

Abstract
Background and purpose:
3,4-Methylenedioxymethamphetamine (MDMA) and cocaine are two widely abused psychostimulant drugs targeting the dopamine transporter (DAT). DAT availability regulates dopamine neurotransmission and uptake of MDMA-derived neurotoxic metabolites. We aimed to determine the effect of cocaine pre-exposure on the acute and long-term effects of MDMA in mice.
Experimental approach:
Mice received a course of cocaine (20 mg·kg−1, ×2 for 3 days) followed by MDMA (20 mg·kg−1, ×2, 3 h apart). Locomotor activity, extracellular dopamine levels and dopaminergic neurotoxicity were determined. Furthermore, following the course of cocaine, DAT density in striatal plasma membrane and endosome fractions was measured.
Key results:
Four days after the course of cocaine, challenge with MDMA attenuated the MDMA-induced striatal dopaminergic neurotoxicity. Co-administration of the protein kinase C (PKC) inhibitor NPC 15437 prevented cocaine protection. At the same time, after the course of cocaine, DAT density was reduced in the plasma membrane and increased in the endosome fraction, and this effect was prevented by NPC 15437. The course of cocaine potentiated the MDMA-induced increase in extracellular dopamine and locomotor activity, following challenge 4 days later, compared with those pretreated with saline.
Conclusions and implications:
Repeated cocaine treatment followed by withdrawal protected against MDMA-induced dopaminergic neurotoxicity by internalizing DAT via a mechanism which may involve PKC. Furthermore, repeated cocaine followed by withdrawal induced behavioural and neurochemical sensitization to MDMA, measures which could be indicative of increased rewarding effects of MDMA.
Keywords: 3,4-methylenedioxymethamphetamine (MDMA, ‘ecstasy’); cocaine; neurotoxicity; dopamine transporter; protein kinase C; trafficking; sensitization
Introduction
The dopamine transporter (DAT) is crucial for the temporal and spatial shaping of dopaminergic neurotransmission in the nigrostriatal and mesocorticolimbic dopaminergic pathways (Giros et al., 1996). It is situated in the pre-synaptic membrane of dopaminergic nerve terminals, and is responsible for the rapid removal of dopamine released into the synaptic cleft upon neuronal stimulation (Amara and Kuhar, 1993; Giros and Caron, 1993; Norregaard and Gether, 2001). As a consequence, the transporter plays a critical role in regulating the availability of dopamine in the synaptic cleft and thus, in modulating the physiological functions of dopamine, including locomotor activity, higher cognitive functions and neuroendocrine systems (Amara and Kuhar, 1993; Giros and Caron, 1993), and the rewarding effects of drugs of abuse. A central concept in drug abuse research is that increased dopamine release in limbic brain regions is associated with the reinforcing effects of drugs (Di Chiara and Imperato, 1988; Koob and Bloom, 1988), although recent studies have described an important role for dopamine release in the dorsal striatum in mediating drug seeking after psychostimulant abstinence (Everitt and Robbins, 2005; See et al., 2007).
3,4-Methylenedioxymethamphetamine (MDMA, ‘ecstasy’) has become a widely used recreational drug of abuse among young people, despite having been shown to be a potent neurotoxin in the brains of rodents and non-human primates (Green et al., 2003). MDMA enters the dopaminergic terminal by diffusion and induces a rapid, Ca2+-dependent release of dopamine (Camarero et al., 2002). MDMA produces dose-dependent and species-specific relatively selective long-term neurotoxic damage. In mice, this neurotoxicity is dopamine specific, reflected by a sustained loss in the concentration of dopamine and its metabolites, in tyrosine hydroxylase (TH) immunoreactivity and in the density of DAT principally in the nigrostriatal pathway (Escobedo et al., 2005; Granado et al., 2008a,b;). MDMA-induced dopaminergic neurotoxicity appears to be produced by a systemically generated metabolite of the drug (Escobedo et al., 2005) that enters the dopaminergic nerve terminal through DAT, as the administration of the cocaine analogue GBR 12,909 immediately before MDMA blocks entry of the metabolite into the terminal and provides protection (O'Shea et al., 2001; Camarero et al., 2002).
Cocaine is another widely used drug of abuse which, in the USA, is consumed by 43.8% of MDMA users (Wish et al., 2006). It is a monoamine uptake inhibitor, and its psychomotor and rewarding effects are attributed primarily to its ability to block the re-uptake of dopamine, thereby causing a marked increase in extracellular dopamine (Ritz et al., 1987; Johanson and Fischman, 1989; Johnson et al., 2006; Tilley and Gu, 2008). Controversy exists about the effect of repeated exposure to cocaine on DAT. In the existing literature, DAT expression/activity has been shown to either increase, decrease or remain unchanged in dorsal and ventral striatum following cocaine exposure (Izenwasser and Cox, 1990; Kula and Baldessarini, 1991; Sharpe et al., 1991; Farfel et al., 1992; Staley et al., 1994; Wilson et al., 1994; Hitri et al., 1996; Letchworth et al., 1997; Samuvel et al., 2008). Differences in administration schedules (continuous infusion vs. intermittent, self-administration vs. passive administration), doses and duration of drug treatment and withdrawal have all contributed to these inconsistent observations in different species and cerebral areas. However, when differences are observed, it appears that trafficking events occur (Jayanthi and Ramamoorthy, 2005), as no changes in total protein DAT have been found (Samuvel et al., 2008).
Thus, considering that both drugs of abuse, cocaine and MDMA, target DAT which in turn plays an important role in drug abuse, we aimed to determine if cocaine pre-exposure and withdrawal might alter MDMA neurotoxicity by modifying DAT density via a protein kinase C (PKC)-mediated mechanism. Furthermore, we sought to evaluate if cocaine pre-exposure and withdrawal altered MDMA-induced locomotor activity and dopamine release in an attempt to determine if cross-sensitization occurred.
Our results showed that repeated treatment with cocaine followed by withdrawal produces DAT internalization which protected against MDMA-induced dopaminergic neurotoxicity by a mechanism which appeared to involve PKC. Furthermore, repeated treatment with cocaine followed by withdrawal induced behavioural and neurochemical sensitization to the acute effects of MDMA.
Methods
Animals, drug administration and experimental design
All animal care and experimental procedures were performed in accordance with the guidelines of the Animal Welfare Committee of the Universidad Complutense de Madrid (following European Council Directive 86/609/EU). Adult male NIH/Swiss mice (Harlan Laboratories Models S.L., Barcelona, Spain) weighing 20–30 g were group housed in conditions of constant temperature (21 ± 2°C) and a 12 h light/dark cycle (lights on: 0800 h) with ad libitum access to food and water.
The animals were injected with cocaine (20 mg·kg−1, i.p.) twice daily (injections separated by 8 h) for 3 days. The dose was chosen based on the work of Koff et al. (1994), and dosing began shortly after the start of the daily light cycle (equivalent to early evening for humans; Schlussman et al., 1998). One or 4 days after finishing cocaine treatment, the animals were either killed for the determination of DAT in plasma membranes or treated with MDMA (20 mg·kg−1, i.p., two injections separated by 3 h) and killed 7 days later for the determination of neurotoxicity. This regimen of MDMA administration has previously been shown to produce approximately 50% reductions in striatal DAT density and dopamine content in NIH/Swiss mice (Colado et al., 2001) parameters which are considered to reflect neurotoxicity (see Colado et al., 2004). For the determination of TH activity, an additional marker of neurotoxicity, 7 days after receiving MDMA some of the mice were given the l-aromatic amino acid decarboxylase inhibitor 3-hydroxybenzylhydrazine (NSD-1015, 100 mg·kg−1, i.p.; Carlsson et al., 1972) 30 min before being killed for the determination of l-3,4-dihydroxyphenylalanine (l-DOPA) concentration which at steady state, can be considered to equate to the rate of dopamine synthesis.
