

Abstract
Upon activation, ERKs translocate from the cytoplasm to the nucleus. This process is required for the induction of many cellular responses, yet the molecular mechanisms that regulate ERK nuclear translocation are not fully understood. We have used a mouse embryo fibroblast ERK1-knock-out cell line expressing green fluorescent protein (GFP)-tagged ERK1 to probe the spatio-temporal regulation of ERK1. Real time fluorescence microscopy and fluorescence correlation spectroscopy revealed that ERK1 nuclear accumulation increased upon serum stimulation, but the mobility of the protein in the nucleus and cytoplasm remained unchanged. Dimerization of ERK has been proposed as a requirement for nuclear translocation. However, ERK1-Δ4, the mutant shown consistently to be dimerization-deficient in vitro, accumulated in the nucleus to the same level as wild type (WT), indicating that dimerization of ERK1 is not required for nuclear entry and retention. Consistent with this finding, energy migration Förster resonance energy transfer and fluorescence correlation spectroscopy measurements in living cells did not detect dimerization of GFP-ERK1-WT upon activation. In contrast, the kinetics of nuclear accumulation and phosphorylation of GFP-ERK1-Δ4 were slower than that of GFP-ERK1-WT. These results indicate that the differential shuttling behavior of the mutant is a consequence of delayed phosphorylation of ERK by MEK rather than dimerization. Our data demonstrate for the first time that a delay in cytoplasmic activation of ERK is directly translated into a delay in nuclear translocation.
Keywords: Cell/Compartmentation, Enzymes/Kinase, Methods/Fluorescence, Nucleus/Nuclear Import, Phosphorylation, Protein/Nuclear Translocation, Signal Transduction/Protein Kinases/MAP
Introduction
Stimulation of numerous cell surface receptors leads to activation of the Raf/MEK7/ERK signaling pathway. In this kinase cascade, Raf phosphorylates only MEK, and MEK phosphorylates only ERK, whereas ERK is able to phosphorylate many substrates in nearly all cell compartments (1). Noncatalytic activation of a few partners by c-Raf is well documented, but the biological outcomes of the ERK pathway are predominantly driven by the kinase activity, as evidenced, for example, by chemical inhibition (reviewed in Ref. 2). ERK is primarily located in the cytoplasm of resting cells, although overexpression results in cytoplasmic and nuclear localization (3). It has long been recognized that in the course of physiological signal transduction, ERK accumulates in the nucleus after acute stimulation of the cell (3, 4). Nuclear translocation of ERK is required for cell cycle entry. Thus, retention of ERK in the cytoplasm alters neither ERK kinase activity nor phosphorylation of cytoplasmic substrates, whereas ERK-dependent transcription and cell proliferation are blocked (5). It has been demonstrated that ERK phosphorylates the Phe-Gly nucleoporins Nup50, Nup153, and Nup154, reducing importin-β-mediated nucleocytoplasmic transport (6). This observation would expand the role of ERK nuclear entry to include the regulation of nucleocytoplasmic transport of certain classes of proteins while crossing the nucleopore.
MEK functions as the cytoplasmic anchor for ERK such that MEK co-overexpression maintains the cytoplasmic localization of overexpressed ERK, whereas saturating levels of the ERK-binding site of MEK abrogates ERK export from the nucleus (7). MEK is sequestered in the cytoplasm as a consequence of active export out of the nucleus mediated by its nuclear export sequence. MEK binds to inactive ERK in resting cells (8). The natural regulation of ERK translocation has also been demonstrated by differential expression of cytoplasmic anchors such as PEA15 (9, 10) or Sef (11) or expression of nuclear anchors such as DUSP5 (12) or Vanishing (13). It has been shown that the stimulation-induced nuclear accumulation of ERK requires the synthesis of short lived nuclear anchors (14). Clearly, regulation of ERK nuclear translocation is an essential feature of the Raf/MEK/ERK signaling cascade.
The precise mechanism of ERK transport across the nuclear pore is not fully understood. ERK lacks a nuclear localization sequence, leading to the suggestion that ERK may enter by a piggyback mechanism via binding to nuclear localization sequence-containing proteins (5). Nuclear localization sequence-dependent mechanisms require energy for Ran-dependent cycling of importins (15). However, reconstituted import assays have shown that ERK can bind directly to FXFG sequences of nucleoporin in the lumen of the nuclear pore complex, indicating that it may enter the nucleus in the absence of energy sources or cytosolic factors (16, 17). Point mutations of ERK revealed that inactive and active ERK interact with nucleoporins via different domains; thus, both active and inactive ERK can be transported across the nuclear pore in an energy-independent fashion (18). However, it has also been proposed that active transport of ERK may also occur and that it requires dimerization of the protein driven by phosphorylation of the TEY activation loop (19). Indeed, in a reconstituted import assay, thiophosphorylated ERK2 import increased in the presence of energy (20). Overall, these observations suggest a role for both an energy-dependent (presumably via dimerization) and an energy-independent mechanism (via direct binding to nucleoporins) in ERK cytoplasmic-nuclear translocation.
Surprisingly, the role of ERK dimerization in nucleocytoplasmic shuttling has proven to be controversial. Two distinct “dimerization mutants” have been used in several studies as follows: the mutation of histidine 176 to glutamic acid plus four leucines to alanines (H176E,L333A,L336A,L341A,L344A or H176E L4A mutant) or the removal of histidine 176 and three adjacent amino acids (deletion 174–177 or Δ4 mutant). Initially, both mutants were shown to not dimerize in vitro and to not accumulate in the nucleus (19), and later it was demonstrated that they remained monomeric in vitro at physiological salt concentrations (21). However, further studies using the H176E L4A mutant indicated its translocation to be normal (unless fused to β-galactosidase) (22, 23), although others found another replacement mutant, H176A L4A, to accumulate even in starved cells (24); this mutant without the glutamic charge was not tested in vitro for its capacity to dimerize. Moreover, dimerization could not be detected in live cells through FRET measurements between co-expressed yellow fluorescent protein-ERK and cyan fluorescent protein-ERK (23). Recently, it has been suggested that ERK dimerization plays a role in the activation of cytoplasmic substrates but not nuclear substrates (25). In parallel investigations focused on the nature and functional role(s) of ERK interactions with mitochondria, it was determined that dimerization of human ERK1 was favored in the mitochondria, also occurred in the nuclei, but was hardly detectable in the cytosol of HeLa cells (26). In view of these and other somewhat disparate assessments of ERK dimerization, we examined in detail the dimerization mutant ERK1-Δ4 because several studies have reported that the related mutation in ERK2 leads to abnormal nuclear translocation (19, 22, 27).
In attempts to retain a normal cytoplasmic localization of ERK transfected to high levels of expression, several studies have resorted to co-expression of MEK (22, 23, 27), although others have imaged cells expressing very low levels of ERK (28). ERK co-expressed with MEK generally displays an abnormally short persistence in the nucleus following stimulation, ranging from 10 to 40 min (23, 27, 29) instead of several hours in the case of endogenous ERK. In our studies, we used a mouse embryo fibroblast ERK1-knock-out cell line (MEFERK1−/− (30)). The lack of endogenous ERK1 allowed us to transfect MEFERK1−/− cells with GFP-ERK1 while maintaining the MEK-ERK balance and ensuring that every ERK1 in the cell is GFP-tagged.
