

Abstract
Mitochondria continuously undergo two opposing processes, fission and fusion. The disruption of this dynamic equilibrium may herald cell injury or death and may contribute to developmental and neurodegenerative disorders. Nitric oxide functions as a signaling molecule, but in excess it mediates neuronal injury, in part via mitochondrial fission or fragmentation. However, the underlying mechanism for nitric oxide–induced pathological fission remains unclear. We found that nitric oxide produced in response to β-amyloid protein, thought to be a key mediator of Alzheimer’s disease, triggered mitochondrial fission, synaptic loss, and neuronal damage, in part via S-nitrosylation of dynamin-related protein 1 (forming SNO-Drp1). Preventing nitrosylation of Drp1 by cysteine mutation abrogated these neurotoxic events. SNO-Drp1 is increased in brains of human Alzheimer’s disease patients and may thus contribute to the pathogenesis of neurodegeneration.
Disrupting the balance between mitochondrial fission and fusion can lead to excessive mitochondrial fragmentation. Fragmentation triggered by dysfunction of the fission-inducing protein Drp1 (dynamin-related protein 1), for example, contributes to synaptic damage and subsequent neuronal loss because of nitrosative/oxidative stress and impaired bioenergetics (1–6). Excessive fission results in abnormally small mitochondria with fragmented cristae (2), as observed in electron microscopy studies of neurons in human Alzheimer’s disease (AD) (7). Drp1 homologs are S-nitrosylated, which regulates their activity (8, 9). Furthermore, β-amyloid protein (Aβ) oligomers induce excessive mitochondrial fission and neuronal damage in a nitric oxide (NO)–mediated fashion (2, 10). We sought to determine whether Drp1 is S-nitrosylated and thereby activated in AD.
Cerebrocortical neurons transfected with the mitochondrial marker mito-DsRed2 were exposed to the NO donor S-nitrosocysteine (SNOC) (11) and morphological changes in mitochondria were monitored by 3D-deconvolution fluorescence microscopy (2). Mitochondria normally displayed an elongated filamentous morphology, but addition of SNOC induced fragmented, smaller mitochondria in a dose-dependent manner, due to fission (Fig. 1, A and B) (2, 11). Using a biotin-switch assay (12), we found that SNOC induced S-nitrosylation of Drp1 (forming SNO-Drp1) in neurons before inducing fission (Fig. 1C).
Fig. 1.

NO induces mitochondrial fission and S-nitrosylation of Drp1. (A) Cortical neurons transfected with mito-DsRed2 were exposed to 200 μM SNOC. Fluorescent images show mitochondrial morphology before and 1 hour after SNOC. (B) SNOC induced mitochondrial fragmentation in a dose-dependent manner. Values are means + SEM (n = 3, *P < 0.05). (C) Cortical neurons were exposed to SNOC or decayed (old) SNOC for <15 min and subjected to biotin-switch assay.
To investigate whether endogenously generated NO can induce SNO-Drp1, we used human embryonic kidney (HEK) 293 cells stably expressing neuronal NO synthase (nNOS). These cells were subjected to biotin-switch assay after incubation with the calcium ionophore A23187 to activate nNOS. Endogenous Drp1 was S-nitrosylated by endogenous NO; this reaction was blocked by the NOS inhibitor N-nitro-L-arginine (NNA; Fig. 2, A and B). SNO-Drp1 was not detected in controls performed without ascorbate to remove NO, thus preventing replacement of NO by biotin (which is detected in this assay), or without biotin-HPDP (N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio)-propionamide).
Fig. 2.