In a separate experiment, the effects of the selective PKC inhibitor 2,6-diamino-N-[(1-(1-oxotridecyl)-2-piperidinyl)methyl] hexanamide dihydrochloride hydrate (NPC 15437; 1 mg·kg−1, i.p., twice daily; Sullivan et al., 1991a,b; Saraiva et al., 2003) were assessed on the neuroprotection afforded by repeated treatment with cocaine finishing 4 days before MDMA. The dose of NPC 15437 was selected based on studies demonstrating 1 mg·kg−1 to be an effective dose in mice with no adverse effects (Mathis et al., 1992). The following groups were tested: (i) control group; (ii) MDMA-treated group; (iii) cocaine finishing 4 days before MDMA; (iv) cocaine + NPC (during cocaine treatment and withdrawal) + MDMA; (v) cocaine + NPC (during cocaine treatment only) + MDMA; (vi) cocaine + NPC (during cocaine withdrawal only) + MDMA; (vii) NPC (during saline pretreatment and withdrawal) + MDMA; and (viii) NPC + saline. In addition, the effect of NPC 15437 given during cocaine treatment and withdrawal was evaluated on DAT density in plasma membrane and endosome fractions 4 days later. Separate groups of mice were pretreated with cocaine and used for the determination of locomotor activity and striatal extracellular dopamine levels following MDMA.
The entire study was carried on dorsal striatum because this structure is the most representative MDMA toxicity target (Granado et al., 2008b), and it plays a critical role in cocaine seeking after abstinence (See et al., 2007).
[3H]WIN 35 428 binding
[3H]WIN 35 428 binding was used as a measure of DAT density. In neurotoxicity studies, a reduction in [3H]WIN 35 428 binding is considered to reflect MDMA-induced dopaminergic terminal loss as confirmed by TH immunohistochemical studies. Following cocaine treatment changes in [3H]WIN 35 428 binding are taken as a measure of DAT trafficking (Little et al., 2002) as cocaine does not produce dopaminergic neurotoxicity (Bennett et al., 1993; Goodman and Sloviter, 1993).
Neurotoxicity studies
[3H]WIN 35 428 binding was measured in striatal membranes by modification of the method described by Segal et al. (2003). Striata from individual animals were sonicated in ice-cold sodium phosphate buffer (20 mM; pH 7.4) containing sucrose (0.32 M). The homogenate was centrifuged at 30 000×g for 15 min at 4°C. The supernatant was discarded, and the wash procedure was repeated twice more. The pellet was finally resuspended in 80 volumes of homogenization buffer.
Transporter trafficking studies
Membrane and endosome fractions were prepared following a modified method of Wang et al. (2005). Striata were sonicated as above and centrifuged at 20 000×g for 20 min at 4°C. The supernatant (endosome fraction) was collected, and the pellet (membrane fraction) was washed twice more and the supernatants joined with the endosome fraction. This endosome fraction was centrifuged at 200 000×g for 60 min at 4°C, and the supernatant was discarded. Both the ‘membrane fraction pellet’ and ‘endosome fraction pellet’ were finally resuspended in 30 volumes of homogenization buffer.
The assay solution contained [3H]WIN 35 428 (5 nM), desipramine (300 nM; to block binding to the noradrenaline transporter) and tissue preparation (approx. 60 µg protein). Non-specific binding was carried out in the presence of cocaine (30 µM). The reaction mixture was incubated for 90 min at 4°C. The assay was terminated by rapid filtration, and radioactivity was counted by scintillation spectrometry. Protein concentrations were measured by the method of Lowry et al. (1951).
Determination of tissue catechols
Striatal catechol concentration was evaluated as a marker of dopamine neurotoxicity, 7 days after MDMA administration. The mice were killed by cervical dislocation and decapitation, the brains rapidly removed and the striatum dissected out on ice. Individual striata were homogenized in HClO4 (0.2 M) containing cysteine (0.1%), sodium metabisulphite (0.1%) and EDTA (0.01%), and centrifuged at 12 000×g for 20 min at 4°C. Dopamine, 3,4-dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA) and l-DOPA were measured byhigh-performance liquid chromatography (HPLC) and electrochemical detection.
Implantation of a microdialysis probe in the striatum and collection of samples
The mice were anaesthetized with sodium pentobarbitone (40 mg·kg−1, Dolethal, Vetoquinol, Madrid, Spain), and secured in a Kopf stereotaxic frame (David Kopf Instruments, Tujunga, CA, USA) coupled to a mouse adapter. A guide cannula (CMA/7 Guide Cannula, CMA Microdialysis AB, Solna, Sweden) was implanted just above the right striatum +0.70 mm anterioposterior and −1.9 mm mediolateral from bregma, and 2.5 mm below the skull surface (Franklin and Paxinos, 1997). The cannula was secured to the skull as described by Izco et al. (2007).
The mice were allowed to recover for at least 24 h before receiving cocaine pretreatment. Four days later, the dialysis probes (membrane: 2 mm × 240 µm; CMA/7) were inserted in the guide cannulae such that the membrane protruded its full length from the end of the probe into the dorsal striatum. Probes were perfused with artificial cerebrospinal fluid (KCl 2.5 mM; NaCl 125 mM; MgCl2.6H2O 1.18 mM; CaCl2.2H2O 1.26 mM; pH 6–7) at a rate of 1 µL·min−1, and samples collected from the freely moving animals at 30 min intervals in tubes containing 5 µL of a solution composed of HClO4 (0.01 M), cysteine (0.2%) and sodium metabisulphite (0.2%). Following a 60 min stabilization period, three 30 min baseline samples were collected before the first MDMA or saline injection. The samples were collected for up to 3 h after the second MDMA injection. Dopamine, DOPAC and HVA were measured by HPLC and electrochemical detection.
At the completion of experiments, the animals were killed by cervical dislocation and decapitation, and the brains were rapidly removed and frozen in dry ice. Coronal sections (25 µm) were cut at the level of the striatum using a cryostat. The sections were mounted on slides and stained with cresyl violet to verify correct probe placement. Only data obtained from probes correctly placed in the striatum were included in the analysis.
Measurement of dopamine and metabolites in striatal tissue and dialysates
The mobile phase consisted of KH2PO4 (0.05 M), octanesulphonic acid (1 mM), EDTA (0.1 mM) and methanol (16%), and was adjusted to pH 3.7 with phosphoric acid, filtered and degassed. The flow rate was 0.3 mL·min−1 for dialysates, and 1 mL·min−1 for tissue. The HPLC system consisted of a pump (Waters 510, Barcelona, Spain) linked to a manual injector (Loop 20 µL, Rheodyne, Rohnert Park, CA, USA) for dialysates, and an automatic sample injector (Loop 200 µL, Waters 717 plus Autosampler) for tissue, a stainless steel reversed-phase column (Spherisorb ODS2, 3 µm, 2.1 × 150 mm for dialysates and 5 µm, 150 × 4.6 mm for tissue; Waters, Chelmsford, MA, USA) with a precolumn and a coulometric detector (Coulochem II, Esa, USA). The working electrode potential was set at 400 mV with a gain of 100 nA or 2 µA (for dopamine in dialysates and tissue, respectively) and 500 nA (for the rest in both types of samples). The current produced was monitored by using integration software (Clarity, DataApex, Prague, Czech Republic).
Measurement of MDMA concentration in striatal tissue
Brain concentrations of MDMA were determined following a previously described method with minor modifications (Sanchez et al., 2001). The striatal tissue was homogenized in ice-cold sodium carbonate–sodium bicarbonate buffer (pH 11.5) using an ultrasonicator. The homogenate was centrifuged at 27 000×g for 20 min at 4°C. The supernatant was applied to a 145 mg C8 end-capped SPE light column (International Sorbent Technology, Waters, Barcelona, Spain). The column was washed with methanol followed by distilled water before applying the sample. The column was washed with water (2 mL) before selective elution of MDMA with methanol (1 mL).
An aliquot (20 µL) of the resulting eluate was injected into a Waters HPLC system which consisted of a pump (Waters 510) linked to a manual sample injector (Loop 20 µL), a stainless steel column (RP 18, 5 µm, 150 × 4.6 mm, XTerra) fitted with a pre-column (RP 18, 5 µm, 20 × 3.9 mm, XTerra) and a UV/visible detector (Waters 2487). The mobile phase consisted of 20 mM potassium dihydrogen phosphate (75%) and acetonitrile (25%), pH 2.5; the flow rate was set to 0.8 mL·min−1, and UV absorption was measured at 235 nm. The current produced was monitored by using integration software (Clarity, DataApex).
Measurement of rectal temperature
Immediately before and up to 6 h after the first MDMA administration, temperature was measured by use of a digital readout thermocouple (BAT-12 thermometer, Physitemp Instruments, Clifton, NJ, USA) with a resolution of 0.1°C and an accuracy of ±0.1°C attached to a RET-3 Rodent Sensor which was inserted 2 cm into the rectum of the mouse, the animal being lightly restrained by holding in the hand. A steady readout was obtained within 10 s of probe insertion.