Using this system, we examined ERK1 localization, cytoplasmic-nuclear translocation, and dimerization in live cells using fluorescence microscopy techniques. Our results demonstrate that the mutant displays delayed kinetics of nuclear entry/shuttling but no differences in overall nuclear accumulation. These real time measurements in ERK1 knock-out cells help to reconcile previous discrepancies between studies with ERK “dimerization mutants,” by emphasizing that the differences in mutant and WT ERK are subtle, and the ability to resolve these differences is dependent on the time scale of the measurements. We also found that mutation of the dimerization motif delayed ERK phosphorylation, suggesting that the mutant is less efficiently phosphorylated by MEK. Our study reveals that dimerization of ERK is not required for nuclear entry but rather that the efficient activation of ERK by MEK is the key determinant of rapid nuclear translocation.
EXPERIMENTAL PROCEDURES
Gene Construction and Sample Preparation
Full-length cDNA of human ERK1 subcloned into pcDNA3 vector (Invitrogen) was subjected to site-directed mutagenesis with the QuikChange site-directed mutagenesis kit (Stratagene) to remove the codons for amino acids Pro193–Asp196 and generate ERK1-Δ4. GFP-ERK1-WT and GFP-ERK1-Δ4 were generated by PCR cloning of human ERK1 or human ERK1-Δ4 into PCR2.1 TOPO (Invitrogen) followed by ligation into the XhoI and BamHI sites of pEGFP-C1 vector from Clontech.
Mouse embryo fibroblast ERK1 knock-out cells (MEFERK1−/− (30)) were cultured in DMEM + 10% fetal calf serum and were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Transient transfections were used within 48 h. For microscopy, cell monolayers were cultured in 8-well Lab-Tek chamber slides (Nunc, Rochester, NY). Cells were starved (0 or 0.1% serum) for 4–12 h. Live cell imaging was carried out in Tyrode's buffer with 20 mm glucose and 0.1% bovine serum albumin, and samples were maintained at 34–36 °C by an objective heater (Bioscience Tools).
For immunofluorescence, transfected cells were plated on coverslips and fixed with methanol at −20 °C at different time points after addition of 10% serum. Anti-phosphorylated ERK1/2 and goat anti-mouse-Cy3 secondary were from Sigma.
Confocal Microscopy
Fluorescence imaging and fluorescence recovery after photobleaching (FRAP) were performed using a Zeiss LSM510 confocal microscope with a 63× 1.2 NA water objective or 63× 1.4 NA oil objective. GFP was excited and bleached using a 488-nm laser line. Photobleaching of a region of interest was achieved by increasing the laser to 95% power. To determine the recovery time for the bleach experiments, images were background-subtracted, and post-bleach curves were fit to Equation 1,
where Io is an offset; A is the amplitude; t is time, and τ is the characteristic recovery time.
Live cell anisotropy measurements for detection of energy migration Förster resonance energy transfer (emFRET or homo-FRET) were carried out with a Zeiss LSM 510-META confocal laser scanning microscope incorporating a polarization beam splitter so as to simultaneously record the parallel (I‖) and perpendicular (I⊥) polarized emission signals. GFP was excited at 488 nm, and emission was collected using 505–530-nm bandpass filters. Images were acquired using a 63× 1.2 NA oil objective with a confocal slice thickness of 1 μm. Anisotropy (r) was calculated from r = (I‖ − GI⊥)/(I‖ + 2GI⊥), where G is the correction factor for the difference in the sensitivity of the two detectors. G = I‖/I⊥ was determined from a sample with r = 0 (in our case, fluorescein in aqueous solution). For more details, see Refs. 31, 32.
Fluorescence Correlation Spectroscopy
Fluorescence correlation spectroscopy (FCS) was performed with an inverted microscope (Eclipse TE200, Nikon) using the 488-nm line (narrow pass interference filter, CVI-Melles Griot) of an argon ion laser (Ion Laser Technology) as the excitation source. The beam intensity was stabilized with a laser amplitude stabilization system (Conoptics). Fluorescence emission was detected with a photon-counting avalanche photodiode (SPCM-AQR, PerkinElmer Life Sciences). Digital signal correlation was performed using a Flex02-03D auto/cross-correlator (Correlator). A 60×, 1.4 NA oil immersion objective (Zeiss) was used. Correlation data at times shorter than that corresponding to the laser stabilization feedback frequency (∼200 kHz) were discarded.
The beam waist, w0, measured using a variation of the knife-edge scan method (33), was 0.266 μm. The axial acceptance was determined to be 4 μm, by scanning a thin fluorescent film in z. FCS correlation curves were fit with MATLAB using the Gaussian acceptance approximation (34) shown in Equation 2,
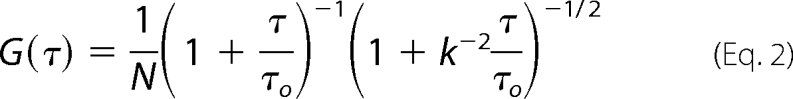 |
where N is the number of diffusing species in the sample volume; τo is the diffusion time, and k is a fixed parameter that accounts for the differing axial and lateral dimensions of the sample volume.
All measurements were performed with a laser irradiance estimated as ∼4 kilowatts/cm2 in the sample plane from measurements with an optical power meter (model S110, Thorlabs) and estimations of the image field. Increasing the laser intensity 2-fold had a negligible effect on τo, demonstrating that neither photophysical saturation nor photobleaching biased the measurements (see supplemental Table S1). This result was also consistent with prior reports of the photobleaching sensitivity of FCS (35, 36). The low illumination intensity used for FCS precluded the population of the enhanced GFP triplet state (37). Brightness (F/N), the average fluorescence intensity (F) divided by the number of molecules in the observation volume (N), a quantity determined from the fluorescence fluctuations, was used to monitor the aggregation state of ERK.
The number of molecules (N) per volume was used to calculate GFP-ERK concentration. Because the axial acceptance of the FCS measurements was larger than the height of the cells, we performed electron microscopy measurements of starved MEFERK1−/− cells, leading to estimates of nuclear and cytoplasmic thickness, 2.2 ± 0.4 and 1.7 ± 0.6 μm, respectively. The calculations of concentration were corrected accordingly.
Western Blot Phosphorylation Analysis
CCL39 (Chinese hamster lung cell) fibroblasts were stably transfected with pcDNA3-GFP-ERK1 or pcDNA3-GFP-ERK1-Δ4. After selection with 500 μg/ml G418 (Invitrogen), cells were grown 3 weeks to stabilize expression at low levels, and GFP-expressing cells were sorted with a FACS-ARIA (BD Biosciences). Cells were grown to confluency in DMEM incubated in 5% CO2 at 37 °C and then for 24 h in serum-free DMEM. Three hours prior to stimulation, cells were incubated at 37 °C in DMEM lacking sodium bicarbonate (catalogue no. 1280, Invitrogen) and supplemented with 25 mm Na-Hepes, pH 7.4. Cells were stimulated with a large volume to reach 10% serum final concentration, and were then washed rapidly in ice-cold phosphate-buffered saline prior to lysis and boiling in Laemmli sample buffer. Proteins from lysates were separated by SDS-PAGE (10% acrylamide/bisacrylamide 29:1 gels), and transferred onto polyvinylidene difluoride (Immobilon-P from Millipore). Phosphorylated ERK1/2 was detected with the monoclonal anti-phospho-ERK1/2 antibody M8159 from Sigma. Total ERK was detected with rabbit serum E1B 1:3000 (38). Secondary antibodies coupled to horseradish peroxidase were purchased from Promega, and chemiluminescence was used to detect immunoreactive bands on autoradiography film to obtain high resolution images and with the Gnome detector from Syngene (United Kingdom) to quantify the data.
