

Abstract
Reversible protein phosphorylation plays a key role in interleukin-2 (IL-2) receptor-mediated activation of Janus tyrosine kinase 3 (JAK3) and signal transducer and activator of transcription 5 (STAT5) in lymphocytes. Although the mechanisms governing IL-2-induced tyrosine phosphorylation and activation of JAK3/STAT5 have been extensively studied, the role of serine/threonine phosphorylation in controlling these effectors remains to be elucidated. Using phosphoamino acid analysis, JAK3 and STAT5 were determined to be serine and tyrosine-phosphorylated in response to IL-2 stimulation of the human natural killer-like cell line, YT. IL-2 stimulation also induced serine/threonine phosphorylation of IL-2Rβ, but not IL-2Rγ. To investigate the regulation of serine/threonine phosphorylation in IL-2 signaling, the roles of protein phosphatase 1 (PP1) and 2A (PP2A) were examined. Inhibition of phosphatase activity by calyculin A treatment of YT cells resulted in a significant induction of serine phosphorylation of JAK3 and STAT5, and serine/threonine phosphorylation of IL-2Rβ. Moreover, inhibition of PP2A, but not PP1, diminished IL-2-induced tyrosine phosphorylation of IL-2Rβ, JAK3, and STAT5, and abolished STAT5 DNA binding activity. Serine/threonine phosphorylation of IL-2Rβ by a staurosporine-sensitive kinase also blocked its association with JAK3 and IL-2Rγ in YT cells. Taken together, these data indicate that serine/threonine phosphorylation negatively regulates IL-2 signaling at multiple levels, including receptor complex formation and JAK3/STAT5 activation, and that this regulation is counteracted by PP2A. These findings also suggest that PP2A may serve as a therapeutic target for modulating JAK3/STAT5 activation in human disease.
Keywords: Cytokines/Interleukins, Phosphorylation/Kinases/Serine-Threonine, Phosphorylation/Kinases/Tyrosine, Phosphorylation/Phosphatases/Serine-Threonine, Phosphorylation/Phosphotyrosine Signals/Receptors, RECEPTORS/Leukocyte/Lymphocyte, Signal Transduction/Jak-STAT, Signal Transduction/Phosphoprotein Phosphatases/PP1/PP2A
Introduction
Interleukin-2 (IL-2)3 is a key regulator of normal immune function and acts on a variety of lymphoid cell types including T, B, and natural killer cells. IL-2 is critical for the activation and subsequent amplification of the immune response following antigenic stimulation, and in promoting the development of regulatory T cells while constraining Th17 cell polarization (1–3). To elicit these biological effects, IL-2 signals through the IL-2 receptor (IL-2R) complex. This complex is comprised of two essential signaling subunits, IL-2Rβ and IL-2Rγ, and one affinity modulating subunit, IL-2Rα. IL-2-induced heterodimerization of IL-2Rβ and IL-2Rγ results in activation of receptor-associated Janus tyrosine kinase (JAK) 1 and JAK3 through trans- or autophosphorylation (4, 5). Subsequent tyrosine phosphorylation of the IL-2Rβ chain provides docking sites for effector molecules including signal transducer and activator of transcription (STAT) 5a and STAT5b via their Src homology 2 domains (6). Human STAT5a and STAT5b are phosphorylated on the conserved tyrosine residues Tyr694 and Tyr699, respectively, which allows for their dissociation from the receptor complex, formation of hetero- or homodimers, and nuclear translocation to bind specific promoter elements that stimulate transcription of target genes that control cell growth and differentiation (7, 8). In addition to tyrosine phosphorylation, IL-2 induces serine phosphorylation of STAT5 within a Pro-Ser-Pro motif localized in the transactivation domain, which also serves to modulate transcription (9–11).
Many studies have focused on the mechanisms driving JAK/STAT activation, however, much less is known about its negative regulation. Members of the suppressor of cytokine signaling family are selectively induced upon cytokine stimulation to provide a classical negative feedback mechanism (12). In addition, protein inhibitor of activated STAT proteins negatively regulate STAT-dependent transcription by inhibiting DNA binding activity, recruiting transcriptional corepressors, and promoting protein sumoylation (13). Negative regulation of JAK/STAT activation is also achieved through dephosphorylation of key tyrosine residues by the tyrosine phosphatases CD45 (14), T cell—protein-tyrosine phosphatase (15), and Src homology 2 domain-containing protein-tyrosine phosphatase 2 (16). Serine/threonine phosphatases may also participate in the regulation of JAK/STAT activation. Indeed, our group and others have shown that inhibition of protein phosphatase types 1 (PP1) and 2A (PP2A) attenuates STAT3 (17) and STAT6 (18, 19) activity; however, their role in controlling IL-2-mediated STAT5 activation has not been determined. The present study was initiated to determine the role of PP1 and PP2A in IL-2-mediated activation of the JAK3/STAT5 signal transduction pathway in human lymphocytes. We provide novel evidence that PP2A, but not PP1, regulates IL-2 signaling at multiple levels, including IL-2R complex formation and downstream activation of JAK3 and STAT5.
EXPERIMENTAL PROCEDURES
Cell Culture and Treatment
The human YT cell line (20) was maintained in RPMI 1640 medium with 10% fetal bovine serum (Atlanta Biologicals) and stimulated in the absence or presence of 10,000 IU of human recombinant IL-2 (NCI Preclinical Repository) at 37 °C for the times indicated. Human peripheral blood mononuclear cells (PBMCs) were obtained from healthy donors, purified by isocentrifugation, and activated with phytohemagglutinin (PHA) (1 μg/ml) for 72 h, as previously described (21). Phosphatase inhibition studies employing calyculin A (CA), okadaic acid (OA), tautomycin (TAU), and cyclosporin A (CSA) (Sigma) were performed at 37 °C using the concentrations and time points indicated.
Antibodies and Other Reagents
The anti-JAK3 (22), anti-STAT5a (23), anti-STAT5b (24), anti-phospho-Ser726 STAT5a and Ser731 STAT5b (α-pS STAT5) (9) polyclonal antibodies were used as previously described. The anti-phosphotyrosine monoclonal 4G10 (α-pY) antibody was purchased from Upstate Biotechnology. The anti-phospho-Tyr694 STAT5a and Tyr699 STAT5b (pY STAT5) monoclonal antibody (Cell Signaling Technology) and the anti-IL-2Rβ/anti-IL-2Rγ chain antibodies (Santa Cruz Biotechnology) were used according to the manufacturer's protocol. The anti-PP2A catalytic subunit monoclonal antibody was purchased from BD Biosciences. Kinase inhibition studies with staurosporine (STS) (Sigma), KT5720 (Sigma), bisindoylymaleimide II (Sigma), PD98059 (New England Biolabs), wortmannin (Calbiochem), or rapamycin (Calbiochem) were performed for 1 h at 37 °C using the concentrations indicated.
Solubilization of Proteins, Immunoprecipitation, and Western Blot Analysis
Cells were pelleted, lysed, and subjected to immunoprecipitation and Western blot analysis as previously reported (25). For all samples, total protein was determined by the bicinchoninic acid method (Pierce). Western blot assays were developed with horseradish peroxidase-conjugated goat anti-mouse immunoglobulin G (IgG; heavy plus light chains) or goat anti-rabbit IgG (heavy plus light chains; KPL) and visualized by using enhanced chemiluminescence and x-ray film. When reblotting, polyvinylidene difluoride membranes were incubated with stripping buffer (100 mm β-mercaptoethanol, 2% SDS, 62.5 mm Tris-HCl, pH 6.7) at 55 °C for 30 min, blocked, and then reprobed.
[32P]Orthophosphate Labeling and Two-dimensional Phosphoamino Acid Analysis
YT cells (1.5 × 107) were metabolically labeled with [32P]orthophosphate (0.5 mCi) (PerkinElmer Life Sciences) for 2 h at 37 °C and left untreated or treated with 100 nm CA for 90 min, followed by stimulation in the absence or presence of 10,000 IU of IL-2 for 15 min. The cells were lysed, and IL-2Rβ, IL-2Rγ, JAK3, or STAT5a/b proteins were immunoprecipitated. All proteins were then separated by SDS-PAGE, transferred to polyvinylidene difluoride membrane, and visualized by Coomassie Blue staining and autoradiography. The corresponding proteins were then excised and subjected to two-dimensional phosphoamino acid analysis using the Hunter Thin Layer Electrophoresis apparatus (C.B.S. Scientific), as previously described (21).
