

Abstract
Immunotherapy targeting of amyloid β (Aβ) peptide in transgenic mouse models of Alzheimer disease (AD) has been widely demonstrated to resolve amyloid deposition as well as associated neuronal, glial, and inflammatory pathologies. These successes have provided the basis for ongoing clinical trials of immunotherapy for treatment of AD in humans. Acute as well as chronic Aβ-targeted immunotherapy has also been demonstrated to reverse Aβ-related behavioral deficits assessing memory in AD transgenic mouse models. We observe that three antibodies targeting the same linear epitope of Aβ, Aβ3–7, differ in their ability to reverse contextual fear deficits in Tg2576 mice in an acute testing paradigm. Reversal of contextual fear deficit by the antibodies does not correlate with in vitro recognition of Aβ in a consistent or correlative manner. To better define differences in antigen recognition at the atomic level, we determined crystal structures of Fab fragments in complex with Aβ. The conformation of the Aβ peptide recognized by all three antibodies was highly related and is also remarkably similar to that observed in independently reported Aβ:antibody crystal structures. Sequence and structural differences between the antibodies, particularly in CDR3 of the heavy chain variable region, are proposed to account for differing in vivo properties of the antibodies under study. These findings provide a structural basis for immunotherapeutic strategies targeting Aβ species postulated to underlie cognitive deficits in AD.
Keywords: Antibodies/Epitope Mapping, Diseases/Alzheimer Disease, Immunology/Humoral Response, Peptides/Conformation, Protein/Structure, Amyloid-Beta Peptide, Immunotherapy
Introduction
Immunotherapy targeting the amyloid β (Aβ)2 peptide via either active (i.e. immunization with Aβ peptide, or fragments derived from it), or passive immunization (i.e. parenteral administration of anti-Aβ antibodies) has been widely demonstrated to be efficacious for modification of AD pathology (1, 2), as well as Aβ-related behavioral deficits (3–5) in transgenic mouse models of Alzheimer disease (AD) (for reviews see Refs. 6, 7). These successes in pre-clinical studies have provided the basis for clinical trials of Aβ immunotherapy for treatment of AD in humans. Results from post-mortem histological evaluation of a limited sampling of patients from clinical trials of active immunotherapy with AN1792 provided initial corroborating evidence of pre-clinical findings with respect to reversal of plaque-associated AD pathology at autopsy in brains of treated patients (8–13). Conclusive evidence for cognitive benefits stemming from reversal of pathology in AD patients undergoing anti-Aβ immunotherapy must await results from adequately powered Phase 3 clinical trial studies. Analysis of cognitive and functional outcomes in patients from Phase 1 and Phase 2 clinical trials provide evidence supporting improvement in some (13, 14), but not all (15) clinical measures of disease.
Aβ-associated behavioral deficits in transgenic mouse models of AD offer a potential surrogate of the cognitive and memory decline seen in AD patients (reviewed in Ref. 16). Arguments in support of this hypothesis stem from the fact that the behavioral deficits are: (a) age related, (b) often concomitant with, or even precede, deposition of Aβ into plaques in the brain; and (c) manifest in behavioral tasks designed to test aspects of memory associated with hippocampal function (17–20), a primary area of Aβ pathology in these mouse models. We have characterized an age-associated deficit in contextual fear conditioning (CFC) in the Tg2576 mouse model of AD. This deficit precedes plaque deposition in the brains of mice and is acutely reversible in animals treated with inhibitors of Aβ production prior to the training session (21). Similar observations demonstrating reversal of behavioral deficits in multiple AD mouse models following acute passive or chronic active immunotherapy have been reported (3–5, 22–25). The reversal of Aβ-associated behavioral deficits in mouse models of AD following immunotherapy, therefore, offers a convenient system to interrogate distinctions among different immunotherapy modalities for efficacy against this end point.
During our investigations into acute reversal of the CFC deficit in Tg2576 mice following peripheral administration of antibodies targeting different epitopes of Aβ,3 we observed differences in potency, as well as overall efficacy, among three antibodies, namely 12A11, 10D5, and 12B4, targeting the same N-terminal epitope of Aβ, specifically Aβ residues 3–7. The in vivo potency of these three monoclonal antibodies does not correlate with an aggregate set of in vitro activities e.g. recognition of soluble monomeric, oligomeric, nor insoluble aggregated Aβ species, in a consistent manner. We undertook comparative structural studies of the three antibodies employing x-ray crystallography of antibody-Fab fragments in complex with Aβ1–40 as well as Aβ1–7 peptide, to gain further insight into the basis for the different in vivo properties. Our results show that all three antibodies recognize Aβ peptide in an extended conformation at the surface of the antibody. The conformation of the Aβ peptide revealed by our x-ray structures is very similar to that observed in complex with three independently derived antibodies recognizing a similar N-terminal epitope of Aβ as reported in two separate studies (26, 27). The comparative studies reported here reveal significant differences in the conformation of the antibody H3 loop, and we postulate that this difference is the primary basis for their differing in vivo activities in the CFC assay. Our findings may be of clinical relevance for immunotherapeutic agents preferentially targeting soluble forms of Aβ for treatment of AD.
EXPERIMENTAL PROCEDURES
Materials
Streptavidin sensor chips and HBS/EP buffer (0.01 m Hepes, pH 7.4, 0.15 m NaCl, 3.0 mm EDTA, 0.005% polysorbate 20 (v/v), 50 mm NaOH) were obtained from Biacore AB (Uppsala, Sweden). Trifluoroacetic acid was purchased from Sigma-Aldrich and added to water (0.1% v/v). The bio-DAE10 peptide (Wyeth), a 24-amino acid peptide containing the N-terminal 10 amino acids of the Aβ peptide, followed by 14 amino acids that constitute a hydrophilic barrel, contains a biotinylated lysine residue in the barrel sequence. The amino acid sequence of bio-DAE10 peptide is: DAEFRHDSGYSGENRSDQK-biotin-GEGGC. The bio-DAE10 peptide was diluted in water to 1 mg/ml and stored at −80 °C as a working stock. Recombinant sAPPα for kinetic studies of antibody binding was purified from HEK293 cells stably transfected with APP695 as described previously (28).
Preparation of Chemically Cross-linked Oligomeric Aβ Species
The preparation of Aβ1–42 consisting of Aβ monomer and oligomers followed the protocol for preparing amyloid-derived diffusible ligands (29) with the addition of a peroxynitrite cross-linking step. Briefly, Aβ1–42 peptide was dissolved to 1 mm in 100% hexafluoroisopropanol then incubated at room temperature for 1 h. The Aβ preparation was divided into 0.5-mg aliquots, and the hexafluoroisopropanol was removed by evaporation followed by lyophilization to remove residual hexafluoroisopropanol. The resultant Aβ peptide residue was stored at −20 °C with dessicant or resuspended in DMSO to a final concentration of 5 mm then added to ice-cold, Ham's F-12 culture media lacking phenol red to bring the peptide to a final concentration of 100 μm. The peptide was incubated at 4 °C for 24 h to produce synthetic Aβ oligomers and then treated with peroxynitrite. For the cross-linking step, a final concentration of 1 mm peroxynitrite (Upstate) in 0.3 n NaOH was added to the 100 μm Aβ preparation, mixed well, and incubated at room temperature for 10 min then followed by immunoprecipitation and Western blot (30).