Antibodies for Immunofluorescence Studies
Monoclonal anti-phospho-ERK was purchased from Sigma (M8159) and used at 1:500. Anti-ERK1 antibody R2 was purchased from Upstate Biotechnology, Inc. (catalogue number 06-182, immunopeptide consisting of the last 35 amino acids of rat ERK1), diluted 1:1000. Secondary antibodies were AlexaFluor-labeled anti-mouse or anti-rabbit from Molecular Probes.
RESULTS
ERK1 Localization
The study of ERK nuclear translocation has classically required fluorescent tagging of the transfected molecules and often the co-expression of MEK to avoid saturation of cytoplasmic anchors. Tagging can alter the mechanism of translocation, and in addition, the co-expression of MEK has often led to a shorter than expected duration of ERK nuclear localization (23, 27, 29). Therefore, we initially characterized ERK1 nuclear translocation using untagged ERK1 in MEFERK1−/− cells devoid of endogenous ERK1, presuming that the endogenous MEK was available to allow normal localization of transfected ERK. Furthermore, we compared translocation of ERK1-WT with that of the dimerization mutant ERK1-Δ4.
The supplemental Fig. S1 demonstrates that anti-ERK1 antibody (R2) bound efficiently to ERK1 in wild type MEF cells, whereas no signals were obtained in MEFERK1−/−. This is the first demonstration of an ERK1-specific antibody that can be used to study ERK1 subcellular localization without cross- labeling of ERK2. Furthermore, ERK1 transfected into MEFERK1−/− cells was detected readily with our anti-ERK1 R2 antibody (supplemental Fig. S2), and the time course of nuclear translocation closely mimicked that of endogenous ERK1 in WT MEF cells (data not shown). Indeed, transfected ERK1-WT could be detected in the nucleus within 5 min after stimulation, and nuclear accumulation persisted for several hours, still being maximal after 2 h of stimulation (supplemental Fig. S2). Surprisingly, ERK1-Δ4 behaved similarly to ERK1-WT. As shown in supplemental Fig. S2, at 10 min the ERK1-Δ4 was already localized in the nucleus, where it was present for at least 2 h, as in the case of ERK1-WT. To probe for differences between WT and mutant, we blocked protein synthesis prior to stimulation to impede synthesis of nuclear anchors. In the presence of cycloheximide, both ERK1-WT and ERK1-Δ4 failed to accumulate in the nucleus by 2 h post-stimulation (supplemental Fig. S3). These results demonstrate that in cells lacking endogenous ERK1, both ERK1-WT and ERK1-Δ4 behave like endogenous ERK1. The “snapshot” immunofluorescence studies did not reveal differences between the behavior of ERK1-WT and ERK1-Δ4. Considering that the differences may have been subtle, and therefore requiring finer temporal resolution, we resorted to GFP fusion proteins of ERK1 for real time microscopy studies.
To verify that adding the 27-kDa GFP to the 44-kDa ERK does not affect the properties of ERK1, we performed control experiments indicating that GFP-ERK1-WT mimicked the nucleocytoplasmic localization patterns of endogenous ERK. Fig. 1, upper row, shows that GFP-ERK1-WT localized to the cytosol in resting cells. As expected, serum addition triggered rapid nuclear translocation within 13 min, and GFP-ERK1-WT was retained in the nucleus for 2 h post-stimulation. This result was in agreement with those gained in experiments tracking endogenous ERK1 in wild type MEF cells and untagged ERK1 transfected in MEFERK1−/− (supplemental Fig. S2). At 3 h post-stimulation, GFP-ERK1-WT nuclear accumulation was markedly diminished (Fig. 1).
FIGURE 1.

Localization of GFP-ERK1 in MEFERK1−/− cells. MEFERK1−/− cells expressing GFP-ERK1 (top row, green in overlay) and endogenous ERK2 were fixed in methanol at −20 °C after starvation for 12 h (time 0) and at different time points after activation by addition of serum. Cells were stained for phosphorylated ERK1/2 (middle row, red in overlay).
We also evaluated the activation of total ERK by following the kinetics of dual phosphorylation by MEK (Fig. 1, 2nd row). Cells were starved for 12 h to avoid decreasing expression of transfected ERK; in these cells, the level of phosphorylated ERK was low in both transfected and untransfected cells (Fig. 1, 0 min, 2nd row). Hence, both endogenous ERK2 and GFP-ERK1-WT were maintained inactive in the cytoplasm in the starved cells. Upon stimulation, phosphorylated ERK increased markedly in all cells and accumulated rapidly in the nucleus (Fig. 1, 13 min, 2nd row). The higher signal in transfected cells at 13 and 60 min reveals that both the short and long term nuclear accumulation of phosphorylated GFP-ERK1-WT closely paralleled the localization of endogenous phospho-ERK2 in the nontransfected MEFERK1−/− cells. As reported previously (14), GFP-ERK1-WT was sequestered in the nucleus even after dephosphorylation had occurred (Fig. 1, 120 and 180 min, 1st row), as evidenced by the persistent nuclear GFP signal and loss of phospho-ERK (120 and 180 min, 2nd row). Collectively, these results indicate that the presence of the GFP tag did not alter either the subcellular localization or the activation patterns of ERK1.
Live Cell Studies of ERK1 Nuclear Accumulation
We studied the real time dynamics of GFP-ERK1 in living cells. Cells overexpressing GFP-ERK1 to a high degree showed the expected accumulation of GFP-ERK1 in the nucleus even in the starved state (as reported previously (3, 8, 28)). However, cells expressing at a physiologically relevant level demonstrated the characteristic retention of GFP-ERK1 in the cytoplasm of unstimulated cells (Fig. 2A and supplemental movie S1). Because the transient transfection resulted in a range of expression levels, we visually selected cells using the criterion that the nucleus be no brighter than the cytoplasm in the starved state. We verified that the expression level of GFP-ERK1 was physiologically relevant by using FCS to determine the upper limit of concentration considered, i.e. when the nucleus and cytoplasm displayed equal intensity in the resting condition. In this case, the GFP-ERK1 concentration was 0.82 ± 0.4 μm in the cytoplasm and 0.77 ± 0.2 μm in the nucleus, values still within endogenous expression levels (39).
FIGURE 2.