Electrophoretic Mobility Shift Assay
Nuclear extracts were prepared and assays conducted as previously published (26). Oligonucleotide sequences corresponding to the β-casein gene promoter for STAT5 (5′-AGATTTCTAGGAATTCAATCC-3′) were obtained from Santa Cruz Biotechnology.
Phosphatase Activity Assay
The PP2A catalytic subunit was immunoprecipitated from YT cell lysate (500 μg) with 4 μg of anti-PP2Ac monoclonal antibody and incubated with 750 μm phosphopeptide KRpTIRR and p-nitrophenyl phosphate Ser/Thr assay buffer (Millipore) at 30 °C for 10 min according to the manufacturer's instructions. Twenty-five microliters from each reaction were mixed with 100 μl of malachite green phosphate detection solution (Millipore) and optical density measured at 650 nm. For each experiment, a standard curve for free phosphate (picomoles) was prepared to determine the amount of released phosphates generated in each sample set.
In Vitro Dephosphorylation Assay
YT cells (2.0 × 107) were incubated without or with 100 nm CA for 60 min prior to solubilization in lysis buffer (1% Triton X-100, 10 mm Tris-HCl pH 8.0, 50 mm NaCl). IL-2Rβ was immunoprecipitated and incubated without or with 0.5 units of purified PP1 or PP2A (Upstate Biotechnology) at 37 °C for 60 min according to the manufacturer's instructions. The reactions were stopped by addition of sample buffer containing 125 mm Tris-HCl, pH 6.8, 10% β-mercaptoethanol, 9.2% SDS, 0.04% bromphenol blue, 20% glycerol, and boiled for 5 min.
RESULTS
Inhibition of PP1/PP2A Activity in YT Cells Results in Serine Phosphorylation of STAT5a/b
Previously, we have identified serine phosphorylation sites within STAT5a and STAT5b that control their function (9, 27). Most of these efforts have focused on characterizing the kinases responsible for these responses, however, little is known about the phosphatases that tightly control this balance. To determine whether STAT5a/b phosphorylation is regulated by PP1 or PP2A activity, phosphoamino acid analysis was performed on STAT5 proteins isolated from YT cells that were metabolically labeled with [32P]orthophosphate and treated without or with the PP1/PP2A inhibitor CA for 60 min prior to stimulation with IL-2 for 10 min. STAT5a/b proteins were individually immunoprecipitated, separated by SDS-PAGE, and visualized by autoradiography (Fig. 1, A and B, upper panels). The STAT5a/b proteins were then excised and subjected to limited acid hydrolysis followed by phosphoamino acid analysis (Fig. 1, A and B, lower panels). This analysis demonstrated that compared with the untreated controls (lane a), CA treatment (lane b) resulted in a significant increase in STAT5a/b phosphorylation that was not further increased by IL-2 treatment (Fig. 1, A and B, upper panels). Phosphoamino acid analysis demonstrated that both proteins were phosphorylated on serine, but not threonine or tyrosine residues (Fig. 1, A and B, lower panels, group b). Additionally, as we previously reported and show herein, IL-2 induced the phosphorylation of both STAT5 proteins on serine and tyrosine, but not threonine, residues (10) (Fig. 1, A and B, group c). Importantly, CA pretreatment inhibited IL-2-induced tyrosine phosphorylation of STAT5a/b (Fig. 1, A and B, group d), which is normally associated with STAT5 activation. These results suggest that STAT5 activation is negatively regulated by serine phosphorylation and that PP1 and/or PP2A activity is involved in the regulation of STAT5 activation during IL-2 signaling.
FIGURE 1.

CA induces serine phosphorylation and inhibits IL-2-mediated tyrosine phosphorylation of STAT5a/b in human lymphocytes. YT cells (1.5 × 107) were labeled with [32P]orthophosphate for 2 h at 37 °C and treated without (lanes a and c) or with 100 nm CA (lanes b and d) for 60 min prior to stimulation in the absence (lanes a and b) or presence of IL-2 (lanes c and d) for 10 min. Cell lysates were then immunoprecipitated (IP) with α-STAT5a (A) or α-STAT5b (B), separated by SDS-PAGE, and subjected to autoradiography (upper panels). The corresponding STAT5 bands were excised and subjected to phosphoamino acid analysis (groups a–d) (lower panels). The position of phosphoserine, -threonine, and -tyrosine standards (pS, pT, and pY) were detected by ninhydrin as indicated (circles). C, YT cells were left untreated (lanes a and j), treated with CA for 60 min (lanes b and k), stimulated with IL-2 for 10 min (lanes c and l), or pretreated with 100 nm CA for 10–150 min prior to stimulation with IL-2 for 10 min (lanes d–i and m–r). STAT5a (upper panel) and STAT5b (lower panel) were immunoprecipitated and Western blotted (WB) with the antibodies indicated. D, YT cells were left untreated (lanes a and h), treated with 100 nm CA for 90 min (lanes b and i), stimulated with IL-2 for 10 min (lanes c and j), or pretreated with 30–200 nm CA for 90 min followed by stimulation with IL-2 for 10 min (lanes d–g and k–n) as indicated. STAT5a (upper panel) and STAT5b (lower panel) proteins were immunoprecipitated and analyzed by Western blot using the antibodies indicated. Representative data from three independent experiments are shown.
Inhibition of PP1/PP2A Activity in YT Cells Decreases IL-2-mediated Tyrosine Phosphorylation of STAT5a/b
To confirm that PP1/PP2A activity regulates IL-2-mediated tyrosine phosphorylation and activation of STAT5a/b, Western blot analysis was performed using STAT5 phosphospecific antibodies (Fig. 1, C and D). YT cells were treated with 100 nm CA for the indicated times before stimulation without or with IL-2. STAT5a (upper panels) and STAT5b (lower panels) were immunoprecipitated from soluble cell lysates, separated by SDS-PAGE, and subjected to Western blot analysis with the antibodies indicated (Fig. 1C). We observed that the IL-2-induced tyrosine phosphorylation of STAT5a (lanes c–i) and STAT5b (lanes l–r) was inhibited by CA pretreatment in a time-dependent manner (Fig. 1C). Specifically, the inhibitory effect of CA upon IL-2-induced STAT5a tyrosine phosphorylation was detectable after a 30-min pretreatment and was maximal after 150 min. Inhibition of STAT5b tyrosine phosphorylation required slightly longer CA pretreatment (60 min) and was also maximal at 150 min. Treatment of YT cells with CA for 60 min also induced the phosphorylation of STAT5a on Ser726 (lanes b) and STAT5b on Ser731 (lanes k). Phosphorylation of these specific serine residues within STAT5a (lanes d-i) and STAT5b (lanes m-r) remained constant throughout the CA treatment time course (Fig. 1C), whereas STAT5a (lane a) and STAT5b (lane j) immunopurified from non-CA and non-IL-2-treated cells did not display detectable levels of tyrosine or serine phosphorylation (Fig. 1C).
Dose-response analysis of CA effects on STAT5a/b phosphorylation was also performed. YT cells were treated with increasing amounts of CA (30–200 nm) for 90 min prior to stimulation with IL-2 for 10 min. Western blot analysis of immunopurified STAT5a (Fig. 1D, upper panel, lanes d–g) and STAT5b (lower panel, lanes k–n) demonstrated that IL-2-mediated tyrosine phosphorylation of the STAT5 proteins was inhibited by CA in a dose-dependent manner (Fig. 1D). The inhibitory effect of CA pretreatment on IL-2-induced STAT5a tyrosine phosphorylation was detectable at 30 nm and maximal at 200 nm, whereas inhibition of STAT5b tyrosine phosphorylation was detectable at 50 nm and maximum at 200 nm. Collectively, these data suggest that PP1/PP2A-regulated serine phosphorylation of STAT5 proteins modulates their tyrosine phosphorylation and activation induced by IL-2.
