

Abstract
p53 can induce apoptosis through mitochondrial membrane permeabilization by interaction of its DNA binding region with the anti-apoptotic proteins BclxL and Bcl2. However, little is known about the action of p53 at the mitochondria in molecular detail. By using NMR spectroscopy and fluorescence polarization we characterized the binding of wild-type and mutant p53 DNA binding domains to BclxL and show that the wild-type p53 DNA binding domain leads to structural changes in the BH3 binding region of BclxL, whereas mutants fail to induce such effects due to reduced affinity. This was probed by induced chemical shift and residual dipolar coupling data. These data imply that p53 partly achieves its pro-apoptotic function at the mitochondria by facilitating interaction between BclxL and BH3-only proteins in an allosteric mode of action. Furthermore, we characterize for the first time the binding behavior of Pifithrin-μ, a specific small molecule inhibitor of the p53-BclxL interaction, and present a structural model of the protein-ligand complex. A rather unusual behavior is revealed whereby Pifithrin-μ binds to both sides of the protein-protein complex. These data should facilitate the rational design of more potent specific BclxL-p53 inhibitors.
Keywords: Apoptosis, Biophysics, Diseases/Cancer, Methods/NMR, Protein/Protein-protein interactions, Protein/Drug Interactions, Tumor/Suppressor/p53
Introduction
Due to its central role in the induction of apoptosis and cell-cycle arrest, p53 is one of the most important checkpoint proteins of cell survival and genomic integrity induced by a variety of different damage stimuli like DNA damage, oncogenic or hypoxic stress (1). p53 can induce apoptosis via transcription-dependent and transcription-independent functions (2). In particular, p53 mediates its pro-apoptotic response via transcriptional activation of pro-apoptotic target genes, such as BAX and PUMA, and trans-repression of pro-survival proteins (for review, see Ref. 3). In addition, death signals are mediated through the transcription-independent p53 death pathway, which acts via p53 protein-mediated direct mitochondrial outer membrane permeabilization (MOMP),2 leading to rapid cytochrome c release.
MOMP is believed to depend on the opening of the dynamic Bax/Bak lipid pore and is triggered by BH3-only proteins (4, 5). In response to a death stimulus, a fraction of stress-stabilized p53 rapidly translocates to mitochondria in primary, immortal, and transformed cells (6). The primary targets of p53 at the mitochondria are pro-survival Bcl2 proteins. Members of the Bcl2 family can be grouped into pro- and anti-apoptotic subfamilies, based on their modular content of BH1, BH2, BH3, and BH4 (Bcl2 homology) domains. The BH1234 domain proteins BclxL and Bcl2 are prototype anti-apoptotic members that stabilize the outer mitochondrial membrane by binding and inhibiting pro-apoptotic members via their hydrophobic BH123 binding pocket (also called the BH3 peptide binding pocket) (7). In contrast, BH123 domain proteins Bax and Bak act as ultimate pro-apoptotic effector proteins. A third class, the BH3-only proteins, like Bad, tBid, or Puma, act as key transducers of death signals and fall into two subgroups, activators and enablers (5). Activator BH3-only proteins such as tBid function by transiently binding to and directly activating Bax and Bak and inducing their oligomerization (5). Enabler BH3-only proteins such as Puma promote MOMP more indirectly. They activate Bax/Bak by forming inhibitory complexes with anti-apoptotic BH1–4 proteins such as Bcl2 and BclxL that normally stabilize the outer mitochondrial membrane. As a consequence, the activator BH3-only proteins or Bax/Bak themselves are liberated from their inhibitory complexes with Bcl2/xL and induce MOMP and the apoptotic cascade. Stress-induced p53 was shown to induce oligomerization of endogenous Bak and permeabilization of the outer mitochondrial membrane, as indicated by a robust cytochrome c release from isolated unstressed mitochondria (6, 8). In contrast, p53 variants most frequently found in cancer cell lines (called “hot spot” mutants) are also deficient in binding to Bcl2/xL proteins at the mitochondria (6, 9). Therefore, these mutants can be called “double hits” because they loose the ability to promote apoptosis through both transcription-dependent and -independent pathways. However, the mechanism how p53 directly induces MOMP at the mitochondria still remains enigmatic.
To address this question we present data on interaction between BclxL and the p53 DNA binding domain (DBD) and extend the previously reported binding model between p53DBD and BclxL (10, 11). We used spin-labeled BclxL to accurately determine the binding interface at the p53DBD by employing paramagnetic relaxation enhancement techniques. For the BclxL side, we used NMR chemical shift perturbation to determine the binding interface with p53DBD. Additionally, using residual dipolar coupling (RDC) techniques we determined the p53 binding competent conformation of BclxL. We found that upon binding, BclxL undergoes slight structural changes both at the p53-binding site but also at the BH123 binding pocket. This pocket forms the main interaction site of BH3-only proteins and therefore regulates MOMP and apoptosis. In addition, we present the first structural data on the binding of Pifithrin-μ, a specific small molecule inhibitor of the p53-BclxL complex. To our surprise, this inhibitor binds to both sides of the complex.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Protein Purification
Residues 94–312 of human p53 encoding for the wild-type, G245S, and R273H mutant p53 DNA binding domain were cloned into a pET28a vector (Novagen) with a His6 tag and a thrombin cleavage site using NdeI/BamHI restriction sites. Unlabeled and uniformly 15N-labeled p53DBD was expressed in Escherichia coli BL21(DE3). The R248W mutant was expressed and purified as described (12). For labeling purposes, 1 g of isotopically enriched 15N-ammonium chloride (Eurisotope, Saarbrücken, Germany) per liter of M9 salts medium was used. Cells were grown at 37 °C to an A600 of 0.7 and shifted to 20 °C after induction with 1 mm isopropyl 1-thio-β-d-galactopyranoside. One hour prior to induction 100 μm ZnSO4 was added to achieve a sufficient Zn2+ concentration for protein expression. Expression was done over 16–20 h and purification was achieved by nickel-nitrilotriacetic acid and size exclusion chromatography. Full-length BclxL shows very low solubility due to its hydrophobic transmembrane helix and the spectral quality can be significantly improved by deletion of the flexible loop without affecting the binding affinity to p53DBD. Therefore, truncated constructs of human BclxL lacking solely the transmembrane domain (BclxLΔTM, residues 1–212) and in addition the flexible loop between helices 1 and 2 (BclxLΔLT, residues 1–44 and 85–212) (7) were cloned into a pET28a expression system with an N-terminal His6 tag and a thrombin cleavage site. If not stated otherwise, BclxLΔLT was used for all in vitro experiments. Buffer exchange for all protein samples to 50 mm potassium phosphate, pH 6.8, 50 mm KCl, 5 mm DTT or 10 mm sodium phosphate, pH 7.2, 1 mm DTT was achieved by passage over G-60 or NAP-5 columns (Amersham Biosciences). The molecular masses and purities of all samples were monitored by SDS-PAGE and samples were concentrated to a final concentration of 0.5–1 mm.
Peptide Synthesis
Peptides were synthesized using standard Fmoc (N-(9-fluorenyl)methoxycarbonyl) chemistry on a tritylchloride-polystyrene resin. Purification was achieved by high pressure liquid chromatography with a preparative C18 reversed phase column and the correct molecular mass was confirmed by ESI and matrix-assisted laser desorption ionization time-of-flight mass spectrometry.
Spin Labeling
Site-directed spin labeling at a single cysteine site at position 151 of BclxLΔLT was performed using the cysteine-selective free radical reagent (1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl)methanethiosulfonate (MTSL, Toronto Research Chemicals) (13). Protein samples were changed to non-reducing buffer conditions using NAP-5 columns (GE Biosciences) and a 10-fold excess of spin label dissolved in acetone was added immediately. The reaction was allowed to proceed for 12–16 h at 4 °C. Excess MTSL was removed by passage over a NAP-5 column pre-equilibrated in NMR sample buffer (50 mm potassium phosphate, 50 mm KCl) without reducing agents.
