

Abstract
Background and purpose:
The glucagon-like peptide-1 receptor (GLP-1R) belongs to Family B of the G protein-coupled receptor superfamily and is a target for treatment of type 2 diabetes. Family B G protein-coupled receptors contain a putative N-terminal signal peptide, but its role in receptor synthesis and trafficking are unclear. Further, the signal peptide is not cleaved in at least one family member.
Experimental approach:
We examined receptor glycosylation and the role of the signal peptide in GLP-1R synthesis and trafficking using constructs containing epitope tags at the N- and/or C-terminus and in which the signal peptide sequence was either present or absent.
Key results:
The signal peptide was absolutely required for GLP-1R synthesis but could be substituted to some extent by increasing positive charge in the N-terminal region of the receptor flanking the signal peptide. The signal peptide is cleaved during synthesis and processing of the receptor. An enhanced GFP-epitope tag at the N-terminus of the receptor permitted synthesis of the receptor but blocked signal peptide cleavage and prevented trafficking to the plasma membrane. Cleavage site mutation allowed synthesis of a full-length receptor, blocked signal peptide cleavage and caused retention within the endoplasmic reticulum.
Conclusions and implications:
Signal peptide cleavage was not essential for receptor synthesis but was obligatory for processing and trafficking of receptors to the plasma membrane. Further, the GLP-1R is subject to N-linked glycosylation and only the mature, fully glycosylated form of the receptor is present in the plasma membrane. Inhibition of glycosylation prevents processing and cell surface expression of the GLP-1R.
Keywords: GPCR, GLP-1, signal peptide, trafficking
Introduction
The glucagon-like peptide-1 receptor (GLP-1R; all receptor nomenclature follows Alexander et al., 2008) is responsible for mediating the effects of the incretin hormone glucagon-like peptide-1 (GLP-1), released from intestinal L cells following nutrient ingestion. A major effect of GLP-1 is to enhance glucose-dependent insulin release from pancreatic β-cells, thereby reducing post-prandial blood glucose elevation. GLP-1 has additional pancreatic and extra-pancreatic effects that contribute to its anti-diabetogenic effects, including increased synthesis of insulin, enhanced pancreatic β-cell mass, inhibition of glucagon secretion and appetite suppression (Baggio and Drucker, 2007; Doyle and Egan, 2007). Enhancing GLP-1R-mediated effects through either inhibition of the rapid breakdown of GLP-1 by dipeptidyl peptidase-IV or by direct activation of the GLP-1R is now a validated therapeutic option for the treatment of type 2 diabetes.
The GLP-1R belongs to the relatively small Family B of the G-protein-coupled receptor (GPCR) superfamily. Despite being integral membrane proteins with seven transmembrane α-helices, a large proportion of GPCRs (∼85%) display no evidence of a signal peptide sequence (Köchl et al., 2002). This is a sequence of approximately 20 amino acids containing a stretch of hydrophobic residues, usually in the N-terminus of the protein, which is often critical for the synthesis and processing of both secreted and integral membrane proteins. However, these signal peptide sequences are removed by signal peptidases within the endoplasmic reticulum. In GPCRs without such a sequence, the first transmembrane domain can function as a signal anchor sequence, promoting post-translational movement of the N-terminus through the translocon into the lumen of the endoplasmic reticulum during which time the nascent peptide may be subject to N-linked glycosylation. Subsequently, signal anchor sequences within the receptor transmembrane domains are thought to regulate further synthesis and insertion into the membrane of the endoplasmic reticulum. Full-length receptors are then processed through the Golgi where further modifications to the glycosylated residues occur before further transport and insertion of the mature protein into the plasma membrane (Wallin and von Heijne, 1995). Within the Golgi, O-linked glycosylation of serine or threonine residues can also occur, although this has been reported for very few GPCRs. Putative signal peptides are present within Family B and Family C GPCRs (the secretin and glutamate receptor families respectively) and also in the relatively small number of glycoprotein hormone receptors of Family A (the rhodopsin family) (Köchl et al., 2002). Thus, signal peptides are associated with receptors in which the N-terminus is either responsible for, or contributes to, ligand binding and which, therefore, is highly structured. Indeed, it has been suggested that a signal peptide is required for GPCRs in which post-translational translocation of the N-terminus across the membrane of the endoplasmic reticulum is not possible due to the presence of stably folded domains (Köchl et al., 2002).
It is, however, unclear whether signal peptides are essential in GPCRs (Köchl et al., 2002). Furthermore, although confirmed in some instances, whether the putative signal peptides of these GPCRs are actually cleaved remains to be experimentally verified, as their presence is often based on predictive algorithms. In a set of 270 recombinant secreted proteins with experimentally verified cleavage sites, these were, however, predicted with <80% accuracy (Zhang and Henzel, 2004). This difficulty in predicting a cleaved signal peptide is illustrated by two Family B GPCRs; the corticotropin-releasing hormone receptor 1 (CRF1 receptor) and CRF2a receptor. Using SignalP 3.0 (Bendtsen, 2004), both receptors have a >98% probability of an N-terminal signal peptide. However, experimental evidence indicates that while the signal peptide of the CRF1 receptor is cleaved (Alken et al., 2005), the putative signal peptide of the CRF2(a) receptor represents a pseudo signal peptide (or uncleaved signal anchor sequence), which forms part of the mature protein (Rutz et al., 2006). While this lack of cleavage appears to be exceptional, this highlights the need for experimental verification of the role of putative signal peptides in proteins, including GPCRs where the presence or absence of cleavage has implications for receptor structure, function and modelling. Further, there is emerging evidence that the signal peptides serve different roles in the synthesis, trafficking and function of different GPCRs (Alken et al., 2005).
Here, we have assessed whether the putative signal peptide of the human (h) GLP-1R is cleaved and determined its role in the synthesis and processing of the receptor. Our results demonstrate that cleavage of the signal peptide is not essential for receptor synthesis, but that it must occur to facilitate effective processing and trafficking of the receptor to the plasma membrane. Further, the GLP-1R undergoes N-linked glycosylation and only the fully glycosylated form is present in the plasma membrane. Inhibition of this glycosylation prevents appropriate processing and cell surface expression.
Methods
Generation of cDNA constructs
The coding sequences of the full-length hGLP-1R containing appropriate restriction sites were cloned by PCR and ultimately ligated into pcDNA3.1(+)-HA (constructs containing the HA-epitope tag at either the N-terminus or C-terminus of the multiple cloning site) and either pEGFP-N1 or pEGFP-C1 (constructs containing the EGFP-epitope tag at the C-terminus and N-terminus respectively). As shown in Figures 1 and 2, sequences were designed to express the N-terminal or C-terminal HA-tagged GLP-1R (HA-GLP-1R and GLP-1R-HA respectively), the C-terminal EGFP-tagged GLP-1R (GLP-1R-EGFP) and, the GLP-1R containing both HA- and EGFP-epitope tags at either N-terminus or C-terminus (HA-GLP-1R-EGFP and EGFP-GLP-1R-HA). The untagged, wild-type (WT) hGLP-1R in pcDNA5-FRT was used as the starting template, the linear sequence of which is given in the NCBI database (Accession, NP_002053; Version, NP_002053.3; GI: 166795283). All constructs were generated to contain an appropriately positioned Kozak sequence and both start and stop codons (Table 1). The HA-epitope tag sequence was generated by annealing two complimentary primers encoding amino acids 98–106 of human influenza haemagglutinin ‘YPYDVPDYA’, which also contained appropriate restriction sites and either the Kozak sequence and start codon (N-terminal tag) or a stop codon (C-terminal tag). Constructs containing the EGFP-epitope tag used either the Kozak sequence and start codon (N-terminal tag) or the stop codon (C-terminal tag) of the EGFP-containing vector. Using SignalP 3.0 (Bendtsen, 2004) and the neural networks algorithm, the signal peptide cleavage site for the hGLP-1R is predicted to be between residues 23 and 24 (alanine-glycine) (note that both the rat and mouse GLP-1Rs have signal peptides predicted to be 21 amino acids in length). Based on the data for the hGLP-1R, constructs were generated in which the coding sequence for the putative signal peptide had been removed (ΔSP) but which retained epitope tag sequences (Figures 2 and 3, Table 1) and an appropriately positioned Kozak sequence and both start and stop codons. Full details of all cloning strategies are available on request. Mutants of the GLP-1R were generated using the QuikChange Site-Directed Mutagenesis Kit based on PCR with slight modifications to the manufacturer's guide. Mutations were at either position −3 of the predicted cleavage site (residue 21: A21R) and based on the HA-GLP-1R-EGFP construct (HA-A21R-GLP-1R-EGFP) or residue 34 of the N-terminus (E34R) and based on both the HA-GLP-1R-EGFP construct (HA-E34R-GLP-1R-EGFP) and the signal peptide-deleted version (HA-ΔSP-E34R-GLP-1R-EGFP). All the recombinant plasmids were then purified with QIAGEN plasmid kit and sequences confirmed by automated sequence analysis (PNACL, University of Leicester, Leicester, UK).
Figure 1.
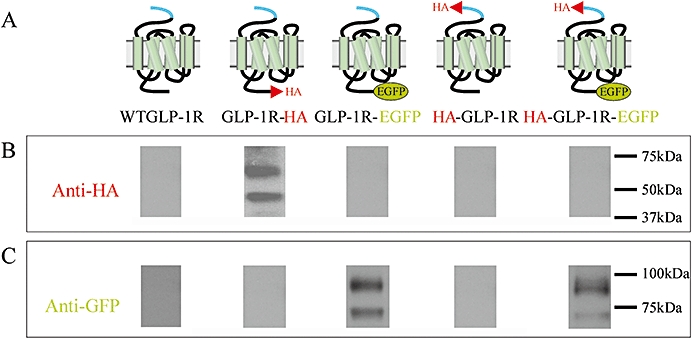
Immunoblotting of cell lysates following transfection of cells with glucagon-like peptide-1 receptor (GLP-1R) constructs. Human embryonic kidney (HEK)-293 cells were transiently transfected with plasmids containing either the WTGLP-1R or various epitope-tagged constructs as indicated (A). After approximately 30 h, cell lysates were prepared and immunoblotted using either an anti-HA antibody (B) or an anti-GFP antibody (C). Data are representative of three independent experiments.