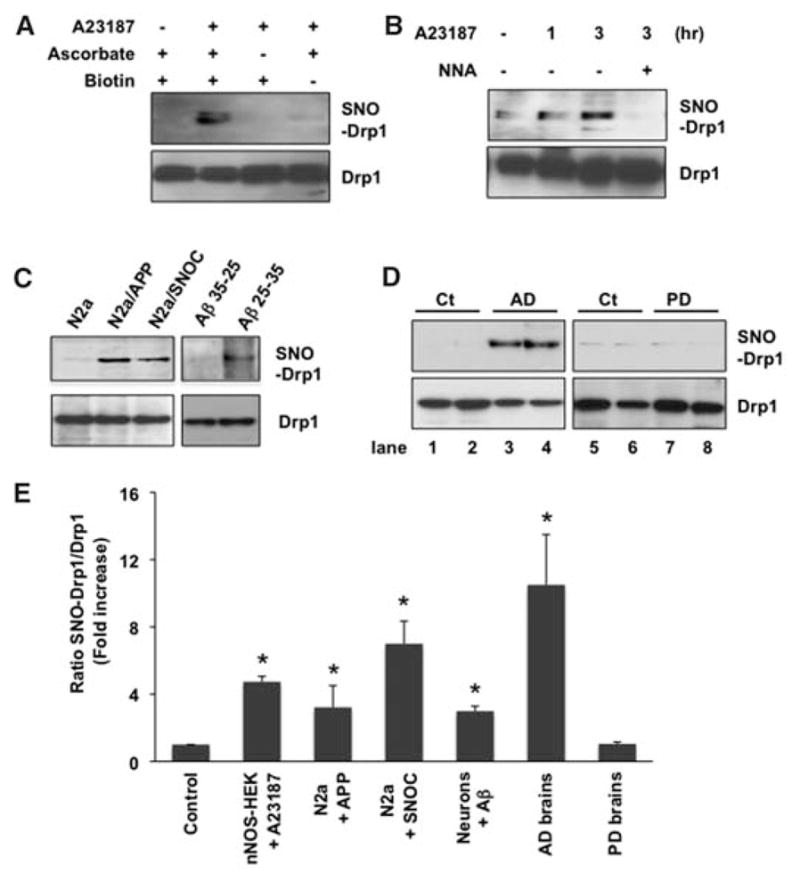
SNO-Drp1 formation in vitro and in vivo. (A and B) Biotin-switch assay of nNOS-HEK293 cells activated with A23187 for 1 to 3 hours. (C) SNO-Drp1 formation by biotin-switch assay in neuronal cells. Left: N2a/APP695 cells were harvested after 30 hours in conditioned medium containing endogenously secreted Aβ soluble oligomers or 30 min after SNOC exposure. Right: Cortical neurons were exposed to 10 μM Aβ25–35 or control Aβ35-25 for 6 hours. (D) SNO-Drp1 detection in vivo by biotin-switch assay of representative human postmortem control (Ct), AD, or Parkinson’s disease (PD) brains. (E) Relative amounts of SNO-Drp1 formation. Biotin-switch assays and immunoblot analyses were quantified by densitometry, and the relative ratio of SNO-Drp1 to total Drp1 was calculated for all samples. Values are means + SEM (n ≥ 4 for each group, *P < 0.02).
Using the same conditions under which Aβ causes mitochondrial fragmentation and consequent neuronal damage (2), we found that Aβ could induce SNO-Drp1 formation. Cerebrocortical neurons were exposed to oligomers of the pathologically active fragment Aβ25–35 or, as a control, reverse-sequence Aβ35-25. Formation of SNO-Drp1 was observed only in Aβ25–35–treated neurons, not in the control (Fig. 2C). Additionally, we tested the effect of endogenously produced Aβ, generated from amyloid precursor protein (APP) in conditioned medium of N2a/APP695 stable neuronal cell lines or CHO cells stably expressing human APP with the Val717 → Phe mutation (designated 7PA2 cells). Exposing N2a cells to SNOC or conditioned medium resulted in SNO-Drp1 formation (Fig. 2C). We also found elevated levels of SNO-Drp1 in vivo in brains of the AD transgenic mouse model Tg2576, which expresses high levels of the Swedish APP mutation (Lys670 → Asn, Met671 → Leu) (fig. S1).
To extend these findings to humans, we examined brains obtained shortly after death from patients manifesting AD (table S1). We found increased SNO-Drp1 levels in 17 of 17 AD brains studied, but not in brains of deceased Parkinson’s disease patients or controls who died of non-CNS causes (Fig. 2, D and E, and fig. S2). To determine whether the level of SNO-Drp1 in AD human brains is of pathophysiological importance, we used a technique to calculate the ratio of SNO-Drp1 (determined by biotin-switch assay) to total Drp1 (from immunoblots) (13). This ratio was comparable to that encountered in our cell-based models manifesting excessive mitochondrial fission and neuronal damage (Fig. 2E), consistent with the notion that pathophysiologically relevant amounts of SNO-Drp1 are present in human AD brains. Thus, SNO-Drp1 can potentially serve as a biomarker for AD.
