

Abstract
Traditional methods of species classification and identification of the organism are based on morphological, physiological, biochemical, developmental and nutritional characteristics. Accurate assignment of taxonomic status to the new biologically active microbial isolates through existing bioinformatics methods is now very essential and also helpful in chemical characterization of the active molecule produced by microorganisms. The bacterial strain M4 (ckm7) was isolated from the pre-treated soil sample collected from the agricultural field of Eastern Uttar Pradesh (U.P.), India and was found to be producing antibacterial and antifungal antibiotics. Taxonomic identification of the isolate belongs to the genus Streptomyces which was done with the help of sequence analysis and later confirmed by biological activity. Sequence comparison study of ckm7 showed 98% identical similarity with 16S rRNA gene sequences of Streptomyces spinichromogenes, Streptomyces triostinicus and Streptomyces capoamus. On the basis of both biological activity and phylogenetic analysis of ckm7, it was concluded that the isolated strain is a new variant of S. triostinicus.
Keywords: Phylogenetics, 16S rRNA gene, ckm7, Streptomyces, biological activity study, antibacterial, antifungal antibiotics
Background
Traditional methods of classification and identification of the organisms are based on morphological, physiological, biochemical developmental and nutritional characteristics. Standard references such as Bergey's Manual of Systematic Bacteriology or the Manual of Clinical Microbiology help in this type of study [1,2]. These traditional methods of bacterial identification have two major drawbacks. Firstly, this method cannot be applicable for noncultivable organisms such as Tropheryma whippelii. Secondly, occasionally biochemical characteristics of some organisms do not fit into patterns of any known genus and species [3]. With the discovery of the polymerase chain reaction (PCR) and DNA sequencing methods, elucidation of closely related taxon with better authenticity has been made successfully in comparison to other conventional methods [4–6]. Beside this, phylogenetic analysis of 16S rRNA gene is presently an important area of evolutionary study and sequence analysis. In addition to analyzing changes that have occurred during the evolution among different organisms, the evolution of a family of sequences may be studied. On the basis of the analysis, sequences that are most closely related can be identified by the places they occupy on neighboring branches of a tree. Phylogenetic analysis of a family of related gene sequences is a method to determine how the family might have been derived during evolution. The evolutionary relationships among the sequences are depicted by placing the sequences as outer branches on a tree. The branching relationships on the inner part of the tree reflect the degree to which different sequences are related. Two sequences that are very much alike will be located as neighboring outside branches and will be joined to a common branch beneath them. The object of phylogenetic analysis is to discover all of the branching relationships in the tree and the branch lengths. One of the most significant uses of phylogenetic analysis of sequences is to make predictions concerning the tree of life. For this purpose, 16S rRNA genes are selected as they are highly conserved in nature and universally present. At the same time it shows enough sequence variation to determine which groups of organisms share the same phylogenetic origin. Ideally, the 16S rRNA gene is also under selection & universal in distribution, meaning that as variation occurs in populations of organisms, certain sequences are not favored with a loss of the more primitive variation. A large number of rRNA sequences from a variety of organisms were aligned and the secondary structure was deduced. Phylogenetic predictions were then made using the distance method [7]. On the basis of rRNA sequence signatures, or regions within the molecule that are conserved in one group of organisms but different in another [7], predicted that early life diverged into three main kingdoms Archaea, Bacteria and Eukarya. Evidence for the presence of additional organisms in these groups has since been found by PCR amplification of environmental samples of RNA [8, 9]. However, few studies so far have been reported on the use of rDNA sequencing for the identification of bacterial isolates in a more systematic fashion [10, 11].