Kinetics of GFP-human ERK1 nuclear accumulation. A, before (0 s) stimulation (cell starved for 12 h), GFP-ERK1-WT (gray) is retained in the cytoplasm and shows the characteristic dark nucleus. B, after stimulation (at 339 s), GFP-ERK1-WT accumulates in the nucleus. C, GFP intensity in the nucleus normalized to initial value as a function of time after serum stimulation. The curves were fit to a sigmoidal function (solid gray line) to extract the half-time to saturation (t½) and the steady-state saturation level (A).
Upon addition of 10% serum, GFP-ERK1 accumulated rapidly in the nucleus (Fig. 2B, supplemental movie 1). Real time measurements of GFP-ERK1 nuclear translocation allowed the quantification of two characteristic parameters (Fig. 2C) as follows: the stimulated steady-state nuclear concentration of GFP-ERK1 relative to the resting concentration (A), and the time after stimulation at which the concentration had increased to half of the maximum (t½ or “half-time”). These real time measurements revealed that in response to activation, mutant ERK1 accumulated in the nucleus to the same extent as the WT protein (Fig. 3A). However, the t½ for nuclear accumulation of GFP-ERK-Δ4 was nearly double that of WT (Fig. 3B). Free GFP was shown to distribute in both the cytoplasm and nucleus, and its subcellular localization did not change upon stimulation (data not shown). The lack of a response of free GFP to stimulation verified that the observed GFP-ERK results were physiologically relevant and that the slower rate of the mutant to reach steady state was specific to ERK function. Nuclear translocation of GFP-ERK1 mimicked endogenous ERK also because leptomycin B (LMB) treatment of resting cells induced the expected nuclear accumulation of GFP-ERK1, although accumulation was to a lower level than when cells were serum-stimulated (data not shown). LMB induces nuclear accumulation in the absence of stimulation by inhibiting the nuclear export protein CRM1 and thereby reducing active nuclear export of MEK·ERK complexes (8).
FIGURE 3.

Characterization of nuclear ERK1 accumulation. Comparison of accumulation levels (A) and t½ (B) for GFP-ERK1-WT and GFP-ERK1-Δ4. Nuclear accumulation was induced by addition of 10% serum.
To determine whether the rate of shuttling of ERK into the nucleus changes with activation, we used a FRAP protocol that bleached the entire nucleus and monitored the recovery of the nuclear GFP intensity as a result of equilibration by exchange between unbleached cytoplasmic and photobleached nuclear GFP-ERK1 (Fig. 4). Both the WT and mutant ERK were slower to recover than free GFP after photobleaching the nucleus in resting cells. This result could reflect sterically hindered diffusion of ERK across the nucleopore or ERK retention in the cytosol by binding protein complexes, or a combination of both processes. During stimulation, the free GFP recovery time was unchanged; however, the GFP-ERK1-WT recovery time was faster than resting, consistent with observations by Costa et al. (28). Similarly, Ando et al. (40) have demonstrated that cell stimulation enhances the rates of ERK nuclear import and export. Whereas GFP-ERK1-Δ4 also displayed an increase in shuttling time during stimulation, it was still slower than for the WT protein (higher τ value). The slower exchange kinetics were consistent with the slower nuclear accumulation of the mutant upon stimulation of resting cells.
FIGURE 4.

FRAP measurements with photobleaching of the entire nucleus to measure nuclear exchange. Comparison of GFP recovery after photobleaching the nucleus for free GFP, GFP-ERK1-WT, and GFP-ERK1-Δ4. In both starved and stimulated state, WT recovery is faster than mutant.
ERK1 Diffusion Is Not Changed by Activation
Using FCS, we determined diffusion coefficients of the ERK constructs in both the cytoplasm and the nucleus, before and after activation. No evidence for systematic changes in diffusion was seen after stimulation, and no significant differences between WT and mutant were detected (Table 1). This result indicates that the acceleration of the nucleocytoplasmic rate of shuttling upon stimulation was not a result of increased mobility in a given compartment. Mobile (free) GFP-ERK1 diffused at a rate one-third that of free GFP. From hydrodynamic considerations, GFP-ERK should have a diffusion constants 60–70% that of free GFP, on the basis of their molecular masses and assuming ellipsoidal molecules with axial ratios not exceeding ∼4 (41). Thus, the slower diffusion of GFP-ERK likely reflects interactions with nuclear and cytoplasmic partner proteins.
TABLE 1.
Results from FCS measurements
Values are given as average ± S.E. “Starved” indicates measurements before serum addition, and “+serum” data were acquired in the cytoplasm from 1 to 10 min after serum addition and in the nucleus from 10 to 15 min. F/N is fluorescence intensity per mobile object (see under “Experimental Procedures”).
| F/N | D | N | |
|---|---|---|---|
| counts/object/s | μm2/s | ||
| GFP cytoplasm | 319 ± 10 | 17.5 ± 1.3 | 7 |
| GFP nucleus | 308 ± 8 | 16.1 ± 0.7 | 18 |
| WT cytoplasm starved | 316 ± 8 | 7.7 ± 0.4 | 31 |
| WT cytoplasm +serum | 308 ± 5 | 6.6 ± 0.3 | 49 |
| WT nucleus starved | 283 ± 14 | 7.1 ± 0.8 | 13 |
| WT nucleus +serum | 299 ± 11 | 6.8 ± 0.6 | 19 |
| Δ4 cytoplasm starved | 346 ± 8 | 6.4 ± 0.4 | 16 |
| Δ4 cytoplasm +serum | 309 ± 9 | 6.7 ± 0.4 | 9 |
| Δ4 nucleus starved | 296 ± 13 | 5.2 ± 0.4 | 17 |
| Δ4 nucleus +serum | 278 ± 10 | 5.7 ± 0.4 | 20 |
Sequestration of ERK in the nucleus has been reported (13, 14), leading to the suggestion that one or more “nuclear anchor” proteins retain ERK in the nucleus. The diffusion coefficient in the nucleus after stimulation did not diminish (Table 1). However, immobile proteins do not contribute to the fluctuation signal and thus do not influence the determination of diffusion constants by FCS. To determine whether ERK1 can bind to anchors that immobilize it in the nucleus, we used a FRAP protocol that involved photobleaching the entire cytoplasm and half of the nucleus, thus allowing a determination of intranuclear GFP-ERK1 redistribution (supplemental movie 2). Fig. 5A shows a typical result from such a photobleaching series. From these experiments, it is clear that ERK1 was not immobilized in the nucleus but rather diffused freely and re-equilibrated within the nucleus in seconds. Both wild type and mutant ERK1 exhibited this property in the resting and activated states, with no reduction in mobility after stimulation (Fig. 5B). Control FRAP experiments showed that free GFP is more mobile than GFP-ERK1 (in qualitative agreement with FCS), as expected because it is smaller and does not interact with binding partners. We conclude that ERK1 either binds only transiently to nuclear targets or that the complexes formed by ERK and nuclear targets are inherently highly mobile.
FIGURE 5.

