

Abstract
Human intestinal lactase-phlorizin hydrolase, LPH, encompasses four homologous domains, which presumably have evolved from two subsequent duplications of one ancestral gene. The profragment, LPHα, comprises homologous domains I and II and functions as an intramolecular chaperone in the context of the brush-border LPHβ region of LPH. Here, we analyze the inter-relationship between homologous domains III and IV of LPHβ and their implication in the overall structure, function, and trafficking of LPH. In silico analyses revealed potential domain boundaries for these domains as a basis for loop-out mutagenesis and construction of deletion or individual domain forms of LPH. Removal of domain IV, which contains lactase, results in a diminished phlorizin hydrolase activity, lack of dimerization in the endoplasmic reticulum (ER), but accelerated transport kinetics from the ER to the Golgi apparatus. By contrast, deletion of domain III, which harbors phlorizin hydrolase, generates a malfolded protein that is blocked in the ER. Interestingly, homologous domain III is transport-competent per se and sorted to the apical membrane in polarized Madin-Darby canine kidney cells. Nevertheless, it neither dimerizes nor acquires complete phlorizin hydrolase activity. Our data present a hierarchical model of LPH in which the homologous domain III constitutes (i) a fully autonomous core domain within LPH and (ii) another intramolecular chaperone besides the profragment LPHα. Nevertheless, the regulation of the trafficking kinetics and activity of domain III and entire LPH including elevation of the enzymatic activities require the correct dimerization of LPH in the ER, an event that is accomplished by the non-autonomous domain IV.
Keywords: Cell/Epithelial, Glycoproteins/Biosynthesis, Glycoproteins/Structure, Protein/Biosynthesis, Protein/Folding, Protein/Intracellular Trafficking, Lactase-Phlorizin Hydrolase, Protein Dimerization
Introduction
Human small intestinal lactase-phlorizin hydrolase (LPH2, EC 3.2.1.23/62), an essential digestive enzyme of the brush-border membrane, contains a large propeptide (1) that acts as an LPH-specific intramolecular chaperone (2). LPH is an integral membrane glycoprotein synthesized as a single chain precursor molecule comprising four homologous domains, which may have arisen by two gene duplications of one ancestral gene (1). Severe reduction of lactase activity, such as in congenital lactase deficiency or adult type hypolactasia, leads to osmotic diarrhea, bloating, cramps, and vomiting after uptake of lactose containing food (3–6). This limits the range of dietary sources as in the case of adult type hypolactasia affecting most adults worldwide with the exception of Caucasians and descendents (7, 8).
The LPHα profragment (Ser20/Arg734) encompasses complete homologous domain I and two-thirds of homologous domain II. It is devoid of sorting signals and catalytic activity and rich in cysteine and hydrophobic amino acid residues (9). LPH is cleaved in the trans-Golgi network by a trypsin-like protease (10–13) at Arg734/Leu735 generating LPHβinitial (160 kDa) (14–16), whereby the profragment LPHα is ultimately degraded (17, 18). Finally, LPHβinitial is sorted in the trans-Golgi network with high fidelity (>90%) to the apical or brush-border membrane of the enterocytes. In the brush-border membrane, LPHβinitial is cleaved by luminal trypsin to LPHβfinal (145 kDa) at Arg868/Leu869 (15, 16), and this form can now exert its digestive function via the two catalytic activities, lactase and phlorizin hydrolase (see Fig. 1A for a schematic presentation of the cleavage sites of LPH). Individual expression of LPHβ (17, 19) leads to its localization in the ER, whereas its co-expression of LPHα generates a mature and transport-competent form of LPHβ (2). Each of the homologous regions of LPH shows similarities in size and sequence with well characterized β-glycosidases from archaea, bacteria, fungi, and plants classified in family 1 of glycoside hydrolases (1, 20, 21).
FIGURE 1.

Schematic presentation of wild type (WT) and LPH deletion mutants. A, main features of intestinal LPH structure. Starting with a cleavable signal sequence (SS; Met1–Gly19) at the N terminus (N), the extracellular region comprises homologous domains I-IV (Ser20–Thr1882). The initial cleavage step takes place between Arg734 and Leu735, generating LPHβinitial in the Golgi apparatus; removal of the polypeptide stretch Leu735/Arg868 occurs by luminal trypsin at Arg868–Ala869, creating brush-border mature enzyme, LPHβfinal (Ala869–Phe1927). MACT, membrane anchor and cytoplasmic tail; C, C terminus. Locations of phlorizin hydrolase (Glu1273) and lactase (Glu1749) activities, respectively, are indicated by stars. B, schematic drawing of domain deletion mutants generated by loop-out mutagenesis. C, structural models of LPH domains III and IV.
The catalytic sites of LPH are localized in homologous domains III and IV (9, 22). This has raised the question whether these domains behave as autonomous regions and attain their enzymatically active conformation independently of each other in the context of the folding of the LPH. Moreover, the question as to which domain is more important concerning trafficking and sorting events is still unanswered. In this study, we investigate the influence of each of the two homologous domains comprised by mature LPHβ, i.e. domains III and IV, on the structural and functional features of LPH by directed change of domain composition.
EXPERIMENTAL PROCEDURES
Materials and Reagents
Tissue culture dishes were obtained from Greiner (Hamburg, Germany). Dulbecco's modified Eagle's medium, minimum essential medium, streptomycin, penicillin, glutamine, fetal calf serum, and trypsin-EDTA were purchased from BioWest (Essen, Germany). DEAE-dextran, pepstatin, leupeptin, antipain, aprotinin, trypsin inhibitor, phenylmethylsulfonyl fluoride, trypsin, Triton X-100, SDS, and molecular weight standards for SDS-PAGE were acquired from Sigma (Deisenhofen, Germany). l-[35S]Methionine (>1000 Ci/mmol) and protein A-Sepharose were obtained from Amersham Biosciences (Freiburg, Germany). Acrylamide, N,N′-methylenebisacrylamide, TEMED, ammonium persulfate, and dithiothreitol were purchased from Carl Roth GmbH (Karlsruhe, Germany). Restriction enzymes were obtained from MBI Fermentas (St. Leon-Rot, Germany), and Isis DNA polymerase was purchased from Qbiogene (Heidelberg, Germany). All other reagents were of superior analytical grade.