MOMP Assays
Release assays for apoptotic activators were performed on mitochondria isolated from freshly harvested mouse liver and cultured cells. Mouse liver mitochondria were gradient-purified (6). Cultured cells were resuspended in mannitol buffer, swollen for 10 min, and Dounce homogenized. Nuclei and unlysed cells were pelleted at 2,000 × g. Mitochondria were recovered from the supernatant by centrifugation at 13,000 × g (14). Mitochondria (350 μg/ml) were incubated with purified p53, tBid (R&D Systems), Type V bovine serum albumin (Sigma), or buffer for 20 min at 30 °C and promptly centrifuged at 4 °C (6). The resulting supernatants and washed mitochondrial pellets were analyzed by immunoblotting. His-tagged human p53 proteins were baculovirally (WT p53) or bacterially expressed (WT p53 and p53 DBD mutants R175H, L194F, G245S, R248W, R249S, R280K, and R282W; previously characterized in Ref. 9. Protein purity was assessed by Coomassie gels and quantitated by comparing against a bovine serum albumin standard.
Immunoprecipitation and Western Blotting
RKO cells were pretreated with 15 μm PFTμ for 1 h, followed by 10 μm Nutlin for an additional 6 h. Immunoprecipitations were performed using 3 μg of anti-p53 antibody (DO-1, Santa Cruz) rotated overnight with 1 mg of total protein lysates at 4 °C. p53 complexes were pulled by protein G-agarose beads (Roche).
Fluorescence Polarization
Fluorescence polarization measurements were performed at room temperature with a BMG Polarstar Galaxy Multi-mode Reader (BMG Lab Systems, Offenburg, Germany). Labeling of BclxLΔLT and BclxLΔTM was done according to the manufacturer's protocol using 5′,6′-carboxyfluorescein succinimidyl ester (Molecular Probes) and the free label was removed by passage over a Superdex-200 size exclusion column (Amersham Biosciences). Labeling yields were between 70 and 80%. 100 nm Fluorescein-labeled BclxLΔLT was titrated with increasing amounts (0.1–100 μm) of p53DBD proteins in 10 mm sodium phosphate, pH 7.2, 1 mm DTT (ionic strength: 20 mm) and 50 mm potassium phosphate, pH 6.8, 50 mm KCl, 5 mm DTT (ionic strength: 150 mm) on a 384-well non-binding surface plate (Corning, Lowell, MA) using a 20-μl volume/well.
For peptide binding assays 750 nm fluorescein-labeled BH3 peptides were incubated with increasing amounts of BclxLΔLT ranging from 0.1 to 20 μm. Buffer conditions were the same as indicated above.
Fluorescence polarization was measured using the appropriate filter for fluorescein excitation and detection (excitation = 485 nm, emission = 520 nm). Five independent measurements were conducted for each titration step and the obtained values were averaged and analyzed with PROFIT 6.1.10 (Quantum Soft, Uetikon am See, Switzerland). Data were fitted to a one-site binding model using Equation 1,
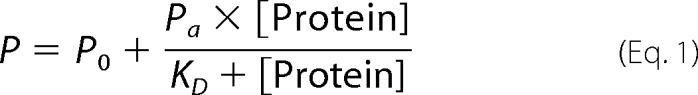 |
where P is the measured polarization value, P0 the initial polarization value, and Pa the polarization amplitude upon binding of the unlabeled protein to fluorescein-labeled BclxL, KD is the dissociation constant, and [Protein] is the concentration of the unlabeled protein. Double-stranded con2×5 DNA (5′-cct aga cat gcc taa t-3′) with two p53 consensus sites was used for competition experiments.
For peptide binding assays a different equation was used taking into account the concentration of the labeled peptide (Equation 2) (15),
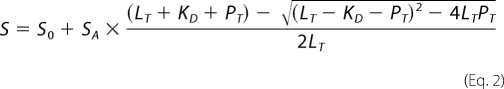 |
where LT is the total ligand (peptide) and PT the total protein concentration at each titration step. S0 and SA are the initial and amplitude in fluorescence polarization value and KD is the dissociation constant. This equation is used because the peptide concentration is close to the expected KD value. As a consequence, the total protein concentration at each titration step cannot be used for curve fitting because a significant amount of protein (>10% of total) is bound to the peptide.
Circular Dichroism (CD) Spectroscopy
CD spectroscopic experiments were performed with a Jasco J-715 spectropolarimeter (Jasco, Groß-Umstadt, Germany). For the determination of protein stability, 10 μm protein solutions in 50 mm potassium phosphate, pH 6.8, 50 mm KCl, 1 mm tris(2-carboxyethyl)phosphine were measured in a 1-mm path length cuvette. Upon heating with a rate of 60 K/h the ellipticity at 218 nm was recorded using a bandwidth of 5 nm and a response of 2 s. The obtained unfolding transitions from 20 to 90 °C were fitted with a Boltzmann function describing the sigmoidal shape of the unfolding process (16).
Nuclear Magnetic Resonance (NMR) Spectroscopy
NMR spectroscopic experiments were measured at 293 K on Bruker DMX600, DMX750, and Avance900 spectrometers (Bruker Biospin, Rheinstetten, Germany), respectively. All titration experiments were done using standard 15N-HSQC experiments with WATERGATE water suppression scheme (17) in either 50 mm potassium phosphate, pH 6.8, 50 mm KCl, 5 mm DTT or 10 mm sodium phosphate, pH 7.2, 1 mm DTT in 95/5% (v/v) H2O/D2O. 256 increments were recorded in the indirect dimension and data were processed using Topspin version 1.3 (Bruker Biospin). For confirming the chemical shift assignments, which was necessary for the larger chemical shift deviations occurring with PFTμ, a 15N-edited NOESY-HSQC (18) experiment was recorded. For paramagnetic relaxation enhancement (PRE) measurements, 150 μm spin-labeled BclxL was titrated into a solution of 150 μm 15N-labeled p53 DBD in 50 mm potassium phosphate, pH 6.8, 50 mm KCl at 293 K and the effect of the spin label after complex formation was monitored with 15N-HSQC spectra. As a reference, a HSQC spectrum was recorded after the reduction of the spin label with a 10-fold molar excess of ascorbic acid for 1 h at room temperature. Chemical shift changes upon ligand titration were calculated as the average of 15N and 1H shift deviations: Δav = (ΔH2 + ΔN2/25)1/2/2 (19), and KD values were determined with Equation 2 using the average chemical shift deviation instead of the fluorescence polarization as an input.
For the determination of dissociation constants using NMR peak volume measurements, the relative fraction of the bound protein (fbound) was determined using the relation: fbound = Vbound/(Vbound + Vfree), where Vbound and Vfree are the peak volumes of the 15N-HSQC signals of the bound and free forms of the corresponding amino acid residues, respectively. For each titration step, fbound was plotted against the concentration of the free ligand LF = LT − Lbound, where LT is the total ligand concentration and Lbound is the concentration of the bound ligand (which is equal to the bound protein concentration). Fitting of the titration curve was done with Equation 1.
HN RDCs were measured in 50 mm potassium phosphate, pH 6.8, 50 mm KCl, 5 mm DTT at 293 K with IPAP-type experiments (20) using 10 mg/ml of Pf1 phage (21) (Hyglos AG, Regensburg, Germany) or 3% C12E5-hexanol mixtures (Otting phases) (22). 256 increments were recorded in the indirect 15N-dimension and RDC values were determined by comparing the splitting of the HN correlation signal in the isotropic and anisotropic case using Sparky (23) and in house scripts. Analysis and back calculation was done with PALES (24). For analysis of RDCs we first generated a minimized energy structure using Xplor-NIH (25) with the coordinates of BclxL (Protein Data Bank code 1lxl) (26). Pairwise spin-spin distances for all heavy atoms were calculated from the crystal structure and used for structure calculation with experimental RDCs for refinement. HN RDCs of BclxL were measured with and without p53DBD. As expected, using the PALES software (24), RDCs back calculated with the resulting structure showed very good agreement with experimental values. For comparison, the experimental HN RDCs of BclxL in complex with p53DBD WT and the p53DBD R273H mutant were fitted against the refined structure using PALES. An identical procedure was done with the experimental RDC values of BclxL in the presence of p53DBD WT. The resulting structures of BclxL refined with each RDC set were aligned with PyMOL (55). Protein-protein and protein-ligand complexes were calculated using the HADDOCK software package (27). All residues, which show chemical shift differences greater than the average chemical shift deviation of all residues plus the standard deviations, were selected as “active” residues. All surrounding surface neighbors were defined as “passive” residues forming the binding site of the ligand/protein together. For PFTμ all atoms were set as active. Ambiguous distance restraints were defined between every active residue of the protein and every active and passive residue of the partner and vice versa. For PFTμ parameter and topology files were generated with PRODRG (28) and xplor2d (29) programs. The actual complex structure calculation was performed using the CNS structure calculation package (30) with a first round rigid body docking generating 2000 structures, a second round simulated annealing step, where the protein side chains and the binding partner are allowed to move. Finally, the best structures were refined in explicit solvent. 200 Water-refined structures were obtained and used for analysis. The quality of the obtained structural models was assessed by the root mean square deviations from the best energy structure and the intermolecular energy term. For visualization, the structures with the lowest values for the intermolecular energy terms were used. The backbone geometry of the best energy model was probed with PROCHECK-NMR (31). Structures were visualized using the PyMOL software package and Chimera (32).