Measurement of locomotor activity
The test was performed in an open arena (26 × 21 × 10 cm) with bottom and sides made of methacrylate covered with a brown non-reflecting material. The behaviour of the mice was recorded in eight arenas run in parallel using a video camera (model CCD-IRIS with a 1:1.4 lens, Sony, Tokyo, Japan) placed above the arenas and connected to a video monitor (model PVM-145E, Sony) and recorder (model AG-5700, Panasonic, Barcelona, Spain). Diffuse lighting was used to minimize shadows in the arenas.
Automated analysis of the videotapes was conducted off-line by the EthoVision programme (version 1.70, Noldus Information Technologies, Barcelona, Spain) which allowed the determination of horizontal distance travelled (cm) during the total observation period (30 min) and in 10 min intervals.
Data analysis and statistical procedures
Locomotor activity and neurochemical data were analysed using a one-way analysis of variance (anova) followed by Newman–Keuls multiple comparison test. Data from brain MDMA concentrations and baseline microdialysis concentrations were analysed using a t-test. Temperature and microdialysis data were analysed by two-way anova with repeated measures using treatment as the between-subjects factor and time as the repeated measure (BMDP/386 Dynamic, BMDP Statistical Solutions, Cork, Ireland).
Materials
(±)-MDMA.HCl was obtained from Ultrafine Chemicals Ltd. (Manchester, UK), and cocaine HCl was supplied by Servicio de Estupefacientes (Ministerio de Sanidad y Consumo, Madrid, Spain). Both were dissolved in 0.9% w/v NaCl (saline) and injected i.p. in a volume of 10 mL·kg−1. Doses are quoted in terms of the base. All control animals were treated with saline. [3H]WIN 35 428 (specific activity = 85.9 Ci·mmol−1) was obtained from PerkinElmer, Spain. Desipramine, NSD-1015, NPC 15437 and HPLC standards were obtained from Sigma-Aldrich (Madrid, Spain). Methanol and HClO4 were obtained from Merck Chemicals (Madrid, Spain). Acetonitrile was obtained from Panreac Quimica (Barcelona, Spain). All other chemical reagents were obtained from Sigma-Aldrich.
Results
Effect of pretreatment with repeated cocaine on MDMA-induced hyperthermia
In order to rule out modifications in MDMA-induced hyperthermia as responsible for alterations in MDMA-induced neurotoxicity, rectal temperature was measured for up to 3 h after the second injection.
MDMA produced a peak hyperthermic response 30–60 min after each injection of between 1 and 2°C which was maintained for 2 h after the second injection (Figure 1A). Pretreatment with cocaine finishing either 1 or 4 days before MDMA did not alter this hyperthermic response.
Figure 1.
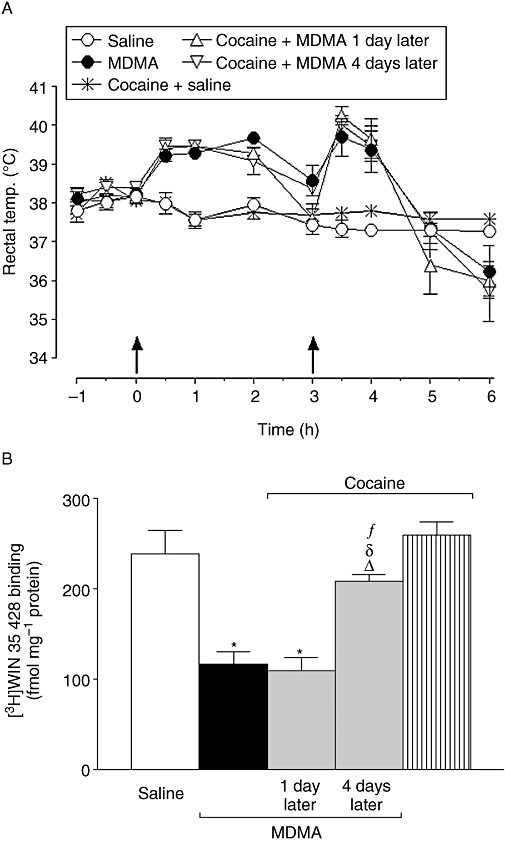
Effect of pretreatment with repeated cocaine (20 mg·kg−1, i.p. twice daily separated by 8 h for 3 days) finishing 1 or 4 days before 3,4-methylenedioxymethamphetamine (MDMA) (20 mg·kg−1, i.p., two injections separated 3 h) on (A) rectal temperature immediately following MDMA administration and (B) [3H]WIN 35 428 binding. Data shown as mean ± SEM, n= 5 − 11. There were no differences between the data obtained from saline controls pretreated 1 or 4 days earlier with cocaine and therefore data were grouped. (A) The MDMA-induced increase in rectal temperature (F1,11= 54.79; P < 0.001) was not modified by pretreatment with cocaine 1 day (F1,13= 0.97; P= 0.32, n.s.) or 4 days (F1,11= 0.93; P= 0.33, n.s.) before. Cocaine pretreatment did not modify the temperature of saline-treated animals (F1,15= 3.64; P= 0.06, n.s.). Two-way analysis of variance (anova). (B) Different from saline: *P < 0.001; different from MDMA: ΔP < 0.001; different from cocaine + MDMA 1 day later: δP < 0.001; different from cocaine + saline: fP < 0.05. One-way anova followed by Newman–Keuls test.
Effect of pretreatment with repeated cocaine on MDMA-induced dopamine neurotoxicity
Seven days after treatment, MDMA produced a marked reduction in the density of [3H]WIN 35 428-labelled DAT (Figure 1B). In addition, MDMA treatment produced a decrease in the concentration of dopamine and its metabolites, DOPAC and HVA, as well as in the concentration of l-DOPA following l-aromatic amino acid decarboxylase inhibition, reflecting a reduction in TH activity (Table 1).
Table 1.
Effect of pretreatment with repeated cocaine (20 mg·kg-1, i.p. twice daily separated by 8 h for 3 days) finishing 1 or 4 days before 3,4-methylenedioxymethamphetamine (MDMA) (20 mg·kg-1, i.p., two injections separated 3 h) on dopamine (DA), DOPAC, HVA and l-DOPA concentration 7 days later
| Treatment | DA | DOPAC | HVA | l-DOPA |
|---|---|---|---|---|
| Saline | 9828 ± 251 | 767 ± 19 | 853 ± 51 | 1386 ± 44 |
| MDMA | 5312 ± 392*** | 330 ± 58*** | 578 ± 49*** | 920 ± 48*** |
| Cocaine + MDMA 1 day later | 5074 ± 517*** | 365 ± 59*** | 572 ± 25*** | 749 ± 66*** |
| Cocaine + MDMA 4 days later | 8342 ± 394*, ΔΔ, δδ, ff | 532 ± 39***, ΔΔ, δδ, ff | 758 ± 61δ, Δ, f | 1137 ± 64**, Δ, δδ, ff |
| Cocaine | 10 259 ± 274 | 717 ± 29 | 931 ± 46 | 1455 ± 54 |
Data represented in ng·(g tissue)−1 and are means ± SEM; n= 5 − 11. There were no differences between the data obtained from saline controls pretreated 1 or 4 days earlier with cocaine and therefore data were grouped. There were no differences between the data obtained from saline controls pretreated 1 or 4 days earlier with cocaine and therefore data were grouped.
Different from saline:
P < 0.05,
P < 0.01,
P < 0.001; different from MDMA:
P < 0.01,
P < 0.001; different from cocaine + MDMA 1 day later:
P < 0.01,
P < 0.001; different from cocaine + saline:
P < 0.05,
P < 0.001. One-way anova followed by Newman–Keuls test.
Cocaine pretreatment finishing 4 days before MDMA administration attenuated the decreases in the dopaminergic parameters produced by MDMA (Figure 1B, Table 1). However, the same regimen of cocaine pretreatment finishing 1 day before MDMA administration failed to protect against MDMA neurotoxicity.
Effect of pretreatment with repeated cocaine on striatal MDMA concentration
Pretreatment with cocaine finishing 1 or 4 days before MDMA administration did not modify striatal concentration of MDMA compared with the concentration obtained in animals pretreated with saline 30 min after MDMA injection (nmol·g−1 tissue: 56.5 ± 3.2 vs. 55.2 ± 2.6 for saline and cocaine pretreated 1 day before MDMA, respectively, P= 0.78, n.s.; 50.7 ± 3.4 vs. 49.8 ± 5.3 for saline and cocaine pretreated 4 days before MDMA, respectively, P= 0.89, n.s.; t-test).