Immunochemical Reagents
Monoclonal antibodies (mAbs) against human intestinal LPH were HBB 1/909 (11) and MLacs 1–10 (23). The polyclonal antibody V496 is directed against the N-terminal part of the LPH prodomain (17). Anti-GFP antibodies were purchased from Invitrogen (Karlsruhe, Germany).
Construction of cDNA Clones
Deletion mutants of intestinal LPH were generated by loop-out mutagenesis PCR with the plasmids pcDNA3-LPH, pLPH-GFP (24), and pLPH-YFP as templates. LPH domains III and IV share a comparable, quite high degree of sequence identity (around 40%) with highly conserved family 1 glycoside hydrolases of known three-dimensional structure from humans (25), plants (26), fungi (27), and bacteria (28, 29). Structure-based sequence alignments with the aforesaid homologous proteins generated by GenTHREADER (30) were used to dissect the LPH domains. Structural alignments were also used to build models of domains III and IV using SWISS-MODEL (31). pcDNA3-LPH was generated by cloning the wild type LPH cDNA in the vector pcDNA3 (Invitrogen) with the EcoRI sites of pLPH (32). LPH was fused to YFP by subcloning the EcoRI/ScaI fragment from pcDNA3-LPH containing full-length LPH in-frame into the EcoRI/SmaI-digested pEYFP-N1 vector (Clontech-Takara, Saint-Germain-en-Laye, France) to create pLPH-YFP. A construct comprising the signal sequence and homologous domain III (D3) of LPH was made by inserting a stop codon into the plasmid pJB20-LPHβfinal with the same system generating plasmid pD3. For the generation of a GFP fusion protein, the cDNA encoding the signal sequence of LPH and homologous domain III was amplified by PCR with pD3 as template and cloned into the EcoRI/AgeI-digested pEGFP-N1 vector (Clontech-Takara). The applied oligonucleotides were purchased from Sigma and are listed in Table 1.
TABLE 1.
A list of the wild type and mutant constructs used in this study

Transient Transfection of COS-1 Cells, Metabolic Labeling of Cells, Immunoprecipitation of Cell Extracts, and SDS-PAGE
COS-1 cells were transiently transfected with DNA using DEAE-dextran essentially as described previously (32). The cells were biosynthetically labeled with 80 μCi of [35S]methionine in methionine-free minimum essential medium. Labeling was performed either continuously or following a pulse- chase protocol where the labeled cells were chased with non-radioactive methionine for different periods of time. In some experiments, the biosynthetically labeled cells were washed with Dulbecco's modified Eagle's medium and treated with 50 μl/ml trypsin for 30 min at 37 °C as described previously by Naim et al. (32). This treatment is intended to probe for cell surface expression of wild type LPH and the deletion mutants. Immunoprecipitation of LPH or the deletion mutants from detergent extracts of the labeled cells was performed according to Naim et al. (12) using a mixture of mAb anti-LPH (HBB 1/909 and MLac1, MLac2, MLac4, MLac6, and MLac10), and V496. This mixture recognizes different conformations of LPH. Immunoprecipitates were treated with endo H or endo F (both from Roche Diagnostics, Mannheim, Germany) where indicated according to Naim et al. (12) followed by analysis using SDS-PAGE. The radioactively labeled protein bands were visualized by phosphorimaging and quantified with Quantity One® software (Bio-Rad, Munich, Germany).
Transfection of MDCK Cells, Generation of a Stable MDCK-D3 Cell Line, and Analysis of Sorting
To generate stable MDCK cell lines expressing domain III (MDCK-D3), the cells were transfected using Metafectene Pro obtained by Biontex (Martinsried, Germany) following the manufacturer's instructions. Selection of stable cells was performed in the presence of 0.3 mg/ml active G418 (Carl Roth GmbH), and after 14–21 days, colonies were isolated and subcultured, and stable transformants were screened by immunoprecipitation. For the analysis of sorting behavior, MDCK-D3 cells were cultured on Transwell filters (obtained from Greiner) and biosynthetically labeled with [35S]methionine. Proteins that have been secreted into the apical and the basolateral medium, respectively, were isolated by adding anti-LPH mAb and protein A-Sepharose to the removed media. Intracellular proteins were isolated by immunoprecipitation after cell lysis. The immunoprecipitates were analyzed by SDS-PAGE and phosphorimaging.
Trypsin Treatment of Immunoprecipitates
To assess the sensitivity of the mutants to trypsin, immunoprecipitated proteins were washed for an additional two times with phosphate-buffered saline containing 0.2% Triton X-100 and were supplemented with 10 μg of bovine serum albumin as a carrier and incubated with 0.33 mg/ml trypsin for the indicated times at 37 °C. The reaction was stopped by boiling for 5 min in SDS-PAGE sample buffer prior to gel electrophoresis.
Cell Lysate Fractionation on Sucrose Density Gradients
COS-1 cells were biosynthetically labeled and solubilized in 6 mm dodecyl-β-m-maltoside (Sigma), 50 mm Tris-HCl, 100 mm NaCl, pH 7.5, and protease inhibitors. The cell extracts were centrifuged at 100,000 × g for 1 h at 4 °C, and the supernatant was loaded onto an 11.5-ml sucrose gradient that consisted of 10–30% or 15–25% sucrose (w/v), 50 mm Tris-HCl, 100 mm NaCl, pH 7.5, 6 mm dodecyl-β-m-maltoside and the same protease inhibitors applied for immunoprecipitation except phenylmethylsulfonyl fluoride. The gradient was centrifuged at 100,000 × g for 18 h at 4 °C and divided into 0.5-ml fractions followed by immunoprecipitation and SDS-PAGE.