Saturation transfer difference experiments (33) with p53DBD and PFTμ were performed at 293 K on a Bruker DMX750 spectrometer. 50 μm p53DBD was mixed with 1 mm PFTμ in 50 mm potassium phosphate, 50 mm KCl, 5 mm DTT, and 5% (v/v) d6-Me2SO. A saturation time of 2 s with low power on-resonance irradiation at 0 ppm was used where protein methyl groups are affected. For the control experiment, saturation was done at −30 ppm where no protein resonance is affected. Subtraction of on- and off-resonance experiments was achieved by appropriate phase cycling.
RESULTS
Determination of the Binding Interface between BclxL and p53DBD
Starting from previous data describing the BclxL-p53DBD protein-protein interface (6, 10), we aimed at getting a more precise structural model of the complex. To this end we used chemical shift perturbation experiments for BclxL and paramagnetic relaxation enhancement data for p53DBD. Significantly shifting signals in the 15N-HSQC spectrum of BclxLΔLT in 10 mm sodium phosphate, pH 7.2, 1 mm DTT (low salt buffer) include residues 20–30, 118–121, and 156–169 (Fig. 1A), which cluster at the negatively charged bottom side of BclxL. The chemical shift deviations were most pronounced in low-salt buffer indicating a strong electrostatic contribution to complex formation. For p53DBD, PRE experiments were performed (Fig. 1B) with MTSL-labeled BclxL in 50 mm potassium phosphate, pH 6.8, 50 mm KCl. MTSL is a nitric oxide radical, which can be selectively attached to cysteine residues by the formation of a disulfide bond. The radical induces enhanced relaxation of the signals in the 15N-HSQC spectrum, leading to a distance-dependent line broadening and reduction in intensity of the signals. This effect is rather long-range with maximal distances up to 25 Å (13). Using 15N-labeled p53DBD and MTSL-labeled BclxL, distances between both proteins were monitored. Upon complex formation a significant effect of the spin label on the signals in the 15N-HSQC spectrum of p53DBD can be observed (Fig. 1B). The PRE value is the ratio between the signal intensities in the 15N-HSQC spectrum of p53DBD with MTSL-labeled BclxLΔLT before and after reduction of the spin label with ascorbic acid. The observed signal intensity pattern reveals three main interaction sites within p53DBD, consisting of residues 110–120, 180–200, 235–255, and residue 275–285, which are also involved in DNA binding. To obtain a model of the complex structure, we used the data shown in Fig. 1, A and B, to identify surface-exposed residues that form the protein-protein interface. Residues with a PRE value of 0.6 and below for p53DBD, and residues with chemical shift differences greater than the average plus the standard deviation (Δav > 0.125) for BclxL were selected as active (supplemental Table S1A). Calculation of the complex structure was done using Haddock1.3 (27) and the CNS structure calculation package (30). The resulting best energy structural models are shown in Fig. 1C and the structural statistics of the best 5 energy models is shown in supplemental Table S1B. The protein-protein interface is formed by the positively charged DNA binding surface of p53DBD and the negatively charged bottom of BclxL, which encompasses BclxL residues 20–30, 118–121, and 156–169 (Fig. 1, C and D). The contact sites on BclxL, residues Ser18, Tyr22, Ser23, Gln26, and Ser28 in helix 1 and 2, Ile114 between helix 3 and 4, and Val155, Asp156, and Glu158 in helix 5 and 6 participate in polar contacts with residues Gly117, Ser121, Cys176, His178, Asn239, Met243, Arg248, Gly279, and Arg280 within p53DBD. As p53DBD interacts with its DNA-binding surface, we reasoned that it should be possible to dissociate the complex with the p53 consensus DNA. Indeed, using fluorescence polarization measurements, the complex between fluorescein-labeled BclxL and p53DBD could be disrupted by 0.5 m eq of p53 consensus DNA (oligonucleotides con2×5 (34)) containing two p53DBD binding sites (Fig. 1E).
FIGURE 1.

Structural model of the p53DBD- BclxL complex. A, chemical shift perturbation of 100 μm BclxLΔLT upon titration with 150 μm unlabeled wild-type p53DBD in 10 mm sodium phosphate, pH 7.2, 1 mm DTT at 293 K. There are three regions in BclxL, which show significant chemical shift differences (above the horizontal line, which indicates the mean value + 1 S.D.) upon p53DBD titration, localizing to BclxL residues 20–35, 100–125, and 150–178. B, 150 μm MTSL-labeled BclxLΔLT was titrated into a solution of 150 μm wild-type 15N-p53DBD in 50 mm potassium phosphate, pH 6.8, 50 mm KCl, 5 mm DTT at 293 K and the intensity ratio of the signals in the 15N-HSQC before and after reduction of the spin label with ascorbic acid was plotted. The positions of three hot spot mutants (G245S, R248W, and R273H) in p53DBD are marked with arrows. Residues showing an intensity ratio of 0.6 (black line) or lower are supposed to be involved in direct binding to BclxL. The lower the intensity ratio, the closer the distance between the residues in p53DBD and the spin label in BclxL. The contact regions within p53 are residues 110–120, 180–200, 235–255, and 275–285. C, structural model of the BclxL-p53DBD complex obtained with NMR data driven docking. Secondary structural elements are labeled. Input data were chemical shift and spin label data shown in A and B. Input data and structural statistics of the best energy structures are shown in supplemental Table S1. p53DBD interacts with its basic DNA-binding site with the acidic bottom of BclxL. D, surface representation of p53DBD. Interacting residues in BclxL are shown as sticks. The hot spot mutants of p53DBD are shown in orange. In BclxL, mostly negatively charged residues take part in the interaction, whereas the whole positively charged DNA-binding surface of p53DBD is covered. Single letter amino acid abbreviations are used. E, the complex between p53DBD and BclxL can be dissociated by the addition of p53 consensus DNA oligonucleotides containing two p53-binding sites (con2×5). Here, 60 μm p53DBD was incubated with 1 μm fluorescein-labeled BclxL in 10 mm sodium phosphate, pH 7.2, 1 mm DTT and con2×5 DNA was titrated in the mixture. The midpoint of the titration curve monitored with fluorescence polarization is 30 μm, indicating that the complex is dissociated by the DNA and exactly two p53DBD molecules bind to the con2×5 oligonucleotide.