ERK1 redistributed rapidly in the nucleus. A, sample trace from photobleaching protocol that photobleached the entire cytoplasm and half the nucleus. GFP-ERK1-WT from the unbleached region (black) quickly redistributed throughout the nucleus, filling in the bleached region (red). B, redistribution times before (in starved cells, t = 0) and after activation with 10% serum. Activated GFP-ERK1-WT (black squares) had a similar redistribution time to nonactivated ERK1 that accumulated in the nucleus due to LMB treatment (red circles at t = 0). No significant change in the ability of GFP-ERK1 to redistribute was observed after serum stimulation. Note that GFP redistribution was so rapid that measurements had to be taken at room temperature, and all GFP-ERK1 measurements were at 30 °C. Measurements of GFP-ERK1 at 35 °C exhibited similar redistribution times (data not shown). Each point represents a measurement from a single cell.
ERK1 Dimerization Is Not Detected in Living Cells
Based on the ability of GFP-ERK1-Δ4 to accumulate in the nucleus, we hypothesized that dimerization is not required for nuclear translocation. Despite the evidence for dimerization from in vitro measurements (19, 21), others have failed to detect dimerization via FRET measurements between cyan fluorescent protein-ERK and yellow fluorescent protein-ERK (23) in living cells. We performed further experiments using emFRET and FCS to confirm that homodimerization was not occurring. MEFERK1−/− cells transfected with GFP-ERK1 were ideal for these studies because all the ERK1 is GFP-tagged, such that even a small percentage of ERK dimers should have been detectable.
FCS experiments were performed with both GFP-ERK1-WT and GFP-ERK1-Δ4. To search for evidence of dimerization, the number of independent mobile objects (N) in the focal volume was determined by FCS, and the normalized fluorescence intensity (F/N) was calculated. Dimerization of ERK would cause an increase in F/N, because the number of diffusing objects would be reduced upon dimerization, whereas the total fluorescence intensity would remain the same. This method was validated using fluorescein-labeled DNA and comparing F/N values for single-stranded and double-stranded DNA. The control experiments yielded the expected ratio of 2 (2.05 ± 0.06) for double/single-labeled molecules (see “Experimental Procedures” and supplemental Table S2). In live cells, no change in the dimerization state (invariant F/N values) of GFP-ERK1 was detected after stimulation with serum (Fig. 6). GFP-ERK1-WT (Fig. 6A) and GFP-ERK1-Δ4 (Fig. 6B) showed the same behavior. In addition, the F/N values for the ERK constructs did not differ significantly from the value for free monomeric GFP expressed in MEFERK1−/− cells (Table 1).
FIGURE 6.

Dimerization was not detected by FCS or emFRET. FCS was used to calculate the fluorescence intensity per mobile object (F/N). No significant change in F/N was observed after stimulation for either WT (A) or mutant ERK1 (B), indicating that ERK1 remained monomeric after activation. Each symbol after activation indicates a single measurement. Connected symbols are measurements from the same cell over time. Values are normalized to the F/N value before stimulation and absolute values are found in Table 1. The gray band represents the single cell standard deviation of prestimulation (t = 0) measurements. C, emFRET measurements in a live cell expressing GFP-ERK1-WT during activation. After serum stimulation, GFP-ERK-WT accumulates in the nucleus (blue line); however, the anisotropy values in both the cytoplasm (black) and nucleus (red) remained unchanged.
In agreement with the FCS determinations, emFRET measurements also failed to detect dimerization of GFP-ERK1 in the cell cytoplasm upon stimulation. emFRET between like fluorophores does not result in changes in the ensemble fluorescence intensity or lifetime. However, it leads to depolarization of the emission because of the lack of correlation between the orientation of the secondarily excited molecule (the “acceptor”) and that of the initially photoselected “donor” (32, 42). Thus, the formation of protein·protein complexes that bring individual GFPs within the emFRET range (<8 nm) can be determined by measurement of the steady-state anisotropy. Note that the rotation of the protein plays essentially no role in depolarization, as the rotational time scale (for any GFP fusion protein) is much longer than the fluorescence lifetime. Thus, binding of the target protein to unlabeled partners will not alter the emFRET signal.
Fig. 6C features anisotropy measurements in a live MEFERK1−/− cell expressing GFP-ERK1-WT. After stimulation with serum, the intensity in the nucleus (Fig. 6C, blue line) increased as GFP-ERK1-WT translocated, yet no change in anisotropy (red and black lines) was detected. We conclude that ERK1 remained monomeric. Anisotropy measurements of unstimulated GFP-ERK1-WT sequestered in the nucleus after LMB treatment or serum-stimulated GFP-ERK1-Δ4 produced similar results (data not shown). The absence of ERK1 homodimerization is consistent with previous ERK2 hetero-FRET measurements (23).
Phosphorylation Kinetics Are Different for Wild Type and Mutant ERK
The inability to detect ERK1 homodimers established that dimerization could not account for the observed differences in nuclear accumulation kinetics between the WT and Δ4 mutant (Fig. 3B). Because the mutant accumulated to the same extent as ERK1-WT (Fig. 3A), we could also rule out gross structural modification as the mechanism. Furthermore, even though the mutant was consistently slower than WT at entering the nucleus, its nuclear entry was still accelerated upon stimulation (Fig. 4). We therefore examined more closely the kinetics of activation of the two proteins, measured by their double phosphorylation by MEK on the TEY sequence. Only a minor proportion of transiently transfected MEF cells expressed GFP-ERKs, and thus the kinetics were compared in stably transfected CCL39 fibroblasts. After selection, cells were cultivated in exponential conditions for about 3 weeks to generate a moderate, stable level of expression and then sorted by fluorescence-activated cell sorter. More than 1000 clones initially resistant to geneticin were combined, thereby averaging out possible differences between individual clonal lines. Starved cells were stimulated for a time course encompassing that of nuclear entry of ERK and phosphorylation quantified by Western blotting. Fig. 7A shows the kinetics of ERK phosphorylation in cells stably transfected with either GFP-ERK1-WT or GFP-ERK1-Δ4. The endogenous ERK in each cell population displayed nearly identical kinetics of ERK phosphorylation, confirming that the samples had been treated equally, and the differences between WT and Δ4 were physiologically relevant (Fig. 7A). GFP-ERK1-WT showed a nearly 2-fold faster phosphorylation kinetics (half-time 1.8 ± 0.03 min) than GFP-ERK1-Δ4 (3.0 ± 0.02 min). Three independent experiments indicated reproducibly that the GFP-ERK1-Δ4 was activated more slowly than GFP-ERK1-WT. The nearly 2-fold delay in phosphorylation correlated well with the nearly equally retarded nuclear accumulation of GFP-ERK1-Δ4.
FIGURE 7.

Phosphorylation kinetics were different for ERK1-WT and ERK1-Δ4. A, plot of phosphorylation over time. Solid lines (cells transfected with mutant ERK) and hatched lines (cells transfected with wild type ERK) are sigmoidal fits to the data to determine the t½ values ± 95% confidence interval (see text). Each trace was normalized to the fit maximum. B, relative phosphorylation levels of ERK1 in cells stimulated for 15 min and then treated with PD184359, blocking MEK activation and allowing the dephosphorylation of ERK.
