

Abstract
Objective
Hyperglycemia is a recognized risk factor for cardiovascular disease in diabetes. Recently we reported that high glucose activates the Ca2+/calcineurin-dependent transcription factor NFAT (Nuclear Factor of Activated T-cells) in arteries ex vivo. Here, we sought to determine if hyperglycemia activates NFAT in vivo and whether this leads to vascular complications.
Methods and results
Intraperitoneal glucose-tolerance test in mice increased NFATc3 nuclear accumulation in vascular smooth muscle. Streptozotocin-induced diabetes resulted in increased NFATc3 transcriptional activity in arteries of NFAT-luciferase transgenic mice. Two NFAT responsive sequences in the osteopontin (OPN) promoter were identified. This proinflammatory cytokine has been shown to exacerbate atherosclerosis and restenosis. Activation of NFAT resulted in increased OPN mRNA and protein in native arteries. Glucose-induced OPN expression was prevented by the ectonucleotidase apyrase, suggesting a mechanism involving the release of extracellular nucleotides. The calcineurin inhibitor cyclos-porin A or the novel NFAT blocker A-285222 prevented glucose-induced OPN expression. Further, diabetes resulted in higher OPN expression, which was significantly decreased by in vivo treatment with A-285222 for 4 weeks or prevented in arteries from NFATc3-/- mice.
Conclusion
These results identify a glucose sensitive transcription pathway in vivo revealing a novel molecular mechanism that may underlie vascular complications of diabetes.
The matrix cytokine osteopontin (OPN) is emerging as a key regulator of chronic inflammatory diseases, including vascular disease. Plasma OPN levels are associated with the presence and extent of coronary artery disease 1, restenosis after balloon angioplasty 2 and human abdominal aortic aneurysm 3. OPN is highly expressed in human atherosclerotic lesions and is not only a marker of inflammation but also an active player in the progression of atherosclerosis and restenosis 4. While OPN deficiency has been shown to result in reduced atherosclerotic lesion areas 5, 6, OPN overexpression is associated with enhanced aortic lesion size 7.
Atherosclerotic vascular disease is a major complication in diabetic patients and the levels of OPN in vivo have been clinically associated with the progression of vascular complications. Plasma levels of OPN significantly correlate to the progression of diabetic nephropathy 8 and OPN levels in the vitreous are enhanced in patients with diabetic retinopathy 9. Further, OPN expression is increased in the media of diabetic arteries 10, 11. Thus, mapping the signaling pathway leading to changes in OPN expression may reveal novel pharmacological targets for prevention of vascular disease.
In the context of vascular remodeling, soluble factors, cytokines, hormones and extracellular nucleotides have been shown to induce OPN expression 12, 13. In particular, UTP has been demonstrated to effectively increase OPN protein production by enhanced transcription and stabilization of OPN mRNA 14, 15. We and others have shown that stimulation of G-protein coupled receptors effectively activates the Ca2+/calcineurin-dependent transcription factor NFAT in arterial smooth muscle 16, 17. More recently, we have shown that high glucose activates NFAT in intact arteries ex vivo by a mechanism involving the release of extracellular nucleotides (i.e. UTP, UDP) acting on P2Y receptors, leading to increased intracellular Ca2+ levels and subsequent activation of the calcineurin/NFAT signaling pathway 18. Elevated extracellular glucose levels have also been shown to induce OPN expression in rat aortic vascular smooth muscle cells (VSMCs) 19. We therefore speculate whether NFAT activation may be involved in the regulation of OPN expression.
The NFAT family consists of four members (NFATc1-c4) originally described in T-cells, but later also demonstrated in cells outside the immune system 20. In cultured VSMCs, NFAT activation has been found to increase migration and proliferation 17, and in a rat carotid artery injury model, blockade of NFAT signaling has been demonstrated to reduce neointima formation 21, 22. However, this being said, downstream gene targets of NFAT are virtually undefined in VSMCs 20.
In this study, we investigate the involvement of the NFAT signaling pathway in the development of vascular complications of diabetes. We show that diabetes-induced hyperglycemia activates NFAT in vascular smooth muscle and that this leads to enhanced expression of OPN, a known marker and promoter of vascular disease.