Structural Changes in BclxL upon Binding to Wild-type p53DBD
We previously showed that oncogenic mutations within the p53DBD impair or abrogate the Bcl2/xL binding ability (6, 9). To investigate these p53DBD mutants in more detail, we performed NMR chemical shift perturbation and fluorescence polarization experiments with BclxL and either wild-type or mutant (G245S, R248W, and R273H) p53DBD. Wild-type p53DBD induced chemical shift changes in the 15N-HSQC spectrum of BclxL (Figs. 2A and supplemental S1) at the p53DBD binding site (see Fig. 1A), but also at the BH3 peptide-binding site comprised of BH123 regions. In Fig. 2A, a section of the 15N-HSQC spectrum of BclxLΔLT with (red) and without (black) p53DBD wild-type is shown. A prototype signal from the direct p53 binding site (Gln19) is labeled in black, signals corresponding to the BH123 region are labeled in red, and unlabeled peaks do not contribute to binding. Mapping of the p53DBD-binding site on BclxL has been previously reported by others (10, 11), and the chemical shift changes shown here are in very good agreement with these data. However, in addition to the chemical shift changes at the direct p53DBD-binding site, we also see changes at the BH123 region. Using the structure of BclxL in complex with a BH3 peptide derived from the BH3-only protein Bad, significant shift differences can be visualized on the structure (Fig. 2B). These effects cluster to the BH123 region in BclxL, with its groove for BH3 peptide (green wire in Fig. 2B) binding. The direct p53DBD binding site is not shown in Fig. 2B but the chemical shifts for that region are of similar magnitude compared with the BH123 region (Fig. 1A). Of note, analysis of the chemical shift differences observed with wild-type p53DBD and structural mutant G245S or DNA contact mutant R273H reveals differences especially in the BH123 region. Only the wild-type p53 protein and to some extent the G245S mutant were able to induce chemical shift changes also in the BH3 peptide binding pocket of BclxL. In contrast, the R273H DNA contact mutant showed only chemical shift changes at the p53-binding interface of BclxL (supplemental Fig. S1). This is directly correlated with a reduced affinity to BclxL as determined by fluorescence polarization at two ionic strengths (see Figs. 2C and supplemental S2, and Table 1). At both, 20 and 150 mm ionic strength, p53DBD wild-type shows the highest affinity for BclxL. Mutants G245S and R273H show significantly lower affinity especially at the 150 mm ionic strength. Therefore, BclxL is not saturated with mutant p53DBD at the sample conditions of the NMR chemical shift perturbation experiments, whereas in the presence of wild-type p53DBD BclxL is fully saturated and shows the most pronounced chemical shift perturbation effects. All investigated p53 mutants are stable at room temperature as shown by CD-detected thermal unfolding transitions (supplemental Fig. S3). The thermal stability of p53DBD wild-type is 43.3 °C, whereas the mutants show lower stability (G245S, 37.2 °C; R273H, 41.3 °C), which is well above the temperature chosen for all measurements (20 °C).
FIGURE 2.

Mutational and functional characterization of the BclxL-p53DBD complex. A, section of a 15N-HSQC spectrum of 15N-labeled BclxLΔLT and unlabeled wild-type p53DBD (black, BclxLΔLT alone; red, +p53DBD wt). Shifting residues are located at the direct p53DBD-binding site (Gln19) or at the BH3-peptide binding site (Ala104, Leu108, and Ala142, red label) of BclxL. B, significant chemical shift changes (Δav > 0.07 ppm) upon titration of wild-type p53DBD, mapped onto the structure of BclxL (PDB code 1g5j). Shifting residues in BclxLΔLT are shown in red. For comparison, the Bad-derived BH3 peptide bound to the BH3 binding pocket is shown in green. C, KD value determination of fluorescein-labeled BclxLΔLT and p53DBD wild-type (●) and mutants G245S (●), R248W (▴), and R273H (▾). Five individual measurements were averaged and standard errors are indicated. Corresponding KD values are shown in Table 1. D and E, isolated mitochondria from unstressed HCT116 human colon carcinoma cells were incubated with the indicated recombinant full-length proteins. Subsequently, aliquots were analyzed for MOMP by immunoblotting. The prototypical BH3-only protein tBid functions exclusively via the Bax/Bak pore and serve as positive control. Only wild-type but not mutant p53 was able to induce MOMP. AIF, apoptosis-inducing factor; BSA, bovine serum albumin; COX, cyclooxygenase.
TABLE 1.
Dissociation constants between BclxL and p53DBD at two different ionic strengths (I)
| p53DBD | Wild-typea | Wild-typeb | G245Sb | R248Wb | R273Hb |
|---|---|---|---|---|---|
| KD (μm) I = 20 mmc | 5.9 ± 0.7d | 5.5 ± 1.3d | 8.8 ± 1.2d | 17.4 ± 2.5d | 17.7 ± 2.4d |
| KD (μm) I = 150 mme | 44 ± 4d | 113 ± 19d | 158 ± 25d |
a Fluorescein-labeled BclxLΔTM was used.
b BclxLΔLT was used.
c Buffer was 10 mm sodium phosphate, pH 7.2, 1 mm DTT (I = 20 mm).
d 5 individual measurements were used for error estimation.
e Buffer was 50 mm potassium phosphate, pH 6.8, 50 mm KCl, 5 mm DTT (I = 150 mm).
To test the impaired ability of p53 mutants to induce MOMP in a cellular assay, we reasoned that tumor-derived p53DBD mutants in the full-length protein should also be compromised in their ability to alter the functional status of Bax and Bak in the outer mitochondrial membrane, and possibly also alter the permeability transition pore components in the inner and outer membrane. As expected, addition of full-length recombinant wild-type p53 protein to mitochondria that were isolated from unstressed human colon carcinoma cells (HCT116) readily released the apoptosis activating factors cytochrome c, Smac, and AIF (apoptosis-inducing factor) into the supernatant (Fig. 2, D and E, and data not shown). In contrast, mutant full-length p53 proteins failed to induce a significant release of these factors, which further correlated with a pronounced decrease to induce oligomerization of endogenous Bax and Bak (8). Moreover, in contrast to full-length wild-type p53, full-length mutants are markedly compromised in their ability to drive voltage-dependent anion channel (a major outer membrane protein and presumed component of the permeability transition pore) into high molecular weight complexes (8).
To gain further information about the mechanism of p53-induced induction of MOMP, we measured RDCs in 50 mm potassium phosphate, 50 mm KCl, 5 mm DTT (high salt buffer). The concentrations of BclxL and p53DBD were 100 and 150 μm, respectively, which is well above the KD between wild-type p53DBD and BclxL in this buffer (see Table 1). RDCs are dependent on the relative orientation of the chemical bonds between two atoms in respect to the preferred orientation of the protein and are therefore a tool for structure validation and refinement. RDCs can be back calculated from the three-dimensional structure and compared with the measured RDCs. We used 15N-BclxLΔLT and measured RDCs between the amide hydrogen and nitrogen in the protein backbone. The measured HN RDC values of free BclxLΔLT agree well with the values back calculated from the crystal structure (PDB code 1lxl, correlation r = 0.96, Fig. 3A, upper left). Structural changes in the BH123 region were probed with a sample containing 15N-BclxLΔLT and wild-type p53DBD. Here, the correlation between measured and calculated RDCs was significantly decreased (r = 0.84, Fig. 3A, lower left). This decrease in correlation (R value) indicates that in the presence of wild-type p53DBD, the structure of BclxL is altered. Looking at the outliers in the correlation plot (Fig. 3A, lower left), residues mostly located in the BH123 region (Phe101, Tyr124, Ser149, Leu154, and Leu198, red label) but also at the direct p53 binding site (Gln19 and Val159, black label) can be identified. The HN bonds of these residues experience a change in their relative orientation, indicating that a conformational change takes place in this region. In contrast, the measured RDCs of the p53DBD R273H mutant in complex with BclxL show better agreement with the structure of free BclxL (r = 0.89) where significant deviations appear only at the direct binding site to p53DBD (data not shown). This difference is caused by the pronounced drop in affinity with this mutant (Table 1).
FIGURE 3.

Structural changes in BclxL upon binding to p53DBD monitored with residual dipolar couplings. A, comparison of the observed and calculated HN residual dipolar couplings of BclxLΔLT (upper left) and BclxLΔLT in the presence of wild-type p53DBD (lower left). Residues showing significant deviations in the BclxL-p53DBD sample are labeled and residues in the BH3-peptide binding site are colored in red. In the presence of wild-type p53DBD, the correlation between observed and back calculated RDCs is decreased from 0.96 to 0.84, indicating a structural change at the BH3 binding groove in BclxL upon complex formation. For back calculation of the RDCs a RDC-refined structure of free BclxL was used. Upper right, HN RDCs of BclxL in its complex with p53DBD (using 10 mg/ml of Pf1 phage for partial alignment) are correlated with HN RDCs back calculated from an RDC-refined structure of BclxL in complex with p53DBD (see B, right panel). The agreement between both sets is very high, yielding a correlation coefficient R of 0.94 (where R ranges from 0 to 1). Da, axial component; Rh, rhomicity of the alignment tensor obtained using single value decomposition with the refined structure of BclxL. Lower right, HN RDCs of the BclxL-p53DBD complex using 3% Otting phases for alignment. For back calculation an RDC-refined structure of BclxL in complex with p53DBD was used (B). Here, the calculated correlation is even better than found for the Pf1 phage case (r = 0.97). B, comparison between BclxL structures in the Bad-peptide bound form (left panel), in the free form (middle panel, PDB code 1lxl), and in the RDC-refined p53DBD-bound form (right panel). In the RDC-refined structure helices 3 and 4 move to a more open position similar to the conformation found when in complex with BH3 peptides and thus seem to facilitate peptide binding. In each structure, the BAD peptide is shown to visualize possible sterical clashes that can be seen in the free conformation of BclxL (yellow circle). C, left, alignment of BH3 peptides. Especially, Bad and Puma show high sequence conservation, whereas the similarity with Bid is less pronounced. Right, affinities between 0.75 μm fluorescein-labeled Puma and Bid BH3 peptides and BclxL measured in 10 mm sodium phosphate, pH 7.2, 1 mm DTT at room temperature.
