

Abstract
Human thiopurine S-methyltransferase (TPMT) is an essential protein in 6-mercaptopurine (6MP) drug metabolism. To understand the pharmacogenetics of TPMT and 6MP, X-ray co-crystal structures of TPMT complexes with S-adenosyl-L-methionine (AdoMet) and 6MP are required. However, the co-crystal structure of this complex has not been reported because 6MP is poorly water soluble. We used molecular dynamics (MD) simulation to predict the structure of the complex of human TPMT-AdoHcy(CH2)6MP, where the sulfur atoms of AdoHcy and 6MP were linked by a CH2 group. After 1300 picoseconds of MD simulation, the trajectory showed that 6MP was stabilized in the TPMT active site by formation of non-bonded interactions between 6MP and Phe40, Pro196 and Arg226 side chains of TPMT. The intersulfur distance between AdoHcy and 6MP as well as the binding modes and the interactions of our TPMT-AdoHcy model are consistent with those observed in the X-ray crystal structure of murine TPMT-AdoHcy-6MP complex. The predicted binding modes of AdoHcy and 6MP in our model are consistent with those observed in murine TPMT X-ray crystal structures, which provides structural insights into the interactions of TPMT, AdoHcy, and 6MP at the atomic level and may be used as a starting point for further study of thiopurine drug pharmacogenetics.
Keywords: methyltransferase, ligand, molecular dynamics simulation, structural changes
Background
Thiopurine drugs including 6-mercaptopurine (6MP), 6- thioguanine, and azathioprine have wide clinical applications ranging from immunosuppressants for preventing graft-rejection in organ transplant patients, to treatment of diseases such as childhood acute lymphoblastic leukemia, autoimmune disorder, and inflammatory bowel disease [1–3]. The toxicity of these drugs is ameliorated by conversion to methylated forms by the thiopurine S-methyltransferase enzyme (TPMT), which uses S-adenosyl-Lmethionine (AdoMet), as a methyl donor. As a result of the transfer of the methyl group, AdoMet is converted to Sadenosylhomocysteine (AdoHcy). Mutations in the TPMT gene that reduce or abolish TPMT functions lead to decreased thiopurine drug methylation, resulting in cytotoxicity and cell death [4–7]. To understand the pharmacogenetics of TPMT and 6MP, X-ray cocrystal structures of TPMT complexes with AdoHcy(CH2)6MP are required. However, the co-crystal structure of this complex has not been reported because 6MP is poorly water soluble [8]. To address this problem, we modeled the AdoHcy(CH2)6MP complex in the human TPMT active site and performed 1300 picoseconds (ps) molecular dynamic (MD) simulations to investigate the atomic interactions and dynamic properties of TPMT, AdoHcy, and 6MP.
Methodology
Preparation of protein initial structure
The X-ray crystal structure of the TPMT-AdoHcy complex was obtained from the Protein Data Bank (PDB identifier: 2H11). Missing atoms and missing residues identified in this structure were corrected using the SwissPdb program [9]. The structure of 6MP was retrieved from the ChemBank Database (ChemBank identifier: 1116).
Preparation of AdoHcy(CH2)6MP complex
The X-ray crystal structure of murine TPMT, which is 77% identical to human TPMT, bound with AdoHcy and 6MP was recently published (PDB identifier: 3BGD) [10]. The intersulfur distance between AdoHcy and 6MP molecules is 3.41 Å, which is approximatly double the 1.85 Å length of a sulfur-carbon single bond, suggesting that the methyl group is directly transferred from the donor to the acceptor by SN2-like mechanism. 6MP interacts with AdoHcy by pointing its sulfur atom towards the sulfur atom of AdoHcy. These two sulfur atoms form a bridge through the CH2 group in the middle. Previous human TPMT-6MP modeling study suggested that 6MP interacts with TPMT by forming water bridges using its nitrogen atoms at the pentagonal and hexagonal rings with the nitrogen atoms of Lys32 and Arg226 side chains [8] (Figures 1A and 1B).
Figure 1.

Possible binding modes and interactions of 6MP against TPMT and AdoMet previously suggested by crystallography [8] (A and B). We modeled the AdoHcy(CH2)6MP complex, where the sulfur atom of 6MP was placed next to the AdoMet structure, forming a bridge with the sulfur atom of AdoMet through the CH2 group (C).
The 6MP structure was manually placed in the human TPMT active site by adaptation of the potential binding modes previously suggested by crystallography [8]. The AdoHcy(CH2)6MP complex was modeled by adding a CH2 group to the sulfur atom of the AdoHcy structure. The same carbon atom was then used as a reference point for placing the 6MP structure. The sulfur atom of the 6MP was placed next to the AdoHcy structure to form a bond with the reference carbon atom. Both structures were arranged on the same plane. Finally, atomic charges were assigned to the complex as shown in Figure 1c. The AdoHcy(CH2)6MP complex was used as an initial ligand structure for MD simulation and as a reference for root mean square deviation (RMSD) calculations for all AdoHcy(CH2)6MP trajectories.
