

Abstract
Background and purpose:
Cannabis is the source of at least seventy phytocannabinoids. The pharmacology of most of these has been little investigated, three notable exceptions being Δ9-tetrahydrocannabinol, cannabidiol and Δ9-tetrahydrocannabivarin. This investigation addressed the question of whether the little-studied phytocannabinoid, cannabigerol, can activate or block any G protein-coupled receptor.
Experimental approach:
The [35S]GTPγS binding assay, performed with mouse brain membranes, was used to test the ability of cannabigerol to produce G protein-coupled receptor activation or blockade. Its ability to displace [3H]CP55940 from mouse CB1 and human CB2 cannabinoid receptors and to inhibit electrically evoked contractions of the mouse isolated vas deferens was also investigated.
Key results:
In the brain membrane experiments, cannabigerol behaved as a potent α2-adrenoceptor agonist (EC50= 0.2 nM) and antagonized the 5-HT1A receptor agonist, R-(+)-8-hydroxy-2-(di-n-propylamino)tetralin (apparent KB= 51.9 nM). At 10 µM, it also behaved as a CB1 receptor competitive antagonist. Additionally, cannabigerol inhibited evoked contractions of the vas deferens in a manner that appeared to be α2-adrenoceptor-mediated (EC50= 72.8 nM) and displayed significant affinity for mouse CB1 and human CB2 receptors.
Conclusions and implications:
This investigation has provided the first evidence that cannabigerol can activate α2-adrenoceptors, bind to cannabinoid CB1 and CB2 receptors and block CB1 and 5-HT1A receptors. It will now be important to investigate why cannabigerol produced signs of agonism more potently in the [35S]GTPγS binding assay than in the vas deferens and also whether it can inhibit noradrenaline uptake in this isolated tissue and in the brain.
Keywords: cannabigerol, CP55940, mouse vas deferens, α2-adrenoceptor, 5-HT1A receptor, CB1 receptor, clonidine, dexmedetomidine, maprotiline, R-(+)-8-hydroxy-2-(di-n-propylamino)tetralin
Introduction
Cannabis sativa is the natural source of a set of at least seventy C21 compounds that are known collectively as phytocannabinoids (see ElSohly and Slade, 2005). To date, pharmacological research has focused primarily on just three of these compounds. One of these is Δ9-tetrahydrocannabinol, the main psychoactive constituent of cannabis, the others being the non-psychoactive phytocannabinoid, cannabidiol and Δ9-tetrahydrocannabivarin, which at low doses can block cannabinoid receptor-mediated actions of Δ9-tetrahydrocannabinol (see Pertwee, 2008). Both Δ9-tetrahydrocannabinol and cannabidiol are present in a currently licensed medicine, Sativex®, and Δ9-tetrahydrocannabivarin has therapeutic potential for the management of disorders such as obesity and drug dependence (see Pertwee, 2008). It is important that more research is directed at exploring the pharmacology of the many other cannabinoids present in cannabis, not least because such research will help to identify any additional therapeutic applications of these phytocannabinoids, compounds that were described in the title of a recent BJP commentary as ‘a neglected pharmacological treasure trove’ (Mechoulam, 2005).
The present investigation focused on the little-studied phytocannabinoid, cannabigerol (Figure 1) which was first detected in cannabis and synthesized by Gaoni and Mechoulam (1964) and subsequently found not to induce Δ9-THC-like psychopharmacological effects in vivo (Grunfeld and Edery, 1969; Mechoulam et al., 1970). Our main objective was to establish whether this compound can activate or block any G protein-coupled receptor as indicated by stimulation of [35S]GTPγS binding to mouse whole brain membranes or by blockade of such stimulation when this is induced by another compound. We also investigated the ability of cannabigerol to displace [3H]CP55940 both from CB1 binding sites in these membranes and from CB2 binding sites in membranes prepared from Chinese hamster ovary (CHO) cells transfected with human CB2 receptors (receptor nomenclature follows Alexander et al., 2008). Some of our experiments were carried out with the mouse isolated vas deferens, a tissue in which cannabinoid receptor agonists can inhibit electrically evoked contractions (Devane et al., 1992; Pertwee et al., 1995). They are thought to do this by targeting prejunctional neuronal cannabinoid CB1 receptors in a manner that inhibits neuronal release of the contractile neurotransmitters, ATP and noradrenaline (Trendelenburg et al., 2000; see von Kügelgen and Starke, 1991; Pertwee, 1997; Schlicker and Kathmann, 2001).
Figure 1.
The chemical structure of cannabigerol.
The results we obtained extend a recent finding that cannabigerol can activate TRPA1 transient receptor potential channels and block the activation of TRPM8 transient receptor potential channels in vitro (De Petrocellis et al., 2008) by providing evidence that it can indeed also target certain G protein-coupled receptors. Some of the results described in this paper have been presented to the International Cannabinoid Research Society (Gauson et al., 2008; 2009; Cascio et al., 2009).
Methods
Animals
All animal care and experimental procedures complied with the UK Animals (Scientific Procedures) Act, 1986 and associated guidelines for the use of experimental animals. MF1 mice aged 6 to 7 weeks and weighing 30 to 35 g were purchased from Harlan UK Ltd. (Blackthorn, UK), whereas C57BL/6 CB1 receptor knockout mice and their wild-type litter mates were obtained from NIH (Rockville, MD, USA). Mice were maintained on a 12/12 h light/dark cycle with free access to food and water. All experiments were performed with tissues obtained from adult male mice.
CHO cells
Chinese hamster ovary cells stably transfected with cDNA encoding human cannabinoid CB2 receptors were maintained at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium nutrient mixture F-12 HAM supplemented with 2 mM L-glutamine, 10% foetal bovine serum, 0.6% penicillin-streptomycin and G418 (600 µg·mL−1). These CHO-hCB2 cells were passaged twice a week using a non-enzymatic cell dissociation solution.
Membrane preparation
Binding assays with [3H]CP55940 and with [35S]GTPγS were performed with mouse whole brain membranes, prepared as described by Thomas et al. (2004) or with CHO-CB2 cell membranes (Ross et al., 1999a). The hCB2 transfected cells were removed from flasks by scraping and then frozen as a pellet at –20°C until required. Before use in a radioligand binding assay, cells were defrosted, diluted in Tris-buffer (50 mM Tris-HCl and 50 mM Tris-Base) and homogenized with a 1-mL-handheld homogenizer. Protein assays were performed using a Bio-Rad Dc kit (Hercules, CA, USA).
Radioligand displacement assay
The assays were carried out with [3H]CP55940 and Tris-binding buffer [50 mM Tris-HCl, 50 mM Tris-Base, 0.1% bovine serum albumin (BSA); pH 7.4], total assay volume 500 µL, using the filtration procedure described previously by Ross et al. (1999b). Binding was initiated by the addition of either brain membranes (33 µg protein per well) or transfected hCB2-cells (25 µg protein per well). All assays were performed at 37°C for 60 min before termination by addition of ice-cold Tris-binding buffer and vacuum filtration using a 24-well sampling manifold (Brandel Cell Harvester) and Brandel GF/B filters that had been soaked in wash buffer at 4°C for at least 24 h (Brandel Inc., Gaitherburg, MD, USA). Each reaction well was washed six times with a 1.2 mL aliquot of Tris-binding buffer. The filters were oven-dried for 60 min and then placed in 5 mL of scintillation fluid (Ultima Gold XR, PerkinElmer). Radioactivity was quantified by liquid scintillation spectrometry. Specific binding was defined as the difference between the binding that occurred in the presence and absence of 1 µM unlabelled CP55940. The concentration of [3H]CP55940 used in our displacement assays was 0.7 nM. Compounds under investigation were stored as stock solutions of 10 mM in dimethyl sulphoxide (DMSO), the vehicle concentration in all assay wells being 0.1% DMSO. The binding parameters for [3H]CP55940, determined by fitting data from saturation-binding experiments to a one-site saturation plot using GraphPad Prism, were 2336 fmol·mg−1 protein (Bmax) and 2.31 nM (Kd) in mouse brain membranes (Thomas et al., 2004) and 215 pmol·mg−1 (Bmax) and 4.3 nM (Kd) in hCB2-transfected cells.