The measured HN RDCs of the BclxL-p53DBD complex can be directly used to obtain a structural model where the basic fold of BclxL is fixed through NOE connectivities and RDCs are included for the refinement to induce local structural changes. Two sets of HN RDCs measured in 10 mg/ml of Pf1 phage and 3% Otting phases were used to probe the conformational changes. As expected, after structure refinement the resulting structure shows much better agreement with the measured RDC values (Fig. 3A, right). The R value increases to 0.94 as compared with 0.84 with the unbound BclxL structure for the phage medium (upper right) and shows an almost perfect agreement for the Otting phase (r = 0.97) (lower right). A comparison between the BclxL structures in the free, the Bad BH3 peptide-bound form (PDB code 1g5j) (35), and the refined p53DBD-bound form indicates differences in the BH3-peptide binding groove of BclxL (Fig. 3B). The biggest differences between the free and p53DBD-bound forms are located within helices 3 and 4. The RDC-refined p53DBD-complexed BclxL structure (right panel in Fig. 3B) shows increased similarity to the BclxL-Bad structure (left panel). In contrast, the structure of free BclxL (middle panel) is in a more closed conformation where peptide binding is hindered due to sterical clashes, which have to be overcome for binding. In the p53DBD-bound form, these clashes between BclxL and the BH3 peptide are not present. Consequently, this should lead to increased affinity because the binding competent conformation of BclxL is already significantly populated.
Influence of BH3 Peptides on Complex Formation between BclxL and p53DBD
The BH3 binding pocket in Bcl2/xL is the central interaction site with pro-apoptotic BH proteins. We used fluorescence-based BH3-peptide binding assays and NMR spectroscopy to monitor the affinity and interaction sites between BclxL and three different BH3 peptides. The sequences of these peptides are shown in Fig. 3C. The affinity between a Puma BH3 peptide and BclxL is at least 7 times higher than the affinity measured with a Bid BH3 peptide (0.14 versus 0.97 μm, Fig. 3C, right). The identical peptide-binding assay with Bid BH3 and BclxL was performed in the presence of increasing amounts of p53DBD wild-type. It could be observed that p53DBD leads to an increased binding affinity for the Bid peptide in a concentration-dependent manner (Table 2). In contrast, for the R273H p53DBD mutant, which shows reduced binding affinity to BclxL, this effect occurs at much higher p53DBD concentrations as compared with the wild-type protein. This correlates well with the observed affinities between BclxL and p53DBD (Table 1). Under these conditions BclxL forms a ternary complex with p53DBD and the BH3 peptide and dissociation of the complex is only possible when at least 150 mm NaCl is added. A control where the Bid peptide was titrated to p53DBD showed no binding even at 60 μm p53DBD concentration. In contrast to the BclxL-p53DBD interaction, the affinity between the Bid BH3 peptide and BclxL is only slightly affected by increasing ionic strength, and the deleted flexible loop between helices 1 and 2 in BclxL does not contribute to peptide binding as shown by peptide-binding assays with BclxLΔTM, and BclxLΔLT at different ionic strengths (supplemental Fig. S4).
TABLE 2.
Affinity between BclxL and a fluorescein-labeled Bid BH3 peptide in the presence of p53DBD
Buffer conditions were: 10 mm sodium phosphate, pH 7.2, 1 mm DTT at room temperature. 750 nm FL-Bid was used.
| [p53DBD variant] |
KD(BclxL-Bid) |
|
|---|---|---|
| p53DBD wild-type | p53DBD R273H | |
| μm | μm | |
| 0 | 0.97 | 0.97 |
| 2 | 0.59 | 1.10 |
| 5 | 0.51 | 1.49 |
| 30 | 0.36 | 0.45 |
In addition to the affinity experiments, 15N-HSQC experiments in low salt buffer were used to characterize the binding site of the BH3 peptides on BclxL (supplemental Fig. S5). These data show that Bid, Bad, and Puma peptides induce strong chemical shift differences at the BH3 binding site but additional chemical shift changes are also observed at the p53-binding site. The chemical shifts for the Bid peptides are smaller as compared with the other ones because of the absence of any aromatic amino acid (Fig. 3C).
Binding of Pifithrin-μ to p53DBD and BclxL
Next we analyzed PFTμ, a recently described specific in vivo inhibitor of the p53DBD-BclxL interaction (36). To this end, we used NMR chemical shift perturbation experiments, molecular modeling approaches, and cell-based assays. After titration of up to 5 mm PFTμ and to 300 μm wild-type p53DBD, chemical shift changes occur at loop 1 and helix 2 (Fig. 4A). Quantitative evaluation using peak integration of the bound and free species for each titration point (see also “Experimental Procedures”) gives a KD value of 0.82 ± 0.2 mm (Fig. 4B). Significant chemical shift perturbations were used for NMR data-driven docking that resulted in 200 water-refined complex structural models. Loop 1 (L1) and helix 2 (H2) are located next to each other and form a hydrophobic pocket upon binding of PFTμ (Fig. 4C). In addition, the sulfonamide moiety of PFTμ is connecting residue Lys120 in loop 1 with residues Gly279 and Arg280 in helix 2 by forming hydrogen bonds (Fig. 4D). This interaction should pull loop 1 closer to helix 2 and restrict the flexibility of loop 1, which is flexible in the μs-ms time scale (37). The 5 best-energy complex structural models are shown in Fig. 4D having a root mean square deviation of 1.15 Å. Saturation transfer difference NMR experiments (33) confirm that the aromatic ring in PFTμ is inserting itself into a binding pocket in p53DBD. Only the parts of the ligand that are in close contact with the protein give rise to a signal (supplemental Fig. S6). The signal at 7.4 ppm can be assigned to the aromatic protons of the phenyl ring of PFTμ. Loop 1 and helix 2 within p53DBD also participate in interaction with BclxL. Thus, distortion of this region causes a reduction in the affinity between both proteins. In support, fluorescence polarization measurements gave evidence for a decrease in the p53-BclxL affinity upon addition of PFTμ. Addition of 1 and 2 mm PFTμ increased the KD value from an average of 5.7 to 9.0 and 9.6 μm, respectively (supplemental Fig. S7). To our surprise, due to its sulfonamide moiety, which is also present in other Bcl2/xL inhibitors, PFTμ also binds to BclxL, the other partner of the complex. As shown in Fig. 5A, chemical shift deviations upon PFTμ titration appear mostly at the BH3 peptide binding pocket, similar to BH3 peptides (supplemental Fig. S5) and other Bcl2/xL inhibitors (38). Quantitative evaluation with NMR and by using Equation 2 for curve fitting gave a dissociation constant of 0.8 ± 0.1 mm (Fig. 5B). Subsequent docking experiments where all significantly shifting residues were used to define the binding interface in BclxL resulted in a structural model where PFTμ is interacting with the BH123 groove in BclxL (supplemental Fig. S8, A and B). The interaction is mostly based on hydrophobic contacts with only one hydrogen bond formed between the amide moiety of PFTμ and the backbone carbonyl of Phe101 in BclxL. The polar sulfone group is pointing outwards to the solvent. The five best-energy models showed a root mean square deviation of 0.96 Å and all calculated models show the same binding mode. A comparison of the obtained chemical shift pattern within BclxL induced by PFTμ with the binding mode of the previously introduced Bcl2/xL inhibitor ABT-737 (38) shows that the binding site of PFTμ is very similar to that of ABT-737 (Fig. 5C). The aromatic ring of PFTμ inserts into one of the two hydrophobic pockets identified by the screening procedure for ABT-737 (see also supplemental Fig. S8). To finally test the effects of PFTμ in a cell-based assay using RKO human colon carcinoma cells harboring wild-type p53, we stabilized p53 by the addition of 10 μm Nutlin, a specific inhibitor of the p53-MDM2 interaction, which prevents proteasomal degradation of p53 (Fig. 5D, bottom panel) (39). The endogenous interaction between p53 and BclxL is shown by immunoprecipitation of p53 with the DO-1 antibody and a subsequent immunoblot detecting BclxL (Fig. 5D, right lane). In the absence of Nutlin, only background levels of BclxL are detectable (Fig. 5D, left lane). Preincubation with 15 μm PFTμ resulted in a loss of the interaction between p53 and BclxL, indicated by a drop back to background levels of BclxL (Fig. 5D, middle lane). A Western blot against p53 after p53 immunoprecipitation serves as the input control.