Figure 2.
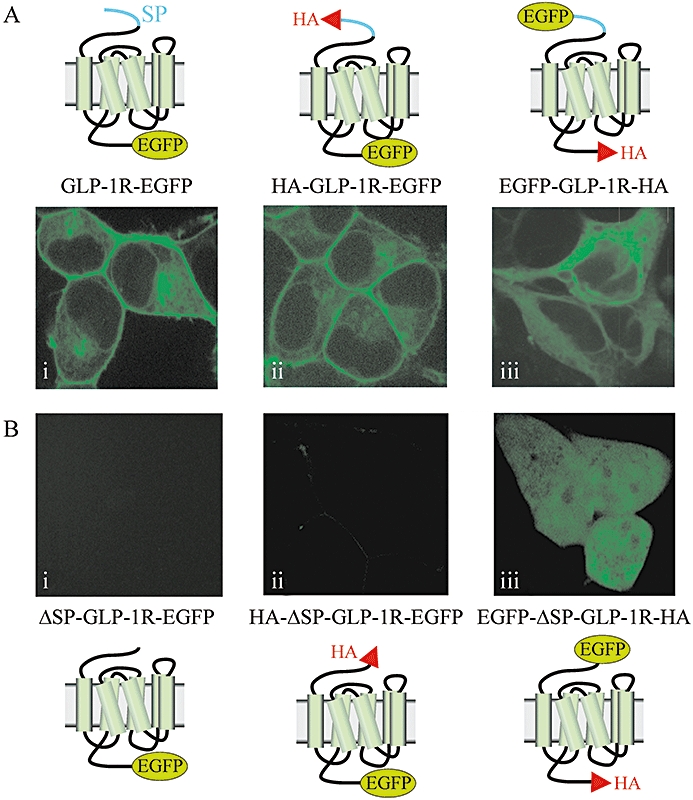
Confocal microscopy of cells transfected with EGFP-tagged glucagon-like peptide-1 receptors (GLP-1Rs) with or without the signal peptide sequence. Human embryonic kidney (HEK)-293 cells were transiently transfected with plasmids containing GLP-1R constructs that had an EGFP-epitope tag at either the C-terminus (GLP-1R-EGFP, HA-GLP-1R-EGFP) or N-terminus (EGFP-GLP-1R-HA) and that either contained the signal peptide sequence (A) or had it deleted [ΔSP; (B)]. After 30–48 h, live cells were examined by confocal microscopy. Images are representative of cells observed in five independent experiments.
Table 1.
Organization of the glucagon-like peptide-1 receptor (GLP-1R) constructs including those with and without the signal peptide sequences
| Construct | Sequence |
|---|---|
| WTGLP-1R | START- AG(4-20)AGPRPQGATVSLWET(36-457) CQASC -STOP |
| HA-GLP-1R | START- HA AG(4-20)AGPRPQGATVSLWET(36-457) CQASC -STOP |
| GLP-1R-HA | START- AG(4-20)AGPRPQGATVSLWET(36-457) CQASCHA -STOP |
| ΔSPGLP-1R-HA | START- RPQGATVSLWET(36-457) CQASCHA -STOP |
| GLP-1R-EGFP | START- AG(4-20)AGPRPQGATVSLWET(36-457) CQASCEGFP-STOP |
| ΔSPGLP-1R-EGFP | START- RPQGATVSLWET(36-457) CQASCEGFP-STOP |
| EGFP-GLP-1R-HA | START-EGFP AG(4-20)AGPRPQGATVSLWET(36-457) CQASCHA -STOP |
| EGFP-ΔSPGLP-1R-HA | START-EGFP RPQGATVSLWET(36-457) CQASCHA -STOP |
| HA-GLP-1R-EGFP | START- HA AG(4-20)AGPRPQGATVSLWET(36-457) CQASCEGFP-STOP |
| HA-ΔSPGLP-1R-EGFP | START- HA RPQGATVSLWET(36-457) CQASCEGFP-STOP |
| HA-A21RGLP-1R-EGFP | START- HA AG(4-20)RGPRPQGATVSLWET(36-457) CQASCEGFP-STOP |
| HA-E34RGLP-1R-EGFP | START- HA AG(4-20)AGPRPQGATVSLWRT(36-457) CQASCEGFP-STOP |
| HA-ΔSPE34RGLP-1R-EGFP | START- HA RPQGATVSLWRT(36-457) CQASCEGFP-STOP |
GLP-1R constructs showing the location of the start and stop codons, the epitope-tags and the sequence of the N-termini around the putative signal peptide sequence. ‘START’ represents the initial methionine residue. HA- and EGFP-epitope tags are shown in shaded boxes; the putative signal peptide sequence is shown underlined and the alanine (A) or glutamic acid (E) to arginine mutations is shown in large, bold lettering.
Cell culture, transfection and stable cell line generation
Human embryonic kidney (HEK)-293 cells were cultured in Gibco MEM supplemented with 10% (v/v) fetal bovine serum, 1% (w/v) L-glutamine and 1% non-essential amino acids at 37°C in a 5% CO2 humidified environment. Cells at approximately 80–90% confluency were transfected using Lipofectamine 2000 following the manufacturer's instructions (24-well plates, 0.5 µg DNA per well; 6-well plates, 2.0 µg DNA per well; 75 cm2 flask or 100 mm dish, 12 µg). Transiently transfected cells were used for experimentation approximately 30–48 h post transfection. A HEK-293 cell line with stable expression of the GLP-1R-EGFP (HEK-293 : GLP-1R-EGFP) was generated following transfection by selection with Geneticin (9418; 1 mg·mL−1) for 20 days starting 30 h post transfection. Clones were isolated using cloning discs and transferred into 24-well plates. The expression of GLP-1R-EGFP was assessed by both immunoblotting of EGFP expression and visualization of EGFP fluorescence by confocal microscopy (see below). Subsequently, cAMP responses to 100 nM GLP-1 were determined (see below) and a clone chosen on the basis of both EGFP fluorescence and cAMP generation. The stable cell line was maintained in media containing 200 µg·mL−1 G418.
Immunoblotting
Cells were cultured in 6-well plates and harvested by addition of lysis buffer [10 mM Tris, pH 7.4; 10 mM EDTA, pH 8.5; 1% (v/v) NP40; 0.1% (w/v) SDS; 150 mM NaCl; 12 mM deoxycholic acid; 0.5 mM phenylmethylsulphonyl fluoride] and left on ice for 15 min. Following addition of an equal volume of 2× sample buffer [80 mM Tris-HCl; 4% (w/v) SDS; 20% (v/v) glycerol; 0.005% (w/v) bromophenol blue; pH 6.8], proteins (4–20 µg; determined by Bradford assay) were separated by 8% (w/v) SDS-PAGE. Proteins were then transferred onto PVDF membranes that were subsequently blocked with TBST [50 mM Tris, pH 7.5, 500 mM NaCl, 0.05% (v/v) Tween-20] containing 5% (w/v) dried milk powder for 1 h at room temperature. After blocking, membranes were incubated either with rat IgG monoclonal anti-HA antibody (1:1000) or with rabbit IgG polyclonal anti-GFP antibody (1:10 000) in TBST-5% (w/v) dried milk powder overnight at 4°C. After washing (3 × 10 min) in TBST, membranes were incubated with either a goat anti-rat IgG, HRP-linked antibody (1:1000) or goat anti-rabbit IgG, HRP-linked antibody (1:3000) for 1 h at room temperature followed by washing (3 × 10 min) in TBST. Visualization was achieved using enhanced chemiluminescence detection and exposure to film. Where required, blots probed with the anti-EGFP antibody were stripped [100 mM 2-mercapotoethanol, 2% (w/v) SDS, 62.5 mM Tris-HCl pH 6.7, 30 min, 50°C] before washing, blocking and re-probing with anti-HA antibody as described above.
Live-cell imaging of EGFP-tagged receptors
HEK-293 cells were plated into 6-well plates (approximately 0.5 × 106 cells per well) containing 25 mm diameter glass coverslips pre-coated with 0.1% (w/v) poly-d-lysine hydrobromide and allowed to adhere overnight. Cells were then transfected with EGFP-tagged GLP-1R constructs as described above. Approximately 30–48 h after transfection, the coverslips with cells were rinsed with pre-warmed (37°C) Krebs-HEPES buffer (KHB; 118.6 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 4.2 mM NaHCO3, 10 mM HEPES, 1.3 mM CaCl2, 11.7 mM glucose, pH 7.4) and mounted in a perfusion chamber containing KHB heated to 37°C with a Peltier unit. The chamber volume was 0.5 mL and where required was perfused at 5 mL·min−1. Cells were imaged using an UltraVIEW confocal microscope (PerkinElmer Life and Analytical Sciences, Beaconsfield, Bucks., UK) with a ×60 oil-immersion objective lens and a 488 nm Kr/Ar laser line. Emitted light was collected above 510 nm and images captured at a rate of approximately 0.4 frames min−1 using a CCD camera. Cells stably expressing the GLP-1R-EGFP construct were similarly visualized 24–48 h following plating on poly-d-lysine coated coverslips. Where required, plasma membrane was stained by addition of 50 µL of 0.05% (w/v) Trypan blue to KHB in the perfusion chamber.
Glycosidase treatments
HEK-293 cells with either transient (30 h post transfection) expression of epitope-tagged (HA or EGFP) GLP-1Rs or with stable expression of GLP-1R-EGFP were cultured to 80–90% confluency in 100 mm dishes. Monolayers were washed with ice-cold PBS (137 mM NaCl, 2 mM KCl, 7.5 mM Na2HPO4, 1.8 mM K2HPO4, pH 7.4) and then harvested with a cell scraper in 2 mL ice-cold PBS. Cells were collected by centrifugation (200×g, 2 min, 4°C) and resuspended in 1 mL of homogenization buffer (1 mM EDTA, 10 mM Tris-HCl, 1 mM phenylmethylsulphonyl fluoride, 200 µg·mL−1 benzamidine, pH 7.4). After incubation on ice for 15 min, cells were sonicated (Sonifier Ultrasonic Cell Disruptor; Branson, CT, USA) at 10% of the maximal amplitude for 3 × 1 s at 1 min intervals. The lysate was centrifuged (300×g, 10 min, 4°C) to pellet nuclei and unbroken cells. The post-nuclear supernatant fractions were used for deglycosylation following the manufacturer's instructions with slight modification. Briefly, the initial denaturation step was performed at room temperature for 30 min and proteins (20 µg) were then treated with 500 units of either peptide N-glycosidase F (PNGase F) or endoglycosidase H (Endo H) for 60 min at 37°C. Reactions were stopped by addition of 5 µL of 5× sample buffer. Proteins were then separated by SDS-PAGE using an 8% resolving gel, transferred to PVDF membrane and immunoblotted using either an anti-HA antibody or an anti-EGFP antibody as described above.