In the studied work, we are reporting the strain identification of our novel isolate ckm7 with the help of 16S rRNA gene sequence analysis. This partial sequencing of 16S rRNA gene was done from Microbial Type Culture Collection (MTCC), IMTECH, Chandigarh, India (http:// www.mtcc.imtech.res.in). Pairwise sequence alignment of 16S rRNA gene sequence was performed to identify closely related homologs with the help of BLAST search tool available at NCBI webserver (http://www.nih.nov.ncbi). Phylogenetic tree was constructed to predict the species level characterization of the studied isolate through distance method based webtool, CLUSTALW (www.ebi.ac.uk/clustalw). Biological activity profiles of the closely related microbial strains and the new isolate have been compared to establish the novelty of the antibiotic produced. Taxonomic position of the isolate belongs to the genus Streptomyces, which was done with the help of in silico sequence analysis and later confirmed by experimental biological activity study. Sequence comparison study of ckm7 showed 98% identity with 16S rRNA gene sequences of S.spinichromogenes, S. triostinicus and S. capoamus. Finally on the basis of both biological activity and phylogenetics study it was concluded that the isolated strain belongs to the genus S. triostinicus.
Methodology
Isolation of biologically active culture
Biologically active culture (M4) was isolated from a phenol (30 °C, 30 min) pre treated soil sample [12] collected from the agricultural field of Eastern U.P., India through serial dilution method. The isolated active culture (M4) was maintained on YMG slants (per liter of distilled water: Yeast extract 4.0g, Malt extract 10 g, Glucose 4.0 g, CaCO3 2.0 g and Agar powder 20 g).
Characterization through conventional method
In conventional method the producer strain was characterized through morphological, cultural and biochemical tests(Figure 1). Cultural characteristics were studied on different ISP media regarding their growth intensity, growth pattern, color of areial mycelia and formation of soluble pigments [13, 14]. Various biochemical tests such as assimilation of sugars, utilization of starch, urea, tyrosine, asparagines and citrate components, nitrate reduction, tolerence range of pH, NaCl and temperature were performed.
Figure 1.

Antibacterial activity (A), antifungal activity (B) and morphological appearance through light microscopy (C) of the producer strain.
Selection of target gene
Since rRNA genes are highly conserved within bacteria therefore 16S rRNA gene sequences are considered as a powerful tool in the study of prokaryotic taxonomic identification. In comparison to 23S rRNA, 5S rRNA genes and ribosomal protein sequences, mostly 16S rRNA was preferred to investigate species relationships within the genus Streptomyces [15–17].
16S rRNA gene analysis
Producer strain was grown on YES (Yeast extract 1%, sucrose 10% and 0.5 mM MgCl2) medium for 24 hr at 28°C and genomic DNA was isolated from the cell mass according to the method described by Saito & Miura [18]. The 16S rRNA gene was selectively amplified by PCR using universal primers. Amplification was carried out with an initial incubation (5 min, 94°C) followed by denaturation (25 cycles of 30 sec at 96°C), annealing (30 sec, 58°C) and elongation (1 min, 72°C). The reaction was terminated with a final extension at 72°C for 5 min. Direct sequencing of PCR product was conducted using 109f and 1115r primers on an AVI 3100 genetic analyzer.(Figure 2)
Figure 2.

Partial gene sequence of 16S rRNA gene of ckm7, sequenced from Microbial Type Culture Collection (MTCC), IMTECH, Chandigarh, India. The length of nucleotide sequence is 1424 base pair.
Pairwise sequence alignment
Closely related homologs were identified through phylogenetic analysis by comparing partial 16S rRNA gene sequence of ckm7 with the nonredundant database of nucleotides sequences deposited at NCBI web server (www.ncbi nlm.nih.gov), through Basic Local Alignment Tool (BLAST) program (http://www.ncbi.nlm.nih.gov/blast/) [19,.
Multiple sequence alignment
After BLAST analysis, a data set of potential orthologs was prepared by considering those database sequences which had >98% sequence identity with the query sequence ckm7. Phylogenetic analysis and nucleotide conservation of the data set sequences were studied through multiple sequence alignment program viz., CLUSTALW v1.83 (http://www.ebi.ac.uk/clustalw). Genetic distances between the strains were calculated using the neighbor joining method.
Phylogenetic analysis
A phylogenetic analysis of ckm7 was performed to determine of how the 16S rRNA sequences of ckm7 and closely related strains might have been derived during evolution. The evolutionary relationships among the sequences were depicted by placing them as outer branches on a phylogenetic tree. The branching relationships on the inner part of the tree reflect the degree to which different sequences are related. Sequences that were very much alike were located as neighboring outside branches and joined to a common branch beneath them. The object of phylogenetic analysis is to find out all of the branching relationships in the tree along with branch lengths. For this dendrogram/phylogram was constructed using distance method of phylogenetic tree construction (e.g. neighbor joining). Distances between the studied sequences helps in understanding the evolutionary distances among the species.