Confocal Fluorescence Microscopy
COS-1 cells grown on coverslips were transfected, and confocal images of living cells were acquired 2 days after transfection on a Leica TCS SP2 microscope with a ×63 water Plan-Apochromat lens (Leica Microsystems, Bensheim, Germany) (34) and processed with the public domain ImageJ software package (available through the National Institutes of Health). For colocalization studies, the cells were co-transfected with GFP- or YFP-tagged LPH cDNA or the mutant LPH cDNA and the protein marker for the ER ER-DsRed and for the Golgi apparatus Golgi-CFP (Clontech-Takara).
Enzymatic Activity Assay
Lactase and phlorizin hydrolase activities were assessed as follows. The 35S-labeled immunoprecipitates were washed with phosphate-buffered saline containing 0.2% Triton X-100 and incubated with 100 μl of this buffer containing lactose or phlorizin at 28 mm final concentrations. The samples were incubated at 37 °C for 2 h, and the amount of released glucose was assessed by high-performance liquid chromatography (HPLC). The quantification of the specific activity was related to the radioactive protein band detected by phosphorimaging and the number of methionines for each construct.
RESULTS
LPH comprises four extracellular regions that contain 38–55% identical residues (1). An interval of about 100 amino acids within each domain is even more homologous, and this internal homology can also be found by comparison of LPH primary sequences of different species. Although the function of domains I and II that constitute the profragment or proregion of LPH, LPHα, has been assessed before and shown to act as an intramolecular chaperone, the individual roles of the two other domains III and IV are poorly understood. The impact of these homologous domains on the generation of a transport-competent configuration of LPH was addressed in conjunction with the question of whether either domain can fold independently. For this, we first generated several cDNA constructs, each lacking the coding region of one homologous domain (Fig. 1B). In silico analysis provided us with potential domain boundaries as a basis for site-directed loop-out mutagenesis. Fig. 1A shows main structural and functional features of human intestinal LPH. Fig. 1C presents homology-based models of domains III and IV showing typical TIM barrel structure of family 1 glycoside hydrolases.
Expression of Wild Type LPH and Domain Deletion Mutants in COS-1 Cells
To examine the contribution of each of the two homologous domains to the structural, functional, and trafficking features of LPH, the LPH deletion mutants were expressed in COS-1 cells, and their characteristics were compared with those of wild type LPH (Fig. 2). Cell lysates were immunoprecipitated, and the precipitated proteins were treated with endo H to determine their glycosylated state as a measure of trafficking capacity (Fig. 2, A and B). LPHΔ4 partially acquired endo H resistance, compatible with complex glycosylation of this deletion mutant in the Golgi apparatus. By contrast, the mutant lacking homologous domain III, LPHΔ3, was not transport-competent, and the introduction of a spacer containing seven glycines to avoid possible sterical hindrances did not alter its trafficking characteristics. Assessment of the proportions of the mannose-rich and complex glycosylated forms after scanning of the gels revealed a substantial increase in the proportion of the complex glycosylated LPHΔ4 as compared with the wild type counterpart (Fig. 2B). This was surprising because it indicated that the deletion of domain IV in LPHΔ4 leads to a more rapid processing of this deletion mutant than the wild type protein.
FIGURE 2.

Glycosylation pattern and subcellular distribution of LPH wild type (WT) and domain deletion mutants in COS-1 cells. A, transiently transfected COS-1 cells were biosynthetically labeled for 8 h with [35S]methionine followed by immunoprecipitation. The immunoprecipitates were divided into two aliquots and treated with endo H or not treated. The proteins were subjected to SDS-PAGE followed by autoradiography. B, densitometric scanning of the endo H treated biosynthetic forms of wild type and mutant LPH shown in A. C, confocal analysis of transfected COS-1 cells. D and E, colocalization of LPH mutants with ER and Golgi markers, respectively, in transfected COS-1 cells. COS-1 cells were co-transfected with GFP-tagged LPH proteins and ER-DsRed- or YFP-tagged LPH proteins and galactosyl transferase-CFP, respectively. Confocal analysis with living cells was performed 48 h after transfection. n, nucleus; arrowheads, cell surface; bars, 20 μm. F, transiently transfected COS-1 cells were biosynthetically labeled for 4 h with [35S]methionine followed by treatment of the intact cells with 50 μg/ml in fetal calf serum-free Dulbecco's modified Eagle's medium for 30 min at 37 °C. The reaction was stopped with cold fetal calf serum and protease inhibitors. Detergent extracts were immunoprecipitated with mAb anti-LPH and subjected to SDS-PAGE followed by autoradiography.
We further investigated the subcellular distribution of the mutant proteins in more detail by confocal laser microscopy. As shown in Fig. 2, LPHΔ3 was retained intracellularly and colocalized with the ER-DsRed marker (Fig. 2D). By contrast, and consistent with the biochemical data, LPHΔ4 colocalized with markers of the ER, the Golgi, and it was also detected at the plasma membrane, similar to the wild type protein (Fig. 2, C–E). The delivery of LPHΔ4 to the cell surface was assessed by probing its accessibility to trypsin in intact cells that were biosynthetically labeled for 4 h. Trypsin cleaves LPH at Arg868/Ala869 to a protein band of 155–160 kDa (LPHβfinal) (Fig. 1A) (see Refs. 15 and 16), and for a review, see Ref. 20), which remains thereafter resistant to this protease. Because LPHΔ4 contains the trypsin site Arg868/Ala869, a cleaved product derived from this mutant should be generated provided that this protein is expressed at the cell surface. This cleavage product should be substantially smaller in size than its trypsin-cleaved wild type counterpart, LPHβfinal, due to the deletion of domain IV. Fig. 2F demonstrates that trypsin has cleaved LPHΔ4 in intact cells to a band of ∼70 kDa. Similarly, wild type LPH was also cleaved at the cell surface to a band of ∼155–160 kDa. In both cases, the wild type LPH and LPHΔ4, the mannose-rich forms were not cleaved, compatible with an intracellular localization of these protein forms and also with the specificity of the trypsin assay in cleaving only those forms that are exposed at the cell surface. Markedly, the majority of the complex forms of both wild type LPH and LPHΔ4 were cleaved by trypsin, implying a rapid transport of these proteins to the cell surface after terminal glycosylation.
