxacin, erythromycin, and telithromycin based on analysis of monophasic action potential duration alternans and cardiac instability. J Pharmacol Exp Ther. 2006;318:352–359. doi: 10.1124/jpet.106.101881. [DOI] [PubMed] [Google Scholar]
- Yeh ETH, Tong AT, Lenihan DJ, Yusuf SW, Swafford J, Champion C, et al. Cardiovascular complications of cancer therapy: diagnosis, pathogenesis, and management. Circulation. 2004;109:3122–3131. doi: 10.1161/01.CIR.0000133187.74800.B9. [DOI] [PubMed] [Google Scholar]