Immunoprecipiation-Western Blot Assay for Recognition of Oligomeric Aβ Species
The peroxynitrited Aβ preparation (∼100 μm) was pre-cleared by incubation with 40–50 μl of Protein-A-Sepharose (CL-4B, Sigma) beads for 1 h at 4 °C. The beads were removed by centrifugation at 3,000 rpm for 10 min, and supernatant containing Aβ preparation was transferred to fresh tubes. Immunoprecipitations were carried out by addition of Protein-A-Sepharose beads (50 μl) and an anti-Aβ3–7-specific antibody (50 μg) to the Aβ preparation (∼1 μg/ml) with overnight sample mixing by rotation at 4 °C. The beads were collected by centrifugation as above, supernatant was discarded, and beads were washed as described before (30). The immunoprecipitated materials were solubilized from washed beads in 50 μl of 2× Tricine sample buffer (Invitrogen) followed by vortex mixing, then boiled for 10 min in water bath. Aliquots from solubilized immunoprecipitates (10 μl per lane) were size-fractionated by SDS-PAGE in 16% Tricine gels (Invitrogen), followed by transfer to nitrocellulose membranes (Amersham Biosciences Hybond-ECL) at 400 mA for 2 h. Transferred proteins were immobilized on membranes by boiling for 10 min in phosphate-buffered saline, and membranes were blocked overnight at 4 °C in a solution of Tris-buffered saline/0.05% Tween 20/5% nonfat dry milk (30). The membranes were washed with 0.05% Tween 20/Tris-buffered saline then incubated with 1 μg/ml 3D6 (36) for 1 h at room temperature. For detection, the membranes were incubated with anti-mouse Ig-horseradish peroxidase (1:10,000 dilution, Amersham Biosciences), developed using ECL-Plus (Amersham Biosciences), and visualized using film. Molecular mass was estimated by SeeBlue Plus2 molecular weight markers (Invitrogen).
Antibody Characterization, Engineering, and Expression of Recombinant Fab Fragments
Antibodies were purified from ascities by Fc affinity followed by size-exclusion and/or ion-exchange chromatography using standard procedures for characterization of in vitro properties. The minimal linear Aβ epitope recognized by antibodies was determined by enzyme-linked immunosorbent assay employing a series of immobilized 10-mer peptides, spanning the Aβ sequence in increments of one residue, and the ability of the monoclonal antibodies to bind the peptide is monitored essentially as described before (31). Antibody V-regions were cloned from the cognate mouse hybridoma lines using either 5′-rapid amplification of cDNA ends (32), or consensus framework primers and mouse constant region primers (33). Following sequence verification, the cloned V-regions were expressed as recombinant mouse antibodies (in IgG2a, and Ck expression vectors) in conditioned media of transiently transfected COS cells. Antigen recognition by the recombinant antibodies, to confirm function of cloned V-regions, was demonstrated by enzyme-linked immunosorbent assay against immobilized Aβ1–42 peptide. Recombinant Fab fragments were expressed following subcloning of antibody V-regions as VH-CH1 and VL-Ck cassettes in UCOE expression vectors CET1018, or CET1019a (Millipore) for generation of stably transfected CHO cell lines. High expressing stable clones were scaled up for production of antibody in bioreactor (12A11 in Wave bioreactor, Amersham Biosciences, 10D5 and 12B4 in Express bioreactor, Sepragen Inc.) conditioned media. Recombinant Fab fragments were purified to >99% purity from conditioned media in a two-step process using an anti-Aβ1–12 affinity column, followed by size-exclusion chromatography. Purity of the recombinant Fab fragments was confirmed by Coomassie Blue stain SDS-PAGE and liquid chromatography-mass spectrometry of the intact Fab using Electrospray-Time of Flight mass spectrometry.
Determination of Antibody Affinity for Aβ
Antibody affinities were determined by kinetic analysis on Biacore 2000, 3000, and T100 instruments (Biacore AB). Biosensor data were analyzed using BIAevaluation software 3.0.2. The affinity constant of the interaction between antibody and bio-DAE10 peptide was calculated from the kinetic rate constants using both 1:1 and bivalent interaction models (34). Streptavidin sensor surfaces were coated with bio-DAE10 peptide as follows. A continuous flow of HBS/EP buffer was maintained over the sensor surface. The streptavidin on the sensor surfaces was conditioned with three injections (1 min each) of a solution containing 1 m NaCl and 25 mm NaOH. The bio-DAE10 peptide was diluted to 0.01 mg/ml in HBS/EP buffer and injected over the surface at a flow of 1 μl/min using a manual injection. Approximately 4–10 resonance units of peptide was immobilized using this protocol. Kinetic experiments for determination of antibody binding and dissociation constants were performed at 25 °C. Antibodies were serially diluted in running buffer (HBS/EP, 200 μg/ml bovine serum albumin, pH 7.4) to working concentrations. All antibodies were injected in triplicate at a flow rate of 50–100 μl/min for 2 min. The reference surface, an unmodified flow cell, was used to correct for systematic noise and instrument drift. Also, prior to the antibodies binding cycle, buffer was injected for nine binding cycles. These “blank” responses were used to double reference the antibody binding data. Dissociation was monitored in HBS/EP buffer for 10 min at the same flow rate. Blank and buffer effects were subtracted for each sensorgram using double referencing (35, 36). Biosensor surfaces were regenerated using 5 μl of 0.1% trifluoroacetic acid at a flow 100 μl/min, followed by HBS/EP. The response was measured in resonance units representing the mass of bound antibody.
Kinetic Determination of Binding to sAPPα
Kinetic estimates were obtained using the ForteBio Octet (Menlo Park, CA). Briefly, the Goat anti-mouse sensors from ForteBio were used to capture the various antibodies onto the kinetic surface of the sensor. After reaching a baseline, sensors were moved to association step containing 275, 137, 69, or 0 nm sAPPα for 600–900 s then dissociated for 900 s. The dissociation of antibody from the goat anti-mouse sensors was compensated for by using the 0 nm sensor as a reference.
Behavioral Testing in the CFC Assay
Heterozygous male Tg2576 mice expressing human amyloid precursor protein with the Swedish mutation or wild-type mice at 20 weeks of age were trained and tested in operant chambers controlled by Med-PC software (Med-Associates, Inc., Burlington, VT) on two consecutive days in the contextual fear conditioning (CFC) paradigm as described before (17, 21). Purified antibodies were administered parenterally by intraperitoneal injection (at doses indicated in Fig. 2), 24 h prior to the training session on the first day.
FIGURE 2.
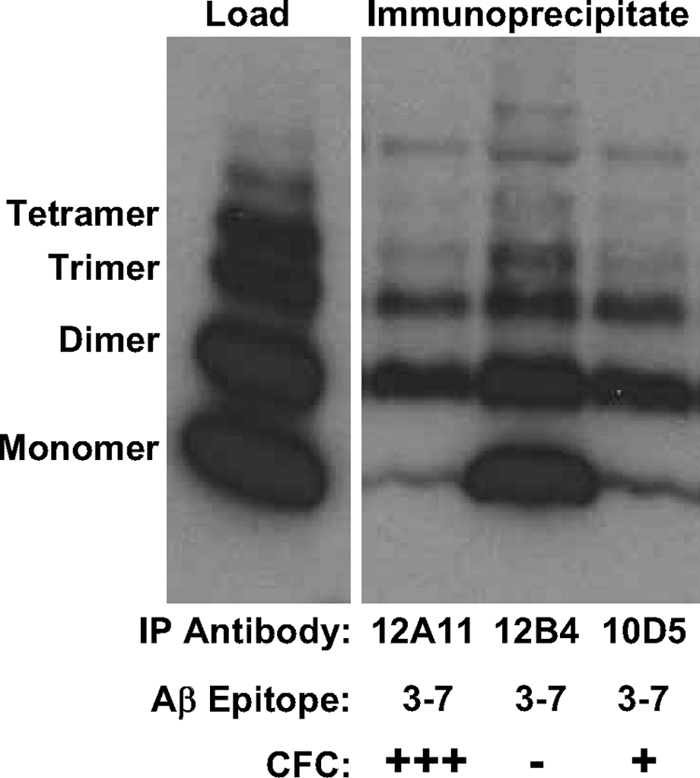
Recognition of chemically cross-linked oligomeric Aβ species by antibodies targeting residues 3–7 of Aβ1–40. Chemically cross-linked Aβ1–40, prepared as described under “Experimental Procedures,” was immunoprecipitated using monoclonal antibodies indicated, and the immunoprecipitates were visualized on Western blots following SDS-PAGE on 18% Tricine gels using antibody 3D6 recognizing residues 1–5 of Aβ. A positive (+) notation indicates potency for reversal of behavioral deficit following treatment with antibody in CFC assay (+++ denotes 1 MPK, and + denotes 30 MPK), and a negative (−) notation indicates a lack of effect following treatment with treatment with the antibody in the CFC assay. Activity in CFC is seen to correlate inversely with recognition of monomeric Aβ species by the respective monoclonal antibody.