Therefore, the differences in the kinetics of trafficking between wild type LPH and LPHΔ4 are restricted to the pathway between the ER and the Golgi. By contrast to wild type LPH and LPHΔ4 no cleavage products could be detected with LPHΔ3 (not shown), supporting the notion that this deletion form is located intracellularly.
Requirements for the LPH Deletion Mutants to Exit the ER
Dimerization of LPH in the ER is absolutely required for LPH to egress this organelle to the Golgi apparatus (35). The differential intracellular distribution and maturation patterns of the deletion mutants as well as the variable proportions of the glycoforms have altogether lead us to examine the quaternary structures of the mutants and assess their relevance to their transport out of the ER. Fig. 3A depicts the results obtained using sucrose density gradients. As has been previously shown, the mannose-rich LPH form was retained in the light as well as dense gradient fractions concomitant with its monomeric and dimeric states, respectively, and indicative of dimerization occurring along the early secretory pathway. The complex glycosylated protein, on the other hand, is detected exclusively in the dense fractions, indicating that the dimerization of the mannose-rich forms of LPH precedes its complex glycosylation and maturation in the Golgi (35). Surprisingly, the transport-competent LPHΔ4 deletion mutant did not require dimerization of its mannose-rich form in the ER prior to ER egress. As shown in Fig. 3A (the second top panel), the mannose-rich form of LPHΔ4 persisted as a monomeric protein, and the complex glycosylated LPHΔ4 initially appeared in the monomeric fractions. The majority of the complex glycosylated molecules were mainly found in the denser gradient fractions. Interestingly, complex glycosylated LPHΔ4 was revealed in two peaks in the gradient, compatible with two quaternary states, a dimeric and presumably a tetrameric state. A tetrameric LPHΔ4 form would be in line with the results obtained by Panzer et al. (36) for the LPH1646MACT mutant lacking 236 amino acids at the C terminus of homologous domain IV. By contrast, LPHΔ3 was exclusively detected in the lighter fractions of the gradients in its mannose-rich glycoform, compatible with retention in the ER as a monomeric protein.
FIGURE 3.

Structural and functional features of LPH deletion mutants in COS-1 cells. A, assessment of the quaternary structure. Transiently transfected COS-1 cells were biosynthetically labeled and solubilized in 6 mm dodecyl-β-m-maltoside. Cell lysates were layered on a sucrose density gradient. After centrifugation for 18 h at 100,000 × g, fractions were collected, immunoprecipitated, and analyzed on SDS-PAGE. WT, wild type. B, transport kinetics of wild type LPH and mutant proteins. Transfected COS-1 cells were pulse-labeled for 1.5 h with [35S]methionine and chased for the indicated periods of time with cold methionine. The immunoprecipitates were treated with endo H or not treated and analyzed by SDS-PAGE on 6% slab gels. C, trypsin sensitivity assay of wild type and LPH mutants. Transiently transfected COS-1 cells were biosynthetically labeled followed by immunoprecipitation of LPH proteins from the cell lysates. The immunoprecipitates were treated with trypsin for different times and analyzed by SDS-PAGE on 7% slab gels.
Transport Kinetics of LPH and Deletion Mutants
We next analyzed the transport kinetics of the mutants in comparison with wild type LPH in pulse-chase experiments. Here, the immunoprecipitated proteins were treated with endo H to clearly discriminate between the mannose-rich and complex glycosylated forms. Complex glycosylated endo H-resistant LPHΔ4 appeared within 1.5 h of chase, and its proportion was substantially higher than its counterpart in the wild type protein (Fig. 3B, compare also Fig. 2, A and B), indicating that it is more efficiently transported to the Golgi apparatus than wild type LPH. By contrast, LPHΔ3 persisted as an endo H-sensitive mannose-rich polypeptide, compatible with ER localization. The Gly spacer containing LPHΔ3-7xGly mutant also revealed similar biosynthetic features as LPHΔ3 (not shown).
Folding of the Deletion Mutants
The variations in the quaternary structure of the deletion mutants as well as in their transport kinetics raised the question of causal folding variations. We therefore examined the folding of these mutants by using three procedures. In the first, the mutants were probed for their protease sensitivity using trypsin; in the second procedure, the enzymatic activities of the mutants were measured; and finally, in the third procedure, reactivity of the mutants with epitope-specific antibodies was assessed.
Trypsin Treatment
The tryptic digestion patterns of the wild type and mutant proteins are depicted in Fig. 3C. Wild type LPH was digested to two main bands corresponding to cleaved mannose-rich and complex glycosylated LPH (see also Ref. 3). This pattern did not change with prolonged digestion times. Similarly, LPHΔ4 pattern was also cleaved to two protein products that correspond to the mannose-rich and complex glycosylated forms. The smaller apparent molecular weights products fit well with a reduction corresponding to the size of the deleted domain IV. In a fashion similar to wild type LPH, the cleaved products of LPHΔ4 were also resistant to trypsin. Importantly, the cleavage of LPHΔ4 to the final products was not preceded by major intermediate cleaved forms, suggesting that one major trypsin site is exposed in the deletion mutant, which is in all likelihood the same as that in wild type LPH. By contrast to wild type LPH and LPHΔ4, LPHΔ3 was completely degraded by trypsin already after 1 min of treatment concomitant with the exposure of several trypsin cleavage sites and thus altered folding in comparison with wild type LPH and LPHΔ4.
Enzymatic Activities of LPH Deletion Mutants
Another approach to examine the folding and maturation pattern of a protein is to assess its biological function. We therefore analyzed the enzymatic activities of lactase and phlorizin hydrolase in these mutants in comparison with their wild type counterparts (Fig. 4). LPHΔ4 revealed slightly reduced activities of phlorizin hydrolase. The lactase activity was as expected absent because the lactase active site is found in residue Glu1749 of domain IV. The lactase activity in LPHΔ3 was not detected. The data provide another support for malfolded LPHΔ3 and correct folding of LPHΔ4.
FIGURE 4.