Effect of PKC inhibition on the neuroprotection against MDMA induced by repeated cocaine
The PKC inhibitor NPC 15437 was co-administered during the cocaine pretreatment and continued to be injected twice daily during the 3-day interval until MDMA administration. Separate groups of animals were treated with the inhibitor only during the 3 days of cocaine pretreatment or only during the 3 days of cocaine withdrawal.
NPC 15437 co-administration during cocaine pretreatment did not alter the hyperthermic response produced by MDMA administration (data not shown). Seven days after MDMA, there was a marked reduction in the striatal density of DAT (Figure 2A) and in the concentration of dopamine (Figure 2B). Cocaine pretreatment 4 days before MDMA administration inhibited the neurotoxic effects of MDMA. The co-administration of the PKC inhibitor NPC 15437 during the cocaine pretreatment and during the cocaine withdrawal period attenuated this protective effect of cocaine (Figure 2A,B). NPC administered only during cocaine pretreatment did not alter the protective effects of cocaine pretreatment. However, NPC administered only during the withdrawal period showed a slight inhibition of the protective effects of cocaine pretreatment.
Figure 2.
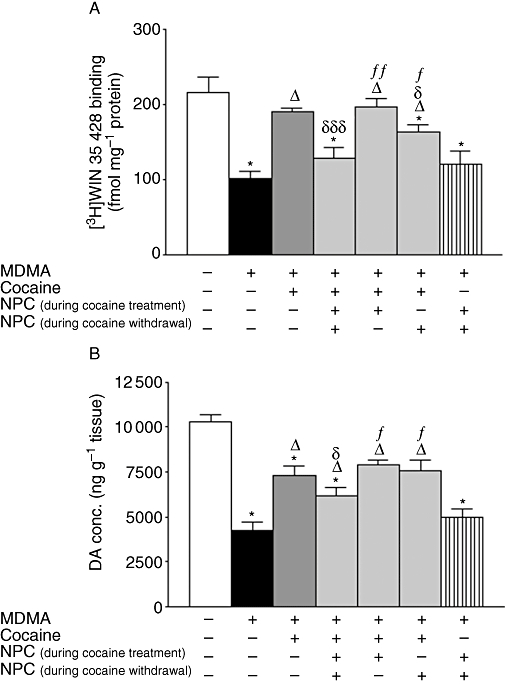
Effect of co-administration of NPC 15437 (1 mg·kg−1, i.p.) and cocaine (20 mg·kg−1, i.p. twice daily separated by 8 h for 3 days) finishing 4 days before 3,4-methylenedioxymethamphetamine (MDMA) (20 mg·kg−1, i.p., two injections separated 3 h) on striatal (A) [3H]WIN 35 428 binding and (B) dopamine (DA) concentration, 7 days later. Data shown as mean ± SEM, n= 6 − 11. Different from saline *P < 0.001; different from MDMA: ΔP < 0.001; different from cocaine + MDMA: δP < 0.05, δδδP < 0.001; different from NPC (during cocaine treatment + withdrawal) + cocaine + MDMA: fP < 0.01, ffP < 0.001. One-way anova followed by Newman–Keuls test.
NPC 15437 administration did not modify MDMA-induced neurotoxicity (Figure 2A,B). Treatment of saline-pretreated animals with NPC 15437 did not modify DAT density nor dopamine content compared with saline-treated controls [for DAT, as fmol·(mg protein)−1; 210 ± 23 vs. 235 ± 14, n= 5–6, P= 0.35, t-test, and for dopamine, as ng·(g tissue)−1: 9259 ± 211 vs. 8811 ± 643, n= 5–6, P= 0.56, t-test].
Effect of repeated treatment with cocaine and PKC inhibition on DAT density in the plasma membrane and endosome fractions
Cocaine treatment produced a reduction in the density of DAT in the plasma membrane (Figure 3A) and a corresponding increase in the endosome fraction (Figure 3B) 4 days after treatment compared with animals treated with saline. This effect was prevented by the PKC inhibitor NPC 15437 administered during the cocaine pretreatment and 3 days withdrawal period. No change was observed in the density of DAT in either fraction, 1 day after finishing cocaine treatment. NPC 15437 administered during saline pretreatment did not modify DAT distribution in control animals [[3H]WIN 35 428 binding, as fmol·(mg protein)−1 for saline- and NPC-treated animals, respectively: in the membrane fraction, 291 ± 31 vs. 326 ± 20 (n= 7, P= 0.35, t-test) and in the endosome fraction 541 ± 78 vs. 508 ± 78 (n= 7, P= 0.77, t-test)].
Figure 3.
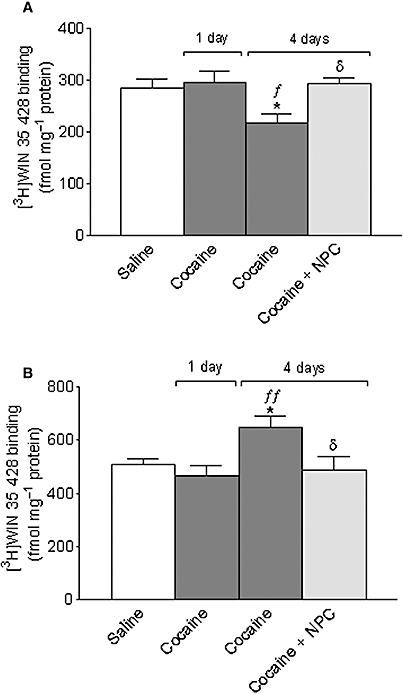
Effect of repeated cocaine (20 mg·kg−1, i.p. twice daily separated by 8 h for 3 days) on striatal plasma membrane (A) and endosome (B) [3H]WIN 35 428 binding 1 or 4 days later. A group of animals were co-administered NPC 15437 (1 mg·kg−1, i.p.) with cocaine and twice daily during the 3-day withdrawal. Data shown as mean ± SEM, n= 7 − 10. Different from saline: *P < 0.01; different from cocaine 1 day before: fP < 0.05, ffP < 0.01; different from cocaine only 4 days before: δP < 0.05. One-way anova followed by Newman–Keuls test.
Effect of pretreatment with repeated cocaine on MDMA-induced locomotor activity
MDMA produced an increase in locomotor activity measured during the 30 min after injection. Cocaine pretreatment finishing 4 days before MDMA treatment potentiated this increase in locomotor activity (Figure 4). Cocaine pretreatment finishing 1 day before MDMA did not modify the MDMA-induced increase in locomotor activity.
Figure 4.
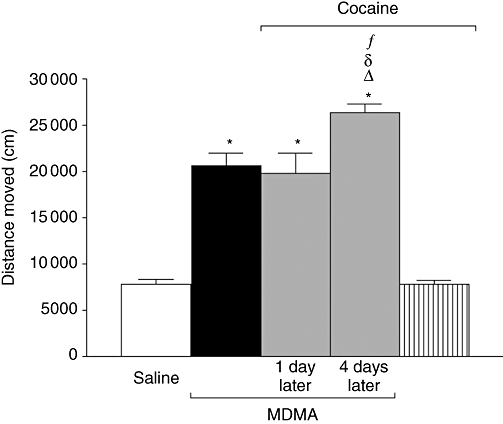
Effect of pretreatment with repeated cocaine (20 mg·kg−1, i.p. twice daily separated by 8 h for 3 days) finishing 1 or 4 days before 3,4-methylenedioxymethamphetamine (MDMA) (20 mg·kg−1, i.p., two injections separated 3 h) on horizontal locomotor activity measured during the 30 min immediately after the second MDMA injection. Data shown as mean ± SEM, n= 5 − 11. There were no differences between the data obtained from saline controls pretreated 1 or 4 days earlier with cocaine and therefore data were grouped. Different from saline: *P < 0.001; different from MDMA: ΔP < 0.001; different from cocaine + MDMA 1 day later: δP < 0.001; different from cocaine + saline: fP < 0.001. One-way anova followed by Newman–Keuls test.
Effect of pretreatment with repeated cocaine before MDMA on extracellular striatal dopamine and metabolite levels
Given the potentiation of MDMA-induced locomotor activity observed 4 days after cocaine pretreatment, extracellular striatal dopamine levels were measured at this time using intracerebral microdialysis. Interestingly, cocaine pretreatment produced a decrease in the basal extracellular dopamine levels (Figure 5 bar graph), although it did not affect basal extracellular metabolite levels (Figure 5 legend).
Figure 5.