In principle, a slower phosphorylation of mutant ERK could be caused by slower MEK activation or perhaps by a general defect of the mutant in its interactions with partners. Because MEK and phosphatases bind to ERK via the same docking site (43), we determined the kinetics of ERK dephosphorylation after chemical inhibition of MEK. Endogenous ERK, GFP-ERK1-WT, and GFP-ERK1-Δ4 dephosphorylated within 3 min and at similar rates after blocking MEK activation with PD184359 (Fig. 7B). The same result was obtained when the cells were stimulated 2 h prior to addition of the MEK inhibitor (data not shown). We conclude that ERK1-Δ4 was dephosphorylated by endogenous phosphatases as efficiently as the wild type protein, implying that the observed differences in nuclear accumulation kinetics between wild type and Δ4 were likely caused by slower MEK phosphorylation, rather than by global changes in the interactions with partners.
DISCUSSION
The outcomes of signaling via the mitogen-activated protein kinase pathway are determined by the regulation of ERK nuclear translocation (5, 10, 44), which is required for activation of many transcription factors. The duration and extent of translocation is dependent on the type of stimulus, and ERK nuclear translocation is required to induce specific responses in the form of gene expression (45). In some cases stimulus-dependent regulation of nuclear translocation may be linked to distinct elevation of calcium concentration triggered by agonists (46). Proteins that regulate ERK nuclear translocation have been identified, including PEA15, which anchors ERK in the cytosol (9) in part by inhibiting the capacity of ERK to bind to nucleoporins (47), and Sef, which blocks the dissociation of MEK·ERK complexes (11).
Because of the rapid nature of ERK nuclear translocation, we fluorescently tagged ERK to study its shuttling dynamics in living cells by quantitative microscopy. To ensure that expression of GFP-ERK1 did not overwhelm the system, we expressed GFP-ERK1 in MEKERK1−/− cells, thus avoiding saturation of ERK1/2 partners. The average concentration of GFP-ERK1 in our experiments was on the order of 1 μm, a value similar to the endogenous levels of total ERK reported recently by Fujioka et al. (39) for HeLa and Cos cells (0.96 and 0.81 μm, respectively.) These authors reviewed the results of other studies, two reporting low concentrations (0.26 and 0.36 μm) and four concentrations ranging from 1 to 2.1 μm. In further confirmation of the physiological relevance of our system, we observed rapid nuclear translocation of GFP-ERK1 within minutes of serum addition. As shown previously for endogenous ERK (14), nuclear localization of GFP-ERK1 lasted for several hours, and it was retained in the nucleus after dephosphorylation. This observation confirms that inactive and active ERK can be sequestered in the nucleus, presumably via binding to nuclear anchoring proteins (14).
Nuclear anchoring, however, is not accompanied by immobilization. GFP-ERK1 was found to be highly mobile, and its mobility did not depend on activation state, indicating that ERK binding partners are also mobile or that interactions are transient. FCS measurements of diffusion constants (Table 1) and FRAP studies showed that GFP-ERK1 redistributed within seconds throughout the nucleus (Fig. 5). The protein mobility was similar for unstimulated and stimulated cells (15 min to 2 h post-stimulation). Free GFP diffusion (Table 1) and nuclear redistribution (Fig. 5) were much faster than for GFP-ERK1, presumably reflecting the smaller size and lack of binding partners. The FCS and emFRET measurements did not detect changes in the aggregation state of ERK1 upon activation in either compartment (Fig. 6 and Table 1), indicating that ERK does not homodimerize before or during nuclear translocation.
By blocking CRM1-dependent nuclear export, accumulation of unstimulated ERK was observed (see Refs. 7, 14 and our data not shown), indicating that the localization of ERK and ERK complexes is actively regulated. Constant shuttling of ERK was further demonstrated by FRAP experiments in resting cells, in which the nuclear pool of GFP-ERK1 recovered after photobleaching the entire nucleus (Fig. 4). Stimulation of the cells led to an increase in nuclear recovery of GFP-ERK1, consistent with previous observations (28) and in support of previous models in which ERK is released from a cytoplasmic anchor upon activation, allowing rapid nuclear accumulation (22). Together with data demonstrating that ERK nuclear entry does not require energy (16, 17), our results suggest that upon release from cytoplasmic anchoring after activation, ERK diffuses to the nucleus while being highly mobile in both compartments at all times.
To gain more insight into the mechanism of ERK nuclear translocation, we examined the behavior of the only ERK mutant shown to display consistently altered nuclear translocation, ERK1-Δ4. To our surprise, ERK1-Δ4 accumulated in the nucleus to the same level as ERK1-WT. This result was observed when nontagged ERK1-Δ4 was expressed in cells lacking endogenous ERK1, and it became obvious following statistical measurements of time-lapse studies of GFP-ERK1-Δ4 serum-induced nuclear translocation. In unstimulated cells, ERK1-Δ4 diffusion in both compartments was similar to that of WT, consistent with the observation that the unphosphorylated form of the other dimerization mutant, ERK2-H176E L4A, translocated normally across the nucleopore in reconstituted assays comparing active and inactive transport (18). FRAP of the nucleus also revealed that GFP-ERK1-Δ4 shuttling was accelerated after stimulation but was not as rapid as GFP-ERK1-WT. Abnormal folding could not explain this phenomenon because the kinase activity of ERK2-Δ4 has been found to be in the range of ERK2 (21).
The dynamics and extent of nuclear accumulation of ERK may reflect import or export rates or both. Indeed, nuclear accumulation can be driven by changes in export alone as demonstrated by treatment of resting cells with LMB. The observation that ERK-Δ4 eventually accumulates to the same extent as ERK-WT implies that, at steady state, the ratio of import to export rates is unaffected by the mutation, provided the mechanisms of import and export are unchanged. The slower accumulation of the mutant could arise from slowed kinetics (import and export) or reflect a delay in the conversion of ERK to an “import-eligible” form, i.e. a delayed phosphorylation. Nuclear exchange measurements indicate a contribution from the former, whereas the sigmoidal shape of the accumulation curves strongly implies the existence of an activation delay as well.
The fact that the mobilities of WT and mutant ERK1 were similar in the resting state, despite a delay in nuclear accumulation subsequent to stimulation, led us to hypothesize that the defect in mutant behavior should be upstream of translocation. In accordance with this supposition, we found that ERK1-Δ4 was phosphorylated more slowly than ERK1-WT and evaluated by double phosphorylation on the TEY activating loop (Fig. 7A). This delay was missed in previous experiments that suggested normal activation of the ERK2 mutant (H176A L4A) in an end point immunoblot assay (5 min of 12-O-tetradecanoylphorbol-13-acetate treatment) (24). The difference between ERK1-Δ4 and ERK1-WT was specific to activation because upon chemical inhibition of MEK, ERK1-Δ4 was dephosphorylated as rapidly as WT (Fig. 7B). This normal dephosphorylation is consistent with the observation that the ERK2 mutant H176A L4A bound normally to the phosphatase MKP3-CS (determined via immunolocalization studies) (24). Abnormal activation is also consistent with failure of ERK2-H176A L4A to co-immunoprecipitate normally with MEK1 (24). Our data suggest a close correlation between activation of ERK and the rate of nuclear accumulation. We conclude that the activation (double phosphorylation) of ERK constitutes the trigger of rapid nuclear translocation.