Enzymatic activity of deletion mutants (Δ-mutants) of LPH. COS-1 cells were transiently transfected. 48 h after transfection, labeled cells were lysed, and proteins were immunoprecipitated. Immunoprecipitates were incubated with lactose and phlorizin, respectively, and the lactase and phlorizin hydrolase activities were measured by determining the concentration of released glucose by HPLC. The enzyme activities of the mutants were compared with those of wild type (WT) LPH. Error bar represents S.E.
Epitope Mapping of Domain Deletion Mutants
The deletion mutants were immunoprecipitated with a panel of mAbs, which are specific in recognizing native or unfolded conformations of LPH (35). The control samples utilized immunoprecipitation of the GFP-tagged mutants with anti-GFP. Fig. 5 shows that LPHΔ4 and LPHΔ3 were isolated with anti-GFP antibody. Surprisingly, none of the mAbs against LPH recognized LPHΔ3, even the two mAbs, MLac6 and MLac10, that recognize unfolded and denatured forms of LPH. LPHΔ4, on the other hand, reacted with all the antibodies utilized with the exception of MLac6 and MLac10. Given that the antibodies were raised against the mature form of LPH, i.e. LPHβ that comprises the two domains III and IV, it is obvious that all antibodies except MLac6 and MLac10 possess epitopes in domain III of LPH. Because MLac6 and MLac10 are directed against unfolded forms of LPH, the results indicate that LPHΔ4 is properly folded, lending a strong support to the protease sensitivity data. LPHΔ3, on the other hand, is malfolded and is therefore not recognized by the antibodies. It is also likely that none of epitopes is found on LPHΔ3. This view is supported by the observation that LPHΔ3 does not react with MLac6 or MLac10, which are directed against malfolded forms of LPH.
FIGURE 5.

Epitope mapping of LPH mutants. COS-1 cells were transfected with DNA coding for GFP-tagged deletion mutants and biosynthetically labeled 48 h after transfection. Cell lysates were divided into equal aliquots and immunoprecipitated with anti-GFP and different anti-LPH mAb. The immunoprecipitated proteins were analyzed by SDS-PAGE.
Domain III Is a Transport-competent and Functional Protein
The data gathered so far strongly suggest that domain III is a central autonomous component of LPH. The next step was therefore to express this domain independently and examine its trafficking and functional properties. As shown in Fig. 6B, domain III expression in COS-1 cells revealed a predominant endo H- and endo F-sensitive protein band in the cell lysates, indicating that it is a mannose-rich glycosylated form of domain III. The cell culture medium contained an endo H-resistant and endo F-sensitive protein, compatible with a complex glycosylated domain III. These results clearly indicate that domain III is secreted into the cell exterior immediately and rapidly upon maturation in the Golgi apparatus. To substantiate the data with a further approach, immunofluorescence images were generated. Fig. 6C shows that domain III was located in the ER, compatible with the major mannose-rich form in the cell lysates. When the cells were subjected to a 20 °C temperature block, domain III was found in the Golgi apparatus. Assessment of the quaternary structure of domain III, performed at 20 °C to analyze mannose-rich and complex glycosylated proteins, revealed monomeric forms of the mannose-rich protein as well as the complex glycosylated form (Fig. 7A). It should be noted that the overall labeling intensity of the complex glycosylated protein in all the lanes as compared with the mannose-rich polypeptide did not comprise more than 10% of total domain III in the cell lysates.
FIGURE 6.

Expression of D3 in COS-1 cells. A, schematic representation of the D3 construct. N, N terminus; C, C terminus; SS, cleavable signal sequence. B, transfected COS-1 cells were biosynthetically labeled, and proteins were immunoprecipitated from cell lysates and, where indicated, from cell culture media. Immunoprecipitates were treated with endo H or endo F or not treated, analyzed by SDS-PAGE, and visualized by autoradiography. C, confocal analysis of GFP-tagged D3 in transfected COS-1 cells. n, nucleus; G, Golgi apparatus.
FIGURE 7.

Structural and functional features of D3. A, assessment of the quaternary structure of D3. Transiently transfected COS-1 cells were biosynthetically labeled at 20 °C to avoid secretion and solubilized in 6 mm dodecyl-β-m-maltoside. Cell lysates were layered on a sucrose density gradient. After centrifugation for 18 h at 100,000 × g, fractions were collected, immunoprecipitated, and analyzed on SDS-PAGE. B, transport kinetics of D3. Biosynthetically labeled proteins were immunoprecipitated from cell lysates and cell culture media after the indicated labeling times and analyzed by SDS-PAGE. lys, lysate; med, medium. C, trypsin sensitivity assay with D3. Transiently transfected COS-1 cells were biosynthetically labeled followed by immunoprecipitation of LPH proteins from cell lysates and cell culture media. The immunoprecipitates were treated with trypsin for different times and analyzed by SDS-PAGE.
Continuous metabolic labeling was performed to determine the transport rate of domain III. As shown in Fig. 7B, domain III appeared in the medium after 90 min of labeling. Finally, we probed the folding of domain III utilizing trypsin sensitivity and measurement of its enzymatic activity. Fig. 7C shows that domain III is predominantly resistant to trypsin. Its phlorizin hydrolase activity is, however, reduced by about 50% (Fig. 8).
FIGURE 8.

Enzymatic activity of D3. COS-1 cells were transiently transfected with wild type (WT) and D3 cDNA, respectively. 48 h after transfection, cell lysates and cell culture media were immunoprecipitated with anti-LPH mAbs. The immunoprecipitates were incubated with phlorizin, and the phlorizin hydrolase activity was measured by determining the concentration of the released glucose by HPLC. The enzyme activity of the mutant was compared with wild type LPH. Error bar represents S.E.; n = 4.