FIGURE 4.

Binding mode of Pifithrin-μ to p53DBD. A, chemical shift perturbation data for the interaction between 300 μm p53DBD and PFTμ measured with 15N-HSQC experiments in 50 mm potassium phosphate, pH 6.8, 50 mm KCl, 5 mm DTT at 293 K. The chemical structure of PFTμ is shown in the inset. Chemical shift differences in the slow exchange regime mostly appear at loop 1 (L1) and helix 2 (H2) (gray boxes). B, titration curve for PFTμ. A KD value of 0.82 mm was obtained by peak volume measurements of the signals corresponding to the free and ligand-bound forms of p53DBD. Five individual residues were used for error estimation. Bars indicate standard error. C, docking result using the chemical shift perturbation data. PFTμ fits into a binding pocket in p53DBD between loop 1 and helix 2. D, overlay of the five best-energy structural models of the complex showing a root mean square deviation of 1.15 Å. The sulfonamide moiety is pointing toward the solvent and forms hydrogen bonds with amide protons of Lys120, Gly279, and Arg280.
FIGURE 5.

Binding of PFTμ to BclxL and effect of PFTμ on the endogenous p53-BclxL complex. A, chemical shift perturbation data for the interaction between 300 μm 15N-BclxLΔLT and PFTμ probed with 15N-HSQC experiments in 10 mm sodium phosphate, pH 7.2, 1 mm DTT at 293 K. There are significant chemical shift changes at the BH123 binding pocket where BH3 peptides of pro-apoptotic BH3-only proteins bind. The BH regions are labeled with boxes. B, titration curve between 300 μm BclxLΔLT and PFTμ. The chemical shift changes upon subsequent titration of PFTμ were measured with 15N-HSQC spectra, normalized, and fitted with Equation 2 (see “Experimental Procedures”). A dissociation constant of 0.8 mm was obtained. Error bars obtained by evaluation of 5 individual residues in BclxL are smaller than the size of the symbols. C, chemical shift perturbations (CSPs) of BclxL in the presence of 10 mm PFTμ mapped onto the BclxL structure. For comparison, ABT-737, a previously published Bcl2/xL inhibitor, is shown in green. A box indicates the binding site for PFTμ obtained by NMR data driven docking (supplemental Fig. S8). D, effect of PFTμ on the endogenous BclxL-p53 interaction. RKO cells were incubated with 15 μm PFTμ and/or 10 μm Nutlin or left untreated. Endogenous p53 was immunoprecipitated with DO-1 antibody from equal amounts of cell lysates and the fraction of complexed BclxL was detected by Western blotting. IP, immunoprecipitation; WB, Western blot.
DISCUSSION
An Allosteric Model of the BclxL-p53 Interaction
As discussed in various review articles (40, 41), currently there are two models used to explain the action of Bcl2 proteins at the mitochondria. The first model, the inactivator model or rheostat model, describes that the primary function of anti-apoptotic proteins like Bcl2 and BclxL is to neutralize the pro-apoptotic effector proteins Bax and Bak by forming heterodimeric complexes. As Bax and Bak are the effector Bcl2 proteins that form lipid pores in the outer mitochondrial membrane, these molecules need to be neutralized. There is a balance between pro- and anti-apoptotic Bcl2 proteins. MOMP is initiated when the anti-apoptotic Bcl proteins are functionally neutralized. This is facilitated by the activation of BH3-only proteins, which bind to anti-apoptotic Bcl2 proteins and liberate the effectors Bax and Bak (42–44). The second model, the direct activator model, is based on the direct activation potential of some BH3-only proteins, i.e. Bid or Bim, which transiently bind to Bax/Bak and induce conformational changes within Bax/Bak required for stable membrane insertion (41, 45). The anti-apoptotic Bcl2 proteins sequester the activator BH3-only proteins and neutralize them. If the pro-apoptotic BH3 only proteins titrate out the anti-apoptotic members, MOMP can occur.
The role of p53 in this network is on the one hand the transcriptional transactivation of BH3-only proteins like Puma and Noxa, both involved in the induction of apoptosis after exposure to γ- and UV-radiation (46) and of Bax, one of the effector Bcl proteins. On the other hand, the role of p53 at the mitochondria is based on protein-protein interaction with the anti-aptoptotic Bcl proteins (6, 9) but also with Bax and Bak (11).
The data presented here add new insights into the action of p53 at the mitochondria. In agreement with previous reports at lower resolution (6, 9, 10), we accurately defined the protein-protein interface between p53DBD and BclxL and presented a structural model of the complex (Fig. 1). Mutations within the NMR-derived protein-protein interface cause weakening of the affinity between both proteins. This was probed by fluorescence polarization measurements with p53DBD mutants most frequently observed in cancer cell lines (so-called hot spot mutants) (Fig. 2C).
In addition to the direct binding site, there are significant structural changes taking place within BclxL located at the BH3 binding region comprised of the BH123 regions (Fig. 3). RDC data used to gain detailed information about the conformational changes taking place upon binding. Finally, we calculated a structural model that satisfies the RDC data obtained with BclxL in complex with p53DBD (Fig. 3B, right panel). In this structure, helices 3 and 4 are in an open conformation also present in the complex with BH3 peptides. In this conformation, binding of a BH3 peptide is not hindered by sterical clashes. The functional relevance of these structural changes on the interaction between BclxL and BH3-only proteins was corroborated by measuring increased affinity between BclxL and a Bid BH3-peptide when p53DBD is present (see Table 2). Therefore p53 may influence and enhance interaction between BclxL and BH3-only proteins in an allosteric mode of action.
The observed affinity changes in the BH3 binding region of BclxL in the presence of p53DBD are in agreement with the rheostat model described above. Because the affinity of BclxL for BH3-only proteins is increased in the presence of p53DBD, p53 might facilitate the dissociation of the reformed BclxL-Bax/Bak complexes by BH3-only proteins. In comparison to the Bid BH3 peptide, Puma BH3 binds with increased affinity to BclxL (0.14 versus 0.97 μm), which reflects the exceptional importance of this BH3-only protein in the dissociation of the p53-BclxL complex and in its action as an “enabler” BH3-only protein (47). p53 stabilizes a conformation in BclxL, which seems to be more binding competent for BH3-only proteins and this conformation might be the solution competent state, which restricts stable membrane insertion of BclxL. Despite the increased affinity of BclxL for BH3-only proteins when p53 is bound, mutations in the BH3 peptide binding groove of BclxL (G138A) cannot be restored by p53 (47).
Binding of Pifithrin-μ to p53DBD and BclxL
We also provide for the first time structural data on the small molecule inhibitor PFTμ that specifically binds to isolated domains of p53DBD and BclxL with weak affinity as determined by NMR. Up to now direct binding could not be shown by others or us for several other known small molecule p53 inhibitors. We tried several compounds including CP-31398 (48) and PFTα (49) but none were able to induce significant chemical shift changes in p53DBD. In addition to the non-active compounds, we used promazine hydrochloride, a heterocyclic aromatic amine, as control to exclude nonspecific hydrophobic interactions. PFTμ is binding to p53DBD at loop 1 and helix 2 and is therefore competing for binding with BclxL (Fig. 4, C and D). The interaction between p53DBD and PFTμ shows a rather low affinity of 0.8 ± 0.2 mm (Fig. 4B). However, the binding mode of PFTμ seems specific because the chemical shift changes are significantly elevated with a maximum value of 0.7 ppm and two signal sets indicating slow exchange between the free and PFTμ-bound form of p53DBD. Despite the low in vitro affinity, the KD values of the BclxL-p53DBD complex upon addition of increasing concentrations of PFTμ change from 5.5 μm in the absence of PFTμ to 9.0 and 9.5 μm in the presence of 1 and 2 mm PFTμ, respectively (supplemental Fig. S7). Of note, p53DBD hot spot mutants lacking apoptotic functionality show similar affinities (see Table 1). Therefore, the slight decrease in affinity of the p53-BclxL complex in the presence of PFTμ might be sufficient to abolish the interaction in vivo. PFTμ contains a sulfonamide moiety, which is also present in some BclxL inhibitors (38). In addition to its binding to p53, titration of 15N-BclxL with PFTμ gave chemical shift changes located in the BH3 peptide-binding groove of BclxL, which binds to pro-apoptotic BH proteins (Fig. 5, A and C). This fact might contribute to the action of PFTμ, because BH3 peptides that bind to that region in BclxL are reported to dissociate or weaken the BclxL-p53DBD complex (9, 10, 47).