PP2A, but Not PP1 or PP2B, Regulates Serine Phosphorylation and IL-2-stimulated Tyrosine Phosphorylation of STAT5 in Human Lymphocytes
To further specify which phosphatase family regulates STAT5 serine phosphorylation, small molecule inhibitors with opposing specificities toward PP1 and PP2A were used. OA preferentially inhibits PP2A (28–30), whereas TAU targets PP1 (29–31). The PP2B inhibitor CSA was used as an additional control. YT cells were left untreated, stimulated with IL-2 for 30 min, or treated with inhibitory concentrations of TAU (1 μm), OA (250 nm), CA (100 nm), or CSA (250 nm) for 60 min before lysis. Both STAT5 proteins were immunoprecipitated with a pan-STAT5 antibody and analyzed by Western blotting for phosphorylation on serine 726/731 (Fig. 2A, upper panel). Compared with the untreated control (lane a), inhibition of PP2A (lane d) and PP1/PP2A (lane e), but not PP1 (lane c) or PP2B (lane f) induced a high level of STAT5 serine phosphorylation that was nearly equivalent to that stimulated by IL-2 (Fig. 2A). The membrane was stripped and reprobed for total STAT5 levels to confirm equivalent loading (Fig. 2A, lower panel). To ensure PP2A catalytic activity was reduced in YT cells treated with the above concentrations of phosphatase inhibitors, a malachite green phosphatase assay employing a synthetic phosphopeptide substrate was performed (Fig. 2B, upper panel). Compared with the untreated control, CA (57.6%) or OA (74.2%), but not TAU (18.7%) or CSA (6.4%) treatment resulted in a significant inhibition of PP2A activity in YT cells. YT cell lysate was analyzed by Western blotting for the catalytic subunit of PP2A to confirm the equivalent protein input into the phosphatase assay (Fig. 2B, lower panel).
FIGURE 2.

PP2A, but not PP1 or PP2B, activity is required for IL-2-induced tyrosine phosphorylation of STAT5. A, YT cells were left untreated (lane a), stimulated with IL-2 for 30 min (lane b), or treated with inhibitory concentrations of the appropriate protein phosphatase uncoupler that included 1 μm TAU (lane c), 250 nm OA (lane d), 100 nm CA (lane e), or 250 nm CSA (lane f) for 60 min. STAT5 proteins were immunoprecipitated (IP), separated by SDS-PAGE, and analyzed by Western blot (WB) using the antibodies indicated. B, YT cells were left untreated or treated with 1 μm TAU, 250 nm OA, 100 nm CA, or 250 nm CSA for 60 min. PP2Ac proteins were immunoprecipitated and activity measured by serine/threonine phosphatase assays (upper panel). Normal mouse antiserum (IgG Cntrl) was used as a negative control for IP. The experiment was performed in triplicate where values are mean ± S.D. of free phosphate levels. Cell lysate was probed by Western blot using anti-PP2Ac to ensure equal protein input (lower panel). C, YT cells were left untreated (lanes a), stimulated with IL-2 for 30 min (lanes b), or treated with increasing concentrations of OA (10–200 nm) for 90 min followed by stimulation in the absence (lanes c–h, upper panel) or presence of IL-2 for 10 min (lanes i–n, upper panel). YT cells were similarly treated with CA (50–200 nm) or TAU (0.5–4 μm) for 90 min followed by stimulation in the absence (lanes c–e and i–k, respectively, lower panel) or presence of IL-2 for 30 min (lanes f–h and l–n, respectively, lower panel). STAT5 proteins were immunoprecipitated and analyzed by Western blot using the antibodies indicated. Representative data from three independent experiments are shown.
These experiments indicate that PP2A, but not PP1, is the primary phosphatase responsible for CA-mediated inhibition of IL-2-induced STAT5 activation. To confirm this, α-pY STAT5 Western blot analysis was performed on YT cells treated with increasing amounts of OA (10–200 nm), CA (100–200 nm), or TAU (0.5–4 μm) for 90 min before IL-2 stimulation for 30 min (Fig. 2C). In these experiments, low concentrations of OA that are not expected to inhibit PP1 (upper panel, lanes c–n) clearly blocked STAT5 tyrosine phosphorylation. CA treatment (lower panel, lanes c–h) resulted in a complete abrogation of IL-2-induced STAT5 tyrosine phosphorylation at all concentrations tested (Fig. 2C). This was expected because CA is equally potent at blocking PP1 and PP2A. There was no detectable inhibition of IL-2-mediated STAT5 tyrosine phosphorylation using the PP1 specific inhibitor TAU (Fig. 2C, lower panel, lanes i–n). Taken together, these results suggest that PP2A is the primary phosphatase responsible for the regulation of IL-2-mediated activation of STAT5 (8).
Treatment of YT Cells with CA Attenuates IL-2-induced STAT5 DNA Binding Activity
Tyrosine-phosphorylated STAT5 proteins translocate to the nucleus where they bind to consensus DNA sequences and regulate the transcription of target genes. To test whether PP2A activity is required for IL-2-induced STAT5 DNA binding activity, electrophoretic mobility shift analysis was performed using a [γ-32P]ATP-labeled STAT5 DNA binding element incubated with nuclear protein extracts isolated from IL-2-stimulated YT cells pretreated with 100 nm CA for 90 min. As shown in Fig. 3, compared with IL-2 stimulation alone, CA pretreatment significantly decreased the formation of the STAT5-DNA complex (lanes d and e). The presence of STAT5 within this complex was confirmed by incubating the extracts with antisera specific for the N or C terminus of STAT5. This resulted in a supershift of the complex, indicating that STAT5 was primarily responsible for its formation (lanes f and g). The specificity of the antisera response was confirmed using normal rabbit serum (lane h), which failed to supershift the STAT5-DNA complex (Fig. 3). Minimal STAT5 DNA binding activity was detected in non-IL-2 stimulated (lane b) or CA treated (lane c) YT cells. A reaction without nuclear extract (free probe, lane a) served as a negative control for DNA binding. Taken together, these data indicate that PP2A-regulated serine phosphorylation negatively impacts IL-2-induced STAT5 activation in YT cells.
FIGURE 3.

STAT5 DNA binding activity is reduced by CA treatment in YT cells. Cells were left untreated (lane b), treated with 100 nm CA for 90 min (lane c), stimulated with IL-2 for 10 min (lanes d and f–h), or pretreated with 100 nm CA for 90 min followed by stimulation with IL-2 for 10 min (lane e) at 37 °C. Nuclear extracts (5 μg) were incubated with a 32P-radiolabeled oligonucleotide probe corresponding to the STAT5 binding site in the β-casein gene promoter. The extracts indicated were coincubated with N-terminal directed α-STAT5 (lane f), C-terminal directed α-STAT5 (lane g), or normal rabbit serum (IgG cntrl) (lane h). Brackets indicate location of nonsupershifted and supershifted STAT5-DNA complexes. Representative data from two independent experiments are shown.
CA Inhibits IL-2-induced Activation of STAT5 and JAK3 in Primary Human PBMCs
To confirm that CA-mediated inhibition of PP1/PP2A affects STAT5 activation in non-transformed cells, tyrosine phosphorylation of STAT5 and its upstream activator JAK3 were assessed in primary human lymphocytes (PBMCs). For this analysis, PBMCs were activated with PHA for 72 h, made quiescent for 24 h, and pretreated with 100 nm CA for 90 min and then stimulated with IL-2 for 10 min. Western blot analysis of soluble cell lysates confirmed that pretreatment with CA significantly inhibited IL-2-mediated tyrosine phosphorylation of STAT5 (Fig. 4A, lanes c and d). Importantly, Western blot analysis of immunopurified JAK3 proteins from PBMC lysates demonstrated that CA treatment significantly reduced IL-2-mediated tyrosine phosphorylation of JAK3 (Fig. 4B, lanes c and d). These data indicate that CA-mediated inhibition of the IL-2 signaling cascade is not limited to the YT tumor cell line but also occurs in primary human lymphocytes. Furthermore, in contrast to previous reports indicating that PP2A acts downstream of JAKs during IL-4 signaling (18), these results suggest that PP1/PP2A control IL-2-stimulated JAK3 activation directly.
FIGURE 4.