It has been demonstrated that at the onset of stimulation ERK is released from MEK (8) and one can expect that a delay in activation leads to a delayed release of ERK. This delayed release would then lead to slower nuclear accumulation because MEK and inactive ERK complexes are retained in the cytosol via the nuclear export sequence of MEK (48). We interpret the delay of shuttling during stimulation as a consequence of the slower re-activation of inactive ERK in complexes with MEK. Indeed, the turnover of ERK activation is very fast, as demonstrated by the total extinction of phospho-ERK within 3 min after blocking MEK activation (Fig. 7B). Furthermore, it has been demonstrated that at peak stimulation only 5% of MEK molecules are active, whereas up to 60% of ERK molecules are active (39). Hence, rapidly inactivated ERK molecules are trapped instantly by the large pool of inactive MEK inside the cytoplasm, and inefficient activation of ERK1-Δ4 would then retard further release from MEK. Our data demonstrate for the first time that a delay in cytoplasmic activation of ERK is immediately translated into a delay of nuclear translocation, highlighting that the constant shuttling of ERK is linked to a rapid turnover of activation. This constant exchange of ERK, regulated by interactions with binding partners, allows nuclear ERK responses to act as immediate sensors of signal strength.
Recently, wild type ERK has been shown to migrate as an ∼80-kDa complex, whereas the ERK dimer mutant H176E L4A did not form an 80-kDa complex or co-precipitate with MEK (25, 49). Surprisingly, the mutant also prevented endogenous ERK from forming the complex. These results were interpreted as demonstrating the formation of homodimers. It is important to note that the presence of cytoplasmic extracts was required to form the 80-kDa complex and that it was observed only after addition of the reducing agent β-mercaptoethanol to reduce higher molecular weight complexes (49). Therefore, although it is clear that the dimerization mutant is impeded in its ability to form high molecular weight complexes, it is unclear whether the 80-kDa complexes represent ERK homodimers. Indeed, prior to our present work, the existence of ERK dimers in vivo was already questioned due to the lack of hetero-FRET between ERKs fused to different fluorescent proteins (23). Interestingly, dimerization in vitro was demonstrated to occur best at an osmolarity higher than that of living cells, likely because of the hydrophobic nature of the interface (21). We have not detected any differences in aggregation state between ERK1-WT and ERK1-Δ4, using both FCS and emFRET measurements in live cells. Our experimental conditions were optimal because all ERK1 molecules expressed were tagged with GFP, and the concentration of GFP-ERKs was in the recently determined range of endogenous ERK expression (39). Our results demonstrate that ERK1-Δ4 activation is less efficient than that of WT ERK, possibly due to abnormal formation of the scaffold complex (24, 25). The ability of low levels of ERK dimerization mutants to displace endogenous ERK from scaffolding proteins such as KSR1 (25) may indicate a greater affinity of the mutants leading to a decrease in the efficiency of activation by MEK.
It was reported recently that phosphorylation of the SPS motif of the kinase insert of ERK is necessary for ERK nuclear translocation (50). Phosphorylation of this SPS motif was shown to be uncoupled from the activating phosphorylation of the TEY motif, precluding an involvement of the SPS in the phenomenon described in this study. However, both mutants share problems with activation as follows: mutation of SPS reduces ERK activation markedly, whereas the Δ4 mutation delays ERK activation. Furthermore, SPS phosphorylation was shown to accelerate movement across the nucleopore (50), whereas ERK dimerization mutants translocate across the nucleopore normally (18).
Our study demonstrates that although ERK is localized to cellular compartments, it is not immobilized. We conclude that the localization of ERK is dictated by the abundance of and affinity for anchoring proteins to which ERK binds. Furthermore, although ERK constantly shuttles between the cytoplasm and nucleus, nuclear exchange increases upon activation. The efficient activation of ERK leads to its release from cytoplasmic anchoring proteins and to rapid nuclear accumulation. Such a regulation of MEK-ERK coupling controlling signal transmission to the nucleus would potentiate a function of nuclear ERK activity in the discrimination of external signal strength.
Acknowledgments
The University of New Mexico Cancer Center Fluorescence Microscopy Facility received support from National Institutes of Health Grants S10 RR14668, S10 RR19287, S10 RR016918, P20 RR11830, and P30 CA118100 and from National Science Foundation Grant MCB9982161. We thank Sheli Ryan for cell culture assistance, Mary Raymond-Stintz for electron microscopy, Marta Vuckovic and Michael Schmidt for assistance with transfection and imaging, and Rainer Heintzmann for helpful discussion. We thank Deborah Rousseau and Jean-Claude Chambard for invaluable help with cell sorting.
This work was supported by European Union FP5 Project QLRT-2000-02278 (MAP Kinase) (to T. M. J. and J. P.), the CNRS, Centre A. Lacassagne, Association pour la Recherche sur le Cancer Contract 3338, and American Cancer Society Grant IRG 192 (to D. S. L.). This work was supported in part by National Institutes of Health Grant DK34128 (to J. W.).
This article was selected as a Paper of the Week.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and S2, Figs. S1–S3, and Movies 1 and 2.
- MEK
- mitogen-activated protein kinase/ERK kinase
- FCS
- fluorescence correlation spectroscopy
- emFRET
- energy migration Förster resonance energy transfer
- FRAP
- fluorescence recovery after photobleaching
- F/N
- fluorescence intensity per mobile object
- ERK
- extracellular signal-regulated kinase
- GFP
- green fluorescent protein
- WT
- wild type
- ERK-Δ4
- human ERK1 deleted from Pro193–Asp196
- MEF
- mouse embryo fibroblasts
- LMB
- leptomycin B
- DMEM
- Dulbecco's modified Eagle's medium.