[35S]GTPγS binding assay
The method for measuring agonist-stimulated [35S]GTPγS binding to cannabinoid CB1 receptors was adapted from the methods of Kurkinen et al. (1997) and Breivogel et al. (2001). The assays were carried out with GTPγS binding buffer (50 mM Tris-HCl, 50 mM Tris-Base, 5 mM MgCl2, 1 mM EDTA, 100 mM NaCl, 1 mM dithiothreitol, 0.1% BSA) in the presence of [35S]GTPγS and GDP, in a final volume of 500 µL. Binding was initiated by the addition of [35S]GTPγS to the wells. Nonspecific binding was measured in the presence of 30 µM GTPγS. The drugs were incubated in the assay for 60 min at 30°C. The reaction was terminated by a rapid vacuum filtration method using Tris-binding buffer as described previously, and the radioactivity was quantified by liquid scintillation spectrometry. In all the [35S]GTPγS-binding assays, we used 0.1 nM [35S]GTPγS, 30 µM GDP and a protein concentration of 5 µg per well. Additionally, mouse brain membranes were preincubated for 30 min at 30°C with 0.5 U·mL−1 adenosine deaminase (200 U·mL−1) to remove endogenous adenosine. Agonists and antagonists were stored at –20°C as 10 mM stock solutions dissolved in distilled water (yohimbine) or DMSO.
Vas deferens experiments
Vasa deferentia were obtained from albino MF1 mice weighing 36 to 53 g. The tissues were mounted vertically in 4 mL organ baths. They were then subjected to electrical stimulation of progressively greater intensity followed by an equilibration procedure in which they were exposed to alternate periods of stimulation (2 min) and rest (10 min) until contractions with consistent amplitudes were obtained (Thomas et al., 2004). These contractions were monophasic and isometric and were evoked by 0.5 s trains of pulses of 110% maximal voltage (train frequency 0.1 Hz; pulse frequency 5 Hz; pulse duration 0.5 ms).
Except in our experiments with phenylephrine, all drug additions were made to the organ baths after the equilibration period and there was no washout between these additions. In most experiments there was an initial application of a potential antagonist or its vehicle. This was followed 28 min later by a 2 min period of electrical stimulation at the end of which the lowest of a series of concentrations of the twitch inhibitors, cannabigerol, clonidine, dexmedetomidine or maprotiline was applied. After a period of rest, the tissues were electrically stimulated for 2 min and then subjected to a further addition of twitch inhibitor. This cycle of drug addition, rest and 2 min stimulation was repeated so as to construct cumulative concentration-response curves. Only one concentration-response curve was constructed per tissue (Pertwee et al., 1996). The rest period was 13 min in the experiments with cannabigerol, dexmedetomidine and maprotiline and 3 min in the clonidine experiments.
In experiments with β,γ-methylene-ATP, no electrical stimuli were applied after the equilibration procedure. Log concentration-response curves of β,γ-methylene-ATP were constructed cumulatively without washout. Cannabigerol was added 30 min before the first addition of β,γ-methylene-ATP, each subsequent addition of which was made immediately after the effect of the previous dose had reached a plateau (dose cycles of 1 to 2 min). Only one addition of phenylephrine was made to each tissue and this was carried out 30 min after the addition of cannabigerol or its vehicle.
Analysis of data
Values have been expressed as means and variability as SEM or as 95% confidence limits. The concentration of the compounds under investigation that produced a 50% displacement of radioligand from specific binding sites (IC50 value) was calculated using GraphPad Prism and the corresponding Ki values were calculated using the equation of Cheng and Prusoff (1973). Net agonist stimulated [35S]GTPγS binding values were calculated by subtracting basal binding values (obtained in the absence of agonist) from agonist-stimulated values (obtained in the presence of agonist) as detailed elsewhere (Ross et al., 1999a). Inhibition of the electrically evoked twitch response of the vas deferens has been expressed in percentage terms, and this has been calculated by comparing the amplitude of the twitch response after each addition of a twitch inhibitor with its amplitude immediately before the first addition of the inhibitor. Contractile responses to phenylephrine and β,γ-methylene-ATP have been expressed as increases in tension (g). Values for EC50, maximal effect (Emax) and SEM or 95% confidence limits of these values have been calculated by nonlinear regression analysis using the equation for a sigmoid concentration-response curve (GraphPad Prism).
Unless stated otherwise, apparent dissociation constant (KB) values for antagonism of agonists by yohimbine in the vas deferens or by yohimbine or cannabigerol in the [35S]GTPγS binding assay have been calculated by Schild analysis (GraphPad Prism). These KB values were calculated only from data obtained in experiments in which yohimbine or cannabigerol produced a right-ward shift in the log concentration response curve of an agonist that was indicated by (2 + 2) dose parallel line analysis to be statistically significant and not to deviate significantly from parallelism (Pertwee et al., 2002). In one set of experiments, the effect of one or other of five concentrations of cannabigerol on the log concentration response curve of 8-OH-DPAT was determined. For these experiments, the KB of cannabigerol was calculated from the intercept on the x-axis (–log KB) of the best-fit straight line of a plot of log (x − 1) against log B constructed by linear regression analysis (GraphPad Prism). The equation for this Schild plot is log (x − 1) = log B − log KB, where x (the ‘concentration ratio’) is the concentration of 8-OH-DPAT that elicits a particular response in the presence of cannabigerol at a concentration, B, divided by the concentration of 8-OH-DPAT that elicits a response of the same size in the absence of cannabigerol. This equation predicts a slope of unity for all receptor-mediated interactions between agonists and antagonists that are competitive and reversible (Tallarida et al., 1979). Log (x − 1) values were determined by (2 + 2) dose parallel line analysis as described previously (Pertwee et al., 2002). Mean values obtained in vitro have been compared with zero using the one-sample t-test and with each other using Student's two-tailed t-test for unpaired data or one-way analysis of variance (ANOVA) followed by Dunnett's test (GraphPad Prism). A P value of 0.05 or less was considered to be significant.
Materials
Cannabigerol was supplied by GW Pharmaceuticals (Porton Down, Wiltshire, UK) and rimonabant (SR141716A) was obtained from Sanofi-Aventis (Montpellier, France). Phenylephrine hydrochloride, β, γ-methyleneadenosine 5′-triphosphate disodium salt (β, γ-methylene-ATP), arachidonoyl ethanolamide (anandamide), clonidine hydrochloride and N-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-N-2-pyridinylcyclohexanecarboxamide maleate (WAY100635) were purchased from Sigma-Aldrich (Poole, Dorset, UK) and R-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo-[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone (R-(+)-WIN55212), (–)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol (CP55940), dexmedetomidine hydrochloride, yohimbine hydrochloride, maprotiline hydrochloride and R-(+)-8-hydroxy-2-(di-n-propylamino)tetralin (8-OH-DPAT) from Tocris (Bristol, UK). For the binding experiments, [3H]CP55940 (160 Ci·mmol−1) and [35S]GTPγS (1250 Ci·mmol−1) were obtained from PerkinElmer Life Sciences Inc. (Boston, MA, USA), GTPγS and adenosine deaminase from Roche Diagnostic (Indianapolis, IN, USA) and GDP from Sigma-Aldrich. Phenylephrine hydrochloride, β, γ-methylene-ATP and maprotiline were dissolved in a 0.9% aqueous solution of NaCl (saline) and yohimbine and clonidine in distilled water. All other compounds were dissolved in pure DMSO. In the vas deferens experiments, all compounds were added to organ baths in a volume of 10 µL.
Results
Cannabigerol is a potent stimulator of [35S]GTPγS binding to brain membranes
In our initial experiments, we investigated the effect of cannabigerol on [35S]GTPγS binding to MF1 mouse brain membranes. We found that at concentrations in the picomolar and low nanomolar range, this cannabinoid produced a concentration-related stimulation of [35S]GTPγS binding to MF1 mouse brain membranes (Figure 2). Experiments performed with brain membranes obtained from either CB1+/+ or CB1−/− C57BL/6J mice yielded similar results (Table 1) and also showed that, as in MF1 mouse brain membranes (Figure 2), cannabigerol at 1 µM had no significant effect on [35S]GTPγS binding and, at 10 µM, produced a marked inhibitory effect.
Figure 2.