Sorting of Domain III in Polarized MDCK Cells
LPH is sorted into the apical membrane in polarized MDCK cells and intestinal cells with high fidelity. Because it is proposed that putative apical sorting signals are located in the ectodomain of the LPH mature form and domain III builds one-half of LPHβ, we analyzed its sorting in a polarized cell line to determine whether or not this region contains putative signals for apical sorting of LPH. Domain III was stably expressed in MDCK cells, and its sorting was analyzed in a membrane filter system as described previously (38). Fig. 9, A and B, demonstrate that domain III is secreted predominantly at the apical surface of MDCK cells. In fact, more than 80% of this protein was found at the apical side, indicating that the sorting of domain III is not as efficient as for wild type LPH. These data suggest that domain IV is most likely devoid of putative apical sorting signals.
FIGURE 9.
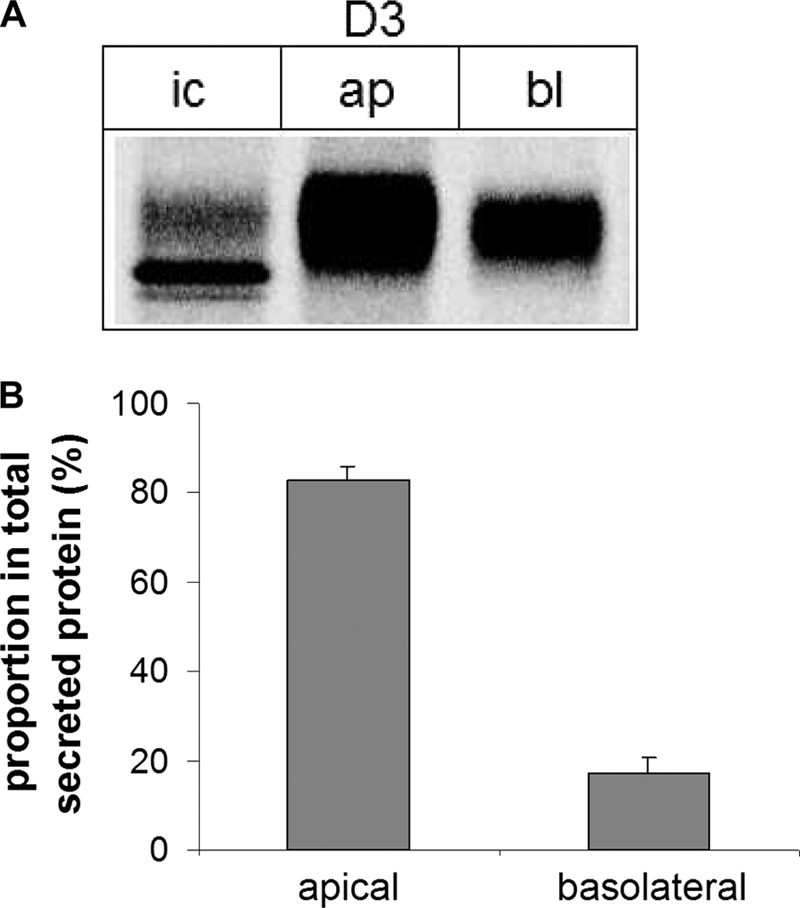
Polarized sorting of D3. A, MDCK-II cells stably expressing D3 were cultured on Transwell filters and biosynthetically labeled with [35S]methionine. Proteins that have been secreted into the apical (ap) and the basolateral (bl) medium, respectively, were isolated by adding anti-LPH mAb and protein A-Sepharose to the collected media. Intracellular (ic) proteins were isolated by immunoprecipitation after cell lysis. The immunoprecipitates were analyzed by SDS-PAGE on 9% slab gels followed by phosphorimaging. B, the quantification of secreted D3 was performed with the Quantity One® software from Bio-Rad. The error bars represent S.E.; n = 5.
DISCUSSION
Characterization of the structure, biosynthesis, and trafficking of the individual subdomains within LPH, an essential brush-border membrane enzyme, constitutes an important step toward understanding its function and impact to the intestinal epithelial cell physiology. Given that the three-dimensional structure of LPH has not been elucidated yet, alternative approaches have to be designed to determine the significance and relevance of the individual subunits to each other in the context of this multiple domain protein.
Lactases, or more properly, β-galactosidases, are grouped within four of the nearly 100 families of glycosyl hydrolases (GHs) that have been characterized (39). Mammalian intestinal lactase (LPH) is classified in the GH1 family, along with enzymes present in a variety of organisms acting against different types of β-glycosides. Although most GH1 enzymes (mostly bacterial) thus far characterized are conformed by a single domain, intestinal lactase is synthesized as a multidomain precursor protein that is encoded by a gene resulting from the fusion of four tandemly arranged repetitions of an ancestor gene (1, 32). Maturation of the enzyme generates the brush-border form comprised by the last two domains, III and IV (2), each with differential activity. Domain III shows specificity toward glycosides, such as phlorizin, whereas domain IV is specifically active against lactose (22). Interestingly, individual expression of domain III, but not domain IV (data not shown), of LPH reveals a correctly folded, transport-competent, and rapidly secreted molecule, thus underscoring its autonomous character that has been conserved from prokaryotes to eukaryotes.
Despite its strong homologies with domain III, domain IV is not a folding-competent, a transport-competent, or an enzymatically active species per se. This domain, however, plays a central regulatory role in the context of the function and trafficking of LPH. It contains the LAC236 stretch that is required for dimerization of LPH (24, 36). The essential function of domain IV within the LPH complex becomes evident when considering its role in the dimerization of LPH. In fact, domain III does not dimerize, and the phlorizin hydrolase activity of this domain is elevated by a factor of 2.5-fold in the dimeric LPH molecule. Additionally, domain IV is rate-limiting along the secretory pathway of LPH from the ER to the Golgi. In fact, LPHΔ4, a deletion mutant that lacks the entire homologous domain IV, acquires more rapidly complex glycosylation than its wild type counterpart, proposing a role of this domain in decelerating LPH processing. Importantly, LPHΔ4 does not dimerize in the ER, lending a strong support to the view that dimerization is initiated by homologous domain IV and supporting previous data that assigned LAC236 an essential role in dimerization. It is very likely therefore that the retarded trafficking of wild type LPH in comparison with LPHΔ4 is due to its dimerization prior to the ER exit, whereas this additional step is not required for LPHΔ4 to acquire transport competence. Interestingly, epitope mapping with a panel of mAbs against the mature brush-border form of LPH (domains III and IV) demonstrated a similar pattern of recognition for LPHΔ4 and LPH, strongly suggesting that the epitopes tested are located in domain III. Given that these antibodies react only with native LPH species, but not with denatured LPH on Western blots, our data strongly suggest that the tertiary structure of LPHΔ4 is comparable with its counterparts in human wild type LPH and that domain III represents the structural core of the mature protein.