We next sought to identify the target cysteine residue for S-nitrosylation on Drp1. Sequence alignment of Drp1 revealed four distinct structural domains: an N-terminal guanosine triphosphatase (GTPase) domain, a dynamin-like middle domain, an insert B domain, and a C-terminal GTPase effector domain (GED). The GED affects both GTPase activity and Drp1 dimer formation (14). In search of S-nitrosylated cysteine residue(s), we mutated each of the nine Drp1 cysteines. To assess S-nitrosylation, we performed a chemical assay using 2,3-diaminonaphthalene (DAN) on immunoprecipitates from HEK293 cells transfected with wild-type or mutant Drp1. This assay monitors release of NO from thiol by conversion of DAN to fluorescent 2,3-naphthyltriazole (NAT). Among the cysteine mutants, only C644A (Cys644 → Ala), located in the GED domain, decreased SNO-Drp1 formation (Fig. 3A). Cys644 was confirmed as the S-nitrosylated residue by biotin-switch assay.
Fig. 3.

S-Nitrosylation of Cys644 in Drp1. (A) HEK293 cells transiently transfected with green fluorescent protein (GFP)–fused constructs for wild-type or representative cysteine mutant Drp1 were exposed to SNOC, lysed, and subjected to immunoprecipitation with antibody to GFP. Precipitated Drp1 was analyzed for S-nitrosylation with the DAN assay by monitoring NO release spectrofluoro-metrically in relative fluorescence units (RFU). Values are means + SEM (n = 3, *P < 0.01). (B) nNOS-HEK293 cells transfected with GFP-fused constructs for wild-type (WT) or representative cysteine mutant Drp1 were exposed to A23187 and subjected to biotin-switch assay. (C) Homology modeling of Drp1 atomic structure: Predicted ribbon structure of human Drp1 showing putative acid/base S-nitrosylation motif around Cys644. Yellow, GTPase domain; green, middle (M) domain; magenta, GED domain; C, Cys; E, Glu; K, Lys.
A23187 exposure, to activate nNOS and produce endogenous NO, resulted in SNO-Drp1 formation, which was virtually eliminated in Drp1 (C644A) but not other cysteine mutant cells (Fig. 3B). Thus, Cys644 was essential for SNO-Drp1 generation. We then constructed an atomic model of Drp1 based on comparative homology using the structure of bacterial dynamin-like protein (PDB ID: 2J68-A) (15). With the software program MODELLER, we found that the Drp1-GED domain contains a consensus motif for S-nitrosylation flanking Cys644 (Fig. 3C) (16). Homology modeling also predicted that Cys644 is exposed to the protein surface, increasing its accessibility to NO and thus facilitating nitrosylation.
Dimer formation of the homologous protein dynamin, a process influenced by residues in the GED domain, increases dynamin GTPase activity (14, 17). We tested whether NO could induce dimer formation of Drp1 and influence its GTPase activity. The Drp1-GED domain is critical for inter-and intramolecular interactions and GTPase activity (14, 18). Indeed, S-nitrosylation of Cys644 in the GED domain led to dimerized SNO-Drp1 (Fig. 4A and fig. S3) and increased GTPase activity (Fig. 4B). In contrast, NO failed to induce dimerization/oligomerization or to increase GTPase activity of the Drp1(C644A) mutant. For this experiment, we measured GTPase activity of purified wild-type or mutant Drp1 proteins after exposure to SNOC or control solutions by monitoring absorbance at 635 nm, representing hydrolysis of GTP by GTPase. The GTPase activity of Drp1(C644A) protein was as reduced as that of dominant-negative Drp1 with a Lys38 → Ala mutation [DN-Drp1(K38A)] (1, 2), consistent with the notion that S-nitrosylation of Drp1 at residue Cys644 increases its GTPase activity.
Fig. 4.

S-Nitrosylation of Drp1 regulates dimerization, GTPase activity, mitochondrial fragmentation, and neuronal damage. (A) HEK cells expressing enhanced GFP (EGFP)–tagged WT-Drp1 or Drp1(C644A) were exposed to 200 μM SNOC or control decayed SNOC. Cell extracts were subjected to SDS–polyacrylamide gel electrophoresis, with or without dithiothreitol (DTT) to reduce disulfide bonds and thus inhibit dimer formation, and immunoblotted. (B) GST-fused WT-Drp1, Drp1(C644A), and DN-Drp1(K38A) were expressed and purified from bacteria, exposed to SNOC, and assayed for GTPase activity 30 min later (*P < 0.01). (C) Atomic-resolution model of Drp1 superimposed onto electron-density map of homologous domains of dynamin dimer: GTPase (yellow), forming the head; middle (green) and GED (magenta) domains, forming the stalk. (D) S-Nitrosylation of Drp1 enhances mitochondrial fission. Cortical neurons transfected with mito-DsRed2 (Ct), mito-DsRed2 plus WT-Drp1, mito-DsRed2 plus DN-Drp1, mito-DsRed2 plus Drp1(C644A), or mito-DsRed2 plus Drp1(C505A) were exposed to SNOC and scored for fragmented mitochondria 1 hour later (*P < 0.05). (E) SNO-Drp1 increases apoptotic cell death. Cortical neurons transfected with plasmid (p)EGFP, pEGFP plus WT-Drp1, pEGFP plus Drp1(C644A), or pEGFP plus DN-Drp1(K38A) were exposed to SNOC, and 18 hours later stained for NeuN (to identify neurons) and Hoechst 33342 (to assess nuclear morphology). Values are means + SEM (n ≥ 3, *P < 0.02). (F) Naturally secreted Aβ decreases dendritic spine density in cortical neurons via NO. Cultured cortical neurons were cotransfected with EGFP and pcDNA3 (Ct), WT-Drp1, or Drp1(C644A) and exposed for 5 days to 7PA2-conditioned medium containing oligomerized Aβ (+) or control (−). At the right are representative images of dendritic spines of neurons transfected with EGFP-Drp1 (n ≥ 7, *P < 0.002).