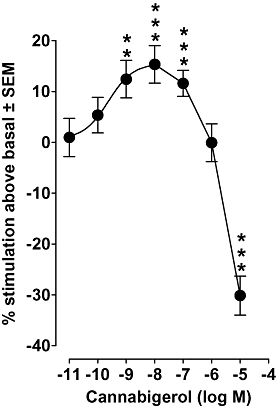
The effect of cannabigerol on [35S]GTPγS binding to whole brain membranes obtained from MF1 mice (n= 7 to 20). Each symbol represents the mean percentage change in binding ± SEM. Asterisks denote values that are significantly different from zero (**P < 0.01; ***P < 0.001; one-sample t-test).
Table 1.
Cannabigerol stimulates [35S]GTPγS binding to brain membranes obtained from MF1, CB1+/+ C57BL/6J and CB1−/− C57BL/6J mice
| Mouse strain | Mean EC50 (95% CL) | Mean Emax (95% CL) | n |
|---|---|---|---|
| MF1 | 0.2 nM (0.006 and 9.0 nM) | 15.5% (8.5 and 22.5%) | 7 to 20 |
| CB1+/+ C57BL/6J | 0.04 nM (0.002 and 0.8 nM) | 17.0% (10.4 and 23.7%) | 8 |
| CB1−/− C57BL/6J | 0.17 nM (0.01 and 2.8 nM) | 11.4% (6.6 and 16.2%) | 8 |
Mean EC50 and Emax values were calculated from data obtained with cannabigerol concentrations of up to 10 nM.
CL, confidence limits.
The mean percentage inhibition of [35S]GTPγS binding was found to be 30.1% ± 3.8 (n= 17) in MF1 mouse brain membranes (Figure 2), 14.2% ± 3.4 (n= 6) in CB1+/+ C57BL/6J mouse brain membranes and 21.8% ± 4.6 (n= 8) in CB1−/− C57BL/6J mouse brain membranes and each of these mean values is significantly less than zero (P < 0.01; one-sample t-test). These results suggest that neither the stimulatory effect nor the inhibitory effect of cannabigerol on [35S]GTPγS binding to mouse brain membranes is CB1 receptor-mediated.
Cannabigerol behaves as an α2-adrenoceptor agonist in the mouse isolated vas deferens
Further evidence that cannabigerol is not a CB1 receptor agonist was obtained from experiments performed with the mouse isolated vas deferens. More specifically, although cannabigerol shared the ability of established cannabinoid receptor agonists such as CP55940 and Δ9-tetrahydrocannabinol (Pertwee et al., 1995) to produce a concentration-related inhibition of electrically evoked contractions (Figure 3A), no right-ward shift in the log concentration response curve of cannabigerol was produced by rimonabant at 100 nM (data not shown), a concentration that equals or exceeds concentrations of this CB1-selective antagonist that have been found previously to antagonize established CB1 receptor agonists in this bioassay (Pertwee et al., 1995; Ross et al., 2001). Cannabigerol inhibited electrically evoked contractions of the vas deferens at concentrations below any found to attenuate contractile responses either to the P2 receptor agonist, β,γ-methylene ATP, or to the α1-adrenoceptor agonist, phenylephrine hydrochloride. Thus it inhibited electrically evoked contractions at concentrations of 100 nM or less (Figure 3A), attenuated contractile responses of the vas deferens to β,γ-methylene ATP, at 1 µM but not at 100 nM (Figure 4) and did not affect contractions induced by phenylephrine hydrochloride even at a concentration of 1 µM (Figure 4). As electrically evoked contractions of the vas deferens are thought to result from the release of ATP and noradrenaline on to postjunctional P2 receptors and α1-adrenoceptors (Introduction), these findings suggest that cannabigerol can inhibit these contractions by acting prejunctionally.
Figure 3.
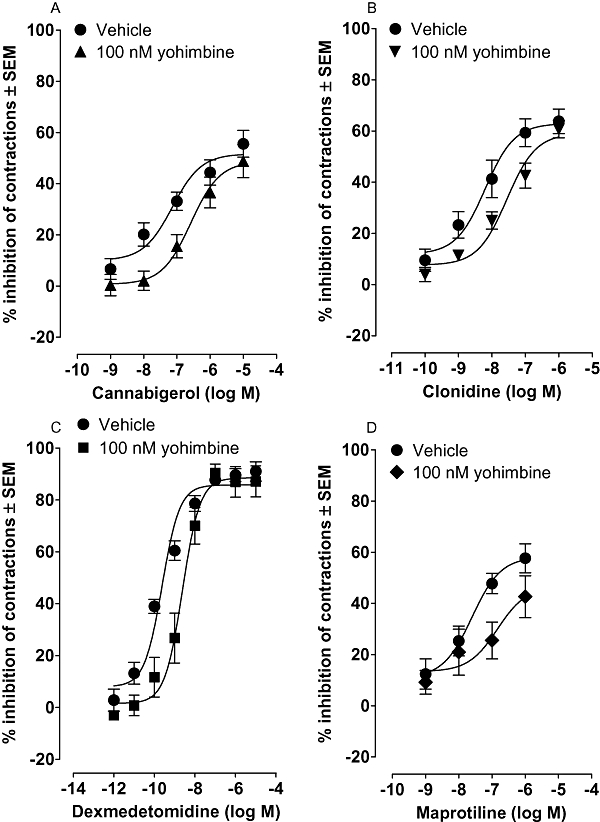
Mean log concentration-response curves of (A) cannabigerol (n= 13), (B) clonidine (n= 7) (C) dexmedetomidine (n= 5) and (D) maprotiline (n= 7) in the MF1 mouse isolated vas deferens constructed in the presence of yohimbine or its vehicle. Each symbol represents the mean value ± SEM for inhibition of electrically evoked contractions expressed as a percentage of the amplitude of the twitch response measured immediately before the first addition of cannabigerol, clonidine or maprotiline to the organ bath. Yohimbine or its vehicle was added 30 min before this first addition and all further additions were made at 5 or 15 min intervals (Methods). Each log concentration response curve was constructed cumulatively. The mean apparent KB value of yohimbine with its 95% confidence limits shown in brackets is 10.1 nM (3.0 and 33.7 nM) against cannabigerol, 14.0 nM (4.9 and 40.6 nM) against clonidine, 8.7 nM (4.3 and 17.8 nM) against dexmedetomidine and 8.2 nM (1.5 and 46.0 nM) against maprotiline. In the absence of yohimbine, electrically evoked contractions were inhibited by cannabigerol, clonidine, dexmedetomidine and maprotiline with mean EC50 values of 72.8 nM (23.4 and 227 nM), 6.3 nM (2.0 and 19.7 nM), 0.24 nM (0.14 and 0.40 nM) and 24.9 nM (5.6 and 111.2 nM) respectively. The 95% confidence limits of these mean values are shown in brackets. The corresponding Emax values are 51.7% (44.3 and 59.1%), 63.0% (54.1 and 71.8%), 85.8% (81.6 and 90.0%) and 58.2% (47.1 and 69.3%) respectively.
Figure 4.
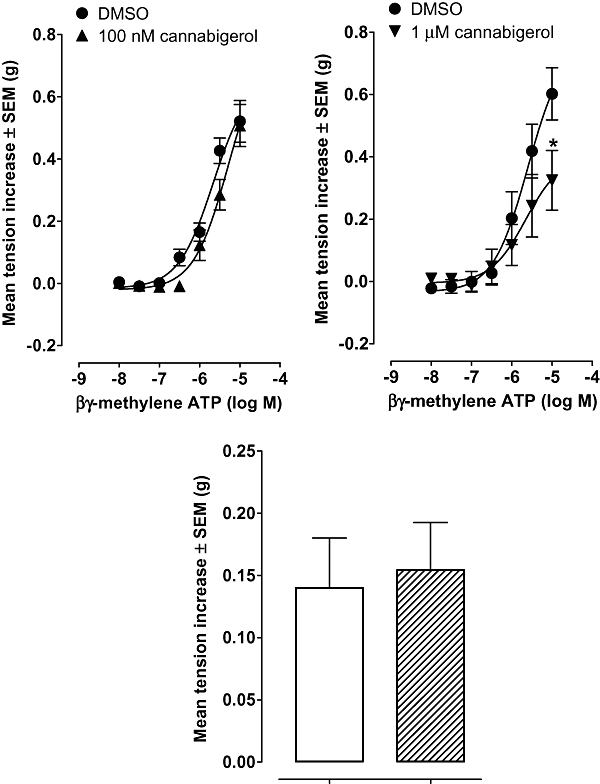
Upper panels: mean increases in tension of the MF1 mouse isolated vas deferens induced by β,γ-methylene ATP in the presence of DMSO (circles) or cannabigerol (triangles). For the construction of log concentration-response curves, β,γ-methylene ATP was first added 30 min (A) after DMSO or 100 nM cannabigerol (n= 6 or 8) or (B) after DMSO or 1 µM cannabigerol (n= 8). The asterisk indicates a significant difference between the contractile response to 10 µM β,γ-methylene ATP in the absence of cannabigerol and the corresponding response in the presence of this cannabinoid (P < 0.05; unpaired t-test). Lower panel: mean increases in tension of the mouse isolated vas deferens induced by 32 µM phenylephrine in the absence or presence of 1 µM cannabigerol. The two mean values are not significantly different (P > 0.05; unpaired t-test). Additions of phenylephrine were made 30 min after DMSO (open columns) or cannabigerol (n= 8). In all panels, mean increases in tension are expressed in grams ± SEM. DMSO, dimethyl sulphoxide.