CA inhibits IL-2-mediated tyrosine phosphorylation of STAT5 and JAK3 in primary human lymphocytes and induces serine phosphorylation of JAK3 in YT cells. A, quiescent PHA-activated normal human PBMCs were left untreated (lane a), treated with 100 nm CA for 60 min (lane b), stimulated with IL-2 for 10 min (lane c), or pretreated with 100 nm CA for 90 min prior to stimulation with IL-2 for 10 min (lane d). Total STAT5 and tyrosine-phosphorylated STAT5 were detected by Western blot (WB) analysis of cell lysates as indicated. B, YT cells were treated as described in A, lysed, and JAK3 was immunoprecipitated (IP) and analyzed by Western blot using the indicated antibodies. C, YT cells (1.5 × 107) were labeled with [32P]orthophosphate for 2 h at 37 °C and treated without (lanes a and c) or with 100 nm CA (lanes b and d) for 60 min prior to stimulation in the absence (lanes a and b) or presence (lanes c and d) of IL-2 for 10 min. Cell lysates were immunoprecipitated with α-JAK3, separated by SDS-PAGE, and subjected to autoradiography (left panel). Phosphoamino acid analysis was performed on the corresponding JAK3 bands (lanes a–d) (right panel). The position of phosphoserine, -threonine, and -tyrosine standards (pS, pT, and pY) were detected by ninhydrin as indicated (circles). D, YT cells were left untreated (lane a), treated with 100 nm CA for 60 min (lane b), stimulated with 100 nm IL-2 for 10 min (lane c), or pretreated with 100 nm CA for 10–150 min followed by stimulation with IL-2 for 10 min (lanes d–i). JAK3 was immunoprecipitated from cell lysates and analyzed by Western blot using the antibodies indicated. Representative data from two independent experiments are shown.
CA Treatment of YT Cells Induces Serine Phosphorylation and Inhibits IL-2-mediated Tyrosine Phosphorylation of JAK3
To substantiate PP1/PP2A regulation of JAK3, its phosphorylation status in YT cells stimulated with IL-2 in the absence or presence of CA was measured. Cells were metabolically labeled with [32P]orthophosphate and treated with 100 nm CA for 60 min prior to stimulation with IL-2 for 10 min. JAK3 was immunoprecipitated, separated by SDS-PAGE, and visualized by autoradiography (Fig. 4C, left panel). Isolated JAK3 was then excised and subjected to phosphoamino acid analysis (Fig. 4C, right panel). Compared with the control, CA treatment increased serine, but not threonine or tyrosine phosphorylation of JAK3 (Fig. 4C, groups a and b). Similar to the results obtained with STAT5 (Fig. 1), IL-2-induced phosphorylation of JAK3 occurred on both serine and tyrosine, but not threonine residues (Fig. 4C, group c), whereas CA pretreatment inhibited IL-2-induced tyrosine phosphorylation (Fig. 4C, group d).
To further investigate the effects of CA on IL-2-mediated JAK3 activation, Western blot analysis using phosphospecific antibodies was performed on YT cells pretreated with CA for increasing periods of time (Fig. 4D). Cells were treated with 100 nm CA for the indicated times before stimulation with IL-2. Endogenous JAK3 tyrosine phosphorylation was then tested by Western blotting (Fig. 4D). Indeed, IL-2-induced tyrosine phosphorylation of JAK3 was inhibited by CA treatment (lanes c–i) in a time-dependent manner (Fig. 4D). Specifically, the inhibitory effect of CA was detectable at 30 min pretreatment and maximal at 150 min. The membrane was stripped and reprobed for total JAK3 to ensure equal loading (Fig. 4D, lower panel). Taken together, these results suggest that the inhibitory effect of PP2A on IL-2 signaling is not limited to the direct regulation of STAT5, but also its upstream activator, JAK3.
CA Inhibits IL-2-induced Tyrosine Phosphorylation of IL-2Rβ in Primary Human PBMCs
To explore whether PP1/PP2A regulation of IL-2 signaling is restricted to JAK3 and STAT5, the effect of CA on IL-2Rβ tyrosine phosphorylation in primary lymphocytes was studied. PBMCs were activated with PHA for 72 h, made quiescent for 24 h, and pretreated with 100 nm CA for 90 min followed by a 10-min stimulation with IL-2. Western blot analysis of immunoprecipitated IL-2Rβ revealed that pretreatment with CA abrogated IL-2-stimulated tyrosine phosphorylation of IL-2Rβ (Fig. 5A, lanes c and d). Intriguingly, CA treatment resulted in a significant reduction in IL-2Rβ mobility during SDS-PAGE (lanes b and d). Retarded gel mobility during SDS-PAGE is a commonly observed consequence of protein phosphorylation (10, 23, 32).
FIGURE 5.

IL-2-mediated tyrosine phosphorylation of IL-2Rβ is inhibited by CA pretreatment in primary human lymphocytes and YT cells. A, quiescent PHA-activated normal human PBMCs were left untreated (lane a), treated with 100 nm CA for 60 min (lane b), stimulated with IL-2 for 10 min (lane c), or pretreated with 100 nm CA for 90 min prior to stimulation with IL-2 for 10 min (lane d). The cells were lysed, the IL-2Rβ was immunoprecipitated (IP) and analyzed by Western blot (WB) using the indicated antibodies. B and C, YT cells (1.5 × 107) were labeled with [32P]orthophosphate for 2 h at 37 °C and treated without (lanes a and c) or with 100 nm CA (lanes b and d) for 60 min prior to stimulation in the absence (lanes a and b) or presence (lanes c and d) of IL-2 for 10 min. IL-2Rβ was immunoprecipitated from cell lysates, separated by SDS-PAGE, and subjected to autoradiography (left panel). Phosphoamino acid analysis was performed on the corresponding IL-2Rβ bands (lanes a–d) (right panel). The position of phosphoserine, -threonine, and -tyrosine standards (pS, pT, and pY) were detected by ninhydrin as indicated (circles). C, YT cells were left untreated (lane a), treated with 100 nm CA for 60 min (lane b), stimulated with 100 nm IL-2 for 10 min (lane c), or pretreated with 100 nm CA for 10–150 min followed by stimulation with IL-2 for 10 min (lanes d–i). IL-2Rβ IPs were assessed by Western blot analysis using the indicated antibodies. Representative data from three independent experiments are shown.
CA Induces Serine and Threonine Phosphorylation of IL-2Rβ and Disrupts Its IL-2-mediated Tyrosine Phosphorylation
To explore PP1/PP2A regulation of IL-2 signaling at the receptor level, the phosphorylation status of the IL-2 receptor subunits in YT cells pretreated with CA were investigated. Cells were metabolically labeled with [32P]orthophosphate and treated without or with 100 nm CA for 60 min prior to stimulation without or with IL-2 for 10 min. IL-2Rβ was immunoprecipitated from corresponding cell lysates, separated by SDS-PAGE, and visualized by autoradiography (Fig. 5B, left panel). The corresponding IL-2Rβ protein was excised and subjected to phosphoamino acid analysis (Fig. 5B, right panel). As seen in Fig. 5B, compared with the control, CA treatment resulted in the incorporation of phosphate onto serine and threonine, but not tyrosine residues of IL-2Rβ (Fig. 5B, groups a and b). IL-2-induced phosphorylation of IL-2Rβ occurred on serine, threonine, and tyrosine residues as previously reported (33) (Fig. 5B, group c). In agreement with our Western blot analysis, CA pretreatment inhibited IL-2-induced tyrosine phosphorylation (Fig. 5B, group d). Phosphorylation of IL-2Rγ was not detected following CA treatment of YT cells (data not shown). This was not surprising because IL-2Rβ, not IL-2Rγ, is the major link to connect signaling pathways to the receptor complex and thus highly phosphorylated in response to IL-2 stimulation (34). Phosphorylation of IL-2Rα was not examined, however, PP1/PP2A action upon this receptor subunit is not predicted due to its minimal intracellular region and negligible role in IL-2R signal transduction (35).
To confirm that CA treatment negatively regulates IL-2-mediated tyrosine phosphorylation of IL-2Rβ, a kinetic analysis of CA effects on this receptor subunit was performed. YT cells were treated with 100 nm CA for the indicated times before stimulation with IL-2 for 10 min. IL-2Rβ was immunoprecipitated from soluble cell lysates, separated by SDS-PAGE, and subjected to Western blot analysis with the antibodies indicated (Fig. 5C). In accordance with the above observations, IL-2-induced tyrosine phosphorylation of IL-2Rβ (lanes c–i) was reduced by CA pretreatment at 10 min and reached maximum inhibition at 90 min (Fig. 5C). The membrane was stripped and reprobed for total IL-2Rβ to ensure equal gel loading. Consistent with the above observations, CA treatment resulted in a significant reduction in IL-2Rβ electrophoretic mobility as compared with the untreated control (Fig. 5C, lanes a and b).