Crystallization and Data Collection
Crystals were grown at 22 °C by vapor diffusion. Crystallization conditions are summarized in Table 1. Crystals were flash frozen in liquid nitrogen for data collection using the cryopreservatives (supplemental Table S3). Diffraction data were measured at the Stanford Synchrotron Radiation Lightsource or the Advanced Light Source (see Table 2). All data were measured at 100 K. Data for 12A11+Aβ1–7 were measured in two runs using different exposure times to avoid saturating the low resolution diffraction. Data for 12A11+Aβ1–40 were collected in three runs (two runs to ensure completeness of high resolution data and one run with shorter exposure time to avoid saturating the low resolution diffraction). Diffraction data were processed with MOSFLM and SCALA (37). Data statistics are summarized in Table 2.
TABLE 1.
Properties of monoclonal antibodies recognizing Aβ3–7
Homology is expressed relative to 12A11 V-regions. Kd was determined by using BIAcore binding to immobilized Aβ1–10, assuming bivalent interaction. Monomer recognition is taken from results shown in Fig. 2.
| mAb | VL homology | VH homology | Kd | Monomer recognition | CFC |
|---|---|---|---|---|---|
| % | % | nm | |||
| 12A11 | 8.4 | + | +++ | ||
| 10D5 | 100 | 83 | 54.5 | + | + |
| 12B4 | 96 | 86 | 24.5 | +++ | − |
TABLE 2.
Crystallographic data statistics
| 12A11Aβ1–7 | 12A11Aβ1–40 | 12B4Aβ1–7 | 10D5Aβ1–7 | |
|---|---|---|---|---|
| Data collection | SSRL 11-1 | ALS 8.2.2 | SSRL 11-1 | SSRL 11-1 |
| Space group | P21 | P21 | P1 | P212121 |
| Unit cell lengths (Å) | a = 43.0 | a = 43.0 | a = 78.9 | a = 96.3 |
| b = 86.0 | b = 87.0 | b = 79.2 | b = 100.0 | |
| c = 57.4 | c = 59.0 | c = 94.1 | c = 104.0 | |
| Angles (°) | β = 94.7 | β = 95.8 | α = 68.7 | |
| β = 65.3 | ||||
| γ = 78.5 | ||||
| Resolution Å (last shell) | 1.50 (1.58) | 1.50 (1.58) | 2.95 (3.03) | 2.15 (2.21) |
| Rsym (last shell)a | 5.2 (12.8) | 4.0 (16.8) | 12.9 (37.9) | 5.8 (23.2) |
| Mean ((I)/sd(I)) | 23.6 (10.9) | 22.8 (4.5) | 7.9 (2.4) | 20.2 (5.7) |
| % completeness (last shell) | 97.8 (88.4) | 98.3 (95.0) | 96.0 (95.2) | 98.6 (88.2) |
| Average multiplicity | 6.6 (5.8) | 4.6 (2.3) | 1.9 (1.9) | 6.2 (4.3) |
| MR solution | ||||
| Model used | PDB ID 2aju | 12A11Aβ1–7 | 12A11Aβ1–40 | 12B4Aβ1–7 |
| Constant domain (12–3.5 Å) CC, R | 5.0, 51.3 | 24.1, 46.8 | 26.6, 46.4 | 28.1, 48.6 |
| Variable domain (12–3.5 Å) CC, R | 19.5, 46.0 | 55.7, 32.6 | 49.8, 36.9 | 53.9, 38.0 |
| No. of monomers in asymmetric unit | 1 | 1 | 4 | 2 |
| Residues included in final model | L:1–218 (219) | L:1–219 (219) | L: 1–218 (219) | L: 1–218 (219) |
| H: 1–43, | H: 1–133, | H: 2–223 (226) | H: 1–136, | |
| 46–136, | 141–222 (222) | Aβ: 2–7b | 144–223 (226) | |
| 141–220 (222) | Aβ: 2–7 | Aβ: 2–7 | ||
| Aβ: 1–7 | ||||
| Rfreea | 20.6 | 21.2 | 26.9 | 21.8 |
| Ra | 17.8 | 18.7 | 23.5 | 16.2 |
| Average B-factor | 15.8 | 20.9 | 27.5 | 33.0 |
| Bond length r.m.s.d. | 0.06 | 0.005 | 0.009 | 0.008 |
| Angle r.m.s.d. | 1.37 | 1.05 | 1.16 | 1.17 |
| Ramachandran plot: (% in preferred/allowed/outliers regions)c | 97.0/2.5/0.5 | 96.4/2.8/0.8 | 91.0/6.6/2.4 | 96.8/2.5/0.7 |
aRsym = ΣhΣi(|Ii(h)| − |〈I(h)〉|)/ΣhΣiIi(h), where Ii(h) = observed intensity, and 〈I(h)〉 = mean intensity obtained from multiple measurements. R and Rfree = Σ‖Fo| − |Fc‖/Σ|Fo|, where |Fo| = observed structure factor amplitude and |Fc| = calculated structure factor amplitude for the working and test sets, respectively.
b In 3 of 4 monomers there is a gap at residues 138–145 in the heavy chain. Residues 2–7 (out of 7) are present for 3 of 4 Aβ peptides in the cell, and residues 2–6 were visible and included in the final model for the last monomer.
c As defined in Coot.
Structure Determination and Refinement
All structures were determined by molecular replacement using Fab domains as search models, using the program COMO (38). Molecular replacement was done in two stages, first finding the rotation and translation solution of the constant domain and later, while fixing the constant domain, finding the solution for the variable domain. When more than one monomer comprises the asymmetric unit, all the constant domains were found and fixed before determining the locations of the variable domains. Models used for molecular replacement and statistics for the molecular replacement procedures are shown in Table 2. Model building was carried out with the program Coot (39). Refinement statistics, including the residues modeled in each case, are shown in Table 2.
The 12A11+Aβ1–7 model was refined using Refmac (37). Aβ peptide residues Asp-1 and Asp-7 are poorly defined in the electron density map, and Asp-7 was modeled as Ala.
The 12A11+Aβ1–40 structure was initially refined using REFMAC (37) and at later stages using Phenix (40) with individual positional and B-factor refinement and 5 TLS (translation libration screw) groups (the heavy and light chains were split into two groups each, at the hinge between the constant and the variable domains, and the peptide as a separate group). Although the unit cell is quite similar to that of 12A11+Aβ1–7, the data sets are not isomorphous as the Fabs have different elbow angles. In this case, Aβ Asp-1 could not be modeled, and no electron density was visible past Asp-7.