One possibility is that the inhibitory effect of cannabigerol on electrically evoked contractions of the mouse vas deferens is mediated by prejunctional α2-adrenoceptors as it is generally accepted that these receptors mediate inhibition of such contractions when activated by endogenously released noradrenaline or by an exogenously added agonist (Pertwee et al., 2005); reviewed in (von Kügelgen and Starke, 1991; Starke, 2001). To test this hypothesis, we investigated whether cannabigerol can be antagonized in the vas deferens by the selective α2-adrenoceptor antagonist, yohimbine. We found not only that yohimbine can indeed antagonize cannabigerol-induced inhibition of electrically evoked contractions but also that the potency with which it produces this antagonism is similar to the potency it displays in the same bioassay as an antagonist of clonidine and dexmedetomidine (Figure 3), both of which are established α2-adrenoceptor agonists (Newman-Tancredi et al., 1998). Yohimbine also antagonized the inhibition of electrically evoked contractions of the vas deferens induced by maprotiline, an inhibitor of noradrenaline uptake (Barbaccia et al., 1986), with a potency that matched the potency with which it antagonized cannabigerol (Figure 3). It seems likely, therefore, that α2-adrenoceptors do indeed mediate cannabigerol-induced inhibition of electrically evoked contractions of the mouse vas deferens.
Cannabigerol also behaves as an α2-adrenoceptor agonist in mouse brain membranes
The results obtained in the vas deferens experiments raised the possibility that cannabigerol-induced stimulation of [35S]GTPγS binding to mouse brain membranes (Figure 2) might also be α2-adrenoceptor-mediated. To investigate this possibility, we first carried out experiments directed at establishing whether dexmedetomidine shares the ability of cannabigerol to stimulate [35S]GTPγS binding to MF1 mouse brain membranes. We found that this α2-adrenoceptor agonist can indeed induce such stimulation and that it is antagonized by yohimbine at 100 nM (Figure 5). This concentration of yohimbine also antagonized cannabigerol-induced stimulation of [35S]GTPγS binding, the data obtained indicating a lack of any significant difference between the apparent KB values of yohimbine for its antagonism of these two compounds (Figure 5).
Figure 5.
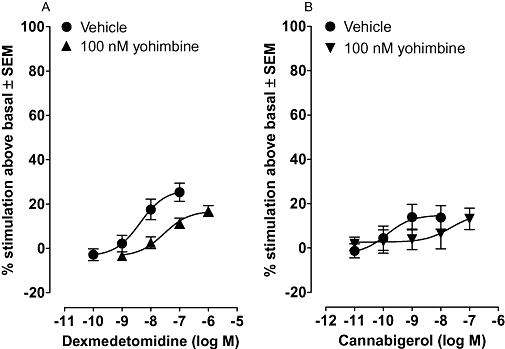
Mean log concentration-response curves of (A) dexmedetomidine (n= 4) and (B) cannabigerol (n= 12) constructed in the absence or presence of 100 nM yohimbine. Each symbol represents the mean percentage change in binding of [35S]GTPγS to MF1 mouse whole brain membranes ± SEM. Mean EC50 values of dexmedetomidine and cannabigerol in the absence of yohimbine with 95% confidence limits shown in brackets are 4.3 nM (0.7 and 25.5 nM) and 0.13 nM (0.004 and 4.4 nM) respectively. The corresponding mean Emax values are 26.5% (17.1 and 36.0%) and 14.8% (5.9 and 23.7%) respectively. The right-ward shifts produced by yohimbine in the log concentration response curves of dexmedetomidine and cannabigerol are significant and do not deviate significantly from parallelism (P > 0.05). The mean apparent KB value of yohimbine for this antagonism, with its 95% confidence limits shown in brackets is 3.9 nM (1.0 and 15.1 nM) against dexmedetomidine and 1.8 nM (0.04 and 90.5 nM) against cannabigerol.
Cannabigerol and cannabinoid receptors
As cannabigerol is a constituent of cannabis, it was of interest to investigate whether it shares the ability of the plant cannabinoids Δ9-tetrahydrocannabinol, Δ9-tetrahydrocannabivarin and cannabidiol to bind to cannabinoid CB1 and CB2 receptors (see Pertwee, 2008). Cannabigerol was able to completely displace [3H]CP55940 from specific binding sites both in mouse brain membranes and in CHO-hCB2 cell membranes, its mean Ki values for this displacement suggesting that it has greater CB1 than CB2 receptor affinity (Figure 6). The binding data obtained with brain membranes suggest that although cannabigerol does bind to cannabinoid CB1 receptors, this is only detectable at concentrations above those at which it stimulates [35S]GTPγS binding to these membranes (Figures 2 and 5). Further experiments were therefore carried out to establish whether a concentration of cannabigerol that produced a clear reduction in specific binding of [3H]CP55940 to brain membranes, also antagonized anandamide or CP55940.
Figure 6.
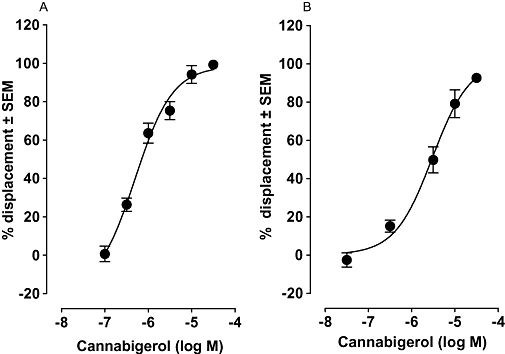
Displacement of [3H]CP55940 by cannabigerol from specific binding sites on (A) MF1 mouse whole brain membranes (n= 4) and (B) CHO-hCB2 cell membranes (n= 8). Each symbol represents the mean percent displacement ± SEM. Mean Ki values with 95% confidence limits shown in brackets are (A) 381 nM (231 and 627 nM) for displacement from brain membranes and (B) 2.6 µM (1.4 and 4.7 µM) for displacement from CHO-hCB2 cell membranes. CHO, Chinese hamster ovary.
The first set of these experiments showed that anandamide-induced stimulation of [35S]GTPγS binding to mouse brain membranes was significantly antagonized by cannabigerol at 10 µM (Figure 7A). This it did with a mean apparent KB value (Table 2) that is significantly less than its mean Ki value for displacement of [3H]CP55940 from brain membranes (Figure 6). However, in line with the ability of cannabigerol by itself to inhibit [35S]GTPγS binding to brain membranes at 10 µM, it also appeared to produce a downward shift in the log concentration-response curve of anandamide. When this component of cannabigerol-induced antagonism that most likely arises from the ability of this compound to inhibit [35S]GTPγS binding was excluded, a significant right-ward shift in the log concentration-response curve of anandamide was still apparent (Figure 7C). Importantly, the mean apparent KB value calculated from this shift (Table 2) did not differ significantly from the mean Ki value of cannabigerol for displacement of [3H]CP55940 from brain membranes (Figure 6). Also, as this right-ward shift did not deviate significantly from parallelism, it is likely that cannabigerol is a competitive antagonist of anandamide. Neither a right-ward shift nor a downward shift in the log concentration-response curve for anandamide-induced stimulation of [35S]GTPγS binding to MF1 mouse brain membranes was induced by cannabigerol at 1 µM (n= 6; data not shown).
Figure 7.