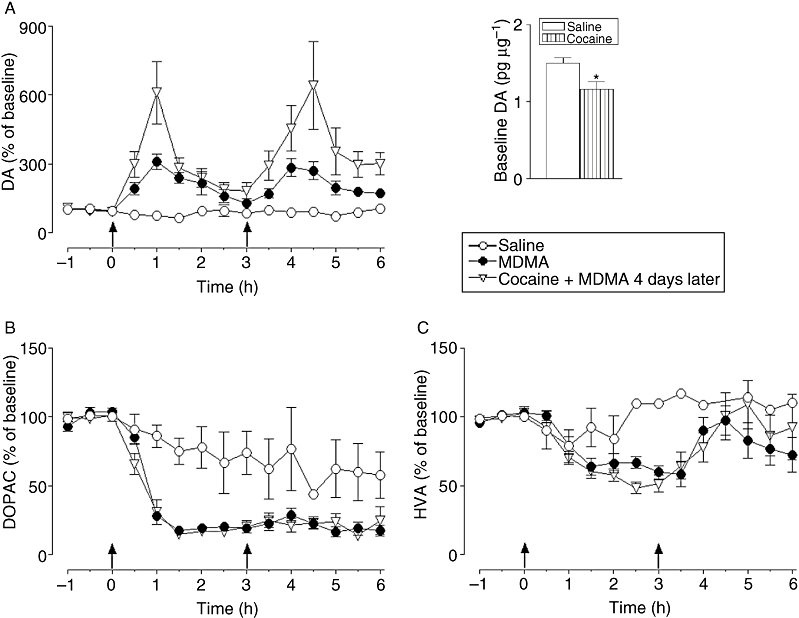
Effect of repeated cocaine (20 mg·kg−1, i.p. twice daily separated by 8 h for 3 days) finishing 4 days before on 3,4-methylenedioxymethamphetamine (MDMA) (20 mg·kg−1, i.p., two injections separated 3 h)-induced changes in extracellular (A) dopamine (DA), (B) DOPAC and (C) HVA concentration in striatum. MDMA increased extracellular levels of dopamine [F1,11= 37.8, P < 0.001] and decreased levels of DOPAC [F1,12= 79.05, P < 0.001] and HVA [F1,10= 3.61, P < 0.05] compared with saline-treated animals. Pretreatment of the animals with cocaine finishing 4 days before MDMA potentiated this increase in extracellular dopamine levels [F1,19= 7.53, P < 0.01], but did not modify levels of DOPAC [F1,18= 0.75, P= 0.36; n.s.] or HVA [F1,17= 0.20, P= 0.64, n.s.]. Data expressed as % of baseline values. Results represent mean ± SEM, n= 4 − 11; two-way anova. Basal dopamine concentrations were lower in mice pretreated with cocaine compared with saline-treated mice (bar graph). Different from saline-pretreated mice: *P < 0.05 (t-test). Basal DOPAC and HVA levels were similar between groups, for DOPAC (pg·µL−1): 182.0 ± 16.2 versus 185.0 ± 20.0 for saline- and cocaine-pretreated mice, respectively (P= 0.91, n.s., t-test) and for HVA (pg·µL−1): 117.7 ± 13.1 versus 118.8 ± 13.5 for saline- and cocaine-pretreated mice, respectively (P= 0.95, n.s., t-test).
MDMA produced a rapid rise in extracellular striatal levels of dopamine of approx. 300% which peaked 60 min after each of the two injections (Figure 5A). Cocaine pretreatment 4 days before MDMA treatment potentiated this increase in extracellular dopamine levels after each of the MDMA injections producing increases of approx. 600% above saline.
In contrast, MDMA treatment produced a rapid and marked reduction in extracellular DOPAC (Figure 5B), and a slightly more delayed and less extensive reduction in extracellular HVA (Figure 5C). Cocaine pretreatment did not modify the MDMA-induced changes in either of the two dopamine metabolites.
Discussion
In the current study, we have shown for the first time that a repeated regimen of cocaine finishing 4 days before MDMA treatment protected against MDMA-induced dopaminergic neurotoxicity in mouse striatum by producing down-regulation of DAT away from the cell surface by a mechanism which appears to involve PKC. In addition, cocaine treatment and withdrawal increased extracellular dopamine concentrations and locomotor activity following challenge with MDMA.
DAT uptake of a systemically derived metabolite is an essential step in the induction of MDMA dopaminergic neurotoxicity in the mouse (O'Shea et al., 2001; Camarero et al., 2002; Escobedo et al., 2005). However, the protection afforded by cocaine 4 days after its administration is unlikely to be due to a prolonged block of DAT as no protection was observed 1 day after its administration. In addition, cocaine and its metabolites are relatively rapidly cleared from the body (Benuck et al., 1987). Moreover, the protection was not due to a change in the disposition of MDMA as striatal levels of the drug were similar in saline- and cocaine-pretreated animals, as was the hyperthermia following MDMA, thus ruling out changes in the hyperthermic response as a possible mechanism of neuroprotection. Rather, the protection seemed to agree with a reduction in the available DAT on the plasma membrane, a fact confirmed by a reduction in [3H]WIN 35 428-labelling of the striatal membranes and a corresponding increase in the endosome fraction, 4 days after finishing repeated cocaine treatment. This effect was not observed in the fractions obtained 1 day after finishing the repeated cocaine treatment.
Considerable evidence indicates a key role of PKC in regulating the activity and availability of DAT at the cell surface (Blakely and Bauman, 2000; Chang et al., 2001; Cervinski et al., 2005). Our results support a role for this kinase in the cocaine-induced decrease in cell surface expression of the transporter as administration of the PKC inhibitor, NPC 15437, in combination with cocaine pretreatment abolished both the protection against MDMA-induced neurotoxicity and prevented DAT trafficking between the plasma membrane and endosome, 4 days after finishing repeated cocaine treatment. Interestingly, the PKC inhibitor had to be administered not only during the time of cocaine exposure, but also during the withdrawal period of 3 days for the reversal of protection to be achieved. Further studies are required to fully explore the relative importance of the signalling pathway during the exposure and withdrawal periods.
PKC regulates DAT internalization by clarithin-mediated and dynamin-dependent cellular mechanisms, resulting in accumulation of the transporter in early endosomes where it is colocalized with transferrin (Daniels and Amara, 1999). Although there is evidence that internalized DAT may be targeted to endosomal/lysomal pathways for degradation (Daniels and Amara, 1999), other studies indicate that internalized DAT is directed to a recycling pool (Melikian and Buckley, 1999). Thus, plasma membrane DAT reductions can result from a combination of accelerated internalization or reduced recycling (Loder and Melikian, 2003). Trafficking to the recycling pool as a result of cocaine seems likely because total DAT protein remains unchanged (Samuvel et al., 2008) and would be in agreement with the observation that DAT density was no longer reduced 11 days after finishing treatment (cocaine group, Figure 1B), perhaps indicating a recovery in the recycling to the plasma membrane.
There are numerous studies in the literature on the effects of cocaine exposure on DAT regulation. In general, the evidence indicates that during repeated cocaine administration and shortly after exposure, a relatively short-lived increase in the number of DAT binding sites and the velocity of dopamine uptake is observed (Koff et al., 1994; Pilotte et al., 1994; Letchworth et al., 1997; Gulley and Zahniser, 2003). This up-regulation is consistent with the more rapid clearance of synaptic dopamine and behavioural tolerance to the effects of cocaine. However, we failed to observe up-regulation 1 day after cocaine, possibly due to the fact that DAT may undergo rapid regulation at even shorter time points, potentially as soon as a few hours after the last cocaine dose, as suggested by the literature. In contrast, at longer times after cessation of repeated cocaine treatment, DAT binding sites and activity are reduced (Sharpe et al., 1991; Kuhar and Pilotte, 1996; Chefer and Shippenberg, 2002). The significance of the reduced cell surface expression of DAT following repeated cocaine treatment is unclear, but could reflect an attempt to maximize the synaptic efficacy of the remaining neurotransmitter during a dopamine-deficient state (Wilson et al., 1994) reflected by a reduction in basal extracellular dopamine levels (Parsons et al., 1991; Robertson et al., 1991; Rossetti et al., 1992; Weiss et al., 1992; Chefer and Shippenberg, 2002; our results).