PP2A, but Not PP1 or PP2B, Inhibition Induces Electrophoretic Mobility Shift of IL-2Rβ
To elucidate the primary phosphatase responsible for regulation of IL-2Rβ phosphorylation, inhibitors with opposing specificity toward PP1 or PP2A were investigated. For this analysis, YT cells were left untreated (lane a) or incubated with inhibitors of PP1 (1 μm TAU) (lane b), PP2A (250 nm OA) (lane c), PP1/PP2A (100 nm CA) (lane d), or PP2B (250 nm CSA) (lane e) for 60 min before being lysed and subjected to IL-2Rβ immunoprecipitation and Western blot analysis (Fig. 6A). Compared with the untreated control, OA and CA, but not TAU or CSA treatment altered the electrophoretic mobility of IL-2Rβ (Fig. 6A). These results suggest that PP2A activity is primarily responsible for regulation of IL-2Rβ serine/threonine phosphorylation in YT cells. To further differentiate the role of PP2A from PP1 in the regulation of IL-2Rβ phosphorylation, an in vitro phosphatase assay was performed. YT cells were pretreated without or with 100 nm CA for 60 min before stimulation in the absence or presence of IL-2 for 10 min. Endogenous IL-2Rβ was immunoprecipitated from soluble cell lysates and subjected to in vitro dephosphorylation using purified PP1 or PP2A enzymes prior to SDS-PAGE and Western blot analysis. As shown in Fig. 6B, compared with the untreated controls (lanes a–c), dephosphorylation using purified PP2A reversed the CA- (lane i) and IL-2-induced (lane h) electrophoretic mobility shift of IL-2Rβ. In contrast, purified PP1 failed to revert the CA-induced altered electrophoretic mobility of IL-2Rβ (Fig. 6B, lanes d–f). Taken together, these data indicate that inhibition of PP2A, but not PP1 or PP2B induces the serine/threonine phosphorylation of IL-2Rβ in vivo and that PP2A directly dephosphorylates IL-2Rβ in vitro.
FIGURE 6.

PP2A directly regulates IL-2Rβ serine phosphorylation by a staurosporine-sensitive kinase. A, YT cells were incubated in the absence (lane a) or presence of TAU (1 μm) (lane b), OA (250 nm) (lane c), CA (100 nm) (lane d), or CSA (250 nm) (lane e) for 60 min. IL-2Rβ was immunoprecipitated (IP), separated by SDS-PAGE, and analyzed by Western blot (WB) as indicated. B, YT cells were incubated in the absence (lanes a, b, d, e, g, and h) or presence (lanes c, f, and i) of CA (100 nm) for 60 min prior to stimulation with IL-2 (lanes b, e, and h) for 10 min. IL-2Rβ was immunoprecipitated and left untreated (lanes a–c) or subjected to dephosphorylation using purified PP1 (lanes d–f) or PP2A (lanes g–i) for 60 min at 37 °C before separation by SDS-PAGE and Western blot analysis as indicated. C, YT cells were left untreated (lanes a and b) or treated with staurosporine (500 nm) (lanes c and d), KT5720 (5 μm) (lanes e and f), bisindoylymaleimide II (BIM) (250 nm) (lanes g and h), PD98059 (250 μm) (lanes i and j), wortmannin (50 μm) (WTMN) (lanes k and l), or rapamycin (100 nm) (RAPA) (lanes m and n) for 60 min at 37 °C prior to treatment with or without CA (100 nm) for an additional 60 min. Cells were lysed and analyzed by Western blot with α-IL-2Rβ. * indicates inhibition of CA altered electrophoretic mobility shift. Representative data from three independent experiments are shown.
STS Inhibits CA-induced Serine/Threonine Phosphorylation of IL-2Rβ
To identify the putative kinase(s) responsible for serine/threonine phosphorylation of the IL-2Rβ subunit, YT cells were incubated with inhibitors of candidate serine/threonine kinases known to participate in cytokine signaling prior to CA treatment. In these assays inhibitors of PKC (250 nm bisindoylymaleimide II) (lanes g and h), MEK1/2 (250 μm PD98059) (lanes i and j), phosphoinositide 3-kinase (PI3K) (50 μm wortmannin) (lanes k and l), and mTOR (100 nm rapamycin) (lanes m and n) had no visible effect on CA induced mobility of IL-2Rβ (Fig. 6C). However, compared with the untreated controls (lanes a and b), preincubation with 500 nm STS (lanes c and d), a broad spectrum inhibitor of serine/threonine kinases (36), blocked the CA-induced mobility shift of IL-2Rβ during SDS-PAGE (Fig. 6C). In addition, incubation with a PKA inhibitor (KT5720, 5 μm) (lanes e and f) also partially blocked the CA-induced electrophoretic mobility shift of IL-2Rβ, suggesting a previously unidentified role for this kinase in IL-2R signaling (Fig. 6C). Taken together, these results indicate that a STS-sensitive serine/threonine kinase is able to phosphorylate IL-2Rβ and possibly uncouple JAK3 and STAT5 activation.
IL-2 Receptor Complex Formation Is Disrupted by CA Treatment in YT Cells
IL-2 binding promotes the formation of a heterotrimeric receptor complex that consists of two essential subunits, IL-2Rβ and IL-2Rγ, and one affinity modulating subunit, IL-2Rα. Failure in the assembly of IL-2Rβ and IL-2Rγ upon stimulation with IL-2 results in the blockade of downstream signaling components (10, 37). Because CA rapidly inhibited IL-2-induced IL-2Rβ tyrosine phosphorylation, we examined the effect of CA treatment on the assembly of the IL-2R complex in YT cells. These cells were treated without or with 100 nm CA for 90 min and then stimulated with IL-2 for 10 min. The IL-2Rβ and IL-2Rγ subunits were then individually immunoprecipitated from soluble cell lysates and probed for the association of the reciprocal receptor subunit and JAK3 by Western blot analysis (Fig. 7). Compared with the untreated control (lane a), IL-2 stimulation (lane c) resulted in co-immunoprecipitation of JAK3 with IL-2Rβ and IL-2Rγ. Importantly, CA pretreatment (lane d) inhibited the association of IL-2Rβ and IL2Rγ, and abolished the binding of JAK3 (Fig. 7A). Similarly, the IL-2-induced co-immunoprecipitation of JAK3 with IL-2Rβ was prevented following pretreatment of YT cells with CA (Fig. 7B, lanes c and d). Membranes were stripped and reprobed for IL-2Rγ (Fig. 7A) and IL-2Rβ (Fig. 7B) to ensure equal loading. Cell lysates were also probed for IL-2Rβ (Fig. 7A) and JAK3 (Fig. 7B) to ensure equal input for the immunoprecipitation assays. These results suggest that PP2A activity is required for the assembly of an active IL-2R complex, in addition to regulating the phosphorylation and activation of JAK3 and STAT5.
FIGURE 7.