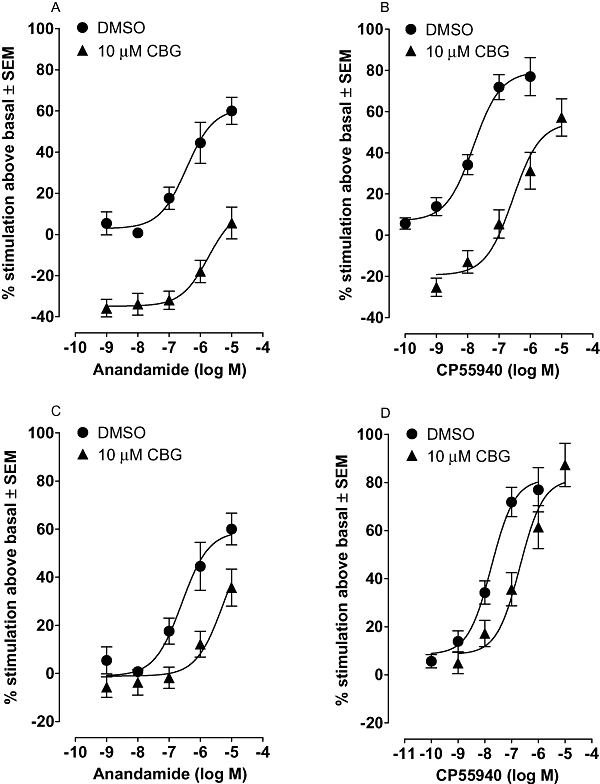
Upper panels: the effect of 10 µM cannabigerol (CBG) on the mean log concentration-response curve of (A) anandamide and (B) CP55940 for stimulation of [35S]GTPγS binding to mouse brain membranes. Lower panels: the effect of 10 µM cannabigerol (CBG) on the mean log concentration-response curve of (C) anandamide and (D) CP55940 for stimulation of [35S]GTPγS binding after subtraction of the inhibitory effect induced by 10 µM cannabigerol on [35S]GTPγS binding in the absence of any other compound (30.1 ± 3.8%; n= 17; Figure 2). This value was subtracted from all values of percent stimulation of [35S]GTPγS binding by anandamide or CP55490 determined in the presence of cannabigerol. Experiments were performed with MF1 mouse whole brain membranes and stimulation of [35S]GTPγS binding is expressed as mean percent stimulation ± SEM (n= 5 or 6). The right-ward shifts produced by cannabigerol in the log concentration response curves of anandamide and CP55940 do not deviate significantly from parallelism (P > 0.05). DMSO, dimethyl sulphoxide.
Table 2.
The mean apparent KB values of cannabigerol for antagonism of anandamide- and CP55940-induced stimulation of [35S]GTPγS binding to MF1 mouse brain membranes
| Agonist | Antagonist (10 µM) | Mean apparent KB (95% CL) | Mean apparent KB (95% CL)1 | n |
|---|---|---|---|---|
| Anandamide | Cannabigerol | 33.1 nM (13.2 and 82.9 nM) | 483 nM (162 and 1445 nM) | 5 |
| CP55940 | Cannabigerol | 53.7 nM (19.4 and 149 nM) | 936 nM (336 and 2606 nM) | 6 |
Calculated after subtraction of the mean inhibitory effect induced by 10 µM cannabigerol on [35S]GTPγS binding in the absence of any other compound (30.1%; Figure 7 legend).
CL, confidence limits.
Also, 10 µM cannabigerol antagonized CP55940-induced stimulation of [35S]GTPγS binding to mouse brain membranes (Figure 7B). Again, cannabigerol appeared to produce both a right-ward and a downward shift in the log concentration response curve of the agonist. After compensating for the downward shift (Figure 7D) it was found that the mean apparent KB value of cannabigerol for antagonism of CP55940 (Table 2) did not differ significantly either from the mean KB value of cannabigerol for antagonism of anandamide or from the mean Ki value of cannabigerol for displacement of [3H]CP55940 from brain membranes (Figure 6). As in the anandamide experiments, the right-ward shift induced by cannabigerol in the log concentration-response curve of CP55940 (Figure 7D) did not deviate significantly from parallelism. Taken together, these findings support to the hypothesis that cannabigerol is a CB1 receptor competitive antagonist, albeit of much lower potency than rimonabant (Thomas et al., 2007).
Cannabigerol and 5HT1A receptors
There is evidence that some established α2-adrenoceptor ligands, including clonidine and yohimbine, target 5-HT1A receptors at concentrations above those at which they activate or block α2-adrenoceptors (Newman-Tancredi et al., 1998). This prompted us to investigate whether cannabigerol interacts with 5-HT1A receptors at concentrations higher than those at which it stimulates [35S]GTPγS binding to MF1 mouse brain membranes.
Initial experiments confirmed that the 5-HT1A-selective antagonist, WAY100635 (Forster et al., 1995), did antagonize the stimulatory effect of 8-OH-DPAT, a 5-HT1A receptor agonist (Forster et al., 1995), on [35S]GTPγS binding to brain membranes (Figure 8A). The mean apparent KB value of WAY100635 for this antagonism was 1 nM. Further experiments showed that, at 1 µM, cannabigerol also antagonized 8-OH-DPAT in this bioassay as indicated by the ability of cannabigerol to produce a parallel right-ward shift in the log concentration response curve of this 5-HT1A-selective agonist (Figure 8B). The data from this experiment and from similar experiments performed either with 10 µM cannabigerol (Figure 8C) or with one or other of three lower concentrations of cannabigerol (data not shown) allowed the construction of a Schild plot (Figure 8D), the slope of which was not significantly different from unity. The mean apparent KB value of cannabigerol for its antagonism of 8-OH-DPAT, as calculated from this Schild plot, was 51.9 nM.
Figure 8.
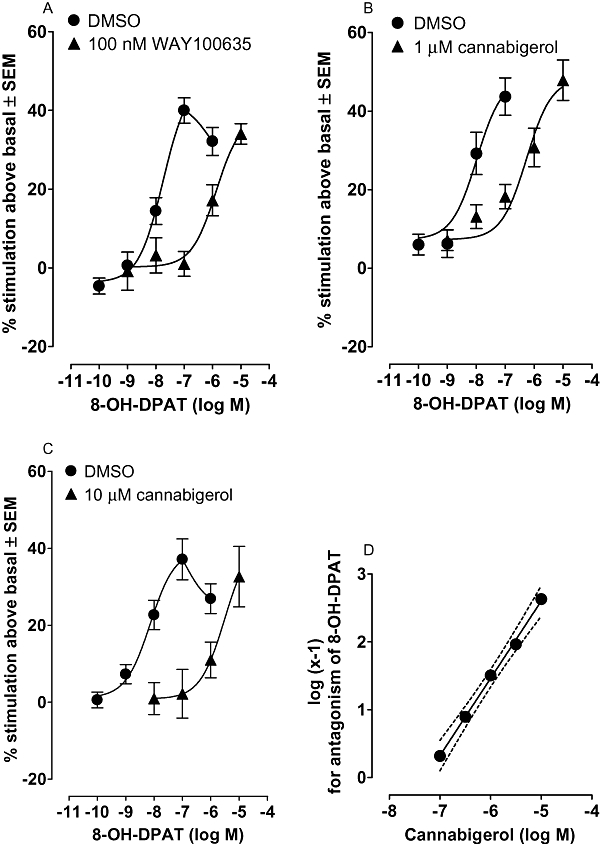
Mean log concentration-response curves of R-(+)-8-hydroxy-2-(di-n-propylamino)tetralin (8-OH-DPAT) constructed in the presence of (A) DMSO or 100 nM WAY100635 (n= 7), (B) DMSO or 1 µM cannabigerol (n= 10) or (C) DMSO or 10 µM cannabigerol (n= 12). Each symbol represents the mean percentage change in binding of [35S]GTPγS to MF1 mouse whole brain membranes ± SEM. Neither the right-ward shift produced by WAY100635 in the log concentration response curve of 8-OH-DPAT nor that produced by 1 or 10 µM cannabigerol deviates significantly from parallelism (P > 0.05). Mean apparent KB values with 95% confidence limits shown in brackets are (A) 1.0 nM (0.5 and 2.3 nM) for WAY100635, (B) 19.6 nM (6.9 and 55.8 nM) for 1 µM cannabigerol and (C) 28.2 nM (7.7 and 102.9 nM) for 10 µM cannabigerol. Panel (D): Schild plot for antagonism of 8-OH-DPAT by 100 nM, 316 nM, 1 µM, 3.16 µM and 10 µM cannabigerol (n= 5 to 12) with the 99% confidence band shown by dotted lines. The mean slope of this best-fit line with 95% confidence limits shown in brackets is 1.1 (1.0 and 1.2) and so does not differ significantly from unity. The mean apparent KB value of cannabigerol calculated from this Schild plot, with 95% confidence limits shown in brackets, is 51.9 nM (37.6 and 68.2 nM). DMSO, dimethyl sulphoxide.