There are 4 Fab molecules in the P1 unit cell of 12B4+Aβ1–7. Therefore, strict non-crystallographic symmetry was applied in the initial stages of refinement, using the program CNS (41). Later stages of refinement were performed with Phenix (40), with two groups of non-crystallographic symmetry restraints. The variable domain and the Aβ peptide were one group, and the constant domain was the second group. Given the moderate resolution of these data (2.95 Å), refinement included individual positional refinement, grouped B-factor and TLS. For the grouped B-factor refinement the heavy and light chains were split into two groups each (at the hinge between the constant and the variable domains), and the peptide was a separate group (5 groups per monomer, 20 groups total). For TLS refinement 3 groups per monomer were selected (variable domain, constant domain, and peptide per monomer; total of 12 groups).
The10D5+Aβ1–7 crystals have two complexes in the asymmetric unit. Restrained non-crystallographic symmetry was initially applied, but was removed in later stages of refinement. Refinement was performed with Phenix (40), using individual positional and B-factor refinement and three TLS groups per monomer (variable domain, constant domain, and peptide). Coordinates and structure factors are being deposited in the Protein Data Bank.
RESULTS
In Vivo and in Vitro Properties of Antibodies Recognizing N-terminal Epitope of Aβ
We have previously demonstrated that DAPT (N-[N-(3,5-diflurophenylacetyl)-l-alanyl]-S-phenylglycine t-butyl ester), a small molecule inhibitor of Aβ production (42), is able to acutely reverse the age- and Aβ-related deficit in CFC, a task that queries hippocampal-dependent memory, in the Tg2576 mouse model of AD (21). We surveyed antibodies targeting different linear epitopes of Aβ peptide for their ability to acutely reverse this CFC deficit following passive parenteral administration ∼24 h prior to the training session. Of particular interest, three antibodies recognizing Aβ residues 3–7, 12A11, 10D5, and 12B4, demonstrated disparate potencies for acute reversal of the CFC deficit. As shown in Fig. 1, 12A11 demonstrates complete reversal of CFC deficit at 1 MPK, 10D5 is efficacious at 30 MPK, and 12B4 is without effect at the highest dose tested (30 MPK).
FIGURE 1.

Different antibodies recognizing the same linear epitope on Aβ, Aβ residues 3–7, demonstrate different in vivo potency for acute reversal of CFC deficit in Tg2576 mice. The antibodies 12A11, 10D5, and 12B4 recognize Aβ residues 3–7 as determined by enzyme-linked immunosorbent assay. All three antibodies were administered by intraperitoneal injection to separate groups of mice at the doses indicated in the figure, ∼ 24 h prior to the training session. In comparison with the isotype control antibody TY11/15, 12A11 is able to restore the freezing behavior of Tg2576 to the levels exhibited by vehicle-treated wild-type mice at doses of 1 MPK and higher. 10D5 restores freezing behavior of Tg2576 mice to wild-type levels at 30 MPK, but not lower doses. The antibody 12B4 is without effect in this assay at the highest dose tested (30 MPK). N/E, no effect; MED, minimum efficacious dose.
The in vivo potencies exhibited by 12A11, 10D5, and 12B4 in the CFC assay are not readily attributable to differences in their primary structure, because the three antibodies are highly conserved in their VL (>95%) and VH (>82%) regions (supplemental Fig. S2, A and B). CFC activity of these antibodies also does not correlate with affinity for immobilized Aβ1–10 peptide (Table 1), avidity for aggregated Aβ1–42, or ability to capture soluble Aβ1–42 (see Table 2 in Ref. 43). Although the higher affinity of 12A11 for immobilized Aβ1–10 (Table 1) and capture of soluble Aβ1–42 (43) is consistent with its potency in CFC, in vitro binding to Aβ by either of these measures does not account for the higher in vivo potency of 10D5 versus 12B4 in the CFC assay. Likewise, 10D5 displays highest avidity to aggregated Aβ1–42 by enzyme-linked immunosorbent assay, followed by 12A11 and then 12B4 (43). Hence, binding to aggregated Aβ also does not predict in vivo activity in the CFC assay.
We compared the three antibodies for recognition of monomeric versus chemically cross-linked oligomeric species of Aβ in a Western blot assay. Our results, shown in Fig. 2, reveal 12B4 displays strongest recognition of monomeric Aβ, whereas 10D5 and 12A11 display roughly equivalent recognition of Aβ monomers. Interestingly, all three antibodies recognize multimers of Aβ peptide to equivalent degrees. The Aβ3–7 epitope is also present in sAPPα. Recognition of circulating human sAPPα in Tg2576 mice could compete with central or peripheral Aβ for antibody binding and contribute to their potency differences in the CFC assay. To address this possibility, we tested the antibodies for binding to sAPPα. We were unable to detect any binding to sAPPα by these antibodies using biolayer interferometry methodology on the Forte Bio instrument (supplemental Fig. S3) over a range of sAPPα concentrations tested. Hence, it is unlikely that recognition of sAPPα is a factor influencing the potencies of the antibodies in the CFC assay.
Structural Basis of Aβ Recognition
To understand differences in antigen recognition associated with their distinct in vivo and in vitro activities at the atomic level, we determined crystal structures of 12A11, 10D5, and 12B4 bound to Aβ. Because these antibodies recognize the N-terminal moiety of the peptide we crystallized their Fab fragments bound to a fragment of Aβ comprising residues 1–7. To assess possible effects that the rest of the Aβ peptide might have on recognition, we also determined the structure of 12A11 bound to the longer Aβ1–40 peptide. Crystallographic data statistics are shown in Table 2.
The Aβ peptide adopts an extended conformation that binds in a groove defined by CDRs H2, H3, L1, and L3 of each Fab (Fig. 3). Residues 2–6 of the peptide could be visualized unambiguously in all four structures. Aβ Asp-1 could be modeled only in the 12A11+Aβ1–7 structure. Likewise, the side chain of Aβ Asp-7 could be modeled in only some of the structures. No electron density is visible for residues past Asp-7 in the 12A11+Aβ1–40 structure. Superposition of the bound peptide from crystals of the two 12A11-Aβ complexes reveals very similar backbone and side-chain conformations for the central 3–6 Aβ residues (supplemental Fig. S1). Hence, the conformation of the peptide revealed in the structure of 12A11 with Aβ1–7 accurately reflects the conformation of the epitope in the context of the longer Aβ antigen. Moreover, the two 12A11 Fab structures show no significant differences, indicating that, at least for this antibody, Aβ residues C-terminal to residue 7 have no effect on binding.
FIGURE 3.

A, overall structure of 12A11 fab with Aβ1–7 peptide. Heavy chain is shown in cyan, light chain in blue, and Aβ peptide in yellow. B, 12A11 with Aβ1–7 peptide. The surface of the heavy chain is shown in cyan and light chain in light blue. Aβ peptide is shown in stick representation with oxygens colored red, nitrogens in blue, and carbons in yellow. C, 12A11 with Aβ1–40 peptide. The 3.0 sigma electron density omit map (calculated while omitting the Aβ peptide from the model for phase calculations) is shown in yellow. Protein and peptide are shown in stick representation with oxygens colored red, nitrogens in blue, and carbons in yellow for the peptides, light blue for the light chain, and cyan for the heavy chain. D, 12A11 with Aβ1–40. Peptide in stick representation with oxygens colored red, nitrogens in blue, and carbons in yellow. Fab in surface representation colored white with the exception of CDRs contacting the peptide. CDR H1 is shown in red, H2 blue, H3 green, L1 magenta, and L3 cyan. Only CDRs contacting the peptide are shown.
The tertiary structures of Aβ1–7 bound to the 12B4 and 10D5 Fabs are very similar to that observed in 12A11, especially the four central residues 3–6 that constitute the recognition epitope (Fig. 4A). The root mean square deviation (r.m.s.d.) for all atoms of Aβ residues 3–6 relative to the 12A11+Aβ1–7 structure ranged from 0.37 to 0.48. Thus, differences in in vivo activity measured by the CFC assay among the three antibodies are not attributable to different conformations of Aβ peptide recognized by the monoclonal antibodies.