Tunicamycin treatment
HEK-293 cells stably expressing the GLP-1R-EGFP construct were cultured to 50% confluency in 75 cm2 flasks before tunicamycin treatment. Cells were washed once with pre-warmed PBS and cultured in fresh media containing 0.1 µg·mL−1 tunicamycin for 80 h with a media change at 40 h. Cells were then harvested and transferred into 6-well plates either with or without coverslips and grown in fresh media containing 0.2 µg·mL−1 tunicamycin for 60 h followed by 0.4 µg·mL−1 tunicamycin for a further 40 h. Cells grown on coverslips were imaged as described above while cells grown without coverslips were lysed and harvested for immunoblotting as described above. This progressive increase in the concentration of tunicamycin was required to prevent the cell death that occurred following immediate exposure to higher concentrations of tunicamycin.
Biotinylation of cell surface proteins
HEK-293 cells stably expressing the GLP-1R-EGFP construct were grown to 90% confluency on 100 mm diameter tissue culture plates and rinsed with ice-cold PBS (3 × 8 mL). Biotinylation was then performed based on a previously described method (Alken et al., 2005). Briefly, plates were incubated with 2 mL of ice-cold PBS containing 0.5 mg·mL−1 EZ-Link Sulfo–NHS–Biotin for 30 min at 4°C with gentle rocking. The biotin solution was replaced by 4 mL ice-cold PBS containing 100 mM glycine and further incubated (10 min, 4°C) with gentle agitation to remove unbound biotin. Cells were then washed with ice-cold PBS and lysed with 2 mL lysis buffer for 20 min at 4°C with vigorous shaking. Lysates were centrifuged (16 000×g, 20 min) to sediment nucleic acids and debris. NeutrAvidin agarose resin (50 µL) was then added to the supernatant and incubated for 1.5 h at room temperature with occasional stirring. Beads were pelleted by centrifugation (16 000×g, 5 s) and washed (3 × 1 mL) initially with buffer 1 [500 mM NaCl, 1 mM EDTA, 0.5% (v/v) Triton X-100, 0.1% (w/v) SDS and 50 mM Tris-HCl, pH 8.0] and subsequently with 1 mL of buffer 2 [500 mM NaCl, 1 mM EDTA, 0.5% (v/v) Triton X-100, 0.1% (w/v) SDS and 50 mM Tris/HCl, pH 7.4]. Proteins were separated from the beads by incubation in 300 µL Laemmli buffer [62.5 mM Tris-HCl, pH 6.8, 2% (w/v) SDS, 20% (v/v) glycerol, 5% (v/v) 2-mercaptoethanol] for 30 min at room temperature with occasional shaking. The solution was centrifuged (16 000×g, 10 min), the supernatant removed and processed for immunoblotting.
Generation and measurement of cAMP
Cell monolayers in 24-well tissue culture plates were washed twice with 1 mL of KHB containing 0.1% (w/v) BSA (KHB-0.1% BSA) and then equilibrated at 37oC for 10 min with 1 mL KHB-0.1% BSA. Buffer was then replaced with 500 µL KHB-0.1% BSA containing agonist at the required concentration. Following a further 10 min incubation at 37oC, buffer was removed and reactions terminated with 400 µL of ice-cold 0.5 M trichloroacetic acid. The cAMP was extracted using a method identical to that for the extraction of inositol 1,4,5-trisphosphate (Willars and Nahorski, 1995) and the cAMP content determined using a radioreceptor assay with binding-protein purified from calf adrenal glands (Brown et al., 1971). The cAMP content was determined by interpolation of a standard curve and related to cellular protein, which was assessed by Bradford assay in separate wells. Concentration–response curves were fitted using Prism (GraphPad Software Inc., San Diego, CA, USA) by a standard four-parameter logistic equation and EC50values determined.
Detemination of ligand binding affinity
The binding affinity of GLP-1 was determined by homologous binding assays on membranes prepared from HEK-293 cells transiently expressing the receptor constructs.
Membrane preparation
Confluent cell monolayers in 75 cm2 flasks were washed and harvested with 2 mL PBS using a cell scraper, collected by centrifugation at 200×g for 2 min at 4°C, and resuspended in 1 mL of homogenization buffer (see under ‘Glycosidase treatments’ above). After incubation on ice for 15 min, cells were sonicated with the Sonifier Ultrasonic Cell Disruptor on ice as described above and centrifuged (20 000×g, 4°C, 10 min). The pellets were resuspended in homogenization buffer at 1 mg mL−1 protein.
Assay
Binding assays were carried out in round-bottomed 96-well plates in a total volume of 200 µL with the component parts diluted in assay buffer [Hank's balanced salt solution composition: 1.26 mM CaCl2; 0.493 mM MgCl2; 20 mM HEPES; 0.1% (w/v) BSA; pH 7.4]. Experiments were conducted such that ligand depletion was <10% and where possible, approximately 2000 c.p.m. were bound maximally. Membranes, [125I]-GLP 7-36 amide (final concentration 0.1 nM) and GLP-1 7-36 amide at a range of concentrations were added to the plates. Binding was allowed to proceed to equilibrium by incubating at room temperature for 4 h. Membranes were then collected on Whatman GF/C glass fibre filters pre-soaked in 0.5% (v/v) polyethyleneimine using a Brandel Cell Harvester washed through with 2% (w/v) BSA. Membranes were washed with ice-cold buffer (composition: 25 mM HEPES, 1.5 mM CaCl2, 1 mM MgSO4, 100 mM NaCl, pH 7.4) and filters allowed to dry. Bound radioactivity was then determined using a TopCount-NXT liquid scintillation counter (PerkinElmer Life and Analytical Sciences, Beaconsfield, Bucks., UK).
Data analysis
Data were fitted using Prism (GraphPad Software Inc., San Diego, CA, USA) by a standard four-parameter logistic equation. Competition binding curves were constructed assuming one class of binding site and Kd values determined using standard analysis of homologous competition binding data (Prism: GraphPad Software Inc., CA, USA).
RT-PCR
At appropriate times following transfection, cells at approximately 90% confluency in 100 mm diameter tissue culture plates were washed with ice-cold PBS and the RNA extraction using TRIzol reagent using slight modifications of the manufacturer's instructions. After adding TRIzol reagent (1 mL), the resulting suspensions were transferred to microfuge tubes and cells fully lysed by trituration (10 times) through a 23-gauge needle. Samples were incubated at room temperature for 5 min to allow complete dissociation of the nucleoprotein complexes. Chloroform (0.2 mL) was then added, tubes shaken vigorously for 15 s and incubated at room temperature for 2–3 min before centrifugation (12 000×g, 15 min, 4°C). The upper aqueous phase was transferred to a clean microfuge tube and the RNA precipitated by addition of isopropyl alcohol (0.5 mL), incubation at room temperature for 10 min and centrifugation (12 000×g, 10 min, 4°C). The RNA pellet was washed [1 mL 75% (v/v) ethanol] and collected by centrifugation (7500×g, 5 min, 4°C). After briefly air drying, the RNA was dissolved in water (100 µL) and treated with DNase I (1000U·mL−1, 30 min, 37°C). The RNA was purified by addition of TRIzol (1 mL), repeating the procedure outline above and finally re-suspending water (100 µL).
Following the manufacturer's instructions, 3 µg of total RNA from each extraction was used as template for reverse-transcription with Transcriptor Reverse Transcriptase using oligonucleotide dT12–18 in a total reaction volume of 20 µL. PCRs were then performed in a 50 µL volume containing 0.5 unit of Taq DNA polymerase. Oligonucleotides for both full-length and signal peptide-deleted version of the receptor were designed to clone the gene encoding the GLP-1R immediately after the putative signal peptide until the end of the receptor: 5′-CGC CCC CAG GGT GCC ACT G-3′ and 5′-GCT GCA GGA GGC CTG GCA AGT-3′. Thermal cycling conditions were: an initial denaturation step at 95°C for 30 s followed by 25 cycles at 95°C for 30 s, annealing at 55°C for 90 s, extension at 72°C for 3 min and a final extension step at 72°C for 10 min. PCR products were analyzed on a 1% agarose gel electrophoresis stained with GelRed.
Materials
GLP-1 7-36 amide was supplied by Bachem (Weil am Rhein, Germany). [3H]-cAMP (42 Ci·mmole−1) and ECL+ reagent were purchased from GE Healthcare (Bucks., UK). Materials for liquid scintillation counting and [125I]-GLP-1 7-36 amide (2000 Ci·mmol−1) were obtained from Perkin Elmer LAS (UK) Ltd. (Bucks., UK). Oligonucleotides, dNTPs mixture (10 mM), DNA ladder (1 kb plus, 100 bp–12 kp range), Lipofectamine 2000, donkey anti-rat IgG Alexa Fluor 594 linked antibody, pcDNA3.1(+), TRIzol reagent, oligonucleotide dT12–18, Geneticin (G418) and all cell culture reagents were purchased from Invitrogen (Paisley, UK). Restriction endonucleases, DNA ligase, Taq, Vent and Phusion High-Fidelity DNA polymerase, PNGase F, Endo H, goat anti-rabbit IgG HRP-linked antibody were all purchased from New England Biolabs (Hitchin, UK). The QuikChange Site-Directed Mutagenesis Kits were from Stratagene (TX, USA). EZ-Link Sulpho-NHS-Biotin and immobilized NeutrAvidin were obtained from Pierce (Northumberland, UK). Pre-stained protein marker (10–250 kDa range) was supplied by BioRad Laboratories (CA, USA). Transcriptor Reverse Transcriptase, DNase I and rat IgG monoclonal anti-HA antibody were from Roche (Basel, Switzerland) and rabbit IgG polyclonal anti-GFP antibody was from Abcam (Cambridge, UK). GelRed was from Cambridge Bioscience (Cambridge, UK). The pEGFP-N1 and pEGFP-C1 plasmids encoding EGFP were from Clontech Laboratories (Saint-Germain-en-Laye, France). Cell culture plastics were obtained from Nalgene (Hereford, UK). Glass coverslips were from VWR International (Lutterworth, UK). DNA plasmid preparation and DNA gel extraction kits were from QIAGEN (Crawley, UK) and agarose powder was from BiozymT BV (Landgraaf, The Netherlands). PVDF transfer membrane was purchased from Millipore (MA, USA). Benzyloxycarbonyl-L-leucyl-L-leucyl-L-leucinal (MG132) and acetyl-L-leucyl-L-leucyl-L-norleucinal (ALLN or MG101) were from Calbiochem (CA, USA). Goat anti-rat IgG, HRP-linked antibody, cloning discs, MEM Non-essential Amino Acid Solution (100×), and all other reagents were obtained from Sigma-Aldrich (Poole, UK).