Psychostimulant challenge following a period of withdrawal after drug exposure produces enhanced neurochemical and behavioural responses – termed sensitization. Thus, cocaine challenge, given days after withdrawal, increases extracellular dopamine and dopamine-mediated behaviours to a greater extent than the same dose of cocaine did initially (Zahniser and Sorkin, 2004). In addition, cross-sensitization may occur following challenge with a drug different to that used for the original exposure. For example, Itzhak and Martin (1999) observed enhanced locomotor activity following ethanol challenge in cocaine-exposed mice after a 10 day withdrawal period. In our study, MDMA-induced locomotor activity was potentiated in mice pretreated with cocaine finishing 4 days before. Locomotor sensitization is often regarded as indirect evidence for hypersensitivity in relevant motivational circuitry, and therefore may be indicative of the addictive wanting for drugs (Robinson and Berridge, 2008). In addition, following challenge with MDMA, which enters the nerve terminal by diffusion and produces dopamine release principally through Ca2+-dependent mechanisms (Camarero et al., 2002), extracellular dopamine concentrations are enhanced in cocaine-pretreated animals consistent with a diminished capacity for dopamine reuptake. Psychostimulant-induced elevations in dopamine levels in the striatum of DAT-deficient mice may be a relevant measure of the addictive value of the drug because studies in DAT–KO mice have shown that cocaine-induced dopamine elevations in the striatum provide the best fit with studies of cocaine-induced place preferences (Shen et al., 2004). Thus, these results might indicate an increase in the MDMA-induced activation of the reward pathways in animals pretreated with cocaine. Consistent with a prominent role of dopamine in the reinforcing properties of MDMA, dopamine D1 receptor blockade with SCH 23390 produced a rightward shift in the dose–response curve for MDMA self-administration in rats (Daniela et al., 2004), although a possible role of 5-HT2C receptors cannot be excluded (Ramos et al., 2005).
Expression of persistent, drug-induced behavioural sensitization has been suggested to contribute to craving and the high relapse rate of cocaine addicts. Specifically, Robinson and Berridge (2003) have suggested that sensitization causes the CNS to attribute greater incentive salience to the drug, resulting in compulsive motivation to take addictive drugs. Brain imaging studies in drug-addicted subjects suggest two abnormalities that would result in decreased output of dopaminergic circuits related to reward: decreases in dopamine D2 receptors and in dopamine release Volkow et al., 2002a. Each would contribute to decreased sensitivity to natural reinforcers (Volkow et al., 2002b), while psychoactive drugs, which produce fast and large increases in extracellular dopamine, would still be able to activate the reward circuits (Volkow et al., 2003). Thus, the relative salience of a drug over a natural stimulus may be amplified in the drug-addicted subject (Volkow et al., 2004), increasing the susceptibility to drug consumption.
Although specific studies were not carried out to determine if the reinforcing properties of MDMA-related reward might change the importance of drug exposure during a period of withdrawal or abstinence is highlighted by recent studies in which MDMA has been shown to reinstate cocaine-seeking behaviour in mice in which cocaine self-administration and contingent cues were previously extinguished (Trigo et al., 2009). Furthermore, pre-exposure to cocaine has been shown to reduce the latency to acquisition of self-administration of MDMA in rats (Schenk et al., 2003).
In conclusion, repeated cocaine followed by a 4 day withdrawal period protects against MDMA-induced dopaminergic neurotoxicity by producing internalization of DAT by a mechanism which appears to involve PKC. However, exposure to this repeated cocaine protocol potentiates MDMA-induced dopamine release and locomotor activity, measures which could be indicative of increased rewarding effects of MDMA.
Acknowledgments
This work was supported by Plan Nacional sobre Drogas (grant no. 3SI/04/01 to E.O.S. and 2008/065 to J.A.L.M.), Santander/Complutense (grant no. PR27/05-13970 to E.O.S.), FIS (grant no. PI070892 to E.O.S.), Red de Trastornos Adictivos (grant no. RD06/0001/006 to M.I.C. and RD06/0001/011 to J.A.L.M.), UCM-CAM (grant no. CCG07-UCM/SAL-2588 to M.I.C.). The authors received predoctoral funding from Universidad Complutense (I.P.), Ministerio de Educación y Ciencia (E.T. and A.M.), Comunidad de Madrid (M.I.) and Red de Trastornos Adictivos (A.L.J.).
Glossary
Abbreviations:
- DAT
dopamine transporter
- DOPAC
3,4-dihydroxyphenylacetic acid
- HVA
homovanillic acid
- MDMA
3,4-methylenedioxymethamphetamine
- PKC
protein kinase C
- TH
tyrosine hydroxylase
Conflict of interest
The authors state no conflict of interest.
References
- Amara SG, Kuhar MJ. Neurotransmitter transporters: recent progress. Annu Rev Neurosci. 1993;16:73–93. doi: 10.1146/annurev.ne.16.030193.000445. [DOI] [PubMed] [Google Scholar]
- Bennett BA, Hyde CE, Pecora JR, Clodfelter JE. Long-term cocaine administration is not neurotoxic to cultured fetal mesencephalic dopamine neurons. Neurosci Lett. 1993;153:210–214. doi: 10.1016/0304-3940(93)90324-e. [DOI] [PubMed] [Google Scholar]
- Benuck M, Lajtha A, Reith ME. Pharmacokinetics of systemically administered cocaine and locomotor stimulation in mice. J Pharmacol Exp Ther. 1987;243:144–149. [PubMed] [Google Scholar]
- Blakely RD, Bauman AL. Biogenic amine transporters: regulation in flux. Curr Opin Neurobiol. 2000;10:328–336. doi: 10.1016/s0959-4388(00)00088-x. [DOI] [PubMed] [Google Scholar]
- Camarero J, Sanchez V, O'Shea E, Green AR, Colado MI. Studies, using in vivo microdialysis, on the effect of the dopamine uptake inhibitor GBR 12909 on 3,4-methylenedioxymethamphetamine (‘ecstasy’)-induced dopamine release and free radical formation in the mouse striatum. J Neurochem. 2002;81:961–972. doi: 10.1046/j.1471-4159.2002.00879.x. [DOI] [PubMed] [Google Scholar]
- Carlsson A, Davis JN, Kehr W, Lindqvist M, Atack CV. Simultaneous measurement of tyrosine and tryptophan hydroxylase activities in brain in vivo using an inhibitor of the aromatic amino acid decarboxylase. Naunyn Schmiedebergs Arch Pharmacol. 1972;275:153–168. doi: 10.1007/BF00508904. [DOI] [PubMed] [Google Scholar]
- Cervinski MA, Foster JD, Vaughan RA. Psychoactive substrates stimulate dopamine transporter phosphorylation and down-regulation by cocaine-sensitive and protein kinase C-dependent mechanisms. J Biol Chem. 2005;280:40442–40449. doi: 10.1074/jbc.M501969200. [DOI] [PubMed] [Google Scholar]
- Chang MY, Lee SH, Kim JH, Lee KH, Kim YS, Son H, et al. Protein kinase C-mediated functional regulation of dopamine transporter is not achieved by direct phosphorylation of the dopamine transporter protein. J Neurochem. 2001;77:754–761. doi: 10.1046/j.1471-4159.2001.00284.x. [DOI] [PubMed] [Google Scholar]
- Chefer VI, Shippenberg TS. Changes in basal and cocaine-evoked extracellular dopamine uptake and release in the rat nucleus accumbens during early abstinence from cocaine: quantitative determination under transient conditions. Neuroscience. 2002;112:907–919. doi: 10.1016/s0306-4522(02)00099-4. [DOI] [PubMed] [Google Scholar]
- Colado MI, Camarero J, Mechan AO, Sanchez V, Esteban B, Elliott JM, et al. A study of the mechanisms involved in the neurotoxic action of 3,4-methylenedioxymethamphetamine (MDMA, ‘ecstasy’) on dopamine neurones in mouse brain. Br J Pharmacol. 2001;134:1711–1723. doi: 10.1038/sj.bjp.0704435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colado MI, OShea E, Green AR. Acute and long-term effects of MDMA on cerebral dopamine biochemistry and function. Psychopharmacology. 2004;173:249–263. doi: 10.1007/s00213-004-1788-8. Berl. [DOI] [PubMed] [Google Scholar]