REFERENCES
- 1.Yoon S., Seger R. (2006) Growth Factors 24, 21–44 [DOI] [PubMed] [Google Scholar]
- 2.Kohno M., Pouyssegur J. (2006) Ann. Med. 38, 200–211 [DOI] [PubMed] [Google Scholar]
- 3.Lenormand P., Sardet C., Pagès G., L'Allemain G., Brunet A., Pouysségur J. (1993) J. Cell Biol. 122, 1079–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen R. H., Sarnecki C., Blenis J. (1992) Mol. Cell. Biol. 12, 915–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brunet A., Roux D., Lenormand P., Dowd S., Keyse S., Pouysségur J. (1999) EMBO J. 18, 664–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kosako H., Yamaguchi N., Aranami C., Ushiyama M., Kose S., Imamoto N., Taniguchi H., Nishida E., Hattori S. (2009) Nat. Struct. Mol. Biol. 16, 1026–1035 [DOI] [PubMed] [Google Scholar]
- 7.Adachi M., Fukuda M., Nishida E. (2000) J. Cell Biol. 148, 849–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fukuda M., Gotoh Y., Nishida E. (1997) EMBO J. 16, 1901–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Formstecher E., Ramos J. W., Fauquet M., Calderwood D. A., Hsieh J. C., Canton B., Nguyen X. T., Barnier J. V., Camonis J., Ginsberg M. H., Chneiweiss H. (2001) Dev. Cell 1, 239–250 [DOI] [PubMed] [Google Scholar]
- 10.Gervais M., Dugourd C., Muller L., Ardidie C., Canton B., Loviconi L., Corvol P., Chneiweiss H., Monnot C. (2006) Mol. Biol. Cell 17, 3940–3951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Torii S., Kusakabe M., Yamamoto T., Maekawa M., Nishida E. (2004) Dev. Cell 7, 33–44 [DOI] [PubMed] [Google Scholar]
- 12.Mandl M., Slack D. N., Keyse S. M. (2005) Mol. Cell. Biol. 25, 1830–1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sur R., Ramos J. W. (2005) Biochem. J. 387, 315–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Volmat V., Camps M., Arkinstall S., Pouysségur J., Lenormand P. (2001) J. Cell Sci. 114, 3433–3443 [DOI] [PubMed] [Google Scholar]
- 15.Terry L. J., Shows E. B., Wente S. R. (2007) Science 318, 1412–1416 [DOI] [PubMed] [Google Scholar]
- 16.Matsubayashi Y., Fukuda M., Nishida E. (2001) J. Biol. Chem. 276, 41755–41760 [DOI] [PubMed] [Google Scholar]
- 17.Whitehurst A. W., Wilsbacher J. L., You Y., Luby-Phelps K., Moore M. S., Cobb M. H. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 7496–7501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yazicioglu M. N., Goad D. L., Ranganathan A., Whitehurst A. W., Goldsmith E. J., Cobb M. H. (2007) J. Biol. Chem. 282, 28759–28767 [DOI] [PubMed] [Google Scholar]
- 19.Khokhlatchev A. V., Canagarajah B., Wilsbacher J., Robinson M., Atkinson M., Goldsmith E., Cobb M. H. (1998) Cell 93, 605–615 [DOI] [PubMed] [Google Scholar]
- 20.Ranganathan A., Yazicioglu M. N., Cobb M. H. (2006) J. Biol. Chem. 281, 15645–15652 [DOI] [PubMed] [Google Scholar]
- 21.Wilsbacher J. L., Juang Y. C., Khokhlatchev A. V., Gallagher E., Binns D., Goldsmith E. J., Cobb M. H. (2006) Biochemistry 45, 13175–13182 [DOI] [PubMed] [Google Scholar]
- 22.Adachi M., Fukuda M., Nishida E. (1999) EMBO J. 18, 5347–5358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burack W. R., Shaw A. S. (2005) J. Biol. Chem. 280, 3832–3837 [DOI] [PubMed] [Google Scholar]
- 24.Wolf I., Rubinfeld H., Yoon S., Marmor G., Hanoch T., Seger R. (2001) J. Biol. Chem. 276, 24490–24497 [DOI] [PubMed] [Google Scholar]
- 25.Casar B., Pinto A., Crespo P. (2008) Mol. Cell 31, 708–721 [DOI] [PubMed] [Google Scholar]
- 26.Galli S., Jahn O., Hitt R., Hesse D., Opitz L., Plessmann U., Urlaub H., Poderoso J. J., Jares-Erijman E. A., Jovin T. M. (2009) PLoS ONE 4, e7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horgan A. M., Stork P. J. (2003) Exp. Cell Res. 285, 208–220 [DOI] [PubMed] [Google Scholar]
- 28.Costa M., Marchi M., Cardarelli F., Roy A., Beltram F., Maffei L., Ratto G. M. (2006) J. Cell Sci. 119, 4952–4963 [DOI] [PubMed] [Google Scholar]
- 29.Furuno T., Hirashima N., Onizawa S., Sagiya N., Nakanishi M. (2001) J. Immunol. 166, 4416–4421 [DOI] [PubMed] [Google Scholar]
- 30.Pagès G., Guérin S., Grall D., Bonino F., Smith A., Anjuere F., Auberger P., Pouysségur J. (1999) Science 286, 1374–1377 [DOI] [PubMed] [Google Scholar]
- 31.Lidke D. S., Nagy P., Barisas B. G., Heintzmann R., Post J. N., Lidke K. A., Clayton A. H., Arndt-Jovin D. J., Jovin T. M. (2003) Biochem. Soc. Trans. 31, 1020–1027 [DOI] [PubMed] [Google Scholar]
- 32.Lidke K. A., Rieger B., Lidke D. S., Jovin T. M. (2005) IEEE Trans. Image Process 14, 1237–1245 [DOI] [PubMed] [Google Scholar]
- 33.Schneider M. B., Webb W. W. (1981) Appl. Optics 20, 1382–1388 [DOI] [PubMed] [Google Scholar]
- 34.Elson E. L., Magde D. (1974) Biopolymers 13, 1–27 [DOI] [PubMed] [Google Scholar]
- 35.Widengren J., Rigler R. (1998) Cell. Mol. Biol. 44, 857–879 [PubMed] [Google Scholar]
- 36.Dittrich P., Malvezzi-Campeggi F., Jahnz M., Schwille P. (2001) Biol. Chem. 382, 491–494 [DOI] [PubMed] [Google Scholar]
- 37.Haupts U., Haupts C., Oesterhelt D. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 3834–3838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lefloch R., Pouysségur J., Lenormand P. (2008) Mol. Cell. Biol. 28, 511–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fujioka A., Terai K., Itoh R. E., Aoki K., Nakamura T., Kuroda S., Nishida E., Matsuda M. (2006) J. Biol. Chem. 281, 8917–8926 [DOI] [PubMed] [Google Scholar]
- 40.Ando R., Mizuno H., Miyawaki A. (2004) Science 306, 1370–1373 [DOI] [PubMed] [Google Scholar]
- 41.Cantor C., Schimmel P. (1980) Biophysical Chemistry Part 2: Techniques for the Study of Biological Structure and Function, Vol. 2, pp. 560–585, W. H. Freeman and Co., San Francisco [Google Scholar]
- 42.Gautier I., Tramier M., Durieux C., Coppey J., Pansu R. B., Nicolas J. C., Kemnitz K., Coppey-Moisan M. (2001) Biophys. J. 80, 3000–3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanoue T., Adachi M., Moriguchi T., Nishida E. (2000) Nat. Cell Biol. 2, 110–116 [DOI] [PubMed] [Google Scholar]
- 44.Robinson M. J., Stippec S. A., Goldsmith E., White M. A., Cobb M. H. (1998) Curr. Biol. 8, 1141–1150 [DOI] [PubMed] [Google Scholar]
- 45.Whitehurst A., Cobb M. H., White M. A. (2004) Mol. Cell. Biol. 24, 10145–10150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chuderland D., Marmor G., Shainskaya A., Seger R. (2008) J. Biol. Chem. 283, 11176–11188 [DOI] [PubMed] [Google Scholar]
- 47.Whitehurst A. W., Robinson F. L., Moore M. S., Cobb M. H. (2004) J. Biol. Chem. 279, 12840–12847 [DOI] [PubMed] [Google Scholar]
- 48.Fukuda M., Gotoh I., Gotoh Y., Nishida E. (1996) J. Biol. Chem. 271, 20024–20028 [DOI] [PubMed] [Google Scholar]
- 49.Philipova R., Whitaker M. (2005) J. Cell Sci. 118, 5767–5776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chuderland D., Konson A., Seger R. (2008) Mol. Cell 31, 850–861 [DOI] [PubMed] [Google Scholar]