On the other hand, domain IV is not an autonomous region. It harbors the lactase catalytic site at Glu1749 (9, 22) and acquires activity only when LPH dimerizes (35). Therefore, domain IV plays a role as a regulatory switch that triggers the dimerization of the LPH molecule, thus activating itself and elevating the phlorizin hydrolase activities in domain III.
Initial cleavage of mature LPH in the Golgi occurs between Arg734 and Leu735 and generates LPHβinitial that is transported with high fidelity to the apical membrane. In the intestinal lumen, LPHβinitial undergoes another cleavage at Arg868/Ala869 by pancreatic trypsin to generate LPHβfinal (15, 16). The significance of the stretch between Leu735 and Arg868 in the context of trafficking and sorting of LPH has been until present obscure. Our data assign a role to this region in association with domain III in the sorting of LPH to the apical membrane. In this respect, it is interesting to note that domain III per se is not as efficiently transported to the apical membrane in polarized MDCK cells as wild type LPH, strongly proposing that the polypeptide stretch Leu735–Arg868 in domain II could be important for the fine-tuning of polarized sorting.
Our data present a hierarchical model of LPH in which the homologous domain III constitutes a fully autonomous core domain within the LPH molecule. In addition, it represents another intramolecular chaperone of LPH besides the profragment LPHα (Fig. 10). This model assumes that the profragment (2) and homologous domain III (this study) attain their native conformation autonomously. Although domain III is transport-competent and enzymatically active per se, it requires homologous domain IV for elevation of its enzymatic activity and regulation of its trafficking kinetics. This occurs via dimerization of the entire LPH molecule, an event that is triggered by domain IV. Nevertheless, domain IV is a non-autonomous domain that cannot fold independently; it requires the profragment as well as domain III as templates for correct folding. Correctly folded domain IV is now capable of triggering the dimerization of LPH in the ER (35), an event that is required for LPH to exit the ER, for regulation of its transport kinetics and elevation of its enzymatic activities. The profragment and domain III function therefore act as switches for correct folding of domain IV, which in turn “pays back” by giving domain III an increased phlorizin hydrolase activity and gaining more activity as the lactase active site. To our knowledge, this is the first example of a mechanism in which a protein has two intramolecular chaperones and is not activated by propeptide cleavage, as described for zymogens, neuropeptides, and prohormones (33, 37), but by intramolecular organization and oligomerization.
FIGURE 10.

Hierarchy model of LPH biosynthesis in the ER. The LPH precursor is synthesized at the ER and translocated in the ER lumen starting with the independently folding N-terminal profragment (A). Subsequently, autonomous domain III is synthesized and acquires correct folding independent of other domains (B), whereas homologous domain IV that associates LPH with the membrane is not capable of folding independently (C) and requires the profragment and domain III as folding templates (D). Finally, the correctly folded monomer LPH interacts with another monomer via the correctly folded domain IV to form a transport-competent LPH dimmer that exits the ER and acquires full lactase and phlorizin hydrolase activities (E).
Acknowledgments
We thank Dr. Hans-Peter Hauri, University of Basel, Switzerland, Dr. Erwin Sterchi, University of Bern, Switzerland, and Dr. Dallas Swallow, University College London, UK for generous gifts of monoclonal anti-LPH antibodies. We also thank Uwe Glockenthör and Jürgen Eikemeyer from the Department of Physiological Chemistry, University of Veterinary Medicine Hannover, Germany for expert technical assistance.
This work was supported by grants from the German Research Foundation Deutsche Forschungsgemeinschaft (Grant Na 331/1-5) and Sonderforschungsbereich (Grant 621) (to H. Y. N.).
- LPH
- lactase-phlorizin hydrolase (all forms)
- D3
- LPH domain III
- endo F
- endoglycosidase F/glycopeptidase F
- endo H
- endoglycosidase H
- ER
- endoplasmic reticulum
- MDCK
- Madin-Darby canine kidney cells
- GFP
- green fluorescent protein
- YFP
- yellow fluorescent protein
- DsRed
- red fluorescent protein
- mAb
- monoclonal antibody
- HPLC
- high-performance liquid chromatography
- GH
- glycosyl hydrolases.