MD simulations
The AMBER v.9 package [11,12] was used to prepare the protein and the ligand files as well as the MD simulations. Topology and parameter files for TPMT, AdoHcy, and 6MP were generated using “ff03” all-atom AMBER force field. The complex was solvated in the octahedron box of TIP3P water with 10 Å distance between the protein surface and the box boundary. Sodium and chloride ions were added to neutralize the system. The SHAKE algorithm [13] was applied to all bonds containing hydrogen atoms with the time step parameter set to 2 femtoseconds.
The energy minimization and MD simulations were carried out using the SANDER program in the AMBER package. Three thousand energy minimization steps (steepest descent for 1000 steps and then conjugate gradient for 2000 steps) were applied to the initial TPMT-AdoHcy(CH2)6MP complex to relieve atomic clashes and bad contacts. After energy minimization, MD simulations were performed using protein-atom restrained simulations for 20 ps and a harmonic potential with a 10 kcal/mol force constant, followed by 1300 ps all-atom unrestrained simulation. The trajectories of the entire system were recorded every 10 ps. The molecular dynamic of the complex was terminated when the simulation reached 1300 ps. The PDB structures of the last 100 ps were extracted from the MD simulated trajectories to generate the average structure, which was subsequently used for 6MP binding mode and the corresponding interaction analysis.
Discussion
After the MD simulation, we calculated RMSDs between Cα of TPMT trajectories recorded every 10 ps and Cα of their X-ray crystal structure. The RMSDs for the trajectories of the AdoHcy(CH2)6MP complex were also calculated using its initial model as a reference structure. The results in Figure 2 show that the RMSDs of both TPMT and AdoHcy(CH2)6MP trajectories were always less than 1.50 Å for the entire simulation. The trajectories were not greatly different from the X-ray crystal structure, with only minor movements of the Cα of TPMT observed. This indicates that our simulation system was stable. The simulation length used in this study was long enough to allow rearrangement of TPMT side chains and the AdoHcy(CH2)6MP complex to find their most stable binding mode.
Figure 2.
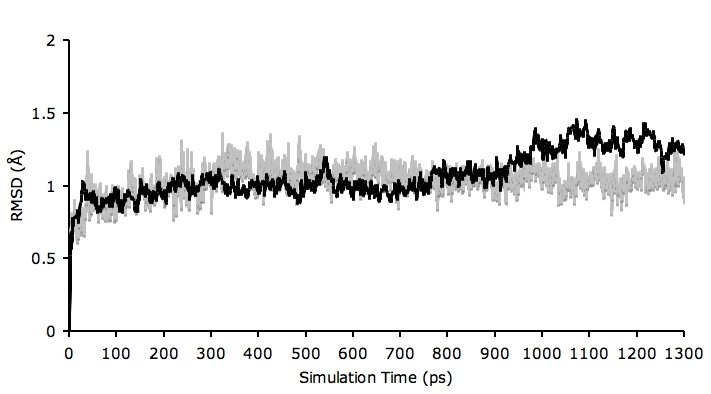
Plot of root mean square deviation (RMSD) of Cα-TPMT (black) and AdoHcy-6MP complex (gray) trajectories against the initial model. The trajectories were captured every 10 ps until the simulation time reached 1300 ps. The RMSD of both structures are always less than 1.50 Å for the entire simulation indicating high stability of the complex.
We verified the quality of our model by using PROCHECK [14]. The results show that all protein residues were in the most favored regions (90.0%) or in the additional allowed regions (10.0%) with the overall PROCHECK score of -0.28, which was in the recommended range. The Ramachandran plot also indicates that our model is reasonable with respect to the overall folds and dihedral angles. The PDB trajectories of the complex may be obtained at http://www.biotec.or.th/GI/publication.
Figure 3A depicts the binding mode of 6MP and its interactions with human TPMT and AdoHcy predicted using MD simulation. The binding modes and the interactions of AdoHcy with the amino acid side chains in the vicinity of the TPMT active site are similar to those observed in the X-ray crystal structures (2H11 and 3BGD). The RMSDs between Cα of our model and the published X-ray crystal structures of human TPMT and murine TPMT (2H11 and 3BGD) are 1.28 Å and 1.24 Å, respectively.
Figure 3.