Results
N-terminal epitope-tags of the GLP-1R are not detected by immunoblotting despite ligand binding and functional coupling
HEK-293 cells were transiently transfected with a number of GLP-1R constructs, each of which contained the signal peptide sequence and which were tagged at either the N- or C-terminus with either an HA- or EGFP-epitope (Figure 1A). Immunoblotting for the epitope tags on whole-cell lysates prepared 30 h following transfection demonstrated immunoreactive bands only for receptor constructs with C-terminal tags. Thus, two bands at ∼47 and ∼64 kDa were detected using an anti-HA antibody in lysates from cells transfected with the GLP-1R containing a C-terminal HA-tag (GLP-1R-HA) (Figure 1B, lane 2). Two bands were also detected, although at ∼66 and ∼93 kDa, using an anti-GFP antibody in lysates from cells transfected with either the GLP-1R containing a C-terminal EGFP-tag (GLP-1R-EGFP) (Figure 1C, lane 3) or the GLP-1R containing both an N-terminal HA-tag and a C-terminal EGFP-tag (HA-GLP-1R-EGFP) (Figure 1C, lane 5). No HA immunoreactivity was observed in lysates from cells transfected with a construct containing an N-terminal HA-tag immediately before the signal peptide (HA-GLP-1R; Figure 1B, lane 4). However, when this N-terminal HA-tagged construct also contained a C-terminal EGFP-tag (HA-GLP-1R-EGFP), two bands of GFP immunoreactivity were present at the same molecular size as those observed with the GLP-1R-EGFP construct (Figure 1C, lane 5).
Each of the epitope-tagged constructs (GLP-1R-HA, GLP-1R-EGFP, HA-GLP-1R and HA-GLP-1R-EGFP) behaved very similarly to the WTGLP-1R as determined by measurement of both ligand binding affinity and agonist potency. Thus, based on homologous competition binding assays, the affinity (Kd) of GLP-1 7-36 amide was in the low nM range for each of the constructs (Table 2). Furthermore, the concentration dependence of GLP-1-mediated cAMP elevation was comparable in the different constructs with similar pEC50 and Emax values (Figure 4, Table 2).
Table 2.
Agonist potency and affinity estimates at the epitope-tagged glucagon-like peptide-1 receptor (GLP-1R) constructs
| WTGLP-1R | GLP-1R-HA | GLP-1R-EGFP | HA-GLP-1R | HA-GLP-1R –EGFP | |
|---|---|---|---|---|---|
| pEC50 | 8.46 ± 0.12 | 8.03 ± 0.07 | 8.34 ± 0.05 | 8.13 ± 0.18 | 8.69 ± 0.16 |
| pKd | 8.71 ± 0.34 | 8.06 ± 0.14 | 8.45 ± 0.38 | 7.83 ± 0.03 | 8.09 ± 0.05 |
Potency estimates (pEC50 values) were derived for cAMP generation mediated by GLP-1 7-36 amide in intact cells transiently transfected with the indicated GLP-1R constructs. Estimates of agonist (GLP-1 7-36 amide) affinity (pKd values) were derived in membranes prepared from cells using homologous competition binding assays. Data are mean ± SEM, n= 3.
Figure 4.

Functional coupling of glucagon-like peptide-1 receptor (GLP-1R) constructs. Human embryonic kidney (HEK)-293 cells were transiently transfected with plasmids containing either the WTGLP-1R or various epitope-tagged constructs. After 30 h, cells were challenged with GLP-1 7-36 amide at the concentrations indicated and cAMP levels determined after 10 min stimulation. Data are mean ± SEM, n= 3. pEC50 values are given in Table 2.
Removal of the signal peptide sequence abolishes receptor expression
Using the GLP-1R-HA, GLP-1R-EGFP, HA-GLP-1R-EGFP and EGFP-GLP-1R-HA constructs as starting points, the putative signal peptide sequences were removed from each to generate the ΔSP-GLP-1R-HA, ΔSP-GLP-1R-EGFP, HA-ΔSP-GLP-1R-EGFP and EGFP-ΔSP-GLP-1R-HA constructs respectively (Table 1). Following transient transfection of HEK-293 cells with GLP-1R-HA, immunoblotting of cell lysates with an anti-HA antibody revealed the characteristic two bands at ∼47 and ∼64 kDa (Figure 3Bi). These bands were entirely absent following transfection with the signal peptide-deleted construct, ΔSP-GLP-1R-HA (Figure 3Bi). Immunoblotting of cell lysates with an anti-GFP antibody revealed the characteristic two bands (∼66 and ∼93 kDa) in the constructs containing both a C-terminal EGFP-tag and the signal peptide sequence (GLP-1R-EGFP and HA-GLP-1R-EGFP) (Figure 3Aii and iii). In the signal peptide-deleted versions of these constructs (ΔSP-GLP-1R-EGFP, HA-ΔSP-GLP-1R-EGFP), there was no evidence of immunoreactivity using either the anti-GFP (Figure 3Aii and iii) or anti-HA antibody (Figure 3Bii and iii). Confocal imaging of live cells transiently transfected with the constructs containing a C-terminal EGFP-tag and a signal peptide sequence (GLP-1R-EGFP and HA-GLP-1R-EGFP) revealed strong plasma membrane fluorescence (Figure 2Ai and ii). In contrast, no fluorescence was visible in cells transfected with the signal peptide-deleted versions of these constructs (Figure 2Bi and ii).
Figure 3.
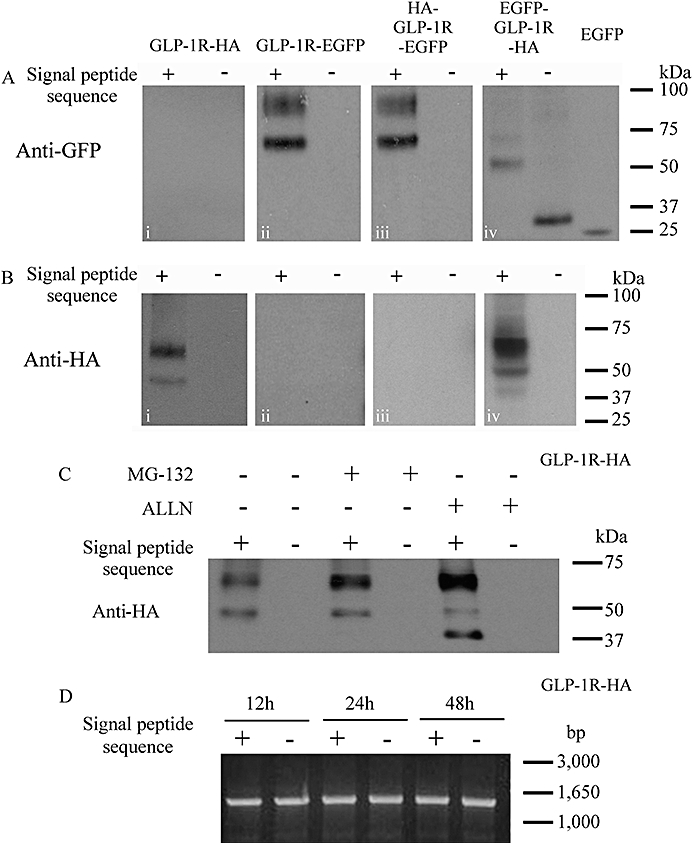
Immunoblotting of cell lysates following transfection of epitope-tagged receptors with or without the signal peptide sequence. Human embryonic kidney (HEK)-293 cells were transiently transfected with plasmids containing various epitope-tagged constructs that either contained the signal peptide sequence (+) or in which it had been deleted (−). After 24–30 h, cell lysates were prepared and immunoblotted using either an anti-HA (A) or an anti-GFP (B) antibody as appropriate. Additionally, groups of cells were transfected with the pEGFP-C1 plasmid to allow expression of EGFP alone. (C) Cells were transfected with either GLP-1R-HA or the signal peptide-deleted equivalent (ΔSP-GLP-1R-HA) and 24 h later treated for a further 6 h with either the proteasome and cathepsin K inhibitor, MG132 (10 µM) or the proteasome and calpain 1 inhibitor, ALLN (MG101; 261 µM/100 µg·mL−1). (D) Cells were transfected with either GLP-1R-HA or ΔSP-GLP-1R-HA and RT-PCR performed at 12, 24 and 48 h using identical forward and reverse primers designed to amplify the sequence representing the full-length receptor but excluding the signal peptide sequence. Data are representative of three independent experiments.