We then superimposed the atomic-resolution model for Drp1 onto a lower-resolution 3D reconstruction representing dimerization of the homologous dynamin molecule (Fig. 4C) (19). Superimposition allowed us to examine the potential effect of NO on Drp1 domain structure. This suggested that NO-induced dimerization triggered a conformation change in the stalk region, composed of the GED and middle domains, thereby regulating GTPase activity analogous to dynamin dimerization (19).
We next examined the effect of S-nitrosylation of Drp1 on mitochondrial fragmentation. An intact Drp1-GTPase domain is required for mitochondrial fission or fragmentation. Knockdown of Drp1 by RNA interference or mutation producing DN-Drp1(K38A), located in the GTPase domain, inhibits excessive mitochondrial fragmentation (1, 2, 20). Because nitrosylation of Drp1 in the GED domain activates GTPase activity, we reasoned that SNO-Drp1 should activate mitochondrial fission. In cortical neurons, NO induced mitochondrial fragmentation (2, 21), and over-expression of wild-type Drp1 slightly enhanced this effect (Fig. 4D). In contrast, expression of Drp1(C644A) or DN-Drp1(K38A) abrogated NO-mediated mitochondrial fission. Among the nine possible Drp1-cysteine mutants, only C644A suppressed NO-induced mitochondrial fragmentation (figs. S4 and S5), consistent with the observation that Cys644 was the predominant Drp1 nitrosylation site. As a control, Drp1(C644A) did not act as a dominant negative to decrease Drp1-mediated mitochondrial fission that was not triggered by NO (fig. S6).
Mitochondrial fragmentation induced by NO or Aβ contributes to neuronal cell injury or death and is abrogated by DN-Drp1(K38A) (2, 21). Because SNO-Drp1 formation was induced here by Aβ, we next examined the effect of nitrosylation of Drp1 on neurotoxicity. Exposure to NO triggers neuronal damage or death in the presence of wild-type Drp1 (2, 21). However, expression of Drp1(C644A) largely protected the neurons (Fig. 4E), consistent with the notion that S-nitrosylation of Drp1 at Cys644 is required for NO-mediated damage. During Aβ-induced neurotoxicity, synaptic damage is reported to represent one of its earliest manifestations (22). Exposure to oligomerized Aβ results in loss of dendritic spines, representing neuronal post-synaptic sites; this form of Aβ-mediated injury is dependent on N-methyl-D-aspartate (NMDA)– type glutamate receptor activity (22). Additionally, dendritic mitochondria localize to developing synaptic sites in a Drp1-dependent manner (4, 5).
Because NMDA receptor stimulation activates nNOS and thus generates NO, we asked whether Aβ-induced synaptic damage might be mediated at least in part by NO. Indeed, Aβ decreased the number of dendritic spines in an NO-dependent manner (Fig. 4F). Transfection with mutant Drp1(C644A), which lacks the nitrosylation site, abrogated the effect of Aβ on spine density, consistent with the notion that S-nitrosylation of Drp1 is involved in synaptic loss after exposure to Aβ.