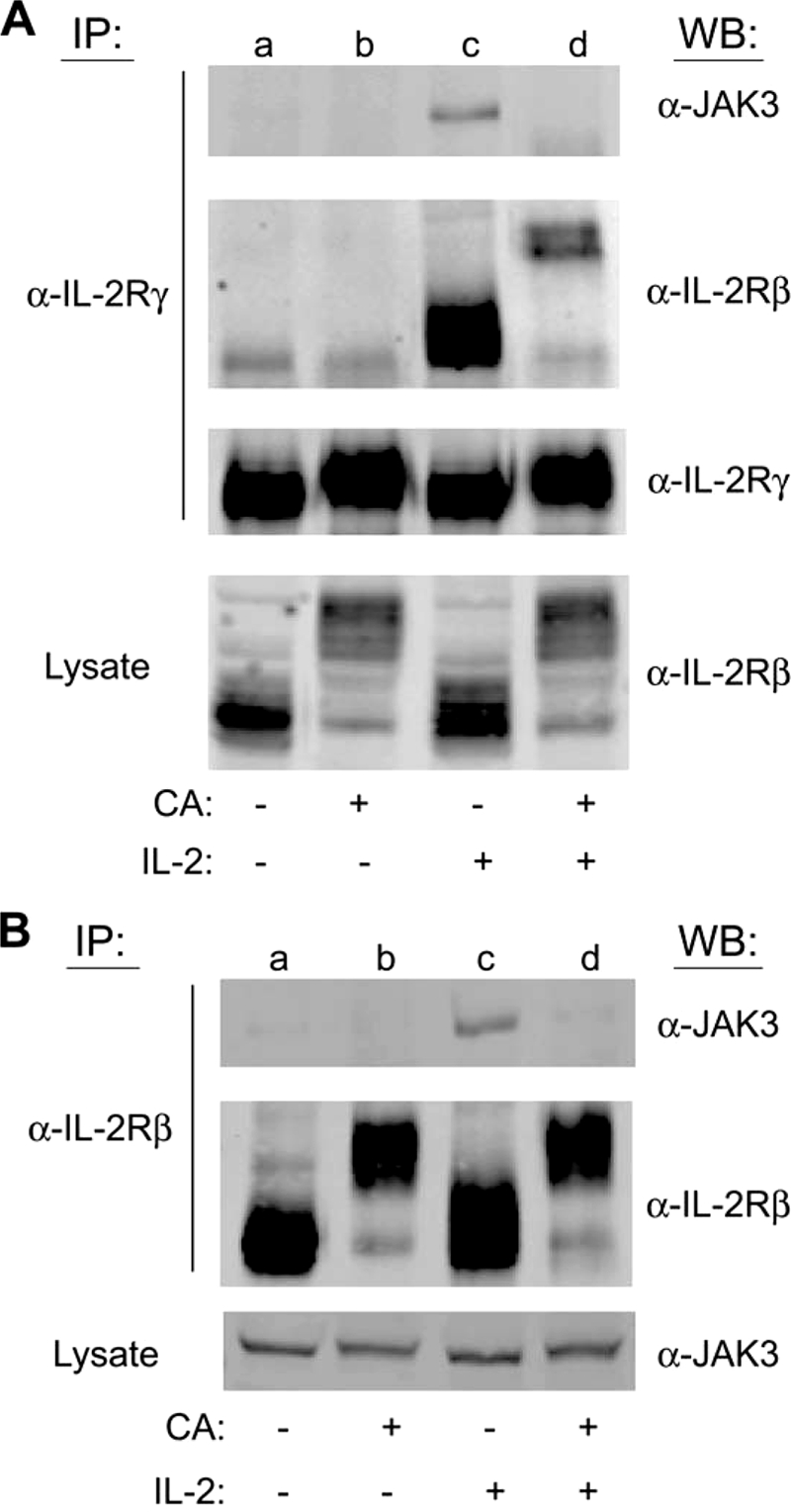
PP2A-regulated serine/threonine phosphorylation uncouples IL-2 receptor complex formation in human lymphocytes. A, YT cells were left untreated (lane a), treated with 100 nm CA for 90 min (lane b), stimulated with IL-2 for 30 min (lane c) or pretreated with 100 nm CA for 90 min followed by stimulation with IL-2 for 30 min (lane d). Cell lysates were then immunoprecipitated (IP) with IL-2Rγ antibody and separated by SDS-PAGE. Co-immunoprecipitation of JAK3 and IL-2Rβ was detected by Western blot (WB) analysis as indicated. Total cell lysate was also probed to determine equal IL-2Rβ input levels (B). YT cell lysates from identical treatments as described in A were immunoprecipitated with IL-2Rβ antibody and separated by SDS-PAGE. Co-immunoprecipitation of JAK3 was detected by Western blot analysis as indicated. Total cell lysate was also probed to determine equal JAK3 input levels. Representative data from two independent experiments are shown.
DISCUSSION
The results presented herein provide direct evidence that serine/threonine phosphorylation functions as an important negative regulator of IL-2 receptor signaling in human lymphocytes and that this is counteracted by the actions of PP2A. Using phosphoamino acid analysis, it was demonstrated that in addition to tyrosine phosphorylation, IL-2 stimulation of YT cells induces serine phosphorylation of JAK3/STAT5 and serine/threonine phosphorylation of IL-2Rβ. Furthermore, inhibition of serine/threonine phosphatase activity by CA treatment of YT cells resulted in serine phosphorylation of JAK3/STAT5 and serine/threonine phosphorylation of IL-2Rβ. CA also attenuated the IL-2-mediated tyrosine phosphorylation of IL-2Rβ, JAK3, and STAT5 in YT and PHA-activated primary human PBMCs. Using pharmalogical inhibitors specific for particular phosphatases and in vitro dephosphorylation assays, PP2A, but not PP1 or PP2B, was ascertained to be primarily responsible for regulating IL-2 receptor signaling in YT cells. Interestingly, serine/threonine phosphorylation of IL-2Rβ was independent of ERK1/2, PI3K, PKC, or mTOR activation and instead mediated, in part, by a STS-sensitive kinase. To delineate the mechanism by which PP2A regulates IL-2-induced activation of JAK3/STAT5 at the receptor level, co-immunoprecipitation experiments were performed to analyze receptor complex formation. Pretreatment of YT cells with CA greatly reduced IL-2-induced association of IL-2Rβ with IL-2Rγ and disrupted the binding of JAK3 to the receptor subunits. Taken together, these findings support the role of PP2A in IL-2R complex formation and JAK3/STAT5 activation, which represents a previously unrecognized negative regulatory mechanism that may reveal novel therapeutic targets to uncouple these critical regulators of lymphocyte proliferation, survival, and function.
Reversible tyrosine phosphorylation is a fundamental mechanism for controlling IL-2 signal propagation via JAK3/STAT5 activation (reviewed in Refs. 38 and 39). Our results indicate that serine/threonine kinases and phosphatases provide additional regulatory mechanisms that modulate IL-2R signal transduction. Although serine phosphorylation has been implicated in the regulation of STAT5 (9, 10, 40), to our knowledge the results presented herein provide the first evidence that JAK3 serine and IL-2Rβ serine/threonine phosphorylation controls their activities in lymphocytes (Fig. 5). Furthermore, IL-2 stimulation of YT cells induces serine phosphorylation of JAK3 and serine/threonine phosphorylation of IL-2Rβ, indicating that this may be a physiological negative feedback mechanism to regulate activation in lymphocytes (Fig. 5). This notion is supported by reports of JAK2 phosphorylation on non-conserved residue Ser523, which was shown to function as a negative regulatory site to dampen the growth hormone and epidermal growth factor response (41).
The roles of many serine/threonine kinases in immune cell development, activation, and effector functions are well established (reviewed in Refs. 42–44), however, these kinases are not currently known to negatively regulate IL-2 receptor signaling directly. To identify the kinase(s) responsible for regulation of IL-2Rβ serine/threonine phosphorylation, we investigated the role of known IL-2-regulated serine/threonine kinases, including ERK1/2, PI3K, PKC, mTOR, and p70S6K (45, 46). However, we did not observe that any of these kinases were responsible for CA-stimulated phosphorylation of IL-2Rβ. Specifically, CA-induced IL-2Rβ phosphorylation proved refractory to kinase inhibition by bisindoylymaleimide II, PD98059, wortmannin, or rapamycin, which inhibit PKC, ERK1/2, PI3K, and mTOR, respectively (Fig. 6). Instead, the findings presented herein indicate that inhibition of PP2A allows for serine/threonine phosphorylation of IL-2Rβ via an as yet unidentified STS-sensitive kinase(s). Interestingly, phosphorylation site prediction analysis of the intracellular region of the IL-2Rβ chain using NetPhos (47) and NetPhosK (48) programs predicts 18 of 30 serine residues and 5 of 12 threonine residues are putative phosphoacceptor sites within human IL-2Rβ. However, a number of kinases are predicted to phosphorylate these residues with overlapping specificity, including PKA, PKC, p38 MAPK, GSK3 (glycogen synthase kinase 3), CKI, CKII, CDC2, CDK5, ATM, and DNA-protein kinase, which suggests a complex interplay among multiple regulatory pathways. Indeed, PKA inhibition partially inhibited the CA-induced electrophoretic mobility shift of IL-2Rβ, suggesting that cAMP-mediated activation of PKA can regulate IL-2R signaling (Fig. 6). It is well established that cAMP is a potent modulator of the immune system (reviewed in Ref. 49), however, its negative regulation of T cell activation is thought to primarily occur via PKA-mediated inhibition of lymphocyte-specific protein-tyrosine kinase activation during T cell receptor engagement (50). These results suggest that PKA is also able to negatively regulate T cell proliferation through inhibition of IL-2R signaling, however, further investigation is required to confirm this hypothesis.