Immunoblotting of the construct containing both an N-terminal EGFP-tag and a C-terminal HA-tag (EGFP-GLP-1R-HA), revealed a band of immunoreactivity at ∼66 kDa using either the anti-GFP antibody (Figure 3Aiv, faint upper band of two bands) or anti-HA antibody (Figure 3Biv) consistent with the size of the lower band in the other constructs containing a C-terminal EGFP-tag (Figure 3Bii and iii). A lower band of ∼52 kDa was also observed with either the anti-GFP antibody (Figure 3Aiv) or anti-HA antibody (Figure 3Biv). Confocal imaging of cells transfected with this construct revealed a cytosolic pattern of fluorescence, with nuclear exclusion and with little or no evidence of plasma membrane fluorescence (Figure 2Aiii). In the signal peptide-deleted version of this construct (EGFP-ΔSP-GLP-1R-HA), GFP immunoreactivity was observed at a low molecular size (∼30 kDa; Figure 3Aiv) that was only slightly greater than the band observed following expression of EGFP alone (∼27 kDa, Figure 3Aiv). Confocal imaging of these cells revealed both cytosolic and nuclear fluorescence (Figure 2Biii). Experiments were performed to assess if the apparent lack of expression of the signal peptide-deleted constructs resulted from synthesis followed by rapid degradation rather than a simple lack of synthesis. Thus, 24 h after transfecting cells with either the GLP-1R-HA or ΔSP-GLP-1R-HA construct, cells were incubated for a further 6 h with either the proteasome and cathepsin K inhibitor, MG132 (10 µM) or the proteasome and calpain 1 inhibitor, ALLN (MG101; 261 µM/100 µg·mL−1) (Willars et al., 2001; Ruvolo et al., 2008). Neither of these inhibitors resulted in the appearance of any HA immunoreactivity for the signal peptide-deleted construct, although both inhibitors tended to increase the intensity of the upper (∼64 kDa) band and cause the appearance of a lower (∼42 kDa) band (although this was only visible at this level of exposure following ALLN treatment) for the construct that included the signal peptide sequence (Figure 3C).
To assess if deletion of the signal peptide sequence resulted in the generation of unstable mRNA, RT-PCR was performed at 12, 24 and 48 h following transfection of cells with either GLP-1R-HA or ΔSP-GLP-1R-HA. This RT-PCR indicated equivalent levels of mRNA at all times and provided no evidence for differences between the construct with or without the signal peptide sequence (Figure 3D).
As shown (Table 2), the epitope-tagged constructs that included the signal peptide sequence (GLP-1R-EGFP and HA-GLP-1R-EGFP) bound GLP-1 7-36 amide with similar affinity to the WT receptor, while stimulation with GLP-1 7-36 amide evoked cAMP accumulation with similar potency and with a similar Emax to that of the WT receptor (Figure 4, Table 2). In cells transiently transfected with these constructs but with the signal peptide sequence removed, there was no detectable binding of GLP-1 7-36 amide (data not shown) and GLP-1 failed to elevate cAMP (Figure 5). In cells transfected with the EGFP-GLP-1R-HA construct there was no binding (data not shown) or function (Figure 5) whether or not the construct contained the signal peptide sequence.
Figure 5.
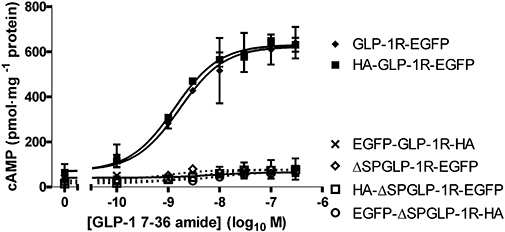
Functional coupling of glucagon-like peptide-1 receptor (GLP-1R) constructs with or without the signal peptide sequence. Human embryonic kidney (HEK)-293 cells transiently transfected with epitope-tagged GLP-1Rs either with or without the signal peptide sequence were assessed for coupling to the generation of cAMP. Cells were challenged with GLP-1 7-36 amide at the concentrations indicated and cAMP levels determined after 10 min stimulation. Data are mean ± SEM, n= 3. pEC50 values for those receptors having the signal peptide sequence and in which cAMP responses were measurable (GLP-1R-EGFP and HA-GLP-1R-EGFP) were similar to those in Table 2.
Lack of a signal peptide sequence can be partially compensated by increasing the positive charge within the N-terminus
As described above, in the absence of a signal peptide sequence the GLP-1R was not expressed. However, replacing the negatively charged, polar glutamic acid residue in the sequence immediately following the signal peptide with a positively charged, polar arginine residue (E34R) facilitated expression in the absence of the signal peptide sequence. As above, immunoblotting of the HA-GLP-1R-EGFP construct with an anti-GFP antibody revealed two bands (Figure 6Ai and v), but no immunoreactivity was observed using the anti-HA antibody (Figure 6Aiii and vii). Removal of the signal peptide sequence (HA-ΔSP-GLP-1R-EGFP) abolished GFP immunoreactivity (Figure 6Ai and v). Immunoreactivity of the E34R mutant (HA-E34R-GLP-1R-EGFP) was identical to that of the construct in the absence of the mutation; that is, two bands were detected using the anti-GFP antibody (Figure 6Aii and vi). In contrast to the HA-GLP-1R-EGFP construct, removal of the signal peptide from the mutated receptor (HA-ΔSP-E34R-GLP-1R-EGFP) did not abolish immunoreactivity. Although the GFP immunoreactivity was relatively weak, two bands were observed, consistent with those seen for both the HA-GLP-1R-EGFP and HA-E34R-GLP-1R-EGFP constructs (Figure 6Aii and vi). An additional lower molecular size band was also observed (∼52 kDa) (Figure 6Aii and vi). Immunoblotting of the HA-ΔSP-E34R-GLP-1R-EGFP construct with an anti-HA antibody also revealed three bands that were of identical molecular size to those detected by the anti-GFP antibody, although the lower band was very faint (Figure 6Aiv and viii). Confocal imaging of live cells transiently transfected with the E34R mutant showed robust plasma membrane fluorescence (Figure 6B). In the absence of the signal peptide (HA-ΔSP-E34R-GLP-1R-EGFP), the predominant fluorescence was cytosolic but overlap with Trypan blue staining indicated some localization at the plasma membrane (Figure 6B). GLP-1 stimulation of cells transfected with the mutated receptor containing the signal peptide sequence (HA-E34R-GLP-1R-EGFP) resulted in a concentration-dependent generation of cAMP similar to that seen in cells expressing HA-GLP-1R-EGFP (Figure 6C). Cells transfected with the signal peptide-deleted construct (HA-ΔSP-GLP-1R-EGFP) barely responded to challenge with GLP-1, consistent with a lack of expression. In cells expressing the mutated, signal peptide-deleted construct (HA-ΔSP-E34R-GLP-1R-EGFP), GLP-1 evoked cAMP generation with similar potency but a lower Emax than in cells expressing either the un-mutated (HA-GLP-1R-EGFP) or mutated epitope-tagged receptor (HA-E34R-GLP-1R-EGFP) (Figure 6C).
Figure 6.
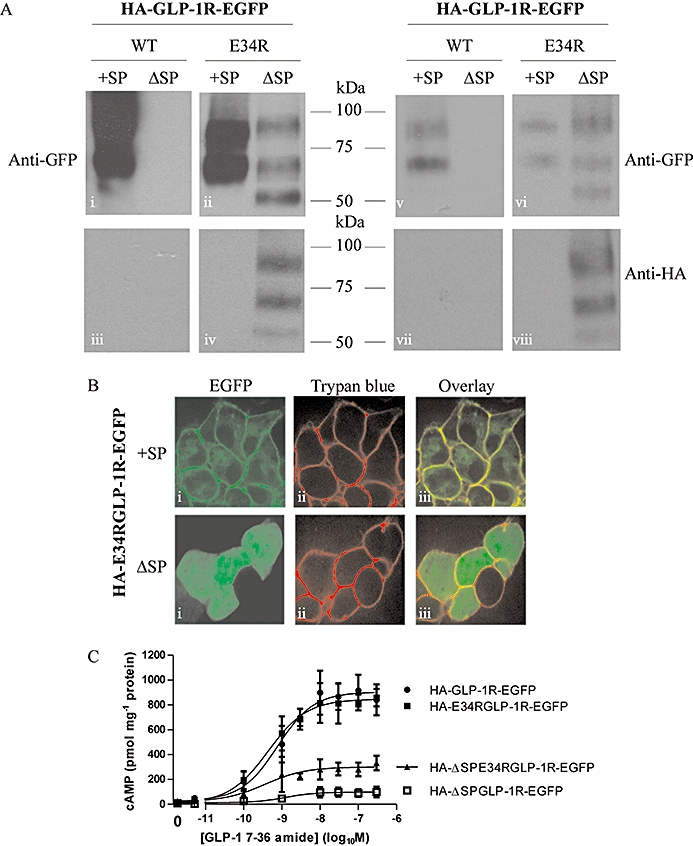
Increasing positive charge within the N-terminus of the receptor partially compensates for the lack of a signal peptide. Human embryonic kidney (HEK)-293 cells were transiently transfected with the glucagon-like peptide-1 receptor (GLP-1R) construct containing an N-terminal HA-tag and C-terminal EGFP tag either with (HA-GLP-1R-EGFP) or without (HA-ΔSP-GLP-1R-EGFP) the signal peptide. We also used similar constructs in which the glutamic acid residue at position 34 had been mutated to an arginine (HA-E34R-GLP-1R-EGFP and HA-ΔSP-E34R-GLP-1R-EGFP). After 24–30 h, cell lysates were prepared and immunoblotted using either an anti-HA or an anti-GFP antibody (A). In the left-hand panel, lanes were equally loaded, showing the relatively poor but nevertheless clear expression of the HA-ΔSP-E34R-GLP-1R-EGFP construct. In the right-hand panels, lanes for the constructs containing the signal peptide sequence (+SP) were loaded with fivefold less protein than lanes for the signal peptide-deleted constructs (ΔSP) to more clearly show the relative patterns of immunoreactive bands. Live cells were examined by confocal microscopy (B) or challenged with GLP-1 7-36 amide at the concentrations indicated and cAMP levels determined after 10 min stimulation (C). Immunoblots and confocal images are representative of three independent experiments while data for cAMP generation are mean ± SEM, n= 3–6. pEC50 values: HA-GLP-1R-EGFP, 9.12 ± 0.24; HA-ΔSP-GLP-1R-EGFP, 9.36 ± 0.15; HA-E34R-GLP-1R-EGFP, 8.74 ± 0.47; HA-ΔSP-E34R-GLP-1R-EGFP, 8.46 ± 0.62.