Binding mode and interactions of 6MP against TPMT and AdoHcy. From our MD simulations, the trajectory shows that 6MP interacts with AdoMet through the CH2 group by pointing its sulfur atom toward the sulfur atom of AdoMet and interacts with Phe40, Pro195, Pro196 and Arg226 of TPMT by non-bonded interactions (A). AdoHcy interacts with amino acid side chains lining in the TPMT active site, i.e. Trp33, Leu69, Cys70, Glu90, Ile91, Ile135 and Arg152, which consistent with those interactions observed in murine TPMTAdoHcy- 6MP X-ray crystal structures (B) [10].
The inter sulfur distance between AdoHcy and 6MP molecules of our model is 3.40 Å (Figure 3A), consistent with that of murine TPMT-AdoHcy-6MP X-ray crystal structure 3BGD (3.41 Å) (Figure 3B) [10]. Our model shows that the location of nitrogen atoms in the pentagonal ring (N3 and N4) of 6MP are positioned 4.00 and 5.24 Å from Pro196 and Arg226 side chains while a nitrogen atom in the hexagonal ring (N1) is positioned 3.60 Å from Phy40 of TPMT (Figure 3A). These distances and the underlying conformation correspond with the murine TPMT-AdoHcy X-ray crystal structures in Figure 3B [10]. The binding modes of 6MP observed in our model and the X-ray crystal structure [10] also indicate that 6MP is stabilized in the TPMT active site by nonbonded interactions with Phe40, Arg152, Pro195, Pro196, Arg226 side chains.
Enzymatic activity and mutagenesis studies of human TPMT mutants have shown that amino acid substitutions in the TPMT active site (Arg at residues 152 and 226 to Ala, Glu or His) alter the Km and Vmax values for 6MP [10]. Mutations of non-active-site residues (for example, residues 80, 132, 180 and 240) also greatly reduce the enzymatic activity of TPMT (to one third of the wild type activity) [8]. This information suggests that the dynamics of TPMT structure and structural distortion resulting from mutations of active-site and non-active-site residues play important roles in formation of TPMT-AdoHcy-6MP complex. The flexible loops in the TPMT structure have been proposed to contribute to structural rearrangements affecting ligand binding, TPMT function as well as enzymatic activity [8,10]. However, loop rearrangement was not observed in our model. The MD simulation performed in this study was considerably too short to observe the movements of these loops.
Conclusion
We used MD simulation to predict the structure of the complex of human TPMT-AdoHcy(CH2)6MP, where the sulfur atoms of AdoHcy and 6MP were linked by a CH2 group. After 1300 ps of MD simulation, the trajectory showed that 6MP was stabilized in the TPMT active site by formation of non-bonded interactions between 6MP and Phe40, Pro196 and Arg226 side chains of TPMT. The intersulfur distance between AdoHcy and 6MP as well as the binding modes and the interactions of our TPMT-AdoHcy model are consistent with those observed in the X-ray crystal structure of murine TPMT-AdoHcy-6MP complex. Our model provides structural insights into the interactions of TPMT, AdoHcy, and 6MP at the atomic level, which may be used as a starting point for further study of thiopurine drug pharmacogenetics.
Acknowledgments
The authors would like to thank Philip Shaw and staff in the Biostatistics and Informatics laboratory for their critical reading and valuable comments.
Footnotes
Citation:Mokmak et al, Bioinformation 4(2): 59-62 (2009)
References
- 1.Coulthard S, Hogarth L. Invest New Drugs. 2005;23:523. doi: 10.1007/s10637-005-4020-8. [DOI] [PubMed] [Google Scholar]
- 2.Lennard L. Eur J Clin Pharmacol. 1992;43:329. doi: 10.1007/BF02220605. [DOI] [PubMed] [Google Scholar]
- 3.Paterson AR, Tidd DM. Springer Verlag. New York: 1975. [Google Scholar]
- 4.Tidd DM, Paterson AR. Cancer Res. 1974;34:738. [PubMed] [Google Scholar]
- 5.Tidd DM, Paterson AR. Cancer Res. 1974;34:733. [PubMed] [Google Scholar]
- 6.Salavaggione OE, et al. Pharmacogenetics and genomics. 2005;15:801. doi: 10.1097/01.fpc.0000174788.69991.6b. [DOI] [PubMed] [Google Scholar]
- 7.Krynetski EY, et al. Proc Natl Acad U S A. 1995;92:949. doi: 10.1073/pnas.92.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu H, et al. Proteins. 2007;67:198. doi: 10.1002/prot.21272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guex N, Peitsch MC. Electrophoresis. 1997;18:2714. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 10.Peng Y, et al. Biochemistry. 2008;47:6216. doi: 10.1021/bi800102x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pearlman DA, et al. Comp Phys Commun. 1995;(91):1. [Google Scholar]
- 12.Case DA, et al. J Comput Chem. 2005;26:1668. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryckaert JP, et al. J Comput Phys. 1977;23:327. [Google Scholar]
- 14.Laskowski RA, et al. J Appl Cryst. 1993;26:283. [Google Scholar]