Cannabigerol (1 µM) was no less effective in producing a parallel right-ward shift in the log concentration response curve of 8-OH-DPAT for stimulating [35S]GTPγS binding to brain membranes when these membranes were obtained from CB1+/+ or CB1−/− C57BL/6J mice (data not shown) rather than from MF1 mice. The mean apparent KB value of cannabigerol with 95% confidence limits shown in brackets is 19.6 nM (6.9 and 55.8; n= 10; Figure 8B) when calculated from the data obtained using MF1 mouse brain membranes and 6.2 nM (2.6 and 14.8 nM; n= 11) and 2.3 nM (0.7 and 7.7 nM; n= 13) respectively when calculated from the CB1+/+ and CB1−/− C57BL/6J mouse brain membrane data.
Discussion
Results from our initial experiments indicated that cannabigerol exhibited significant potency both as a stimulator of [35S]GTPγS binding to mouse brain membranes (Figure 2) and as an inhibitor of electrically evoked contractions of the mouse isolated vas deferens (Figure 3). Neither of these effects appeared to be mediated by cannabinoid CB1 receptors. Thus, cannabigerol displayed no less potency or efficacy as a stimulator of [35S]GTPγS binding to CB1−/− mouse brain membranes than to CB1+/+ mouse brain membranes and its inhibitory effect on electrically evoked contractions of the vas deferens was not antagonized by the CB1-selective antagonist, rimonabant, when this was administered at 100 nM, a concentration higher than that required to antagonize the established cannabinoid receptor agonists, CP55940, R-(+)-WIN55212 and Δ9-tetrahydrocannabinol, in the same bioassay (Pertwee et al., 1995; Ross et al., 2001). For reasons yet to be established, it was only within a range of very low concentrations that cannabigerol produced concentration-related increases in [35S]GTPγS binding to CB1+/+ or CB1−/− mouse brain membranes. Thus, within the concentration range 0.01 nM to 1 µM, its log concentration response curve was bell-shaped and indeed, at 10 µM, cannabigerol markedly inhibited [35S]GTPγS binding to brain membranes.
It is unlikely that cannabigerol inhibited electrically evoked contractions of the vas deferens by acting postjunctionally to block the actions of the two neurotransmitters that are thought to induce these contractions: ATP acting on postjunctional P2X receptors and noradrenaline acting on postjunctional α1-adrenoceptors (von Kügelgen and Starke, 1991; Trendelenburg et al., 2000). Thus, cannabigerol inhibited electrically evoked contractions at concentrations at which it did not significantly affect the amplitude of contractions induced in the vas deferens either by the P2X agonist, β,γ-methylene-ATP, or by the α1-adrenoceptor agonist, phenylephrine. Cannabigerol, therefore, differs from another plant cannabinoid, Δ9-tetrahydrocannabivarin, which in the MF1 mouse vas deferens does reduce the amplitude of contractions induced by 10 µM β,γ-methylene-ATP or 32 µM phenylephrine when it is applied at the lowest of the concentrations at which it inhibits electrically evoked contractions of this tissue (Thomas et al., 2005). The concentrations of β,γ-methylene-ATP and phenylephrine used in these previous experiments with Δ9-tetrahydrocannabivarin were the same as those used in our cannabigerol experiments.
Results obtained with the selective α2-adrenoceptor antagonist, yohimbine, suggest that both the inhibitory effect of cannabigerol on electrically evoked contractions of the vas deferens and its stimulatory effect on [35S]GTPγS binding to mouse brain membranes are mediated by α2-adrenoceptors. Thus, yohimbine antagonized both these effects of cannabigerol at a concentration (100 nM) at which it is expected to display selectivity as an α2-adrenoceptor antagonist (Newman-Tancredi et al., 1998). Moreover, the mean apparent KB values of yohimbine for this antagonism in vas deferens and brain membranes (10.1 and 1.8 nM respectively) were not significantly different either from each other (Results), or from a previously reported Ki value (5.8 nM) for its binding to human α2A-adrenoceptors (Newman-Tancredi et al., 1998). The hypothesis that cannabigerol can activate α2-adrenoceptors is also supported by data obtained with yohimbine and two selective α2-adrenoceptor agonists, dexmedetomidine and clonidine. These data show that yohimbine possesses similar potency as an antagonist of cannabigerol-, dexmedetomidine- and clonidine-induced inhibition of electrically evoked contractions of the vas deferens (Figure 3) and as an antagonist of cannabigerol and dexmedetomidine, in the [35S]GTPγS binding assay (Figure 5). The evidence that cannabigerol targets a prejunctional site to inhibit electrically evoked contractions of the vas deferens (see previous paragraph) and that prejunctional α2-adrenoceptors can mediate such contractions lends further support to the hypothesis that cannabigerol can activate α2-adrenoceptors. Additional experiments are now required first to establish whether the inhibitory effect of cannabigerol in the vas deferens and its stimulatory effect in the [35S]GTPγS binding assay are mediated by α2A-, α2B- and/or α2C-adrenoceptors, and second to investigate why the potency that cannabigerol displays in these two bioassays is so different (Table 1 and Figures 2 and 3). In the meantime, it is noteworthy that there is evidence that electrically evoked neuronal release of noradrenaline from the vas deferens can be inhibited by the activation not only of α2A-adrenoceptors but also of α2C-adrenoceptors (Scheibner et al., 2001), particularly when the frequency of the electrical stimulation is relatively low as it was in this investigation.
There is also a need for further experiments directed at establishing whether cannabigerol shares the ability of two other plant cannabinoids, Δ9-tetrahydrocannabinol and cannabidiol, to inhibit the neuronal uptake of noradrenaline (see Pertwee, 2008). Thus, our experiments showed first that the noradrenaline uptake inhibitor, maprotiline, can suppress electrically evoked contractions of the mouse isolated vas deferens, and second that yohimbine antagonizes cannabigerol and maprotiline in this bioassay with similar potency. It remains possible, therefore, that cannabigerol inhibits the twitch response of the vas deferens at least in part by blocking the reuptake of released noradrenaline in a manner that causes this catecholamine to accumulate at prejunctional α2-adrenoceptors and so inhibit both its evoked release and that of ATP. If cannabigerol can indeed inhibit noradrenaline reuptake, this would be consistent with a preliminary report that it increases struggling behaviour in the mouse tail suspension test, an indication that it may possess antidepressant activity (Musty and Deyo, 2006). Importantly though, cannabigerol could not have acted in this way to stimulate [35S]GTPγS binding to the mouse brain membranes we used in this investigation, leaving open the possibility that it might be both a potent direct α2-adrenoceptor agonist and an inhibitor of noradrenaline reuptake. It is also noteworthy that it is unlikely that any elevation in extracellular concentration of noradrenaline induced by inhibition of its neuronal uptake would produce much α1-adrenoceptor mediated augmentation of electrically evoked contractions of the vas deferens. Thus, there is evidence that under the stimulation conditions used in this investigation, the contraction amplitude of the vas deferens is determined much more by ATP-induced activation of postjunctional P2X purinoceptors than by noradrenaline-induced activation of postjunctional α1-adrenoceptors (Pertwee et al., 2002).
Whereas cannabigerol was found to display significantly less potency and efficacy than dexmedetomidine as an inhibitor of electrically evoked contractions of the vas deferens (Figure 3), in the [35S]GTPγS binding assay in which both compounds displayed high potency but rather low efficacy (Figure 5), no such differences were detected. Why this should be remains to be established, possible explanations being that different α2-adrenoceptor subtypes, or indeed different types of receptor, mediate the effects of cannabigerol in brain and vas deferens and/or, as just discussed, that inhibition of noradrenaline reuptake may play a significant part in cannabigerol-induced inhibition of evoked contractions of the vas deferens.