Materials and Methods
The current study uses mice of the following strains: FVBN 9×-NFAT-luciferase reporter (NFAT-luc), BalB/c NFATc3-/- and wild-type littermates (NFATc3+/+) and the combination of both strains (NFAT-luc/NFATc3+/+, NFAT-luc/NFATc3+/- and NFAT-luc/NFATc3-/-); C57Bl/6 OPN-/- and wild-type littermates; adult NMRI, C57Bl/6 and BalB/c. Aortas, cerebral arteries and plasma were used. Hyperglycemia was induced by intraperitoneal glucose tolerance test (IP-GTT) or by streptozotocin treatment. NFATc3 nuclear accumulation was assessed by confocal immunofluorescence and NFAT-dependent transcriptional activity was assayed in arteries of NFAT-luc transgenic mice as previously described 18, 23. Electrophoretic mobility shift assay (EMSA) was used to show binding of NFAT to the OPN promoter and chromatin immunoprecipitation (ChIP) to measure NFAT enrichment on the OPN promoter as previously described 24, 25. OPN expression was measured by quantitative RT-PCR, western blotting, confocal immunofluorescence and ELISA. Glucagon and insulin were measured by radioimmunoassay. Please see the supplemental materials for expanded Materials and Methods section (available online at http://atvb.ahajournals.org).
Results
Hyperglycemia induces NFATc3 nuclear accumulation in vivo
Raising the blood glucose from 68.8 mg/dL to 316.5 mg/dL (3.8 mmol/L to 17.6 mmol/L), as measured 18 min after i.p. glucose tolerance test (IP-GTT), significantly increases NFATc3 nuclear accumulation in cerebral arteries in vivo (Figure 1). Insulin is normally secreted after a glucose challenge such as the one performed here, hence a potential effect of insulin on NFAT activation could be anticipated. However, insulin does not promote NFATc3 nuclear accumulation in intact cerebral arteries (Supplemental Figure I, available online at http://atvb.ahajournals.org) and thus, it is more likely that the NFATc3 activation observed after IP-GTT was caused by hyperglycemia rather than by insulin.
Figure 1. Hyperglycemia increases NFATc3 nuclear accumulation in vivo.
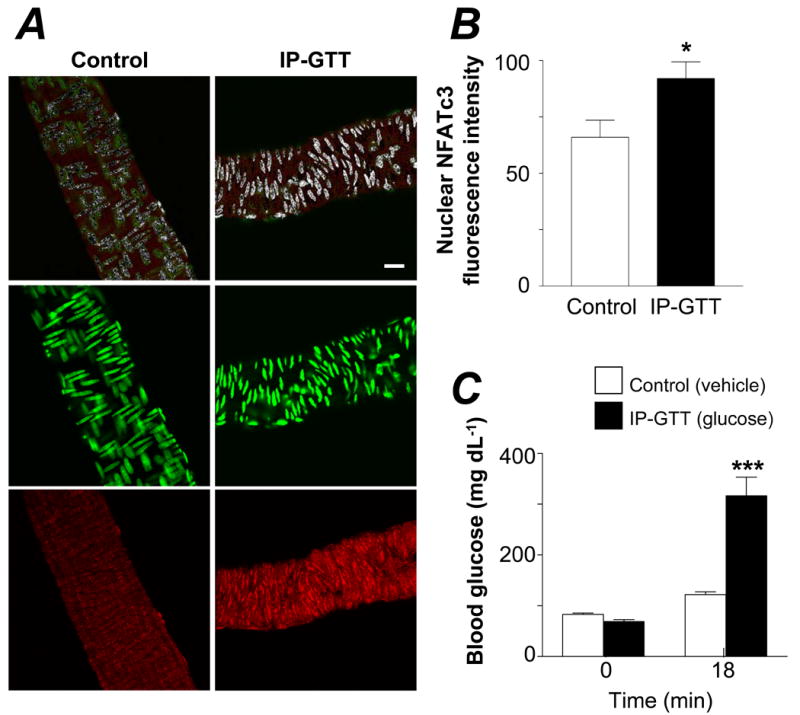
A. Confocal images showing NFATc3 nuclear accumulation in cerebral arteries from hyperglycemic (IP-GTT) and normoglycemic (control) C57Bl/6 mice. Upper panels are pseudo colored images showing nuclear NFATc3 in white; middle and lower panels are original images showing the DNA-binding dye SYTOX Green (green) and NFATc3 (red), respectively. Bar=20 μm. B. Summarized data from experiments in A showing fluorescence intensity of nuclear NFATc3 (N=6 in each group, 147 and 181 images per group, *P < 0.05). C. Glucose levels before and after IP-GTT, corresponding to experiments in A and B (***P < 0.001 vs. all groups).