Criteria for species identification
Identification of species through sequence similarity basis was performed according to criteria used by Bosshard et al., [6] which states following selection parameters:
when the percentage similarity of the query sequence and the reference sequence is 99% or above, the unknown isolate would be assigned to reference species;
when percentage similarity is between 99 95 %, the unknown isolate would be assigned to the corresponding genus;
when percentage similarity is less than 95 %, the unknown isolate would be assigned to a family.
Discussion
Biological importance of the strain
The isolate showed broad-spectrum antibacterial and antifungal activity (Table 1 and 2 in supplementary material). The active crude was purified using normal phase silica gel column chromatography and finally on reverse phase high pressure liquid chromatography (HPLC). Molecular weights of antibacterial (M4B1A) and antifungal (M4B3) compounds were found to be 1269 and 1331 respectively as deduced by mass spectroscopy. Both the active compounds are colored (M4B1A: dark red; M4B3: light yellow), indicating the presence of chromophore groups in their structure. UV absorption of both the compounds, in methanol, produced maximum absorbance near 208 nm suggested the presence of one or more amido chromophoric groups in the structure. Presence of phenoxazone nucleus in the antibacterial compound was confirmed by the presence of absorption hump near 443 nm [20] whereas consecutive bands at 320, 337 and 356 nm confirmed the polyene nature of antifungal compound [21]. Details of the chemical characterization of the compounds is not presented here and being published elsewhere. Survey of Active compounds produced by the closely related microorganisms of the phylogenetic tree through literature concluded that these microbes produced various compounds with diverse chemical structure and biological activity. Quinoxaline antibiotics which is a chromopeptide lactones is reported by Streptomyces triostinicus [22], suggests the phylogenetic similarity between ckm7 and Streptomyces triostinicus.
Comparison with the active compounds produced by the other members of clad-1 reveals that Streptomyces roseochromogenes is reported to produce nojirimycin, a piperidinose sugar antibiotic [23] and clorobiocin, containing aminocoumarin ring [24] whereas Streptomyces griseovariabilis is reported as a Bandunamide, a cyclopeptide [25] and chlorobiocin [26] producer. All the compounds have molecular weights in the range of 500 to 1100 whereas quinoxaline antibiotics [27] showed higher molecular weight i.e. greater than1800 does not coincide with the molecular weight of the M4 isolates 1269 and 1331. On the basis of molecular weight and activity profile study it could be assumed that isolated compounds may be of some other group since the studied compounds showed no chemical similarities with the compounds produced by our isolate.
Sequence analysis of CKM7
Identification of species was further confirmed by computational analysis. Gene sequence of 16S rRNA of M4 was compared with nucleotide database of NCBI webserver through BLAST tool. Result showed that query sequence ckm7 was best pairwise aligned with 16S rRNA gene sequence of Streptomyces spinichromogenes, Streptomyces triostinicus and Streptomyces capoamus with similar sequence similarity and identity of 98 %, but with variable BLAST score value i.e. 2696, 2688 and 2674 bits respectively. BLASTn result concluded that ckm7 is a partial 16S rRNA gene sequence of Streptomyces spinichromogenes origin but later through phylogenetic analysis we found that ckm7 was actually a partial 16S rRNA gene sequence of Streptomyces triostinicus only. For phylogenetic analysis a new data set of 16S rRNA gene sequences was prepared after removing the sequences of non-cultured and repeating strains from the BLAST result. Sequence comparison and phylogenetic tree construction of new data set sequences was made through multiple sequence alignment tool i.e. ClustalW.