The stimulation of [35S]GTPγS binding to mouse brain membranes produced by concentrations of cannabigerol in the low nanomolar range does not appear to be cannabinoid CB1 receptor-mediated. However, evidence was obtained that at higher concentrations, this phytocannabinoid can target the CB1 receptor as an antagonist. Thus, in experiments performed with MF1 mouse brain membranes, cannabigerol was found to reduce specific binding of [3H]CP55940 to MF1 mouse brain membranes (Ki= 381 nM) and, at 10 µM, to antagonize the cannabinoid receptor agonists, anandamide and CP55940, in the [35S]GTPγS binding assay. No significant antagonism of anandamide was induced by 1 µM cannabigerol.
Although the effect of cannabigerol on [35S]GTPγS binding to MF1 mouse brain membranes is stimulatory at 1, 10 and 100 nM, it was found to be insignificant at 1 µM and inhibitory at 10 µM. Why the stimulatory effect of cannabigerol on [35S]GTPγS binding to brain membranes disappears and then changes to an inhibitory effect as its concentration is progressively increased remains to be established. It does seem likely, however, that neither this concentration-dependent loss of the stimulatory effect of cannabigerol nor the inhibitory effect it produces at 10 µM are CB1 receptor-mediated, as both effects were also detectable in CB1−/− C57BL/6J mouse brain membranes. The inhibitory effect produced by 10 µM cannabigerol most probably explains why this concentration of this phytocannabinoid seemed to produce downward as well as right-ward shifts in the log concentration-response curves of anandamide and CP55940. When the component of cannabigerol-induced antagonism that seemed to arise from its ability to inhibit [35S]GTPγS binding to MF1 mouse brain membranes in a seemingly CB1 receptor-independent manner was excluded, significant right-ward shifts in the log concentration response curves of anandamide and CP55940 were still apparent (Figure 8). Importantly, the mean apparent KB values calculated from these dextral shifts did not differ significantly from the mean Ki value of cannabigerol for its displacement of [3H]CP55940 to MF1 mouse brain membranes, lending further support to the hypothesis that cannabigerol is a CB1 receptor antagonist. Cannabigerol reduced specific binding of [3H]CP55940 not only to brain membranes but also to membranes obtained from CHO cells transfected with human CB2 receptors. It did this in manner that suggests it to possess less affinity for CB2 than CB1 receptors. Further experiments are now required to determine whether cannabigerol is a CB2 receptor agonist or antagonist.
Cannabigerol was also found to antagonize the 5-HT1A selective agonist, R-(+)-8-hydroxy-2-(di-n-propylamino)tetralin (8-OH-DPAT), in the [35S]GTPγS binding assay in a seemingly competitive manner. This it could do at concentrations above those at which it induced an apparent α2-adrenoceptor mediated stimulation of [35S]GTPγS binding to mouse brain membranes but below the concentration at which it produced detectable antagonism of anandamide and CP55940 in this bioassay. Cannabigerol therefore resembles the selective α2-adrenoceptor agonists, clonidine and dexmedetomidine, both of which also target 5-HT1A receptors at concentrations above those at which they activate α2-adrenoceptors. However, it differs from these other two compounds in blocking rather than activating the 5-HT1A receptor (Newman-Tancredi et al., 1998). The antagonism of 8-OH-DPAT induced by cannabigerol was presumably CB1 receptor-independent as it was produced with similar potency in experiments performed with CB1−/− C57BL/6J mouse brain membranes, as in experiments performed with CB1+/+ C57BL/6J mouse brain membranes. Interestingly, in contrast to its effects on the log concentration-response curves of anandamide and CP55940, cannabigerol failed to produce a detectable downward displacement of the 8-OH-DPAT log concentration-response curve when it was administered at a concentration (10 µM) that by itself produced a marked inhibition of [35S]GTPγS binding to brain membranes. The reason for this difference remains to be established. The absence of any such downward displacement does, however, suggest that cannabigerol is a neutral 5-HT1A receptor antagonist that does not interact with this receptor as an inverse agonist.
In conclusion, we have obtained evidence from in vitro experiments that cannabigerol is a potent α2-adrenoceptor agonist. This was unexpected as the structure of this plant cannabinoid is unlike that of any established α2-adrenoceptor ligand and as no other cannabinoid has been reported to behave in this way. We have also obtained evidence that cannabigerol can block 5-HT1A and cannabinoid CB1 receptors albeit with a potency lower than that with which it appears to activate α2-adrenoceptors. Further experiments are now required to investigate whether cannabigerol targets any particular subtype of α2-adrenoceptor and whether it inhibits the neuronal uptake of noradrenaline. It will also be important to establish first whether cannabigerol possesses high potency and significant efficacy as an α2-adrenoceptor agonist when administered in vivo, and second whether it displays significant potency in vivo as a 5-HT1A receptor antagonist. It would be of interest as well to establish whether cannabigerol can block CB1 receptors in vivo, although there is already evidence from experiments with rhesus monkeys that at one dose at least (16.5 µg·kg−1 i.v.) cannabigerol does not alter the ability of Δ9-THC to induce in vivo effects that are presumably CB1 receptor-mediated (Mechoulam et al., 1970). In addition, it would be of interest to investigate whether any ability cannabigerol has to activate α2-adrenoceptors in vivo ceases to be detectable at higher doses as happens in vitro. It will also be important to identify which pharmacological actions of cannabigerol are responsible for its reported ability to increase struggling behaviour in the mouse tail suspension test (Musty and Deyo, 2006) or to inhibit human keratinocyte proliferation (Wilkinson and Williamson, 2007), as these effects indicate that cannabigerol may have therapeutic potential as an antidepressant and/or for the treatment of psoriasis. The possibility that cannabigerol has other clinical applications, for example for the production of α2-adrenoceptor-mediated analgesia (Tryba and Gehling, 2002; Giovannoni et al., 2009), also merits investigation.
Acknowledgments
This investigation was supported by grants from GW Pharmaceuticals and the National Institutes of Health (DA-03672).
Glossary
Abbreviations:
- 8-OH-DPAT
R-(+)-8-hydroxy-2-(di-n-propylamino)tetralin
- anandamide
arachidonoyl ethanolamide
- BSA
bovine serum albumin
- β,γ-methylene-ATP
β,γ-methylene adenosine 5′-triphosphate disodium salt
- CHO
Chinese hamster ovary
- CP55940
(–)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol
- DMSO
dimethyl sulphoxide
- GTPγS
guanosine-5′-O-(3-thiotriphosphate)
- R-(+)-WIN55212
R-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo-[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate
- rimonabant
N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide hydrochloride
- WAY100635
N-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-N-2-pyridinylcyclohexanecarboxamide maleate
Conflict of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edn. Br J Pharmacol. 2008;153(Suppl 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbaccia ML, Ravizza L, Costa E. Maprotiline: an antidepressant with an unusual pharmacological profile. J Pharmacol Exp Ther. 1986;236:307–312. [PubMed] [Google Scholar]
- Breivogel CS, Griffin G, Di Marzo V, Martin BR. Evidence for a new G protein-coupled cannabinoid receptor in mouse brain. Mol Pharmacol. 2001;60:155–163. [PubMed] [Google Scholar]
- Cascio MG, Gauson LA, Stevenson LA, Ross RA, Pertwee RG. Cannabigerol behaves as a potent alpha-2-adrenoceptor partial agonist. Symposium on the Cannabinoids. Burlington, Vermont, International Cannabinoid Research Society. 2009. p. 73.
- Cheng Y-C, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Vellani V, Schiano-Moriello A, Marini P, Magherini PC, Orlando P, et al. Plant-derived cannabinoids modulate the activity of transient receptor potential channels of ankyrin type-1 and melastatin type-8. J Pharmacol Exp Ther. 2008;325:1007–1015. doi: 10.1124/jpet.107.134809. [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- ElSohly MA, Slade D. Chemical constituents of marijuana: the complex mixture of natural cannabinoids. Life Sci. 2005;78:539–548. doi: 10.1016/j.lfs.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Forster EA, Cliffe IA, Bill DJ, Dover GM, Jones D, Reilly Y, et al. A pharmacological profile of the selective silent 5-HT1A receptor antagonist, Way-100635. Eur J Pharmacol. 1995;281:81–88. doi: 10.1016/0014-2999(95)00234-c. [DOI] [PubMed] [Google Scholar]
- Gaoni Y, Mechoulam R. Hashish II. The structure and synthesis of cannabigerol, a new hashish constituent. Proc Chem Soc (London) 1964:82. [Google Scholar]
- Gauson LA, Cascio MG, Thomas A, Ross RA, Pertwee RG. Some pharmacological effects of five little-investigated plant cannabinoids. Symposium on the Cannabinoids. Burlington, Vermont, International Cannabinoid Research Society. 2008. p. 143.