In contrast to the millimolar NMR-based affinities in vitro, we observed that in vivo 15 μm PFTμ is sufficient to break the p53-BclxL complex (Fig. 5D), in agreement with other reports (36, 50). The difference is likely caused by other simultaneous effects of PFTμ toward BclxL/2 that weaken the complex with p53 in cells. Specifically, as shown by Steele et al. (50), 15 μm PFTμ also up-regulates Noxa, and this BH3-only protein competes with p53 for BclxL/2 binding. Nutlin and other p53-dependent cytotoxic agents induce formation of a p53-BclxL/2 complex. In contrast, PFTμ not only directly interferes with the p53-BclxL/2 complex but also indirectly does so by inducing a competitive BclxL/2-Noxa complex (50). Of particular note, PFTμ also breaks the Bim-BclxL/2 complex (50), which is in excellent agreement with our chemical shift perturbation data that indicate that PFTμ also interferes with the BH3 binding pocket of BclxL (Fig. 5, A and C). Thus, these data suggest that PFTμ only appears to be highly effective in disturbing the p53-BclxL/2 complex in vivo. However, in a cellular context PFTμ exerts a composite effect of p53-directed and p53-independent yet undefined cellular actions, all centering on BclxL/2. This provides a basis for the difference in PFTμ affinities as determined in vitro and in vivo. In addition, we have to keep in mind that the NMR-based affinities were obtained with monomeric recombinant proteins, whereas in the experiments in RKO cells endogenous full-length p53, which is a tetramer, was used. Drastically increased ligand binding effects due to multimerization are well known in the literature (51, 52). Moreover, the physiologic concentration of p53 and BclxL in vivo is much lower than that used in our in vitro assays, and the presence of a (mitochondrial) membrane might also influence the binding behavior between both proteins in vivo.
For designing a more p53-specific mode of action of PFTμ, a stepwise optimization might be achieved by either focusing on p53DBD or BclxL. With characterization of the binding sites of PFTμ at hand (Figs. 4 and 5), this task should now be feasible. At first glance, BclxL seems to be the better suited target because its BH3 peptide-binding groove is an ideal case for structure-based drug design, as previously reported by using the structure-activity relationship by NMR approach (53), which resulted in a high affinity Bcl2/xL ligand (38). Optimization of PFTμ for the p53DBD side is likely to be much more challenging because of the lack of a defined binding pocket for small molecule ligands. In addition, a ligand targeting the DNA binding interface in p53 most likely influences the DNA binding properties of p53, as it was observed with Pifithrin-α (49). On the other hand, a high-affinity p53-binding small molecule would assure that a p53-specific pathway is targeted.
In conclusion, we presented structural and biophysical data on the influence of p53 on the conformational state of BclxL. We provide evidence for an allosteric structural change within the BH3 peptide-binding groove in BclxL that occurs upon binding of p53 to the “bottom” side of BclxL. This groove opens up and adopts a conformation similar to that found in BclxL-BH3-peptide complexes. In addition, the affinity of BclxL for BH3-peptides increases in the presence of p53DBD. This allows us to expand the existing model by proposing that p53 partly fulfils its function at the mitochondria by facilitating the interaction between BclxL and BH3-only proteins, which in turn displaces Bax/Bak from the anti-apoptotic Bcl-proteins and consequently promotes MOMP. Furthermore, we showed that Pifithrin-μ binds to p53DBD as well as to BclxL. Despite the discrepancy between its in vivo activity and the rather low affinities in vitro, which in some cases has been reported already (54), we provide clear evidence for specific binding of PFTμ to p53DBD and BclxL and were able to determine its binding mode to both proteins. These data should aid further optimization of small molecule ligands targeting this essential protein-protein interaction.
Supplementary Material
Acknowledgments
We thank York Tomita (Georgetown University) for the kind gift of plasmids and protein samples, Alexander Dehner for initial experiments, Stephan Lagleder (TUM) for protein samples, Klaus Kaluza (Roche Diagnostics) for fluorescence polarization measuring time, Michael Sattler and Johannes Buchner (TUM) for helpful discussions, and Murray Coles (MPI Tübingen) for providing software tools for structure calculation and analysis.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 CA60664 from the NCI (to U. M. M.). This work was also supported by the Center for Integrated Protein Science Munich and Deutsche Forschungsgemeinschaft Grant SFB594 (to H. K.) and a grant from Elitenetzwerk Bayern, CompInt (to F. H.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S8 and Table S1.
- MOMP
- mitochondrial outer membrane permeabilization
- BH
- Bcl2 homology
- BclxLΔLT
- human BclxL with missing transmembrane helix and missing flexible loop (residues 1–44, 85–212)
- BclxLΔTM
- human BclxL with missing transmembrane helix (residues 1–212)
- DBD
- DNA binding domain
- MTSL
- (1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl)methanethiosulfonate
- RDC
- residual dipolar coupling
- PRE
- paramagnetic relaxation enhancement
- PTFμ
- Pifithrin-μ
- DTT
- dithiothreitol
- WT
- wild-type
- HSQC
- heteronuclear single quantrum coherence.
REFERENCES
- 1.Römer L., Klein C., Dehner A., Kessler H., Buchner J. (2006) Angew. Chem. Int. Ed. Engl. 45, 6440–6460 [DOI] [PubMed] [Google Scholar]
- 2.Johnstone R. W., Ruefli A. A., Lowe S. W. (2002) Cell 108, 153–164 [DOI] [PubMed] [Google Scholar]
- 3.Fuster J. J., Sanz-González S. M., Moll U. M., Andrés V. (2007) Trends Mol. Med. 13, 192–199 [DOI] [PubMed] [Google Scholar]
- 4.Kuwana T., Bouchier-Hayes L., Chipuk J. E., Bonzon C., Sullivan B. A., Green D. R., Newmeyer D. D. (2005) Mol. Cell 17, 525–535 [DOI] [PubMed] [Google Scholar]
- 5.Chipuk J. E., Bouchier-Hayes L., Green D. R. (2006) Cell Death Differ. 13, 1396–1402 [DOI] [PubMed] [Google Scholar]
- 6.Mihara M., Erster S., Zaika A., Petrenko O., Chittenden T., Pancoska P., Moll U. M. (2003) Mol. Cell 11, 577–590 [DOI] [PubMed] [Google Scholar]
- 7.Sattler M., Liang H., Nettesheim D., Meadows R. P., Harlan J. E., Eberstadt M., Yoon H. S., Shuker S. B., Chang B. S., Minn A. J., Thompson C. B., Fesik S. W. (1997) Science 275, 983–986 [DOI] [PubMed] [Google Scholar]
- 8.Wolff S., Erster S., Palacios G., Moll U. M. (2008) Cell Res. 18, 733–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomita Y., Marchenko N., Erster S., Nemajerova A., Dehner A., Klein C., Pan H., Kessler H., Pancoska P., Moll U. M. (2006) J. Biol. Chem. 281, 8600–8606 [DOI] [PubMed] [Google Scholar]