Mutating the residue at position −3 of the predicted signal peptide cleavage site blocks both cleavage and plasma membrane expression
Expression of a construct containing an N-terminal EGFP tag and a C-terminal tag (EGFP-GLP-1R-HA) revealed an immunoblotting pattern and cellular distribution consistent with the synthesis of a full-length receptor, from which the signal peptide was not cleaved and which was not trafficked to the plasma membrane (see above). To distinguish the role of signal peptide cleavage from the presence of the EGFP-epitope tag on receptor trafficking we generated a construct containing a mutation in the vicinity of the cleavage site, which would be predicted to reduce the likelihood of cleavage. Using the neural networks algorithm, SignalP 3.0 (Bendtsen, 2004) predicts the signal peptidase 1 cleavage site of the human GLP-1R to lie between proline 23 and arginine 24 (C-score 0.528). Such cleavage sites are characterized by small, uncharged residues at positions −1 and −3 (von Heijne, 1990). Replacing the alanine residue at −3 with a positively charged and large arginine residue (A21R) was predicted to markedly reduce the probability of signal peptide cleavage (C-score 0.267). As above, immunoblotting of the wild type epitope-tagged receptor (HA-GLP-1R-EGFP) transiently expressed in HEK-293 cells revealed two bands with the anti-GFP antibody and no bands with the anti-HA antibody (Figure 7A) consistent with the synthesis of a full-length receptor from which the signal peptide (and HA-tag) was cleaved. In contrast, immunoreactivity of the cleavage site mutant (HA-A21R-GLP-1R-EGFP) was observed using both anti-GFP and anti-HA antibodies, although in each case only one band at the lower molecular size was observed (Figure 7A). Confocal imaging of cells transiently transfected with the A21R mutant demonstrated cytosolic fluorescence with no evidence of fluorescence at the plasma membrane (Figure 7B). Challenge of cells expressing the A21R mutant barely evoked cAMP generation in comparison with the response in cells expressing the HA-GLP-1R-EGFP construct (Figure 7C).
Figure 7.
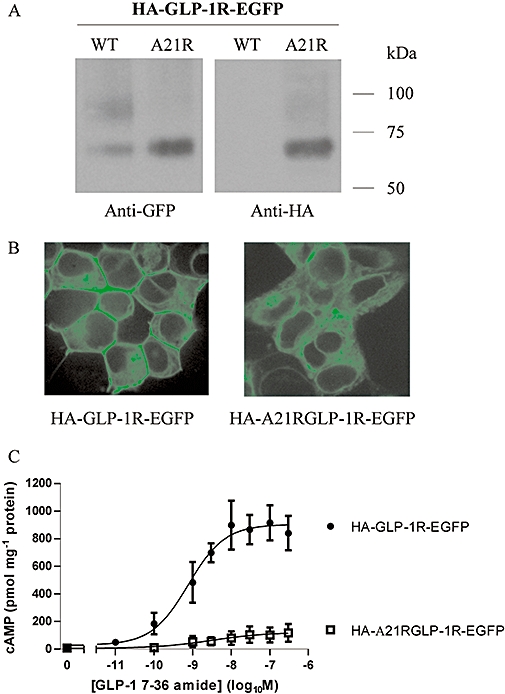
Mutation of the predicted signal peptide cleavage site blocks both cleavage and plasma membrane expression of the glucagon-like peptide-1 receptor (GLP-1R). Human embryonic kidney (HEK)-293 cells were transiently transfected with either HA-GLP-1R-EGFP or this construct containing an alanine to arginine mutation at position −3 relative to the predicted cleavage site (HA-A21R-GLP-1R-EGFP). After 24–30 h, cell lysates were prepared and immunoblotted using either an anti-HA or an anti-GFP antibody (A). Live cells were examined by confocal microscopy (B) or challenged with GLP-1 7-36 amide at the concentrations indicated and cAMP levels determined after 10 min stimulation (C). Immunoblots and confocal images are representative of three independent experiments while data for cAMP generation are mean ± SEM, n= 3. pEC50 value for HA-GLP-1R-EGFP, 9.09 ± 0.22.
Glycosylation of the GLP-1R
Immunoblotting of GLP-1R constructs containing a C-terminal HA- or EGFP-tag revealed two distinct bands (Figure 8 and elsewhere). For the HA-tagged receptor these were ∼47 and ∼64 kDa, whereas for the EGFP-tagged receptor these were approximately ∼66 and ∼93 kDa. The predicted molecular sizes of the HA- and EGFP-tagged GLP-1R constructs (with no signal peptide) are ∼51 and ∼78 kDa respectively, suggesting that the bands may represent differentially glycosylated forms of the receptor. This was assessed by determining the effect of the treatment of cell lysates with either Endo H or PNGase F. Endo H removes N-linked, high-mannose oligosaccharides added in the endoplasmic reticulum to protein domains that will become extracellular. Subsequent processing of these glycosylations to complex oligosaccharides during Golgi processing results in resistance to Endo H. However, both high-mannose oligosaccharides and complex oligosaccharides can be removed by PNGase F. Treatment with Endo H had no effect on the position of the upper band (∼64 kDa in GLP-1R-HA and ∼93 kDa in both GLP-1R-EGFP and HA-GLP-1R-EGFP) but reduced the molecular size of the lower band from ∼47 to ∼42 Da in GLP-1R-HA and from ∼66 to ∼52 kDa in both GLP-1R-EGFP and HA-GLP-1R-EGFP (Figure 8). In contrast, treatment of cell lysates with PNGase F resulted in a marked or complete loss of the upper bands in either HA- or EGFP-tagged receptors and the appearance of a lower band consistent with the position of the lower band in each construct following treatment with Endo H (Figure 8).
Figure 8.
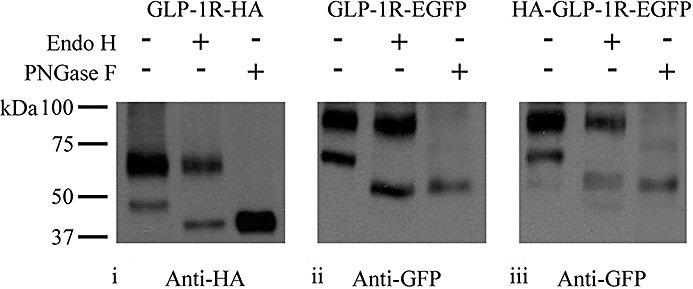
Glycosylation of the glucagon-like peptide-1 receptor (GLP-1R). Human embryonic kidney (HEK)-293 cells were transiently transfected with plasmids containing epitope-tagged GLP-1Rs as indicated. After 48 h, cell lysates were prepared and either untreated or subjected to treatment with either Endo H or PNGase F as described in Methods. Lysates were then processed and immunoblotted using either an anti-HA (i) or anti-GFP (ii and iii) antibody as indicated. Data are representative of three independent experiments.
In the HEK-293 : GLP-1R-EGFP cell line EGFP fluorescence was predominantly at the plasma membrane (Figure 9A). The pattern of GFP staining in immunoblots was entirely consistent with that in cells transiently expressing this construct (compare Figure 9Bi, lane 2 with e.g. Figure 1C, lane 3). Furthermore, the potency of GLP-1 7-36 amide was similar to those of transiently expressed WT and epitope-tagged receptors (pEC50 8.47 ± 0.05, n= 3; compare with data in Table 2). Treatment of lysates from the cells with stable expression of GLP-1R-EGFP with either Endo H or PNGase F also altered the pattern of immunoreactivity in a manner entirely consistent with that seen following transient expression. Thus, Endo H treatment reduced the molecular size of the lower band only (from∼66 kDa to ∼52 kDa; Figure 9Bi, lane 3), while treatment with PNGase F resulted in only one band at the molecular size of the lower band seen following treatment with Endo H (∼52 kDa; Figure 9Bi, lane 4). Immunoblotting with an anti-GFP antibody of cell surface proteins captured following biotinylation of intact cells with stable expression of GLP-1R-EGFP revealed only the higher molecular size (∼93 kDa) band (Figure 9Bi, lane 1). Culture of cells in the presence of tunicamycin to inhibit glycosylation resulted in only one immunoreactive band coincident with the lower of the two bands observed in untreated cells (∼66 kDa; compare Figure 9Ci, lane 1 with Figure 9Bi, lane 2). Treatment of lysates from cells cultured in the presence of tunicamycin with either Endo H or PNGase F reduced the molecular size of this one band to ∼52 kDa (Figure 9Ci, lanes 2 and 3). Confocal imaging of cells with stable expression of GLP-1R-EGFP revealed strong fluorescence at the plasma membrane (Figure 9A and Bii), whereas tunicamycin treatment abolished this fluorescence and produced a pattern consistent with a cytosolic distribution (Figure 9Cii).
Figure 9.
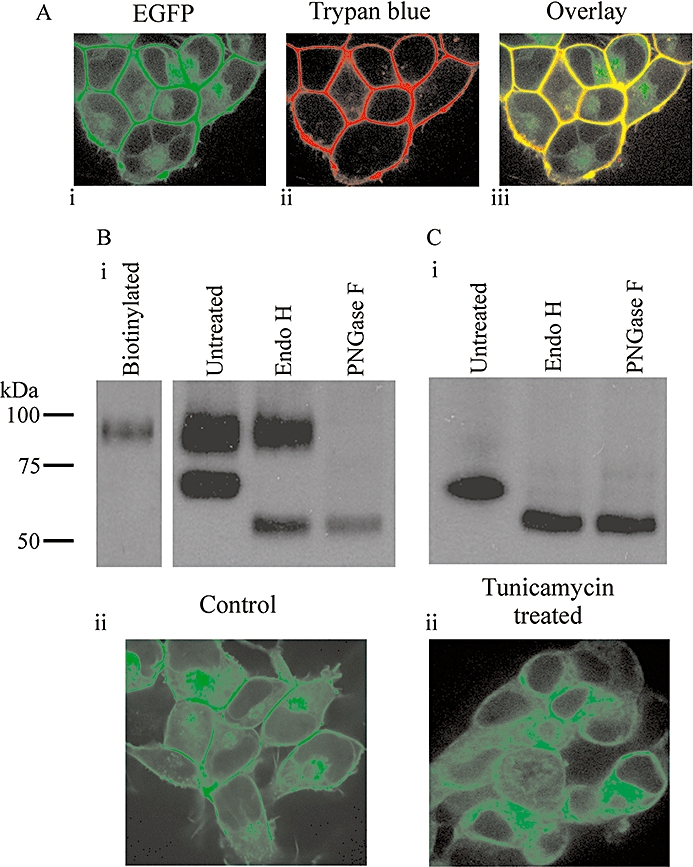
Influence of the glycosylation inhibitor, tunicamycin, on the glycosylation pattern and subcellular distribution of the glucagon-like peptide-1 receptor (GLP-1R). Human embryonic kidney (HEK)-293 cells with stable expression of the C-terminal, EGFP-tagged GLP-1R (GLP-1R-EGFP) were either untreated (A and B) or treated with tunicamycin (C) for 7.5 days as described in Methods. Cells were then examined by confocal microscopy (A, Bii and Cii), including following membrane staining with trypan blue (A) or lysates prepared and either untreated or subjected to treatment with either Endo H or PNGase F as described. Lysates were then processed and immunoblotted using an anti-GFP antibody (Bi and Ci). Alternatively, proteins at the plasma membrane of intact cells were biotinylated, captured using streptavidin and subject to immunoblotting using the anti-GFP antibody (Bi; ‘biotinylated’). Data are representative of six coverslips and three immunoblots.