FIGURE 4.

Aβ peptide and antibodies are shown in stick representation with oxygens colored red, nitrogens in blue, and carbons in pink for 12A11, yellow for 10D5, and cyan for 12B4. A, superposition of the Aβ peptide. The superposition was done using Cα atoms of residues 3–6 of Aβ. B–F, regions in the Fabs that differ in sequence between 12A11, 10D5, and 12B4, as described in the text.
Despite the similarity in the tertiary structure of the bound peptide, differences in the CDR sequences of the three antibodies (Table 3) give rise to distinct interactions with the bound peptide. To compare the interactions, we show residue-by-residue interactions between Aβ and 12A11, the structures determined at the highest resolution, in Fig. 5. Antibody CDR residues contacting antigen for all three antibodies are summarized in supplemental Table S4. In all three antibodies, Aβ residues Ala-2 and Glu-3 interact principally with the light chain CDRs L3 and L1, whereas Arg-5 and Asp-7 interact with CDRs H2 and H3, respectively. Aβ Phe-4 and His-6 are the most deeply buried in the groove, with their side chains interacting with both the light and the heavy chain (Fig. 3, B and D). Both Phe-4 and His-6 are in direct contact with CDRs L1 and L3. Phe-4 is also in direct contact with heavy chain CDR H2 and residue His-6 contacts CDR H3 (Fig. 5, Table 3, and supplemental Table S4).
TABLE 3.
CDR homology of light chain variable regions (L1, L2, and L3), and heavy chain variable regions (H1, H2, and H3)
CDR residues contacting the antigen are highlighted in boldface, and CDR3 of 12A11 VH domain displays unique 3- to 5-residue deletions relative to the other antibodies shown.
| Antibody | L1 | L2 | L3 | H1 | H2 | H3 |
|---|---|---|---|---|---|---|
| 12A11 | 24RSSQNIIHSNGNTYLE | 55KVSNRFS | 94FQSSHVPLT | 31TSGMSVG | 52HIWWDDDKYYNPSLKS | 100RTTT-----ADYFAY |
| 10D5 | RSSQNIIHSNGNTYLE | KVSNRFS | FQGSHVPLT | TSGMGVS | HIYWDDDKRYNPSLKS | RPITPVL--VDAMDY |
| 12B4 | RSSQNIVHSNGNTYLE | KVSNRFS | FQGSHVPLT | TNGMGVS | HIYWDEDKRYNPSLKS | RRIIYDV--EDYFDY |
| PFA1 | RSSQSIVHSNGNTYLE | KVSNRFS | FQGSHVPLT | TSGMGVG | HIWWDDDKSYNPSLKS | RAHTTVL--GDWFAY |
| PFA2 | RSSQSIVHSNGNTYLE | KVSNRFS | FQGSHVPLT | TSGMGVG | HIWWDDDKNYNPSLKS | RAHNVVL--GDWFAY |
| WO2 | RSSQSIVHSNGHTYLE | QVSTRFS | FQASLVPLT | TSKVGVS | HIYWDDDKRYNPSLES | RGFYGRKYEVNHFDY |
FIGURE 5.

12A11 interaction with Aβ1–7 and Aβ1–40. For clarity representative antibody contacts per residue of Aβ peptide are shown. A–E, 12A11:Aβ1–7; F, 12A11:Aβ1–40. Protein and peptide are shown in stick representation with oxygens colored red, nitrogens in blue, and carbons in yellow for the peptides, light blue for the light chain, and cyan for the heavy chain.
12A11 residues H-Trp-54 and H-Tyr-60 are replaced by Tyr and Arg, respectively, in both 12B4 and 10D5 (Fig. 4B and Table 3, H2 sequence). Both 12A11 H-Trp-54 and 12B4/10D5 H-Tyr-54 interact with Aβ Arg-5 (Fig. 4B), but the phenolic OH of the 12B4/10D5 Tyr-54 side chain also contacts Aβ Phe-4 (Fig. 4C). This OH group also forms a hydrogen bond with the backbone carbonyl of Aβ Arg-5, instead of a water molecule found in this position in the 12A11 structure (Fig. 4C). At position 60, 12A11 H-Tyr-60 and one of the two crystallographically independent copies of 10D5 H-Arg-60 contact Aβ Arg-5, but 12A11 H-Tyr-60 also packs against Aβ Phe-4 while 10D5 H-Arg-60 forms a water-mediated hydrogen bond to the backbone of Aβ Glu-3 (Fig. 4D). In 12B4 and the other copy of 10D5, H-Arg-60, is hydrogen-bonded directly to Aβ Glu-3 backbone (Fig. 4D).
In the 12A11+Aβ1–40 structure, Aβ Arg-5 forms an internal salt bridge with Asp-7 (Fig. 5F). In all three structures determined with the shorter Aβ1–7, the C-terminal carboxylate forms an equivalent internal salt bridge (Figs. 4E and 5F). Presumably, this is an artifact of using the shorter peptide, but the presence of the salt bridge in all of the structures strongly suggests that the side chain-side chain interaction between Arg-5 and Asp-7 is a real feature of the longer peptide bound to these antibodies.
The only other position that interacts with the bound peptide that differs among the antibody sequences is residue 96 of the light chain, which is Ser in 12A11 and Gly in 10D5 and 12B4 (Table 3, L3 sequence). This difference does not seem to alter the binding to the peptide, as Aβ interacts with the backbone carbonyl (Fig. 4F).
Comparison to Other Antibodies That Recognize the N Terminus of Aβ
The conformation of the Aβ peptide revealed from our crystals of 12A11, 10D5, and 12B4 is very similar to that observed in crystals of three independently derived antibodies that recognize the N-terminal epitope of Aβ: PFA1, PFA2, and WO2 reported previously (26, 27). Superposition of the peptide in the complexes reported by Gardberg et al. (26) and Miles et al. (27), with Aβ1–7 bound to 12A11, gives an average r.m.s.d. of 0.68 Å for all atoms of residues 3–6 (Fig. 6A). The similarity in Aβ conformation is reflected in the strong sequence conservation in the CDR loops L1, L2, L3, H1, and H2, including residues that directly bind the peptide and others that position the CDRs for binding the extended peptide (Table 3). The few differences include CDR-L3 residue 96 (12A11 numbering, Table 3), which is a Gly, Ser, or Ala, but as noted above this difference is probably insignificant as the peptide interacts with the main chain at this position. In WO2, CDR-H2 residues 54 and 60 are Tyr and Arg, as in 12B4 and 10D5. The conformation for WO2 H-Arg-60 is similar to the conformation seen in the first monomer in the asymmetric unit of 10D5 (Fig. 4D). For PFA1 and PFA2, heavy chain residue 54 is Trp as in 12A11, and residue 60 is Ser or an Asn. In PFA1, H-Ser-60 packs against Aβ Phe4. In PFA2 the equivalent Asn does not interact directly with the peptide, but it forms a hydrogen bond with H-Trp-54, which is a peptide-binding residue.
FIGURE 6.

PFA1, pdb id 2IPU (red); PFA2, pdb id 2R0W (green); and WO2, pdb id 3BKJ (blue), and antibodies 12A11 (pink), 10D5 (yellow), and 12B4 (cyan). A, superposition of Cα atoms of residues 2–6 of the peptide showing similarity in this region. B, superposition of the Cα atoms of the variable domains of the structures showing differences in the region of heavy chain CDR-3. For clarity only Aβ peptide from the structure of 12A11 is shown in stick representation. C, H-Asp-108 (12A11, 10D5, 12B4, PFA1, and PFA2) and H-Asn-110 (WO2) binding Aβ-His-6 (numbering system as in Table 3). D, H-His-102 bridging Aβ Asp-7 and Aβ Arg-5 in PFA1 and PFA2 (red and green). Aβ Asp-7 salt bridging Aβ Arg-5 in 12A11 (pink).