Nucleotides conservation
For better understanding of new species identification, nucleotides level conservation was evaluated by comparing 16S rRNA sequences of S. spinichromogenes, S. capoamus, S. triostinicus and ckm7 through multiple sequence alignment analysis. Result of sequence alignment showed that conservation of nucleotides was much higher in S. triostinicus and ckm7 than S. spinichromogenes and S. capoamus (Figure 3). Fragment of 16S rRNA gene sequence of Streptomyces triostinicus from position 500 to 1300 was identical to the ckm7 sequence without any substitution or mutation whereas in the case of S. spinichromogenes and S. capoamus there were differences in nucleotide level conservation at regular interval. Although the BLAST score value of S. spinichromogenes was higher than S. triostinicus but on the basis of multiple sequence alignment and phylogenetics study, ckm7 gene sequence showed much higher nucleotide conservation and close evolutionary relationship with S. triostinicus. Thus local pairwise sequence alignment through BLAST program is not enough for computational identification of new species.
Figure 3.

Conservation of nucleotides in the 16S rRNA sequences of S. spinichromogenes, S. capoamus, S. triostinicus and ckm7. In the diagram arabic numbers i.e. 1 to 14 indicates range of nucleotides from 100 to 1400 while roman numbers on the top of each row except ckm7 indicates difference in number of nucleotides with ckm7 sequence.
Phylogenetics of CKM7
Constructed phylogenetic tree of ckm7 was categorized into four major clads on the basis of their evolutionary distances calculated through neighborjoining method (Figure 4). First clad was further divided into two subclads viz., subclad 1.1 and 1.2. The subclad 1.1 leads ckm 7 alone whereas subclad 1.2 further divided into 1.2.1 and 1.2.2 and so on and ultimately form a branched group of four gene sequences of Streptomyces triostinicus (AB184519.1), Streptomyces griseovariabilis (AY654298.1), Streptomyces ginsengisoli (AB245392.1) and Streptomyces roseochromogenes (AB184512.1). On observing the evolutionary distances among the species as shown in phylogenetic tree, it is clear that Streptomyces triostinicus have evolutionary distance of 0.00315 which was closely related to ckm7 evolutionary distance i.e. 0.00403. Further the above clad revealed common ancestral origin, in terms of 16S ribosomal gene sequence homology with another clad 2 comprising of S. spinichromogenes (AB184534.1), S. antibioticus (EF063450.1), S. cinnabarinus (AB184266.1) and S. kagawaensis (AB184496.1). On the other hand clad 3 divided into subclad 3.1 and 3.2, where subclad 3.1 further sub divided into 3.1.1 covering S. capoamus (AB184385.1) and while 3.1.2 further subdivided twice and form a cluster of three genes of S. bungoensis (AB184696.1), S. longwoodensis (AB184580.1) and S. galbus (AB184201.1). Later subclad 3.2 divided into 3.2.1 showing S. coeruleorubidus (AB184841.1) and 3.2.2 with S. cyaneus (AJ399471.1). Clad 4 shows 3 closely related 16S rRNA gene sequences of S. olivaceoviridis (AB184288.1), S. corchorusii (AB184267.1) and S. hygroscopicus (AB184725.1). Gene sequences of species of clad 3 and 4 showed distant relationship with ckm7 sequence, thus assumed putative distant orthologs.
Figure 4.
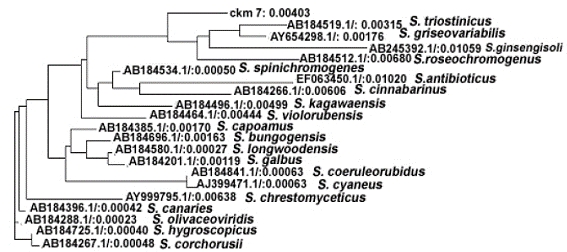
Unrooted phylogenetic tree showing evolutionary relationship of ckm7 with other members of Streptomyces along with their evolutionary distances.
Conclusion
Sequence comparisons of specific genes were applied successfully to investigate the relationship among the members of genus Streptomyces on the basis of their nature of product and other characteristics such as antibiotic producers and pathogenic Streptomyces. We showed that molecular weight and activity profile study of compounds produced by our isolate (ckm7) is not sufficient for the identification of the isolated strain since the isolated compounds showed no chemical similarities with related compounds produced by other microorganisms. Thus, gene sequence level comparison was employed in this study. BLAST results revealed higher sequence similarity of ckm7 with 16S rRNA partial gene sequence of S. spinichromogenes. Results of multiple sequence alignment analysis using CLUSTALW showed higher level of nucleotide conservation of ckm7 with S. triostinicus. This is further confirmed through evolutionary tree study using neighborjoining distance method of phylogenetic analysis. BLAST analysis showed 98%99% sequence similarity between ckm7 and other Streptomyces members. Hence, the isolated strain was designated as S. triostinicus.