- 10.Petros A. M., Gunasekera A., Xu N., Olejniczak E. T., Fesik S. W. (2004) FEBS Lett. 559, 171–174 [DOI] [PubMed] [Google Scholar]
- 11.Sot B., Freund S. M., Fersht A. R. (2007) J. Biol. Chem. 282, 29193–29200 [DOI] [PubMed] [Google Scholar]
- 12.Klein C., Hesse F., Dehner A., Engh R. A., Schwaiger M., Hansen S. (2004) Biol. Chem. 385, 95–102 [DOI] [PubMed] [Google Scholar]
- 13.Battiste J. L., Wagner G. (2000) Biochemistry 39, 5355–5365 [DOI] [PubMed] [Google Scholar]
- 14.Jürgensmeier J. M., Xie Z., Deveraux Q., Ellerby L., Bredesen D., Reed J. C. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 4997–5002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kenakin T. P. (1993) Pharmacological Analysis of Drug-Receptor Interaction, Raven, New York [Google Scholar]
- 16.Privalov P. L. (1979) Adv. Protein Chem. 33, 167–241 [DOI] [PubMed] [Google Scholar]
- 17.Piotto M., Saudek V., Sklenár V. (1992) J. Biomol. NMR 2, 661–665 [DOI] [PubMed] [Google Scholar]
- 18.Jahnke W., Bauer C., Gemmecker G., Kessler H. (1995) J. Magn. Reson. Ser. B 106, 86–88 [Google Scholar]
- 19.Dehner A., Furrer J., Richter K., Schuster I., Buchner J., Kessler H. (2003) ChemBioChem 4, 870–877 [DOI] [PubMed] [Google Scholar]
- 20.Ottiger M., Delaglio F., Bax A. (1998) J. Magn. Reson. 131, 373–378 [DOI] [PubMed] [Google Scholar]
- 21.Hansen M. R., Mueller L., Pardi A. (1998) Nat. Struct. Biol. 5, 1065–1074 [DOI] [PubMed] [Google Scholar]
- 22.Rückert M., Otting G. (2000) J. Am. Chem. Soc. 122, 7793–7797 [Google Scholar]
- 23.Goddard T. D., Kneller D. G. (2004) SPARKY3, University of California, San Francisco, CA [Google Scholar]
- 24.Zweckstetter M., Bax A. (2000) J. Am. Chem. Soc. 122, 3791–3792 [Google Scholar]
- 25.Schwieters C. D., Kuszewski J. J., Tjandra N., Clore G. M. (2003) J. Magn. Reson. 160, 65–73 [DOI] [PubMed] [Google Scholar]
- 26.Muchmore S. W., Sattler M., Liang H., Meadows R. P., Harlan J. E., Yoon H. S., Nettesheim D., Chang B. S., Thompson C. B., Wong S. L., Ng S. L., Fesik S. W. (1996) Nature 381, 335–341 [DOI] [PubMed] [Google Scholar]
- 27.Dominguez C., Boelens R., Bonvin A. M. (2003) J. Am. Chem. Soc. 125, 1731–1737 [DOI] [PubMed] [Google Scholar]
- 28.Schüttelkopf A. W., van Aalten D. M. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 1355–1363 [DOI] [PubMed] [Google Scholar]
- 29.Kleywegt G. J., Jones T. A. (1997) Methods Enzymol. 277, 525–545 [DOI] [PubMed] [Google Scholar]
- 30.Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 31.Laskowski R. A., Rullmann J. A., MacArthur M. W., Kaptein R., Thornton J. M. (1996) J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 32.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 33.Mayer M., Meyer B. (2001) J. Am. Chem. Soc. 123, 6108–6117 [DOI] [PubMed] [Google Scholar]
- 34.Dehner A., Klein C., Hansen S., Müller L., Buchner J., Schwaiger M., Kessler H. (2005) Angew. Chem. Int. Ed. Engl. 44, 5247–5251 [DOI] [PubMed] [Google Scholar]
- 35.Petros A. M., Nettesheim D. G., Wang Y., Olejniczak E. T., Meadows R. P., Mack J., Swift K., Matayoshi E. D., Zhang H., Thompson C. B., Fesik S. W. (2000) Protein Sci. 9, 2528–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strom E., Sathe S., Komarov P. G., Chernova O. B., Pavlovska I., Shyshynova I., Bosykh D. A., Burdelya L. G., Macklis R. M., Skaliter R., Komarova E. A., Gudkov A. V. (2006) Nat. Chem. Biol. 2, 474–479 [DOI] [PubMed] [Google Scholar]
- 37.Cañadillas J. M., Tidow H., Freund S. M., Rutherford T. J., Ang H. C., Fersht A. R. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 2109–2114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oltersdorf T., Elmore S. W., Shoemaker A. R., Armstrong R. C., Augeri D. J., Belli B. A., Bruncko M., Deckwerth T. L., Dinges J., Hajduk P. J., Joseph M. K., Kitada S., Korsmeyer S. J., Kunzer A. R., Letai A., Li C., Mitten M. J., Nettesheim D. G., Ng S., Nimmer P. M., O'Connor J. M., Oleksijew A., Petros A. M., Reed J. C., Shen W., Tahir S. K., Thompson C. B., Tomaselli K. J., Wang B., Wendt M. D., Zhang H., Fesik S. W., Rosenberg S. H. (2005) Nature 435, 677–681 [DOI] [PubMed] [Google Scholar]
- 39.Vassilev L. T., Vu B. T., Graves B., Carvajal D., Podlaski F., Filipovic Z., Kong N., Kammlott U., Lukacs C., Klein C., Fotouhi N., Liu E. A. (2004) Science 303, 844–848 [DOI] [PubMed] [Google Scholar]
- 40.Chipuk J. E., Green D. R. (2008) Trends Cell Biol. 18, 157–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Youle R. J. (2007) Science 315, 776–777 [DOI] [PubMed] [Google Scholar]
- 42.Chipuk J. E., Kuwana T., Bouchier-Hayes L., Droin N. M., Newmeyer D. D., Schuler M., Green D. R. (2004) Science 303, 1010–1014 [DOI] [PubMed] [Google Scholar]
- 43.Chittenden T., Flemington C., Houghton A. B., Ebb R. G., Gallo G. J., Elangovan B., Chinnadurai G., Lutz R. J. (1995) EMBO J. 14, 5589–5596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Willis S. N., Fletcher J. I., Kaufmann T., van Delft M. F., Chen L., Czabotar P. E., Ierino H., Lee E. F., Fairlie W. D., Bouillet P., Strasser A., Kluck R. M., Adams J. M., Huang D. C. S. (2007) Science 315, 856–859 [DOI] [PubMed] [Google Scholar]
- 45.Kuwana T., Mackey M. R., Perkins G., Ellisman M. H., Latterich M., Schneiter R., Green D. R., Newmeyer D. D. (2002) Cell 111, 331–342 [DOI] [PubMed] [Google Scholar]
- 46.Villunger A., Michalak E. M., Coultas L., Müllauer F., Böck G., Ausserlechner M. J., Adams J. M., Strasser A. (2003) Science 302, 1036–1038 [DOI] [PubMed] [Google Scholar]
- 47.Chipuk J. E., Bouchier-Hayes L., Kuwana T., Newmeyer D. D., Green D. R. (2005) Science 309, 1732–1735 [DOI] [PubMed] [Google Scholar]
- 48.Foster B. A., Coffey H. A., Morin M. J., Rastinejad F. (1999) Science 286, 2507–2510 [DOI] [PubMed] [Google Scholar]
- 49.Komarov P. G., Komarova E. A., Kondratov R. V., Christov-Tselkov K., Coon J. S., Chernov M. V., Gudkov A. V. (1999) Science 285, 1733–1737 [DOI] [PubMed] [Google Scholar]
- 50.Steele A. J., Prentice A. G., Hoffbrand A. V., Yogashangary B. C., Hart S. M., Lowdell M. W., Samuel E. R., North J. M., Nacheva E. P., Chanalaris A., Kottaridis P., Cwynarski K., Wickremasinghe R. G. (2009) Blood 114, 1217–1225 [DOI] [PubMed] [Google Scholar]
- 51.Schottelius M., Laufer B., Kessler H., Wester H. J. (2009) Acc. Chem. Res. 42, 969–980 [DOI] [PubMed] [Google Scholar]
- 52.Carlson C. B., Mowery P., Owen R. M., Dykhuizen E. C., Kiessling L. L. (2007) ACS Chem. Biol. 2, 119–127 [DOI] [PubMed] [Google Scholar]
- 53.Shuker S. B., Hajduk P. J., Meadows R. P., Fesik S. W. (1996) Science 274, 1531–1534 [DOI] [PubMed] [Google Scholar]
- 54.Davey P. G., Barza M. (1987) J. Antimicrob. Chemother. 20, 639–644 [DOI] [PubMed] [Google Scholar]
- 55.DeLano W. L. (2008) The PyMOL Molecular Graphics System, DeLano Scientific, Palo Alto, CA [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.