- Daniela E, Brennan K, Gittings D, Hely L, Schenk S. Effect of SCH 23390 on (±)-3,4-methylenedioxymethamphetamine hyperactivity and self-administration in rats. Pharmacol Biochem Behav. 2004;77:745–750. doi: 10.1016/j.pbb.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Daniels GM, Amara SG. Regulated trafficking of the human dopamine transporter. Clathrin-mediated internalization and lysosomal degradation in response to phorbol esters. J Biol Chem. 1999;274:35794–35801. doi: 10.1074/jbc.274.50.35794. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobedo I, O'Shea E, Orio L, Sanchez V, Segura M, de la Torre R, et al. A comparative study on the acute and long-term effects of MDMA and 3,4-dihydroxymethamphetamine (HHMA) on brain monoamine levels after i.p. or striatal administration in mice. Br J Pharmacol. 2005;144:231–241. doi: 10.1038/sj.bjp.0706071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci. 2005;8:1481–1489. doi: 10.1038/nn1579. [DOI] [PubMed] [Google Scholar]
- Farfel GM, Kleven MS, Woolverton WL, Seiden LS, Perry BD. Effects of repeated injections of cocaine on catecholamine receptor binding sites, dopamine transporter binding sites and behavior in Rhesus monkey. Brain Res. 1992;578:235–243. doi: 10.1016/0006-8993(92)90252-5. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. San Diego, CA: Academic Press; 1997. [Google Scholar]
- Giros B, Caron MG. Molecular characterization of the dopamine transporter. Trends Pharmacol Sci. 1993;14:43–49. doi: 10.1016/0165-6147(93)90029-j. [DOI] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- Goodman JH, Sloviter RS. Cocaine neurotoxicity and altered neuropeptide Y immunoreactivity in the rat hippocampus; a silver degeneration and immunocytochemical study. Brain Res. 1993;616:263–272. doi: 10.1016/0006-8993(93)90217-b. [DOI] [PubMed] [Google Scholar]
- Granado N, Escobedo I, O'Shea E, Colado I, Moratalla R. Early loss of dopaminergic terminals in striosomes after MDMA administration to mice. Synapse. 2008a;62:80–84. doi: 10.1002/syn.20466. [DOI] [PubMed] [Google Scholar]
- Granado N, O'Shea E, Bove J, Vila M, Colado MI, Moratalla R. Persistent MDMA-induced dopaminergic neurotoxicity in the striatum and substantia nigra of mice. J Neurochem. 2008b;107:1102–1112. doi: 10.1111/j.1471-4159.2008.05705.x. [DOI] [PubMed] [Google Scholar]
- Green AR, Mechan AO, Elliott JM, O'Shea E, Colado MI. The pharmacology and clinical pharmacology of 3,4-methylenedioxymethamphetamine (MDMA, ‘ecstasy’) Pharmacol Rev. 2003;55:463–508. doi: 10.1124/pr.55.3.3. [DOI] [PubMed] [Google Scholar]
- Gulley JM, Zahniser NR. Rapid regulation of dopamine transporter function by substrates, blockers and presynaptic receptor ligands. Eur J Pharmacol. 2003;479:139–152. doi: 10.1016/j.ejphar.2003.08.064. [DOI] [PubMed] [Google Scholar]
- Hitri A, Little KY, Ellinwood EH., Jr Effect of cocaine on dopamine transporter receptors depends on routes of chronic cocaine administration. Neuropsychopharmacology. 1996;14:205–210. doi: 10.1016/0893-133X(95)00090-Z. [DOI] [PubMed] [Google Scholar]
- Itzhak Y, Martin JL. Effects of cocaine, nicotine, dizocipline and alcohol on mice locomotor activity: cocaine-alcohol cross-sensitization involves upregulation of striatal dopamine transporter binding sites. Brain Res. 1999;818:204–211. doi: 10.1016/s0006-8993(98)01260-8. [DOI] [PubMed] [Google Scholar]
- Izco M, Marchant I, Escobedo I, Peraile I, Delgado M, Higuera-Matas A, et al. Mice with decreased cerebral dopamine function following a neurotoxic dose of MDMA (3,4-methylenedioxymethamphetamine, ‘ecstasy’) exhibit increased ethanol consumption and preference. J Pharmacol Exp Ther. 2007;322:1003–1012. doi: 10.1124/jpet.107.120600. [DOI] [PubMed] [Google Scholar]
- Izenwasser S, Cox BM. Daily cocaine treatment produces a persistent reduction of [3H]dopamine uptake in vitro in rat nucleus accumbens but not in striatum. Brain Res. 1990;531:338–341. doi: 10.1016/0006-8993(90)90797-f. [DOI] [PubMed] [Google Scholar]
- Jayanthi LD, Ramamoorthy S. Regulation of monoamine transporters: influence of psychostimulants and therapeutic antidepressants. AAPS J. 2005;7:E728–E738. doi: 10.1208/aapsj070373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johanson CE, Fischman MW. The pharmacology of cocaine related to its abuse. Pharmacol Rev. 1989;41:3–52. [PubMed] [Google Scholar]
- Johnson MA, Rajan V, Miller CE, Wightman RM. Dopamine release is severely compromised in the R6/2 mouse model of Huntington's disease. J Neurochem. 2006;97:737–746. doi: 10.1111/j.1471-4159.2006.03762.x. [DOI] [PubMed] [Google Scholar]
- Koff JM, Shuster L, Miller LG. Chronic cocaine administration is associated with behavioral sensitization and time-dependent changes in striatal dopamine transporter binding. J Pharmacol Exp Ther. 1994;268:277–282. [PubMed] [Google Scholar]
- Koob GF, Bloom FE. Cellular and molecular mechanisms of drug dependence. Science. 1988;242:715–723. doi: 10.1126/science.2903550. [DOI] [PubMed] [Google Scholar]
- Kuhar MJ, Pilotte NS. Neurochemical changes in cocaine withdrawal. Trends Pharmacol Sci. 1996;17:260–264. doi: 10.1016/0165-6147(96)10024-9. [DOI] [PubMed] [Google Scholar]
- Kula NS, Baldessarini RJ. Lack of increase in dopamine transporter binding or function in rat brain tissue after treatment with blockers of neuronal uptake of dopamine. Neuropharmacology. 1991;30:89–92. doi: 10.1016/0028-3908(91)90047-f. [DOI] [PubMed] [Google Scholar]
- Letchworth SR, Daunais JB, Hedgecock AA, Porrino LJ. Effects of chronic cocaine administration on dopamine transporter mRNA and protein in the rat. Brain Res. 1997;750:214–222. doi: 10.1016/s0006-8993(96)01384-4. [DOI] [PubMed] [Google Scholar]
- Little KY, Elmer LW, Zhong H, Scheys JO, Zhang L. Cocaine induction of dopamine transporter trafficking to the plasma membrane. Mol Pharmacol. 2002;61:436–445. doi: 10.1124/mol.61.2.436. [DOI] [PubMed] [Google Scholar]
- Loder MK, Melikian HE. The dopamine transporter constitutively internalizes and recycles in a protein kinase C-regulated manner in stably transfected PC12 cell lines. J Biol Chem. 2003;278:22168–22174. doi: 10.1074/jbc.M301845200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Mathis C, Lehmann J, Ungerer A. The selective protein kinase C inhibitor, NPC 15437, induces specific deficits in memory retention in mice. Eur J Pharmacol. 1992;220:107–110. doi: 10.1016/0014-2999(92)90020-5. [DOI] [PubMed] [Google Scholar]
- Melikian HE, Buckley KM. Membrane trafficking regulates the activity of the human dopamine transporter. J Neurosci. 1999;19:7699–7710. doi: 10.1523/JNEUROSCI.19-18-07699.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norregaard L, Gether U. The monoamine neurotransmitter transporters: structure, conformational changes and molecular gating. Curr Opin Drug Discov Devel. 2001;4:591–601. [PubMed] [Google Scholar]
- O'Shea E, Esteban B, Camarero J, Green AR, Colado MI. Effect of GBR 12909 and fluoxetine on the acute and long term changes induced by MDMA (‘ecstasy’) on the 5-HT and dopamine concentrations in mouse brain. Neuropharmacology. 2001;40:65–74. doi: 10.1016/s0028-3908(00)00106-4. [DOI] [PubMed] [Google Scholar]
- Parsons LH, Smith AD, Justice JB., Jr Basal extracellular dopamine is decreased in the rat nucleus accumbens during abstinence from chronic cocaine. Synapse. 1991;9:60–65. doi: 10.1002/syn.890090109. [DOI] [PubMed] [Google Scholar]