REFERENCES
- 1.Mantei N., Villa M., Enzler T., Wacker H., Boll W., James P., Hunziker W., Semenza G. (1988) EMBO J. 7, 2705–2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jacob R., Peters K., Naim H. Y. (2002) J. Biol. Chem. 277, 8217–8225 [DOI] [PubMed] [Google Scholar]
- 3.Behrendt M., Keiser M., Hoch M., Naim H. Y. (2009) Gastroenterology 136, 2295–2303 [DOI] [PubMed] [Google Scholar]
- 4.Büller H. A., Grand R. J. (1990) Annu. Rev. Med. 41, 141–148 [DOI] [PubMed] [Google Scholar]
- 5.Digeon B., Walker-Smith J. A. (1986) Dig. Dis. 4, 139–146 [DOI] [PubMed] [Google Scholar]
- 6.Phillips T., Macdonald I., Keyser A. (1978) Proc. Nutr. Soc. 37, 24A. [PubMed] [Google Scholar]
- 7.Harvey C. B., Pratt W. S., Islam I., Whitehouse D. B., Swallow D. M. (1995) Eur. J. Hum. Genet. 3, 27–41 [DOI] [PubMed] [Google Scholar]
- 8.Wang Y., Harvey C. B., Pratt W. S., Sams V. R., Sarner M., Rossi M., Auricchio S., Swallow D. M. (1995) Hum. Mol. Genet. 4, 657–662 [DOI] [PubMed] [Google Scholar]
- 9.Zecca L., Mesonero J. E., Stutz A., Poirée J. C., Giudicelli J., Cursio R., Gloor S. M., Semenza G. (1998) FEBS Lett. 435, 225–228 [DOI] [PubMed] [Google Scholar]
- 10.Skovbjerg H., Danielsen E. M., Noren O., Sjöström H. (1984) Biochim. Biophys. Acta. 798, 247–251 [DOI] [PubMed] [Google Scholar]
- 11.Hauri H. P., Sterchi E. E., Bienz D., Fransen J. A., Marxer A. (1985) J. Cell Biol. 101, 838–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naim H. Y., Sterchi E. E., Lentze M. J. (1987) Biochem. J. 241, 427–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Danielsen E. M., Skovbjerg H., Norén O., Sjöström H. (1984) Biochem. Biophys. Res. Commun. 122, 82–90 [DOI] [PubMed] [Google Scholar]
- 14.Yeh K. Y., Yeh M., Holt P. R. (1991) Am. J. Physiol. 260, G379–384 [DOI] [PubMed] [Google Scholar]
- 15.Wüthrich M., Grünberg J., Hahn D., Jacob R., Radebach I., Naim H. Y., Sterchi E. E. (1996) Arch. Biochem. Biophys. 336, 27–34 [DOI] [PubMed] [Google Scholar]
- 16.Jacob R., Radebach I., Wüthrich M., Grünberg J., Sterchi E. E., Naim H. Y. (1996) Eur. J. Biochem. 236, 789–795 [DOI] [PubMed] [Google Scholar]
- 17.Naim H. Y., Jacob R., Naim H., Sambrook J. F., Gething M. J. (1994) J. Biol. Chem. 269, 26933–26943 [PubMed] [Google Scholar]
- 18.Ouwendijk J., Peters W. J., van de Vorstenbosch R. A., Ginsel L. A., Naim H. Y., Fransen J. A. (1998) J. Biol. Chem. 273, 6650–6655 [DOI] [PubMed] [Google Scholar]
- 19.Oberholzer T., Mantei N., Semenza G. (1993) FEBS Lett. 333, 127–131 [DOI] [PubMed] [Google Scholar]
- 20.Naim H. Y. (2001) Histol. Histopathol. 16, 553–561 [DOI] [PubMed] [Google Scholar]
- 21.Cantarel B. L., Coutinho P. M., Rancurel C., Bernard T., Lombard V., Henrissat B. (2009) Nucleic Acids Res. 37, D233–D238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arribas J. C., Herrero A. G., Martín-Lomas M., Cañada F. J., He S., Withers S. G. (2000) Eur. J. Biochem. 267, 6996–7005 [DOI] [PubMed] [Google Scholar]
- 23.Maiuri L., Raia V., Potter J., Swallow D., Ho M. W., Fiocca R., Finzi G., Cornaggia M., Capella C., Quaroni A. (1991) Gastroenterology 100, 359–369 [DOI] [PubMed] [Google Scholar]
- 24.Jacob R., Weiner J. R., Stadge S., Naim H. Y. (2000) J. Biol. Chem. 275, 10630–10637 [DOI] [PubMed] [Google Scholar]
- 25.Hayashi Y., Okino N., Kakuta Y., Shikanai T., Tani M., Narimatsu H., Ito M. (2007) J. Biol. Chem. 282, 30889–30900 [DOI] [PubMed] [Google Scholar]
- 26.Chuenchor W., Pengthaisong S., Robinson R. C., Yuvaniyama J., Oonanant W., Bevan D. R., Esen A., Chen C. J., Opassiri R., Svasti J., Cairns J. R. (2008) J. Mol. Biol. 377, 1200–1215 [DOI] [PubMed] [Google Scholar]
- 27.Nijikken Y., Tsukada T., Igarashi K., Samejima M., Wakagi T., Shoun H., Fushinobu S. (2007) FEBS Lett. 581, 1514–1520 [DOI] [PubMed] [Google Scholar]
- 28.Sanz-Aparicio J., Hermoso J. A., Martínez-Ripoll M., Lequerica J. L., Polaina J. (1998) J. Mol. Biol. 275, 491–502 [DOI] [PubMed] [Google Scholar]
- 29.Isorna P., Polaina J., Latorre-García L., Cañada F. J., González B., Sanz-Aparicio J. (2007) J. Mol. Biol. 371, 1204–1218 [DOI] [PubMed] [Google Scholar]
- 30.McGuffin L. J., Jones D. T. (2003) Bioinformatics 19, 874–881 [DOI] [PubMed] [Google Scholar]
- 31.Arnold K., Bordoli L., Kopp J., Schwede T. (2006) Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 32.Naim H. Y., Lacey S. W., Sambrook J. F., Gething M. J. (1991) J. Biol. Chem. 266, 12313–12320 [PubMed] [Google Scholar]
- 33.Steiner D. F., Docherty K., Carroll R. (1984) J. Cell. Biochem. 24, 121–130 [DOI] [PubMed] [Google Scholar]
- 34.Jacob R., Naim H. Y. (2001) Curr. Biol. 11, 1444–1450 [DOI] [PubMed] [Google Scholar]
- 35.Naim H. Y., Naim H. (1996) Eur. J. Cell Biol. 70, 198–208 [PubMed] [Google Scholar]
- 36.Panzer P., Preuss U., Joberty G., Naim H. Y. (1998) J. Biol. Chem. 273, 13861–13869 [DOI] [PubMed] [Google Scholar]
- 37.Barr P. J. (1991) Cell 66, 1–3 [DOI] [PubMed] [Google Scholar]
- 38.Jacob R., Brewer C., Fransen J. A., Naim H. Y. (1994) J. Biol. Chem. 269, 2712–2721 [PubMed] [Google Scholar]
- 39.Coutinho P. M., Henrissat B. (1999) J. Mol. Microbiol. Biotechnol. 1, 307–308 [PubMed] [Google Scholar]