Differences in Heavy Chain CDR-3
The greatest divergence in sequence and tertiary structure among the antibodies occurs in CDR3 of the heavy chain (Figs. 4E and 6B and Table 3, H3 sequence). This region is located near the C terminus of the Aβ1–7 peptide, and the different antibodies show some variation in their interactions with this portion of Aβ. H-Asp-108 (10D5 and 12B4 numbering, Table 3) is present in all the antibodies except WO2, and it anchors the loop on one side by binding to Aβ His-6 (Fig. 6C and Table 3). In WO2, the equivalent H-Glu-108 interacts with Aβ Asp-7, and Aβ His-6 is instead bound to H-Asn-110. Other residues of the loop are not conserved, and some of them interact with Aβ residues 6–7. For example, in PFA1 and PFA2 the internal salt bridge between Aβ Arg-5 and Asp-7 is broken by H-His-102, which packs against Aβ Arg-5 and hydrogen bonds to Aβ Asp-7 (Fig. 6D), whereas the Arg-5-Asp-7 interaction is present in WO2. This and other differences due to variations in CDR-H3 may explain the variation in the position of Aβ Asp-7 and, when visible, Ser-8.
DISCUSSION
We investigated the structural basis underlying antigen recognition to identify distinguishing features among three antibodies (12A11, 10D5, and 12B4) with differing potencies for acute reversal of CFC deficits in Tg2576 mice. These antibodies recognize the same linear epitope of Aβ, and their in vivo potencies are not readily explained by an aggregate set of in vitro properties. The in vitro properties we interrogated include differences in affinity for immobilized Aβ1–10 (Table 1), avidity for immobilized aggregated Aβ (43), capture of total soluble Aβ (43), recognition of Aβ monomers versus chemically cross-linked multimers (Fig. 2), and binding to sAPPα (supplemental Fig. S3). To further investigate differences in antigen recognition by the three antibodies, we solved x-ray crystal structures of Fab fragments in complex with Aβ peptide.
Our comparative structural analysis revealed the three antibodies under study recognize the same conformation of Aβ peptide, with strong conservation of antigen-contacting residues in the antibody CDRs, excepting H2 and H3. The basis for different potencies in the CFC assay among the three antibodies is suggested by differences in residues contacting peptide in the H2 and H3 loops, and distinct H3 loop conformations. Assuming Aβ-related behavioral deficits in Tg mouse models of AD are mediated by soluble oligomeric Aβ species, and behavioral deficits offer preclinical surrogates of cognitive decline in AD, our findings may be of clinical relevance for AD immunotherapies specifically targeting soluble Aβ. Each of these points is discussed in turn below.
Our comparative structural studies employed co-crystallization of recombinant Fab fragments of 12A11, 10D5, and 12B4 with primarily Aβ1–7 peptide. The structure of 12A11 with Aβ1–40 peptide was also solved to very high resolution (Table 2). Superposition of the bound peptide reveals excellent agreement between Aβ1–7 and Aβ1–40 in the conformation of the main chain as well as orientation of the side chains (supplemental Fig. S1), demonstrating that the conformation of the peptide revealed in the structure of 12A11 with Aβ1–7 accurately reflects the conformation of the native Aβ1–40 antigen recognized by this antibody. Furthermore, antibody:antigen contacts are preserved between the crystal structure of 12A11 with Aβ1–7 and Aβ1–40. Therefore, it is reasonable to assume the antigen conformation, and antibody:antigen contacts observed in the structures of 10D5:Aβ1–7 and 12B4:Aβ1–7 are reflective of the respective antibodies recognition of Aβ1–40 peptide. Our assumption is supported by the strong conservation between antigen conformation and antibody:antigen contacts reported here and the structures of PFA1 and PFA2 with Aβ1–8 peptide (26), as well as the structure of WO2 antibody with Aβ1–16 and Aβ1–28 (27). Finally, the antibodies in our study have been demonstrated to recognize Aβ in plaques as well as soluble Aβ species and are efficacious in plaque reduction in passive immunotherapy studies (43–46), suggesting that the conformation of the epitope observed in crystal structures reflects at-least one conformation adopted by N terminus of Aβ under physiological conditions.
The structural data presented here suggest that antibodies that recognize the same elongated form of the peptide may be specific to different molecular species of Aβ. Because the heavy chain CDR3 loop is positioned in the region where a longer peptide would be located, the different interactions and Aβ conformations observed in this region may be important in binding to distinct forms of Aβ. CDR-H3 of 12A11 is three amino acids shorter than that of 12B4 and 10D5. This loop is compact and adopts an “open” position that might permit binding to a longer peptide in conformations that would be disallowed by the extended loop present in 12B4 and 10D5. Although Aβ residues C-terminal to Asp-7 are not expected to directly bind these antibodies (as demonstrated by the structure of 12A11+Aβ1–40), the CDR-H3 loop may favor or disfavor binding of antibody to certain species of Aβ peptide (different folds, oligomerization, isomerization, or racemization species), thereby acting as a “blocking” moiety. For example, 12A11 preferentially recognizes chemically cross-linked multimers of Aβ versus monomeric Aβ (Fig. 2), suggesting that 12A11 CDR-H3 loop conformation correlates with preferential binding to multimers, whereas the CDR-H3 loop found in 12B4 (Fig. 6B) correlates with equivalent binding to Aβ monomers and multimers. Likewise, chemical isomerization is enhanced in Aβ peptides purified from AD brains (47–49), and one of the residues that may undergo such a change is Aβ Asp-7, which was found changed to l-iso-aspartate in an in vitro aging study (50). Because the CDR-H3 loop is close to Aβ Asp-7, it may interact differently with the different Asp-7 isomers, giving one antibody preference over another. Overall, we postulate differences in sequence and structure of CDR-H3 contribute to the different in vivo activities observed in the CFC assay between 12A11, 10D4, and 12B4.
In addition to the highly conserved conformation of Aβ peptide observed between our co-crystals and that reported by Gardberg et al. (26), and Miles et al. (27), antibody residues in the involved CDR regions show strong conservation of primary structure with equally remarkable conservation of residues contacting antigen in CDRs L1, L3, and H2 (Table 3). The greatest divergence in primary and tertiary structure is observed in CDR3 of the heavy chain (Fig. 6 and Table 3). This suggests that the H3 loop underlies the disparate in vivo activities of 12A11, 10D5, and 12B4 in the CFC assay, as discussed above. The dominance of the heavy chain variable region for conferring antibody specificity, owing to CDR3 diversity arising from V(D)J junctional hypervariability, is well recognized (51). The impact of the divergent H3 loops in PFA1, PFA2, and WO2 relative to 12A11 remains uncertain, because in vivo properties of these later three antibodies have not been reported. Finally, the highly conserved V-region primary structure among five independently derived mouse monoclonal antibodies (supplemental Fig. S2, A and B) reveals a restricted V-region repertoire utilization elicited against human Aβ3–7 in mouse.
The molecular basis underlying neural and memory loss, and consequent dementia, in AD patients remains an area of active investigation. Behavioral deficits in transgenic mouse models of AD arguably represent surrogate models for investigating the pathogenic factors responsible for memory and cognitive decline in AD patients (6, 23, 52). Principal arguments used to support this assumption are based on the fact that the deficits in mouse models are age- and Aβ-related (18–20, 53), albeit without the same extent of robust neural loss seen in AD. The observation that behavioral and cognitive deficits in many mouse models temporally precede frank deposition and accumulation of Aβ in plaques, combined with the fact that these deficits are reversible in the presence of plaque burden in the animals (5, 22), has been interpreted as indicating a primary role for soluble Aβ species as drivers of cognitive decline in mouse models (52, 54). Consequently, the role of soluble oligomeric Aβ species in driving neural loss and functional deficits has become a primary focus of investigation (52, 53, 55–60). Ultimately, clinical trials targeting Aβ via immunotherapy or secretase inhibitors for treatment and/or prevention of AD will provide a critical test of the amyloid cascade hypothesis and how well behavioral deficits testing memory in transgenic mouse models of AD translate to memory decline seen in AD.