- Pilotte NS, Sharpe LG, Kuhar MJ. Withdrawal of repeated intravenous infusions of cocaine persistently reduces binding to dopamine transporters in the nucleus accumbens of Lewis rats. J Pharmacol Exp Ther. 1994;269:963–969. [PubMed] [Google Scholar]
- Ramos M, Goni-Allo B, Aguirre N. Administration of SCH 23390 into the medial prefrontal cortex blocks the expression of MDMA-induced behavioral sensitization in rats: an effect mediated by 5-HT2C receptor stimulation and not by D1 receptor blockade. Neuropsychopharmacology. 2005;30:2180–2191. doi: 10.1038/sj.npp.1300735. [DOI] [PubMed] [Google Scholar]
- Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219–1223. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- Robertson MW, Leslie CA, Bennett JP., Jr Apparent synaptic dopamine deficiency induced by withdrawal from chronic cocaine treatment. Brain Res. 1991;538:337–339. doi: 10.1016/0006-8993(91)90451-z. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. Addiction. Annu Rev Psychol. 2003;54:25–53. doi: 10.1146/annurev.psych.54.101601.145237. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. Review. The incentive sensitization theory of addiction: some current issues. Philos Trans R Soc Lond B Biol Sci. 2008;363:3137–3146. doi: 10.1098/rstb.2008.0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetti ZL, Hmaidan Y, Gessa GL. Marked inhibition of mesolimbic dopamine release: a common feature of ethanol, morphine, cocaine and amphetamine abstinence in rats. Eur J Pharmacol. 1992;221:227–234. doi: 10.1016/0014-2999(92)90706-a. [DOI] [PubMed] [Google Scholar]
- Samuvel DJ, Jayanthi LD, Manohar S, Kaliyaperumal K, See RE, Ramamoorthy S. Dysregulation of dopamine transporter trafficking and function after abstinence from cocaine self-administration in rats: evidence for differential regulation in caudate putamen and nucleus accumbens. J Pharmacol Exp Ther. 2008;325:293–301. doi: 10.1124/jpet.107.130534. [DOI] [PubMed] [Google Scholar]
- Sanchez V, Camarero J, Esteban B, Peter MJ, Green AR, Colado MI. The mechanisms involved in the long-lasting neuroprotective effect of fluoxetine against MDMA (‘ecstasy’)-induced degeneration of 5-HT nerve endings in rat brain. Br J Pharmacol. 2001;134:46–57. doi: 10.1038/sj.bjp.0704230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraiva L, Fresco P, Pinto E, Gonçalves J. Isoform-selectivity of PKC inhibitors acting at the regulatory and catalytic domain of mammalian PKC-alpha, -betaI, -delta, -eta and -zeta. J Enzyme Inhib Med Chem. 2003;18:475–483. doi: 10.1080/14756360310001603158. [DOI] [PubMed] [Google Scholar]
- Schenk S, Gittings D, Johnstone M, Daniela E. Development, maintenance and temporal pattern of self-administration maintained by ecstasy (MDMA) in rats. Psychopharmacology (Berl) 2003;169:21–27. doi: 10.1007/s00213-003-1407-0. [DOI] [PubMed] [Google Scholar]
- Schlussman SD, Ho A, Zhou Y, Curtis AE, Kreek MJ. Effects of ‘binge’ pattern cocaine on stereotypy and locomotor activity in C57BL/6J and 129/J mice. Pharmacol Biochem Behav. 1998;60:593–599. doi: 10.1016/s0091-3057(98)00047-1. [DOI] [PubMed] [Google Scholar]
- See RE, Elliott JC, Feltenstein MW. The role of dorsal vs ventral striatal pathways in cocaine-seeking behavior after prolonged abstinence in rats. Psychopharmacology (Berl) 2007;194:321–331. doi: 10.1007/s00213-007-0850-8. [DOI] [PubMed] [Google Scholar]
- Segal DS, Kuczenski R, O'Neil ML, Melega WP, Cho AK. Escalating dose methamphetamine pretreatment alters the behavioral and neurochemical profiles associated with exposure to a high-dose methamphetamine binge. Neuropsychopharmacology. 2003;28:1730–1740. doi: 10.1038/sj.npp.1300247. [DOI] [PubMed] [Google Scholar]
- Sharpe LG, Pilotte NS, Mitchell WM, De Souza EB. Withdrawal of repeated cocaine decreases autoradiographic. Eur J Pharmacol. 1991;203:141–144. doi: 10.1016/0014-2999(91)90804-y. [DOI] [PubMed] [Google Scholar]
- Shen HW, Hagino Y, Kobayashi H, Shinohara-Tanaka K, Ikeda K, Yamamoto H, et al. Regional differences in extracellular dopamine and serotonin assessed by in vivo microdialysis in mice lacking dopamine and/or serotonin transporters. Neuropsychopharmacology. 2004;29:1790–1799. doi: 10.1038/sj.npp.1300476. [DOI] [PubMed] [Google Scholar]
- Staley JK, Hearn WL, Ruttenber AJ, Wetli CV, Mash DC. High affinity cocaine recognition sites on the dopamine transporter are elevated in fatal cocaine overdose victims. J Pharmacol Exp Ther. 1994;271:1678–1685. [PubMed] [Google Scholar]
- Sullivan JP, Connor JR, Shearer BG, Burch RM. 2,6-Diamino-N-([1-oxotridecyl)-2-piperidinyl]methyl)hexanamide (NPC 15437): a selective inhibitor of protein kinase C. Agents Actions. 1991a;34:142–144. doi: 10.1007/BF01993261. [DOI] [PubMed] [Google Scholar]
- Sullivan JP, Connor JR, Tiffany C, Shearer BG, Burch RM. NPC 15437 interacts with the C1 domain of protein kinase C. An analysis using mutant PKC constructs. FEBS Lett. 1991b;285:120–123. doi: 10.1016/0014-5793(91)80739-p. [DOI] [PubMed] [Google Scholar]
- Tilley MR, Gu HH. Dopamine transporter inhibition is required for cocaine-induced stereotypy. Neuroreport. 2008;19:1137–1140. doi: 10.1097/WNR.0b013e3283063183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trigo JM, Orejarena MJ, Maldonado R, Robledo P. MDMA reinstates cocaine-seeking behaviour in mice. Eur Neuropsychopharmacol. 2009;19:391–397. doi: 10.1016/j.euroneuro.2008.12.010. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ. Role of dopamine in drug reinforcement and addiction in humans: results from imaging studies. Behav Pharmacol. 2002a;13:355–366. doi: 10.1097/00008877-200209000-00008. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ. Positron emission tomography and single-photon emission computed tomography in substance abuse research. Semin Nucl Med. 2003;33:114–128. doi: 10.1053/snuc.2003.127300. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ, Swanson JM. Dopamine in drug abuse and addiction: results from imaging studies and treatment implications. Mol Psychiatry. 2004;9:557–569. doi: 10.1038/sj.mp.4001507. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Logan J, Jayne N, Franceschi D, et al. ‘Nonhedonic’ food motivation in humans involves dopamine in the dorsal striatum and methylphenidate amplifies this effect. Synapse. 2002b;44:175–180. doi: 10.1002/syn.10075. [DOI] [PubMed] [Google Scholar]
- Wang X, Baumann MH, Xu H, Morales M, Rothman RB. ±)-3,4-Methylenedioxymethamphetamine administration to rats does not decrease levels of the serotonin transporter protein or alter its distribution between endosomes and the plasma membrane. J Pharmacol Exp Ther. 2005;314:1002–1012. doi: 10.1124/jpet.105.088476. [DOI] [PubMed] [Google Scholar]
- Weiss F, Markou A, Lorang MT, Koob GF. Basal extracellular dopamine levels in the nucleus accumbens are decreased during cocaine withdrawal after unlimited-access self-administration. Brain Res. 1992;593:314–318. doi: 10.1016/0006-8993(92)91327-b. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Nobrega JN, Carroll ME, Niznik HB, Shannak K, Lac ST, et al. Heterogeneous subregional binding patterns of 3H-WIN 35 428 and 3H-GBR 12 935 are differentially regulated by chronic cocaine self-administration. J Neurosci. 1994;14:2966–2979. doi: 10.1523/JNEUROSCI.14-05-02966.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wish ED, Fitzelle DB, O'Grady KE, Hsu MH, Arria AM. Evidence for significant polydrug use among ecstasy-using college students. J Am Coll Health. 2006;55:99–104. doi: 10.3200/JACH.55.2.99-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahniser NR, Sorkin A. Rapid regulation of the dopamine transporter: role in stimulant addiction? Neuropharmacology. 2004;47:80–91. doi: 10.1016/j.neuropharm.2004.07.010. [DOI] [PubMed] [Google Scholar]