Supplementary material
Acknowledgments
We acknowledge Council of Scientific & Industrial Research (CSIR), New Delhi for financial support as a SRF at Division of Fermentation Technology, Central Drug Research Institute, Lucknow (INDIA).
Footnotes
Citation:Singh et al, Bioinformation 4(2): 53-58 (2009)
References
- 1.Krieg NR. In: Holt JG, editor. Baltimore, MD: The Williams & Wilkins Co; 1984. [Google Scholar]
- 2.Murray PR, et al., editors. 7. Washington, D.C: ASM Press; 1999. [Google Scholar]
- 3.Woo PCY, et al. Mol Pathol. 2000;53:211. doi: 10.1136/mp.53.4.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lane DJ, et al. PNAS. 1985;82:6955. doi: 10.1073/pnas.82.20.6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clarridge III JE. Clinical Microbiology Reviews. 2004;17:840. doi: 10.1128/CMR.17.4.840-862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bosshard PP, et al. J Clin Microbiol. 2003;41:4134. doi: 10.1128/JCM.41.9.4134-4140.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woese CR. Microbiol Rev. 1987;51:221. doi: 10.1128/mr.51.2.221-271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boettger EC. FEMS Microbiol Lett. 1989;65:171. doi: 10.1016/0378-1097(89)90386-8. [DOI] [PubMed] [Google Scholar]
- 9.Edwards UT, et al. Nucleic Acids Res. 1989;17:7843. doi: 10.1093/nar/17.19.7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cloud JL, et al. J Clin Microbiol. 2002;40:400. doi: 10.1128/JCM.40.2.400-406.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drancourt M, et al. J Clin Microbiol. 2000;38:3623. doi: 10.1128/jcm.38.10.3623-3630.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goodfellow M. The Prokaryotes. 2. New York: SpringerVerlag; 1992. [Google Scholar]
- 13.Sardi P. Appl Environ Microbiol. 1992;58:2691–2693. doi: 10.1128/aem.58.8.2691-2693.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shirling EB, Gottlieb D. Int J Syst Bacteriol. 1996;61:313. [Google Scholar]
- 15.Mehling A, et al. FEMS Microbiol Lett. 1995;128:119. doi: 10.1111/j.1574-6968.1995.tb07510.x. [DOI] [PubMed] [Google Scholar]
- 16.Liao D, Dennis PP. J Mol Evol. 1994;38:405. doi: 10.1007/BF00163157. [DOI] [PubMed] [Google Scholar]
- 17.Stackebrandt E, et al. Gene. 1992;115:255. doi: 10.1016/0378-1119(92)90567-9. [DOI] [PubMed] [Google Scholar]
- 18.Saito H, Miura KI. Biochem Biophys Acta. 1963;72:619. [PubMed] [Google Scholar]
- 19.Altschul SF, et al. Nucleic Acids Res. 1997;25:3389. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paradkar VR, et al. World J Microbiol Biotechnol. 1998;14:705. [Google Scholar]
- 21.Tai JC, et al. Biochemistry. 1990;29:3522. [Google Scholar]
- 22.Niwa T, et al. Agric Biol Chem. 1970;34:966. [Google Scholar]
- 23.Galm U, et al. Antimicrob Agents Chemother. 2004;48:1307. doi: 10.1128/AAC.48.4.1307-1312.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tian XS, et al. Chinese Chemical Letters. 2003;14:1255. [Google Scholar]
- 25.Gauze GF, et al. Antibiotiki. 1979;24:643. [PubMed] [Google Scholar]
- 26.Takeuchi T, et al. Int J Syst Bacteriol. 1996;46:476. doi: 10.1099/00207713-46-2-476. [DOI] [PubMed] [Google Scholar]
- 27.Bramwell PA, et al. Lett Appl Microbiol. 1998;27:255. doi: 10.1046/j.1472-765x.1998.t01-11-00449.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.