Supplementary Material
Acknowledgments
We thank Jayakumar Rajadas for facilitating the initial contacts between the groups at Elan and Stanford, Nanhua Yan for valuable discussions and input, and Gergely Toth for expert assistance with molecular modeling.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3 and Tables S3 and S4.
J. S. Jacobsen, et al., manuscript in preparation.
- Aβ
- amyloid β
- AD
- Alzheimer disease
- CDR
- complementarity determining region
- CFC
- contextual fear conditioning
- H#
- heavy chain CDR number
- IP
- immunoprecipitation
- L#
- light chain CDR number
- MPK
- milligrams per kilogram of body weight
- SA
- streptavidin
- sAPPα
- soluble amyloid precursor protein α
- Tg
- transgene
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
REFERENCES
- 1.Schenk D., Barbour R., Dunn W., Gordon G., Grajeda H., Guido T., Hu K., Huang J., Johnson-Wood K., Khan K., Kholodenko D., Lee M., Liao Z., Lieberburg I., Motter R., Mutter L., Soriano F., Shopp G., Vasquez N., Vandevert C., Walker S., Wogulis M., Yednock T., Games D., Seubert P. (1999) Nature 400, 173–177 [DOI] [PubMed] [Google Scholar]
- 2.Wilcock D. M., DiCarlo G., Henderson D., Jackson J., Clarke K., Ugen K. E., Gordon M. N., Morgan D. (2003) J. Neurosci. 23, 3745–3751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Janus C., Pearson J., McLaurin J., Mathews P. M., Jiang Y., Schmidt S. D., Chishti M. A., Horne P., Heslin D., French J., Mount H. T., Nixon R. A., Mercken M., Bergeron C., Fraser P. E., St George-Hyslop P., Westaway D. (2000) Nature 408, 979–982 [DOI] [PubMed] [Google Scholar]
- 4.Morgan D., Diamond D. M., Gottschall P. E., Ugen K. E., Dickey C., Hardy J., Duff K., Jantzen P., DiCarlo G., Wilcock D., Connor K., Hatcher J., Hope C., Gordon M., Arendash G. W. (2000) Nature 408, 982–985 [DOI] [PubMed] [Google Scholar]
- 5.Dodart J. C., Bales K. R., Gannon K. S., Greene S. J., DeMattos R. B., Mathis C., DeLong C. A., Wu S., Wu X., Holtzman D. M., Paul S. M. (2002) Nat. Neurosci. 5, 452–457 [DOI] [PubMed] [Google Scholar]
- 6.Brody D. L., Holtzman D. M. (2008) Annu. Rev. Neurosci. 31, 175–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schenk D., Hagen M., Seubert P. (2004) Curr. Opin. Immunol. 16, 599–606 [DOI] [PubMed] [Google Scholar]
- 8.Bayer A. J., Bullock R., Jones R. W., Wilkinson D., Paterson K. R., Jenkins L., Millais S. B., Donoghue S. (2005) Neurology 64, 94–101 [DOI] [PubMed] [Google Scholar]
- 9.Bombois S., Maurage C. A., Gompel M., Deramecourt V., Mackowiak-Cordoliani M. A., Black R. S., Lavielle R., Delacourte A., Pasquier F. (2007) Arch. Neurol. 64, 583–587 [DOI] [PubMed] [Google Scholar]
- 10.Masliah E., Hansen L., Adame A., Crews L., Bard F., Lee C., Seubert P., Games D., Kirby L., Schenk D. (2005) Neurology 64, 129–131 [DOI] [PubMed] [Google Scholar]
- 11.Nicoll J. A., Barton E., Boche D., Neal J. W., Ferrer I., Thompson P., Vlachouli C., Wilkinson D., Bayer A., Games D., Seubert P., Schenk D., Holmes C. (2006) J. Neuropathol. Exp. Neurol. 65, 1040–1048 [DOI] [PubMed] [Google Scholar]
- 12.Nicoll J. A., Wilkinson D., Holmes C., Steart P., Markham H., Weller R. O. (2003) Nat. Med. 9, 448–452 [DOI] [PubMed] [Google Scholar]
- 13.Gilman S., Koller M., Black R. S., Jenkins L., Griffith S. G., Fox N. C., Eisner L., Kirby L., Rovira M. B., Forette F., Orgogozo J. M. (2005) Neurology 64, 1553–1562 [DOI] [PubMed] [Google Scholar]
- 14.Hock C., Konietzko U., Streffer J. R., Tracy J., Signorell A., Müller-Tillmanns B., Lemke U., Henke K., Moritz E., Garcia E., Wollmer M. A., Umbricht D., de Quervain D. J., Hofmann M., Maddalena A., Papassotiropoulos A., Nitsch R. M. (2003) Neuron 38, 547–554 [DOI] [PubMed] [Google Scholar]
- 15.Holmes C., Boche D., Wilkinson D., Yadegarfar G., Hopkins V., Bayer A., Jones R. W., Bullock R., Love S., Neal J. W., Zotova E., Nicoll J. A. (2008) Lancet 372, 216–223 [DOI] [PubMed] [Google Scholar]
- 16.Kobayashi D. T., Chen K. S. (2005) Genes Brain Behav. 4, 173–196 [DOI] [PubMed] [Google Scholar]
- 17.Jacobsen J. S., Wu C. C., Redwine J. M., Comery T. A., Arias R., Bowlby M., Martone R., Morrison J. H., Pangalos M. N., Reinhart P. H., Bloom F. E. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 5161–5166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsiao K., Chapman P., Nilsen S., Eckman C., Harigaya Y., Younkin S., Yang F., Cole G. (1996) Science 274, 99–102 [DOI] [PubMed] [Google Scholar]
- 19.Dineley K. T., Xia X., Bui D., Sweatt J. D., Zheng H. (2002) J. Biol. Chem. 277, 22768–22780 [DOI] [PubMed] [Google Scholar]
- 20.Westerman M. A., Cooper-Blacketer D., Mariash A., Kotilinek L., Kawarabayashi T., Younkin L. H., Carlson G. A., Younkin S. G., Ashe K. H. (2002) J. Neurosci. 22, 1858–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Comery T. A., Martone R. L., Aschmies S., Atchison K. P., Diamantidis G., Gong X., Zhou H., Kreft A. F., Pangalos M. N., Sonnenberg-Reines J., Jacobsen J. S., Marquis K. L. (2005) J. Neurosci. 25, 8898–8902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kotilinek L. A., Bacskai B., Westerman M., Kawarabayashi T., Younkin L., Hyman B. T., Younkin S., Ashe K. H. (2002) J. Neurosci. 22, 6331–6335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen G., Chen K. S., Kobayashi D., Barbour R., Motter R., Games D., Martin S. J., Morris R. G. (2007) J. Neurosci. 27, 2654–2662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartman R. E., Izumi Y., Bales K. R., Paul S. M., Wozniak D. F., Holtzman D. M. (2005) J. Neurosci. 25, 6213–6220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oddo S., Vasilevko V., Caccamo A., Kitazawa M., Cribbs D. H., LaFerla F. M. (2006) J. Biol. Chem. 281, 39413–39423 [DOI] [PubMed] [Google Scholar]