PP2A is a heterotrimeric enzyme that achieves specificity in the cell through the actions of its regulatory/targeting subunits (51). The PP2A heterotrimer is composed of a catalytic (C) subunit bound to a structural A subunit and a regulatory B subunit (52). There are a large number of regulatory subunits that can bind to this core A/C dimer, and their actions control the activity, specificity, selectivity, and cellular localization of PP2A (53, 54). Indeed, through different combinations of these subunits, the cell is capable of assembling over 200 biochemically distinct PP2A complexes (55–57). In addition, phosphorylation of the C subunit of PP2A on Tyr307 by tyrosine kinases such as SRC, lymphocyte-specific protein-tyrosine kinase, epidermal growth factor receptor, insulin receptor, and JAK2 can actively inhibit PP2A phosphatase activity (55, 56, 58). Interestingly, it has been reported that IL-2 stimulation of T cells results in the down-regulation of PP2A activity (59), suggesting that IL-2R signaling can modulate the function of PP2A. Therefore, it is tempting to speculate that IL-2 induced activation of JAK3 results in tyrosine phosphorylation and functional inhibition of PP2A, which in turn promotes serine phosphorylation of JAK3, STAT5, and IL-2Rβ. This would provide a negative feedback mechanism for IL-2R signaling.
Others have previously shown that CA treatment of HepG2 cells promotes the phosphorylation and degradation of the IL-6 family cytokine receptor gp130 (60). However, this does not hold true for IL-2Rβ in YT cells (Fig. 4C). We propose that serine/threonine phosphorylation of IL-2Rβ may result in conformational and electrostatic changes that destabilize the IL-2 receptor signaling complex, thus preventing the dimerization of IL-2 receptor subunits and their association with JAK3 and possibly JAK1. The resulting defect in IL-2 receptor complex assembly prevents the formation of docking sites for JAK3 and subsequent activation of downstream proteins such as STAT5. It is important to note that although global PP2A-regulated phosphorylation attenuated IL-2 activation of JAK3/STAT5, we cannot rule out that individual phosphoserine/threonine residues may positively regulate this pathway under certain conditions. To more fully elucidate this mechanism, challenging future work will be required to identify and functionally evaluate the PP2A regulatory sites targeted in IL-2Rβ, JAK3, and STAT5 as well as identify the regulatory subunit(s) required for PP2A dephosphorylation of these acceptor sites.
In summary, the results presented herein provide direct evidence that in addition to tyrosine phosphorylation, serine/threonine kinases and phosphatases act in concert to regulate IL-2 activation of JAK3/STAT5 in human lymphocytes. Specifically, PP2A-regulated serine/threonine phosphorylation negatively regulated IL-2 signaling at multiple levels, from IL-2R complex formation to downstream activation of JAK3 and STAT5. Although several molecules have been shown to negatively control JAK/STAT activation, these proteins are almost exclusively dependent upon tyrosine phosphorylation. These results further define the intricate balance that serine/threonine/tyrosine phosphorylation provides to modulate signaling pathways in lymphocytes. Through better understanding of these cross-talk pathways novel therapeutic strategies are likely to arise that can be used to alter lymphocyte function and disease progression.
Acknowledgment
We thank Steven Martinez for excellent technical assistance.
This work was supported, in whole or in part, by grants from the Lizanell and Colbert Coldwell Foundation (to R. A. K.) and the National Center for Research Resources (5G12RR008124), a component of the National Institutes of Health, and the University of Texas at El Paso. This work was also supported by National Institutes of Health Grant 5RO1CA116356-02 (to J. A. F.).
- IL
- interleukin
- CA
- calyculin A
- CSA
- cyclosporin A
- ERK
- extracellular signal-regulated kinase
- IL-2R
- interleukin-2 receptor
- JAK
- Janus tyrosine kinase
- MEK
- mitogen-activated protein kinase-extracellular signal-regulated protein kinase kinase
- mTOR
- mammalian target of rapamycin
- OA
- okadaic acid
- p70S6k
- p70 ribosomal protein S6 kinase
- PBMC
- peripheral blood mononuclear cells
- PHA
- phytohemagglutinin
- PKC
- protein kinase C
- PP1
- protein phosphatase 1
- PP2A
- protein phosphatase 2A
- STS
- staurosporine
- STAT
- signal transducers and activators of transcription
- TAU
- tautomycin.
REFERENCES
- 1.Laurence A., Tato C. M., Davidson T. S., Kanno Y., Chen Z., Yao Z., Blank R. B., Meylan F., Siegel R., Hennighausen L., Shevach E. M., O'Shea J. J. (2007) Immunity 26, 371–381 [DOI] [PubMed] [Google Scholar]
- 2.Rochman Y., Spolski R., Leonard W. J. (2009) Nat. Rev. Immunol. 9, 480–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson B. H. (2004) J. Immunol. 172, 3983–3988 [DOI] [PubMed] [Google Scholar]
- 4.Witthuhn B. A., Silvennoinen O., Miura O., Lai K. S., Cwik C., Liu E. T., Ihle J. N. (1994) Nature 370, 153–157 [DOI] [PubMed] [Google Scholar]
- 5.Nelson B. H., Willerford D. M. (1998) Adv. Immunol. 70, 1–81 [DOI] [PubMed] [Google Scholar]
- 6.Leonard W. J. (1996) Nat. Med. 2, 968–969 [DOI] [PubMed] [Google Scholar]
- 7.Lin J. X., Mietz J., Modi W. S., John S., Leonard W. J. (1996) J. Biol. Chem. 271, 10738–10744 [PubMed] [Google Scholar]
- 8.Lin J. X., Leonard W. J. (2000) Oncogene 19, 2566–2576 [DOI] [PubMed] [Google Scholar]
- 9.Nagy Z. S., Wang Y., Erwin-Cohen R. A., Aradi J., Monia B., Wang L. H., Stepkowski S. M., Rui H., Kirken R. A. (2002) J. Leukocyte Biol. 72, 819–828 [PubMed] [Google Scholar]
- 10.Kirken R. A., Malabarba M. G., Xu J., DaSilva L., Erwin R. A., Liu X., Hennighausen L., Rui H., Farrar W. L. (1997) J. Biol. Chem. 272, 15459–15465 [DOI] [PubMed] [Google Scholar]
- 11.Decker T., Kovarik P. (2000) Oncogene 19, 2628–2637 [DOI] [PubMed] [Google Scholar]
- 12.Greenhalgh C. J., Hilton D. J. (2001) J. Leukocyte Biol. 70, 348–356 [PubMed] [Google Scholar]
- 13.Shuai K., Liu B. (2005) Nat. Rev. Immunol. 5, 593–605 [DOI] [PubMed] [Google Scholar]
- 14.Irie-Sasaki J., Sasaki T., Matsumoto W., Opavsky A., Cheng M., Welstead G., Griffiths E., Krawczyk C., Richardson C. D., Aitken K., Iscove N., Koretzky G., Johnson P., Liu P., Rothstein D. M., Penninger J. M. (2001) Nature 409, 349–354 [DOI] [PubMed] [Google Scholar]
- 15.Simoncic P. D., Lee-Loy A., Barber D. L., Tremblay M. L., McGlade C. J. (2002) Curr. Biol. 12, 446–453 [DOI] [PubMed] [Google Scholar]