Discussion and conclusion
In GLP-1R constructs containing an HA-tag located N-terminal to the putative signal peptide, no HA immunoreactivity could be detected, although EGFP immunoreactivity was detected with HA-GLP-1R-EGFP, confirming receptor synthesis. Further, following transfection of either HA-GLP-1R or HA-GLP-1R-EGFP, GLP-1 7-36 amide both bound to cells and stimulated cAMP generation, as did the WTGLP-1R. Thus, unlike the CRF2a receptor (Rutz et al., 2006) but similar to other Family B GPCRs, including the CRF1 receptor (Alken et al., 2005), the VPAC1 receptor for vasoactive intestinal peptide and pituitary adenylyl cyclase activating peptide (Couvineau et al., 2004) and the parathyroid hormone receptor (Shimada et al., 2002), the GLP-1R signal peptide is cleaved during synthesis. Thus, incorporation of the signal peptide structure within models of the extracellular, N-terminal domain of the GLP-1R (Lin and Wang, 2009) is not required. Using predictive programmes, the signal peptidase cleavage site is consistently predicted between residues 23 and 24. For example, SignalP 3.0 (Bendtsen, 2004) indicates this cleavage site with the neural networks algorithm, although the hidden Markov model indicates cleavage between positions 21 and 22 (alanine-glycine) is a possibility.
Many proteins contain a signal peptide thought to play a critical role in synthesis but which is absent from the mature protein. For GPCRs it has been suggested that a signal peptide is required where the N-terminal domains are highly structured (Köchl et al., 2002). The recent crystal structure of this region in the GLP-1R indicates that it is indeed highly structured (Runge et al., 2008). Although clearly critical in some circumstances, the full roles of signal peptides are unclear and in some instances they are not obligatory. For example, the Na+-Ca2+-exchanger contains a cleaved signal peptide, but removal influences neither plasma membrane expression nor function (Loo et al., 1995). Thus, another internal sequence must fulfil the role of a signal anchor sequence. The Family A endothelin ETB GPCR also has a cleaved signal peptide and although synthesis is unaffected by removal this causes retention within the endoplasmic reticulum. This indicates that while another region, possibly the first transmembrane domain, provides a signal anchor sequence, the N-terminal domain cannot be post-translationally translocated across the endoplasmic reticulum membrane (Köchl et al., 2002). Similarly, the Family B VPAC1 receptor is synthesized but not trafficked to the plasma membrane in the absence of its signal peptide (Couvineau et al., 2004). For the Family B CRF1 receptor, the signal peptide is not essential for expression of a functional receptor despite promoting an early step of receptor biogenesis and greatly enhancing expression (Alken et al., 2005). We found no evidence that lack of the signal peptide sequence either reduced the stability of the mRNA or resulted in synthesis of a protein that was subject to rapid proteasomal degradation. Thus, in contrast to the examples above, a full-length GLP-1R was not synthesized following removal of the signal peptide sequence, indicating that other sequences are unable to act as signal anchors. Under these circumstances synthesis would be expected to proceed in the cytoplasm until the hydrophobic first transmembrane region, which would generate a soluble fragment of ∼12 kDa. In the signal peptide-deleted construct EGFP-ΔSP-GLP-1R-HA, EGFP immunoreactivity was observed at ∼30 kDa, which is greater than EGFP (27 kDa) but considerably less than a fusion containing EGFP and the GLP-1R N-terminal domain. This suggests that EGFP is synthesized with a small part of the receptor N-terminus but that this cannot be post-translationally translocated across the endoplasmic reticulum membrane. Indeed, the protein sequence following the signal peptide of the GLP-1R is relatively hydrophobic (G27ATVSLWETVQKW39) and this may be sufficient for recognition by the signal recognition particle and subsequent elongation arrest (Hatsuzawa et al., 1997). Recently, it has been shown that a similar region of the endothelin ETB receptor (Q28-W54) is an important domain in the trafficking of this receptor by facilitating translocon gating at the membrane of the endoplasmic reticulum (Alken et al., 2009). However, in the GLP-1R there may be insufficient positive charge in this region for membrane integration (Kida et al., 2000), thereby preventing further synthesis and resulting in an apparently unrestricted subcellular distribution. This was confirmed by mutation of the negatively charged glutamic acid at position 34 to a positively charged arginine (E34R). Thus, even in the absence of a signal peptide, this construct was synthesized and at least a proportion trafficked to the plasma membrane where it coupled GLP-1 to cAMP generation.
Interestingly, when the GLP-1R, including the signal peptide, had an N-terminal EGFP and C-terminal HA-epitope (EGFP-GLP-1R-HA), immunoblotting indicated a full-length construct containing both epitope-tags. The subcellular distribution of this was consistent with trapping in the endoplasmic reticulum, indicating that EGFP does not prevent the signal peptide facilitating translocation into the endoplasmic reticulum. However, this suggests either that EGFP is sufficient to block receptor trafficking or that signal peptide cleavage is required for export. It is unclear whether in this construct EGFP blocks trafficking, but other similarly tagged GPCRs, including the vasopressin V1B/V3 and CB1 cannabinoid receptors, are expressed at the plasma membrane (Robert et al., 2005; Tappe-Theodor et al., 2007). Thus, it is likely that in the EGFP-GLP-1R-HA, either EGFP or an associated conformational change around the cleavage site prevents signal peptidase access and this lack of cleavage blocks further receptor trafficking. This was confirmed by an alanine to arginine mutation at the −3 position of the predicted cleavage site (A21R). Consistent with the requirement for small uncharged residues at −3 and −1 (von Heijne, 1990), this was predicted to inhibit cleavage. Indeed, the mutation allowed synthesis of a full-length receptor but prevented both signal peptide cleavage and trafficking to the plasma membrane. Thus, signal peptide is essential for receptor synthesis but cleavage is not. Cleavage is, however, a prerequisite for trafficking from endoplasmic reticulum to plasma membrane.
Consistent with previous studies on the recombinant GLP-1R in CCL39 fibroblasts (Widmann et al., 1995), the current data show that the two bands detected by immunoblotting represent different N-linked glycosylation states. Thus, Endo H reduced the molecular size of the lower of the two bands, indicating that this represents receptor glycosylated with high-mannose oligosaccharides in the endoplasmic reticulum and which has not been processed further. PNGase F reduced the molecular size of both bands, indicating that the upper band represents the mature, fully glycosylated receptor. Biotinylation experiments demonstrated that only this form is present in the plasma membrane and which, therefore, would bind agonist. Indeed, of the two immunoreactive bands in the rat GLP-1R, only the higher-molecular-sized band binds GLP-1 (Widmann et al., 1995).
In the EGFP-GLP-1R-HA construct, only the lower molecular size band was apparent and detected by both anti-GFP and anti-HA antibodies. Thus, this construct, which is resistant to signal peptidase activity, had immunoreactivity consistent with glycosylation in the endoplasmic reticulum but no further processing. This was supported by the confocal imaging of EGFP fluorescence. Incubation of cells stably expressing the GLP-1R-EGFP construct with tunicamycin also produced subcellular fluorescence consistent with endoplasmic reticulum retention, highlighting the importance of glycosylation to receptor processing and trafficking. Immunoblotting of tunicamycin-treated cells also revealed only one band of EGFP immunoreactivity, consistent with the molecular size of the lower band in untreated cells. Interestingly, this band was still sensitive to both Endo H and PNGase F treatment, which reduced the size of the band to that following such treatment in the control (tunicamycin-untreated cells). This suggests that tunicamycin may not be a fully effective inhibitor of the initial step in N-linked glycosylation. Alternatively, the band may represent receptor synthesized and glycosylated with high-mannose oligosaccharides prior to inhibition of glycosylation and which cannot be further processed due to either the direct effects of tunicamycin on the glycosylation process or more general effects such as induction of the unfolded protein response.
Glycosylation sites in the sequons Asn-Xaa-Ser/Thr are predicted in the N-terminus of the GLP-1R at N63, N82 and N115 using NetNGlyc 1.0 Server (http://www.cbs.dtu.dk/services/NetNGlyc/). Our data indicate that full glycosylation of the receptor is required for expression of the mature functional receptor at the plasma membrane. Interestingly, N63 is an important residue within the NRTFD sequence (representing residues 63–67 of the GLP-1R) that may act as an endogenous agonist (Dong et al., 2008). Thus, the exogenously added NRTFD pentapeptide is a full agonist at recombinant GLP-1Rs whereas mutation of the asparagine residue to either leucine or isoleucine markedly reduces potency. However, the exogenous peptide was not glycosylated and it is unclear how glycosylation of N63 would affect any agonism mediated by this region in the intact receptor.