- 26.Gardberg A. S., Dice L. T., Ou S., Rich R. L., Helmbrecht E., Ko J., Wetzel R., Myszka D. G., Patterson P. H., Dealwis C. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 15659–15664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miles L. A., Wun K. S., Crespi G. A., Fodero-Tavoletti M. T., Galatis D., Bagley C. J., Beyreuther K., Masters C. L., Cappai R., McKinstry W. J., Barnham K. J., Parker M. W. (2008) J. Mol. Biol. 377, 181–192 [DOI] [PubMed] [Google Scholar]
- 28.Esch F. S., Keim P. S., Beattie E. C., Blacher R. W., Culwell A. R., Oltersdorf T., McClure D., Ward P. J. (1990) Science 248, 1122–1124 [DOI] [PubMed] [Google Scholar]
- 29.Dahlgren K. N., Manelli A. M., Stine W. B., Jr., Baker L. K., Krafft G. A., LaDu M. J. (2002) J. Biol. Chem. 277, 32046–32053 [DOI] [PubMed] [Google Scholar]
- 30.Walsh D. M., Tseng B. P., Rydel R. E., Podlisny M. B., Selkoe D. J. (2000) Biochemistry 39, 10831–10839 [DOI] [PubMed] [Google Scholar]
- 31.Lee M., Bard F., Johnson-Wood K., Lee C., Hu K., Griffith S. G., Black R. S., Schenk D., Seubert P. (2005) Ann. Neurol. 58, 430–435 [DOI] [PubMed] [Google Scholar]
- 32.Frohman M. A., Dush M. K., Martin G. R. (1988) Proc. Natl. Acad. Sci. U.S.A. 85, 8998–9002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones S. T., Bendig M. M. (1991) Biotechnology 9, 88–89 [DOI] [PubMed] [Google Scholar]
- 34.Karlsson R., Fält A. (1997) J. Immunol. Methods 200, 121–133 [DOI] [PubMed] [Google Scholar]
- 35.Karlsson R., Michaelsson A., Mattsson L. (1991) J. Immunol. Methods 145, 229–240 [DOI] [PubMed] [Google Scholar]
- 36.Myszka D. G. (1999) J. Mol. Recognit. 12, 279–284 [DOI] [PubMed] [Google Scholar]
- 37.Collaborative Computational Project, N. (1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–76315299374 [Google Scholar]
- 38.Jogl G., Tao X., Xu Y., Tong L. (2001) Acta Crystallogr. D. Biol. Crystallogr. 57, 1127–1134 [DOI] [PubMed] [Google Scholar]
- 39.Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 40.Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 41.Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 42.Dovey H. F., John V., Anderson J. P., Chen L. Z., de Saint Andrieu P., Fang L. Y., Freedman S. B., Folmer B., Goldbach E., Holsztynska E. J., Hu K. L., Johnson-Wood K. L., Kennedy S. L., Kholodenko D., Knops J. E., Latimer L. H., Lee M., Liao Z., Lieberburg I. M., Motter R. N., Mutter L. C., Nietz J., Quinn K. P., Sacchi K. L., Seubert P. A., Shopp G. M., Thorsett E. D., Tung J. S., Wu J., Yang S., Yin C. T., Schenk D. B., May P. C., Altstiel L. D., Bender M. H., Boggs L. N., Britton T. C., Clemens J. C., Czilli D. L., Dieckman-McGinty D. K., Droste J. J., Fuson K. S., Gitter B. D., Hyslop P. A., Johnstone E. M., Li W. Y., Little S. P., Mabry T. E., Miller F. D., Audia J. E. (2001) J. Neurochem. 76, 173–181 [DOI] [PubMed] [Google Scholar]
- 43.Bard F., Barbour R., Cannon C., Carretto R., Fox M., Games D., Guido T., Hoenow K., Hu K., Johnson-Wood K., Khan K., Kholodenko D., Lee C., Lee M., Motter R., Nguyen M., Reed A., Schenk D., Tang P., Vasquez N., Seubert P., Yednock T. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 2023–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bacskai B. J., Kajdasz S. T., Christie R. H., Carter C., Games D., Seubert P., Schenk D., Hyman B. T. (2001) Nat. Med. 7, 369–372 [DOI] [PubMed] [Google Scholar]
- 45.Bacskai B. J., Kajdasz S. T., McLellan M. E., Games D., Seubert P., Schenk D., Hyman B. T. (2002) J. Neurosci. 22, 7873–7878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bard F., Cannon C., Barbour R., Burke R. L., Games D., Grajeda H., Guido T., Hu K., Huang J., Johnson-Wood K., Khan K., Kholodenko D., Lee M., Lieberburg I., Motter R., Nguyen M., Soriano F., Vasquez N., Weiss K., Welch B., Seubert P., Schenk D., Yednock T. (2000) Nat. Med. 6, 916–919 [DOI] [PubMed] [Google Scholar]
- 47.Kuo Y. M., Emmerling M. R., Woods A. S., Cotter R. J., Roher A. E. (1997) Biochem. Biophys. Res. Commun. 237, 188–191 [DOI] [PubMed] [Google Scholar]
- 48.Roher A. E., Lowenson J. D., Clarke S., Wolkow C., Wang R., Cotter R. J., Reardon I. M., Zürcher-Neely H. A., Heinrikson R. L., Ball M. J., et al. (1993) J. Biol. Chem. 268, 3072–3083 [PubMed] [Google Scholar]
- 49.Shimizu T., Watanabe A., Ogawara M., Mori H., Shirasawa T. (2000) Arch. Biochem. Biophys. 381, 225–234 [DOI] [PubMed] [Google Scholar]
- 50.Zirah S., Kozin S. A., Mazur A. K., Blond A., Cheminant M., Ségalas-Milazzo I., Debey P., Rebuffat S. (2006) J. Biol. Chem. 281, 2151–2161 [DOI] [PubMed] [Google Scholar]
- 51.Nemazee D. (2006) Nat. Rev. Immunol. 6, 728–740 [DOI] [PubMed] [Google Scholar]
- 52.Walsh D. M., Selkoe D. J. (2007) J. Neurochem. 101, 1172–1184 [DOI] [PubMed] [Google Scholar]
- 53.Lesné S., Koh M. T., Kotilinek L., Kayed R., Glabe C. G., Yang A., Gallagher M., Ashe K. H. (2006) Nature 440, 352–357 [DOI] [PubMed] [Google Scholar]
- 54.Walsh D. M., Selkoe D. J. (2004) Neuron 44, 181–193 [DOI] [PubMed] [Google Scholar]
- 55.Cheng I. H., Scearce-Levie K., Legleiter J., Palop J. J., Gerstein H., Bien-Ly N., Puoliväli J., Lesné S., Ashe K. H., Muchowski P. J., Mucke L. (2007) J. Biol. Chem. 282, 23818–23828 [DOI] [PubMed] [Google Scholar]
- 56.Gong Y., Chang L., Viola K. L., Lacor P. N., Lambert M. P., Finch C. E., Krafft G. A., Klein W. L. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 10417–10422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kayed R., Head E., Thompson J. L., McIntire T. M., Milton S. C., Cotman C. W., Glabe C. G. (2003) Science 300, 486–489 [DOI] [PubMed] [Google Scholar]
- 58.Lacor P. N., Buniel M. C., Chang L., Fernandez S. J., Gong Y., Viola K. L., Lambert M. P., Velasco P. T., Bigio E. H., Finch C. E., Krafft G. A., Klein W. L. (2004) J. Neurosci. 24, 10191–10200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Townsend M., Shankar G. M., Mehta T., Walsh D. M., Selkoe D. J. (2006) J. Physiol. 572, 477–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cleary J. P., Walsh D. M., Hofmeister J. J., Shankar G. M., Kuskowski M. A., Selkoe D. J., Ashe K. H. (2005) Nat. Neurosci. 8, 79–84 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.