- 16.Chen Y., Wen R., Yang S., Schuman J., Zhang E. E., Yi T., Feng G. S., Wang D. (2003) J. Biol. Chem. 278, 16520–16527 [DOI] [PubMed] [Google Scholar]
- 17.Woetmann A., Nielsen M., Christensen S. T., Brockdorff J., Kaltoft K., Engel A. M., Skov S., Brender C., Geisler C., Svejgaard A., Rygaard J., Leick V., Odum N. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 10620–10625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woetmann A., Brockdorff J., Lovato P., Nielsen M., Leick V., Rieneck K., Svejgaard A., Geisler C., Ødum N. (2003) J. Biol. Chem. 278, 2787–2791 [DOI] [PubMed] [Google Scholar]
- 19.Wang Y., Malabarba M. G., Nagy Z. S., Kirken R. A. (2004) J. Biol. Chem. 279, 25196–25203 [DOI] [PubMed] [Google Scholar]
- 20.Yodoi J., Teshigawara K., Nikaido T., Fukui K., Noma T., Honjo T., Takigawa M., Sasaki M., Minato N., Tsudo M., et al. (1985) J. Immunol. 134, 1623–1630 [PubMed] [Google Scholar]
- 21.Ross J. A., Nagy Z. S., Kirken R. A. (2008) J. Biol. Chem. 283, 4699–4713 [DOI] [PubMed] [Google Scholar]
- 22.Malabarba M. G., Rui H., Deutsch H. H., Chung J., Kalthoff F. S., Farrar W. L., Kirken R. A. (1996) Biochem. J. 319, 865–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamashita H., Xu J., Erwin R. A., Farrar W. L., Kirken R. A., Rui H. (1998) J. Biol. Chem. 273, 30218–30224 [DOI] [PubMed] [Google Scholar]
- 24.Behbod F., Nagy Z. S., Stepkowski S. M., Karras J., Johnson C. R., Jarvis W. D., Kirken R. A. (2003) J. Immunol. 171, 3919–3927 [DOI] [PubMed] [Google Scholar]
- 25.Cheng H., Ross J. A., Frost J. A., Kirken R. A. (2008) Mol. Cell. Biol. 28, 2271–2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagy Z. S., Rui H., Stepkowski S. M., Karras J., Kirken R. A. (2006) J. Immunol. 177, 5032–5040 [DOI] [PubMed] [Google Scholar]
- 27.Kirken R. A., Malabarba M. G., Xu J., Liu X., Farrar W. L., Hennighausen L., Larner A. C., Grimley P. M., Rui H. (1997) J. Biol. Chem. 272, 14098–14103 [DOI] [PubMed] [Google Scholar]
- 28.Honkanen R. E., Codispoti B. A., Tse K., Boynton A. L., Honkanan R. E. (1994) Toxicon 32, 339–350 [DOI] [PubMed] [Google Scholar]
- 29.Ishihara H., Martin B. L., Brautigan D. L., Karaki H., Ozaki H., Kato Y., Fusetani N., Watabe S., Hashimoto K., Uemura D., et al. (1989) Biochem. Biophys. Res. Commun. 159, 871–877 [DOI] [PubMed] [Google Scholar]
- 30.Bialojan C., Takai A. (1988) Biochem. J. 256, 283–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacKintosh C., Klumpp S. (1990) FEBS Lett. 277, 137–140 [DOI] [PubMed] [Google Scholar]
- 32.Park S. H., Yamashita H., Rui H., Waxman D. J. (2001) Mol. Endocrinol. 15, 2157–2171 [DOI] [PubMed] [Google Scholar]
- 33.Asao H., Kumaki S., Takeshita T., Nakamura M., Sugamura K. (1992) FEBS Lett. 304, 141–145 [DOI] [PubMed] [Google Scholar]
- 34.Gaffen S. L. (2001) Cytokine 14, 63–77 [DOI] [PubMed] [Google Scholar]
- 35.Giri J. G., Ahdieh M., Eisenman J., Shanebeck K., Grabstein K., Kumaki S., Namen A., Park L. S., Cosman D., Anderson D. (1994) EMBO J. 13, 2822–2830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karaman M. W., Herrgard S., Treiber D. K., Gallant P., Atteridge C. E., Campbell B. T., Chan K. W., Ciceri P., Davis M. I., Edeen P. T., Faraoni R., Floyd M., Hunt J. P., Lockhart D. J., Milanov Z. V., Morrison M. J., Pallares G., Patel H. K., Pritchard S., Wodicka L. M., Zarrinkar P. P. (2008) Nat. Biotechnol. 26, 127–132 [DOI] [PubMed] [Google Scholar]
- 37.Howard O. M., Kirken R. A., Garcia G. G., Hackett R. H., Farrar W. L. (1995) Biochem. J. 306, 217–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ross J. A., Nagy Z. S., Cheng H., Stepkowski S. M., Kirken R. A. (2007) Arch. Immunol. Ther. Exp. 55, 231–245 [DOI] [PubMed] [Google Scholar]
- 39.Shuai K., Liu B. (2003) Nat. Rev. Immunol. 3, 900–911 [DOI] [PubMed] [Google Scholar]
- 40.Yamashita H., Nevalainen M. T., Xu J., LeBaron M. J., Wagner K. U., Erwin R. A., Harmon J. M., Hennighausen L., Kirken R. A., Rui H. (2001) Mol. Cell. Endocrinol. 183, 151–163 [DOI] [PubMed] [Google Scholar]
- 41.Mazurkiewicz-Munoz A. M., Argetsinger L. S., Kouadio J. L., Stensballe A., Jensen O. N., Cline J. M., Carter-Su C. (2006) Mol. Cell. Biol. 26, 4052–4062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matthews S. A., Cantrell D. A. (2006) Curr. Opin. Immunol. 18, 314–320 [DOI] [PubMed] [Google Scholar]
- 43.Matthews S. A., Cantrell D. A. (2009) Immunol. Rev. 228, 241–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Juntilla M. M., Koretzky G. A. (2008) Immunol. Lett. 116, 104–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karnitz L. M., Burns L. A., Sutor S. L., Blenis J., Abraham R. T. (1995) Mol. Cell. Biol. 15, 3049–3057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winston L. A., Hunter T. (1996) Curr. Biol. 6, 668–671 [DOI] [PubMed] [Google Scholar]
- 47.Blom N., Gammeltoft S., Brunak S. (1999) J. Mol. Biol. 294, 1351–1362 [DOI] [PubMed] [Google Scholar]
- 48.Blom N., Sicheritz-Pontén T., Gupta R., Gammeltoft S., Brunak S. (2004) Proteomics 4, 1633–1649 [DOI] [PubMed] [Google Scholar]
- 49.Schillace R. V., Carr D. W. (2006) Crit. Rev. Immunol. 26, 113–131 [DOI] [PubMed] [Google Scholar]
- 50.Vang T., Torgersen K. M., Sundvold V., Saxena M., Levy F. O., Skålhegg B. S., Hansson V., Mustelin T., Taskén K. (2001) J. Exp. Med. 193, 497–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Virshup D. M., Shenolikar S. (2009) Mol. Cell 33, 537–545 [DOI] [PubMed] [Google Scholar]
- 52.Shenolikar S., Nairn A. C. (1991) Adv. Second Messenger Phosphoprotein Res. 23, 1–121 [PubMed] [Google Scholar]
- 53.Millward T. A., Zolnierowicz S., Hemmings B. A. (1999) Trends Biochem. Sci. 24, 186–191 [DOI] [PubMed] [Google Scholar]
- 54.Goldberg Y. (1999) Biochem. Pharmacol. 57, 321–328 [DOI] [PubMed] [Google Scholar]
- 55.Chen J., Martin B. L., Brautigan D. L. (1992) Science 257, 1261–1264 [DOI] [PubMed] [Google Scholar]
- 56.Chen J., Parsons S., Brautigan D. L. (1994) J. Biol. Chem. 269, 7957–7962 [PubMed] [Google Scholar]
- 57.Guo H., Damuni Z. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 2500–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yokoyama N., Reich N. C., Miller W. T. (2001) J. Interferon Cytokine Res. 21, 369–378 [DOI] [PubMed] [Google Scholar]
- 59.Brockdorff J., Nielsen M., Dobson P., Geisler C., Röpke C., Svejgaard A., Odum N. (1997) Tissue Antigens 49, 228–235 [DOI] [PubMed] [Google Scholar]
- 60.Mitsuhashi S., Shima H., Tanuma N., Sasa S., Onoe K., Ubukata M., Kikuchi K. (2005) Mol. Cell. Biochem. 269, 183–187 [DOI] [PubMed] [Google Scholar]