Our results show that pathophysiologically relevant amounts of SNO-Drp1 form in human AD brains. Exposure of cerebrocortical neurons in culture to Aβ results in the formation of amounts of SNO-Drp1 relatively similar to those found in human AD brains. The S-nitrosylation of Drp1 causes dimer formation and increased GTPase activity, thus accelerating the process of mitochondrial fragmentation and contributing to neuronal synaptic damage or cell death. This relationship between nitrosative stress and mitochondrial fragmentation suggests that Drp1 can be a target of S-nitrosylation in AD. Drp1 is nitrosylated via a redox-mediated pathway in response to Aβ oligomers, causing mitochondrial fission and synaptic damage. This reaction may be facilitated by increased levels of copper, as observed in human AD brains (23). Copper(II)-mediated catalysis is responsible for generating bioactive S-nitrosothiols (24), such as SNO-Drp1. Thus, the redox consequences of nitrosative stress, resulting in Drp1 activation, may provide a mechanistic link among the actions of oligomeric Aβ, mitochondrial fission, and neuronal damage. Drp1 may represent a drug target to antagonize the progression of neurodegenerative disorders in which nitrosative stress and mitochondrial dysfunction play a key role.
Supplementary Material
Acknowledgments
We thank T. Newmeyer, J. Zaremba, J. Russo, E. Masliah, H. Xu, and Y. Zhang for help or reagents. Supported by the Japan Society for the Promotion of Science (T.N.) and NIH grants P01 HD29587, R01 EY05477, P01 ES016738, and P30 NS057096 (S.A.L.).
Footnotes
www.sciencemag.org/cgi/content/full/324/5923/102/DC1
Materials and Methods
SOM Text
Figs. S1 to S6
Table S1
References
References and Notes
- 1.Youle RJ, Karbowski M. Nat Rev Mol Cell Biol. 2005;6:657. doi: 10.1038/nrm1697. [DOI] [PubMed] [Google Scholar]
- 2.Barsoum MJ, et al. EMBO J. 2006;25:3900. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Nat Rev Neurosci. 2008;9:505. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li H, et al. Proc Natl Acad Sci USA. 2008;105:2169. [Google Scholar]
- 5.Li Z, Okamoto K, Hayashi Y, Sheng M. Cell. 2004;119:873. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Wang X, et al. Proc Natl Acad Sci USA. 2008;105:19318. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baloyannis SJ. J Alzheimers Dis. 2006;9:119. doi: 10.3233/jad-2006-9204. [DOI] [PubMed] [Google Scholar]
- 8.Kang-Decker N, et al. J Cell Sci. 2007;120:492. doi: 10.1242/jcs.03361. [DOI] [PubMed] [Google Scholar]
- 9.Wang G, Moniri NH, Ozawa K, Stamler JS, Daaka Y. Proc Natl Acad Sci USA. 2006;103:1295. doi: 10.1073/pnas.0508354103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan H, et al. Cell Death Differ. 2007;14:462. doi: 10.1038/sj.cdd.4402046. [DOI] [PubMed] [Google Scholar]
- 11.See supporting material on Science Online.
- 12.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Nat Cell Biol. 2001;3:193. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 13.Uehara T, et al. Nature. 2006;441:513. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- 14.Zhu PP, et al. J Biol Chem. 2004;279:35967. doi: 10.1074/jbc.M404105200. [DOI] [PubMed] [Google Scholar]
- 15.Low HH, Lowe J. Nature. 2006;444:766. doi: 10.1038/nature05312. [DOI] [PubMed] [Google Scholar]
- 16.Stamler JS, Toone EJ, Lipton SA, Sucher NJ. Neuron. 1997;18:691. doi: 10.1016/s0896-6273(00)80310-4. [DOI] [PubMed] [Google Scholar]
- 17.Ramachandran R, et al. EMBO J. 2007;26:559. doi: 10.1038/sj.emboj.7601491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pitts KR, McNiven MA, Yoon Y. J Biol Chem. 2004;279:50286. doi: 10.1074/jbc.M405531200. [DOI] [PubMed] [Google Scholar]
- 19.Chen YJ, Zhang P, Egelman EH, Hinshaw JE. Nat Struct Mol Biol. 2004;11:574. doi: 10.1038/nsmb762. [DOI] [PubMed] [Google Scholar]
- 20.Estaquier J, Arnoult D. Cell Death Differ. 2007;14:1086. doi: 10.1038/sj.cdd.4402107. [DOI] [PubMed] [Google Scholar]
- 21.Yuan H, et al. Cell Death Differ. 2007;14:462. doi: 10.1038/sj.cdd.4402046. [DOI] [PubMed] [Google Scholar]
- 22.Shankar GM, et al. J Neurosci. 2007;27:2866. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bush AI, Masters CL, Tanzi RE. Proc Natl Acad Sci USA. 2003;100:11193. doi: 10.1073/pnas.2135061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Nat Rev Mol Cell Biol. 2005;6:150. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.