- Gauson LA, Cascio MG, Ross RA, Pertwee RG. Cannabigerol displays significant potency as a 5-HT1A receptor antagonist. Symposium on the Cannabinoids. Burlington, Vermont, International Cannabinoid Research Society. 2009. p. 25.
- Giovannoni MP, Ghelardini C, Vergelli C, Dal Piaz V. α2-agonists as analgesic agents. Med Res Rev. 2009;29:339–368. doi: 10.1002/med.20134. [DOI] [PubMed] [Google Scholar]
- Grunfeld Y, Edery H. Psychopharmacological activity of the active constituents of hashish and some related cannabinoids. Psychopharmacologia. 1969;14:200–210. doi: 10.1007/BF00404218. [DOI] [PubMed] [Google Scholar]
- von Kügelgen I, Starke K. Noradrenaline-ATP co-transmission in the sympathetic nervous system. Trends Pharmacol Sci. 1991;12:319–324. doi: 10.1016/0165-6147(91)90587-i. [DOI] [PubMed] [Google Scholar]
- Kurkinen KMA, Koistinaho J, Laitinen JT. 35S]GTP autoradiography allows region-specific detection of muscarinic receptor-dependent G-protein activation in the chick optic tectum. Brain Res. 1997;769:21–28. doi: 10.1016/s0006-8993(97)00663-x. [DOI] [PubMed] [Google Scholar]
- Mechoulam R. Plant cannabinoids: a neglected pharmacological treasure trove. Br J Pharmacol. 2005;146:913–915. doi: 10.1038/sj.bjp.0706415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R, Shani A, Edery H, Grunfeld Y. Chemical basis of hashish activity. Science. 1970;169:611–612. doi: 10.1126/science.169.3945.611. [DOI] [PubMed] [Google Scholar]
- Musty RE, Deyo RA. A cannabigerol extract alters behavioral despair in an animal model of depression. Symposium on the Cannabinoids. Burlington, Vermont, International Cannabinoid Research Society. 2006. p. 32.
- Newman-Tancredi A, Nicolas J-P, Audinot V, Gavaudan S, Verrièle L, Touzard M, et al. Actions of α2 adrenoceptor ligands at α2A and 5-HT1A receptors: the antagonist, atipamezole, and the agonist, dexmedetomidine, are highly selective for α2A adrenoceptors. Naunyn-Schmiedeberg's Arch Pharmacol. 1998;358:197–206. doi: 10.1007/pl00005243. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol Ther. 1997;74:129–180. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Δ9-tetrahydrocannabinol, cannabidiol and Δ9-tetrahydrocannabivarin. Br J Pharmacol. 2008;153:199–215. doi: 10.1038/sj.bjp.0707442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, Griffin G, Lainton JAH, Huffman JW. Pharmacological characterization of three novel cannabinoid receptor agonists in the mouse isolated vas deferens. Eur J Pharmacol. 1995;284:241–247. doi: 10.1016/0014-2999(95)00318-f. [DOI] [PubMed] [Google Scholar]
- Pertwee RG, Fernando SR, Griffin G, Ryan W, Razdan RK, Compton DR, et al. Agonist-antagonist characterization of 6′-cyanohex-2′-yne-Δ8-tetrahydrocannabinol in two isolated tissue preparations. Eur J Pharmacol. 1996;315:195–201. doi: 10.1016/s0014-2999(96)00631-0. [DOI] [PubMed] [Google Scholar]
- Pertwee RG, Ross RA, Craib SJ, Thomas A. (-)-Cannabidiol antagonizes cannabinoid receptor agonists and noradrenaline in the mouse vas deferens. Eur J Pharmacol. 2002;456:99–106. doi: 10.1016/s0014-2999(02)02624-9. [DOI] [PubMed] [Google Scholar]
- Pertwee RG, Thomas A, Stevenson LA, Maor Y, Mechoulam R. Evidence that (-)-7-hydroxy-4′-dimethylheptyl-cannabidiol activates a non-CB1, non-CB2, non-TRPV1 target in the mouse vas deferens. Neuropharmacology. 2005;48:1139–1146. doi: 10.1016/j.neuropharm.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Ross RA, Brockie HC, Stevenson LA, Murphy VL, Templeton F, Makriyannis A, et al. Agonist-inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656 and AM630. Br J Pharmacol. 1999a;126:665–672. doi: 10.1038/sj.bjp.0702351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RA, Gibson TM, Stevenson LA, Saha B, Crocker P, Razdan RK, et al. Structural determinants of the partial agonist-inverse agonist properties of 6′-azidohex-2′-yne-Δ8-tetrahydrocannabinol at cannabinoid receptors. Br J Pharmacol. 1999b;128:735–743. doi: 10.1038/sj.bjp.0702836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RA, Gibson TM, Brockie HC, Leslie M, Pashmi G, Craib SJ, et al. Structure-activity relationship for the endogenous cannabinoid, anandamide, and certain of its analogues at vanilloid receptors in transfected cells and vas deferens. Br J Pharmacol. 2001;132:631–640. doi: 10.1038/sj.bjp.0703850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibner J, Trendelenburg A-U, Hein L, Starke K. Stimulation frequency-noradrenaline release relationships examined in α2A-, α2B- and α2C-adrenoceptor-deficient mice. Naunyn-Schmiedeberg's. Arch Pharmacol. 2001;364:321–328. doi: 10.1007/s002100100432. [DOI] [PubMed] [Google Scholar]
- Schlicker E, Kathmann M. Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol Sci. 2001;22:565–572. doi: 10.1016/s0165-6147(00)01805-8. [DOI] [PubMed] [Google Scholar]
- Starke K. Presynaptic autoreceptors in the third decade: focus on α2-adrenoceptors. J Neurochem. 2001;78:685–693. doi: 10.1046/j.1471-4159.2001.00484.x. [DOI] [PubMed] [Google Scholar]
- Tallarida RJ, Cowan A, Adler MW. pA2 and receptor differentiation: a statistical analysis of competitive antagonism. Life Sci. 1979;25:637–654. doi: 10.1016/0024-3205(79)90505-8. [DOI] [PubMed] [Google Scholar]
- Thomas A, Ross RA, Saha B, Mahadevan A, Razdan RK, Pertwee RG. 6″-Azidohex-2″-yne-cannabidiol: a potential neutral, competitive cannabinoid CB1 receptor antagonist. Eur J Pharmacol. 2004;487:213–221. doi: 10.1016/j.ejphar.2004.01.023. [DOI] [PubMed] [Google Scholar]
- Thomas A, Stevenson LA, Wease KN, Price MR, Baillie G, Ross RA, et al. Evidence that the plant cannabinoid Δ9-tetrahydrocannabivarin is a cannabinoid CB1 and CB2 receptor antagonist. Br J Pharmacol. 2005;146:917–926. doi: 10.1038/sj.bjp.0706414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A, Baillie GL, Phillips AM, Razdan RK, Ross RA, Pertwee RG. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br J Pharmacol. 2007;150:613–623. doi: 10.1038/sj.bjp.0707133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trendelenburg AU, Cox SL, Schelb V, Klebroff W, Khairallah L, Starke K. Modulation of 3H-noradrenaline release by presynaptic opioid, cannabinoid and bradykinin receptors and β-adrenoceptors in mouse tissues. Br J Pharmacol. 2000;130:321–330. doi: 10.1038/sj.bjp.0703305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tryba M, Gehling M. Clonidine – a potent analgesic adjuvant. Curr Opin Anaesthesiol. 2002;15:511–517. doi: 10.1097/00001503-200210000-00007. [DOI] [PubMed] [Google Scholar]
- Wilkinson JD, Williamson EM. Cannabinoids inhibit human keratinocyte proliferation through a non-CB1/CB2 mechanism and have a potential therapeutic value in the treatment of psoriasis. J Dermatol Sci. 2007;45:87–92. doi: 10.1016/j.jdermsci.2006.10.009. [DOI] [PubMed] [Google Scholar]