In summary, the putative signal peptide of the GLP-1R was cleaved during synthesis and processing of the receptor. Further, while cleavage was not essential for receptor synthesis, it was obligatory for effective processing and trafficking to the plasma membrane. In contrast to a number of other proteins, including some related GPCRs, the signal peptide was an absolute requirement for synthesis of the GLP-1R. This requirement could be overcome to some extent by increasing the positive charge in the N-terminal region adjacent to the signal peptide. We also confirmed that the GLP-1R was subject to N-linked glycosylation and that it was only the mature, fully glycosylated receptor that was present in the plasma membrane. Further, inhibition of glycosylation prevented appropriate processing and expression of the GLP-1R. Interestingly, two single-amino-acid mutations within the calcium-sensing receptor signal peptide sequence reduce both intracellular and plasma membrane expression and result in parathyroid dysfunction (Pidasheva et al., 2005). Whether such changes underlie altered GLP-1 function in any human disease remains to be established.
Acknowledgments
This study was funded by the Biotechnology and Biological Sciences Research Council through a Doctoral Training Grant with additional support from AstraZeneca, Alderely Park, Macclesfield, UK.
Glossary
Abbreviations:
- ALLN
acetyl-L-leucyl-L-leucyl-L-norleucinal
- CRF
corticotropin-releasing hormone
- EGFP
enhanced GFP
- EGFP-GLP-1R-HA
GLP-1R containing an N-terminal EGFP-tag and a C-terminal HA-tag
- WTGLP-1R
untagged (wild-type) GLP-1R
- Endo H
endoglycosidase H
- GFP
green fluorescent protein
- GLP-1
glucagon-like peptide-1
- GLP-1R
GLP-1 receptor
- GLP-1R-EGFP
GLP-1R containing a C-terminal EGFP-tag
- GLP-1R-HA
GLP-1R containing an C-terminal HA-tag
- GPCR
G protein-coupled receptor
- HA
sequence corresponding to amino acids 98-106 of human influenza hemagglutinin
- HA-A21RGLP-1R-EGFP
epitope-tagged construct with A21R mutation
- HA-E34RGLP-1R-EGFP
epitope-tagged construct with E34R mutation
- HA-GLP-1R
GLP-1R containing an N-terminal HA-tag immediately before the signal peptide
- HA-GLP-1R-EGFP
HA-GLP-1R with an additional C-terminal EGFP-tag
- HA-ΔSPE34RGLP-1R-EGFP
epitope-tagged E34R mutant lacking the putative signal peptide sequence
- HEK-293
human embryonic kidney
- HEK-293 : GLP-1R-EGFP
HEK-293 cell line with stable expression of the GLP-1R-EGFP
- KHB
Krebs-HEPES buffer
- MG132
benzyloxycarbonyl-L-leucyl-L-leucyl-L-leucinal
- PNGase F
peptide N-glycosidase F
- ΔSP-GLP-1R-HA, ΔSP-GLP-1R-EGFP
HA-ΔSP-GLP-1R-EGFP and EGFP-ΔSP-GLP-1R-HA, constructs from which the putative signal peptide sequences were removed
- VPAC1 receptor
receptor for vasoactive intestinal peptide and pituitary adenylyl cyclase activating peptide
Conflicts of interest
G.F.W. is an employee of AstraZeneca. G.B.W. and Y.H. have received funding from AstraZeneca to support this work.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 3rd edition (2008 revision) Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alken M, Rutz C, Köchl R, Donalies U, Oueslati M, Furkert J. The signal peptide of the rat corticotropin-releasing factor receptor 1 promotes receptor expression but is not essential for establishing a functional receptor. Biochem J. 2005;390:455–464. doi: 10.1042/BJ20050113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alken M, Schmidt A, Rutz C, Furkert J, Kleinau G, Rosenthal W, et al. The sequence after the signal peptide of the G protein-coupled endothelin B receptor is required for efficient translocon gating at the endoplasmic reticulum membrane. Mol Pharmacol. 2009;75:801–811. doi: 10.1124/mol.108.051581. [DOI] [PubMed] [Google Scholar]
- Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–2157. doi: 10.1053/j.gastro.2007.03.054. [DOI] [PubMed] [Google Scholar]
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Brown BL, Albano JDM, Ekins RP, Sgherzi AM, Tampion W. A simple and sensitive saturation assay method for the measuremant of adenosine 3'5'-cyclic monophosphate. Biochem J. 1971;121:561–562. doi: 10.1042/bj1210561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couvineau A, Rouyer-Fessard C, Laburthe M. Presence of a N-terminal signal peptide in class II G protein-coupled receptors: a crucial role for expression of the human VPAC1 receptor. Regul Pept. 2004;123:181–185. doi: 10.1016/j.regpep.2004.06.025. [DOI] [PubMed] [Google Scholar]
- Dong M, Gao F, Pinon DI, Miller LJ. Insights into the structural basis of endogenous activation of Family B G protein-coupled receptors. Mol Endocrinol. 2008;22:1489–1499. doi: 10.1210/me.2008-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle ME, Egan JM. Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacol Ther. 2007;113:546–593. doi: 10.1016/j.pharmthera.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatsuzawa K, Tagaya M, Mizushima S. The hydrophobic region of signal peptides is a determinant for SRP recognition and protein translocation across the ER membrane. J Biochem. 1997;121:270–277. doi: 10.1093/oxfordjournals.jbchem.a021583. [DOI] [PubMed] [Google Scholar]
- von Heijne G. The signal peptide. J Membr Biol. 1990;115:195–201. doi: 10.1007/BF01868635. [DOI] [PubMed] [Google Scholar]
- Kida Y, Sakaguchi M, Fukuda M, Mikoshiba K, Mihara K. Membrane topogenesis of a type I signal-anchor protein, mouse synaptotagmin II, on the endoplasmic reticulum. J Cell Biol. 2000;150:719–729. doi: 10.1083/jcb.150.4.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köchl R, Alken M, Rutz C, Krause G, Oksche A, Rosenthal W, et al. The signal peptide of the G protein-coupled human endothelin B receptor is necessary for translocation of the N-terminal tail across the endoplasmic reticulum membrane. J Biol Chem. 2002;277:16131–16138. doi: 10.1074/jbc.M111674200. [DOI] [PubMed] [Google Scholar]
- Lin F, Wang R. Molecular modeling of the three-dimensional structure of GLP-1R and its interactions with several agonists. J Mol Model. 2009;15:53–65. doi: 10.1007/s00894-008-0372-2. [DOI] [PubMed] [Google Scholar]
- Loo TW, Ho C, Clarke DM. Expression of a functionally active human renal sodium-calcium exchanger lacking a signal sequence. J Biol Chem. 1995;270:19345–19350. doi: 10.1074/jbc.270.33.19345. [DOI] [PubMed] [Google Scholar]
- Pidasheva S, Canaff L, Simonds WF, Marx SJ, Hendy GN. Impaired cotranslational processing of the calcium-sensing receptor due to signal peptide missense mutations in familial hypocalciuric hypercalcemia. Hum Mol Genet. 2005;14:1679–1690. doi: 10.1093/hmg/ddi176. [DOI] [PubMed] [Google Scholar]
- Robert J, Clauser E, Petit PX, Ventura MA. A novel C-terminal motif is necessary for the export of the vasopressin V1b/V3 receptor to the plasma membrane. J Biol Chem. 2005;280:2300–2308. doi: 10.1074/jbc.M410655200. [DOI] [PubMed] [Google Scholar]
- Runge S, Thogersen H, Madsen K, Lau J, Rudolph R. Crystal structure of the ligand-bound glucagon-like peptide-1 receptor extracellular domain. J Biol Chem. 2008;283:11340–11347. doi: 10.1074/jbc.M708740200. [DOI] [PubMed] [Google Scholar]
- Rutz C, Renner A, Alken M, Schulz K, Beyermann M, Wiesner B, et al. The corticotropin-releasing factor receptor type 2a contains an N-terminal pseudo signal peptide. J Biol Chem. 2006;281:24910–24921. doi: 10.1074/jbc.M601554200. [DOI] [PubMed] [Google Scholar]
- Ruvolo VR, Kurinna SM, Karanjeet KB, Schuster TF, Martelli AM, McCubrey JA, et al. PKR regulates B56α-mediated BCL2 phosphatase activity in acute lymphoblastic leukemia-derived REH cells. J Biol Chem. 2008;283:35474–35485. doi: 10.1074/jbc.M800951200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada M, Chen X, Cvrk T, Hilfiker H, Parfenova M, Segre GV. Purification and characterization of a receptor for human parathyroid hormone and parathyroid hormone-related peptide. J Biol Chem. 2002;277:31774–31780. doi: 10.1074/jbc.M204166200. [DOI] [PubMed] [Google Scholar]
- Tappe-Theodor A, Agarwal N, Katona I, Rubino T, Martini L, Swiercz J, et al. A molecular basis of analgesic tolerance to cannabinoids. J Neurosci. 2007;27:4165–4177. doi: 10.1523/JNEUROSCI.5648-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallin E, von Heijne G. Properties of N-terminal tails in G-protein coupled receptors: a statistical study. Protein Eng. 1995;8:693–698. doi: 10.1093/protein/8.7.693. [DOI] [PubMed] [Google Scholar]
- Widmann C, Dolci W, Thorens B. Agonist-induced internalisation and recycling of the glucagon-like peptide-1 receptor in transfected fibroblasts and in insulinomas. Biochem J. 1995;310:203–214. doi: 10.1042/bj3100203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willars GB, Nahorski SR. Quantitative comparisons of muscarinic and bradykinin receptor-mediated Ins(1,4,5)P3 and Ca2+ signalling in human neuroblastoma cells. Br J Pharmacol. 1995;114:1113–1142. doi: 10.1111/j.1476-5381.1995.tb13325.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willars GB, Royall JE, Nahorski SR, El Gehani F, Everest H, McArdle CA. Rapid down-regulation of the type I inositol 1,4,5-trisphosphate receptor and desensitization of gonadotropin-releasing hormone-mediated Ca2+ responses in alpha T3-1 gonadotropes. J Biol Chem. 2001;276:3123–3129. doi: 10.1074/jbc.M008916200. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Henzel WJ. Signal peptide prediction based on analysis of experimentally verified cleavage sites. Protein Sci. 2004;13:2819–2824. doi: 10.1110/ps.04682504. [DOI] [PMC free article] [PubMed] [Google Scholar]