Streptozotocin-induced hyperglycemia increases NFAT-dependent transcriptional activity
NFAT-luciferase reporter (NFAT-luc) mice were made diabetic by streptozotocin (STZ) treatment and luciferase activity measured in aortas. In these mice, blood glucose levels gradually increased (Figure 2A) and this was accompanied by an increase in NFAT-dependent transcriptional activity (Figure 2C). Luciferase activity correlated significantly to blood glucose values (r = 0.661, p <0.05, Figure 2B).
Figure 2. Streptozotocin-induced hyperglycemia increases NFAT-dependent transcriptional activity.

A. Blood glucose levels in NFAT-luc transgenic mice treated with STZ or vehicle. *P < 0.05 and ***P < 0.001 vs. vehicle. B. Correlation between luciferase activity (RLU μg-1) and blood glucose levels at day 16 (N=12). C. NFAT- luciferase activity in aortas treated as in A, measured at day 8, 12 and 16 after the first STZ injection, normalized to vehicle. Luciferase activity in vehicle mice did not differ between days 8, 12 and 16. N=12-16 for each time point. *P < 0.05 vs. vehicle. D. Luciferase activity in aortas from diabetic NFAT-luc/NFATc3+/+, NFAT-luc/NFATc3+/- and NFAT-luc/NFATc3-/- mice, measured at day 12 after the first STZ injection. Values are normalized to non-diabetic NFAT-luc/NFATc3+/+ vehicle (dotted line). N=6-9 mice in each group. *P < 0.05 vs. vehicle.
Similar experiments were performed in NFAT-luc mice that had been backcrossed with NFATc3-/- mice, yielding NFAT-luc/NFATc3+/+, NFAT-luc/NFATc3+/- and NFAT-luc/NFATc3-/- mice. Diabetes resulted in increased NFAT-dependent transcriptional activity in NFATc3 competent mice, whereas no changes were detected in heterozygous or NFATc3 null mice (Figure 2D). The response to hyperglycemia was more pronounced in NFAT-luc/NFATc3+/+ mice than in NFAT-luc mice (Figure 2C vs. Figure 2D), probably reflecting strain differences (FVBN vs. BalB/c).
Regulation of NFAT activity leads to changes in OPN expression
Culture of intact cerebral arteries in 25 mmol/L glucose for 2 days significantly increased OPN expression in VSMCs when compared to arteries cultured in 5 mmol/L glucose (Figure 3B). Mannitol had no effect on OPN expression, ruling out a possible osmotic effect of glucose. Glucose-induced OPN expression was prevented by the novel NFAT blocker A-285222 23. We have previously shown that release of extracellular nucleotides acting on P2Y receptors mediates glucose-induced NFATc3 activation and that this is prevented by incubation of arteries with the ecto-nucleotidase apyrase 18. Here we show that apyrase (0.32 U/mL) prevents glucose-induced OPN expression. Also, that culture of intact cerebral arteries with UTP results in increased OPN expression (Figures 3A and C), which is prevented by A-285222. A whole set of parallel experiments were performed in mouse aortas, demonstrating that regulation of NFAT activity leads to changes in OPN expression not only in resistance arteries but also in a large conduit artery (Supplemental Figure IV).
Figure 3. Regulation of NFAT activity leads to changes in OPN protein expression in intact arteries.
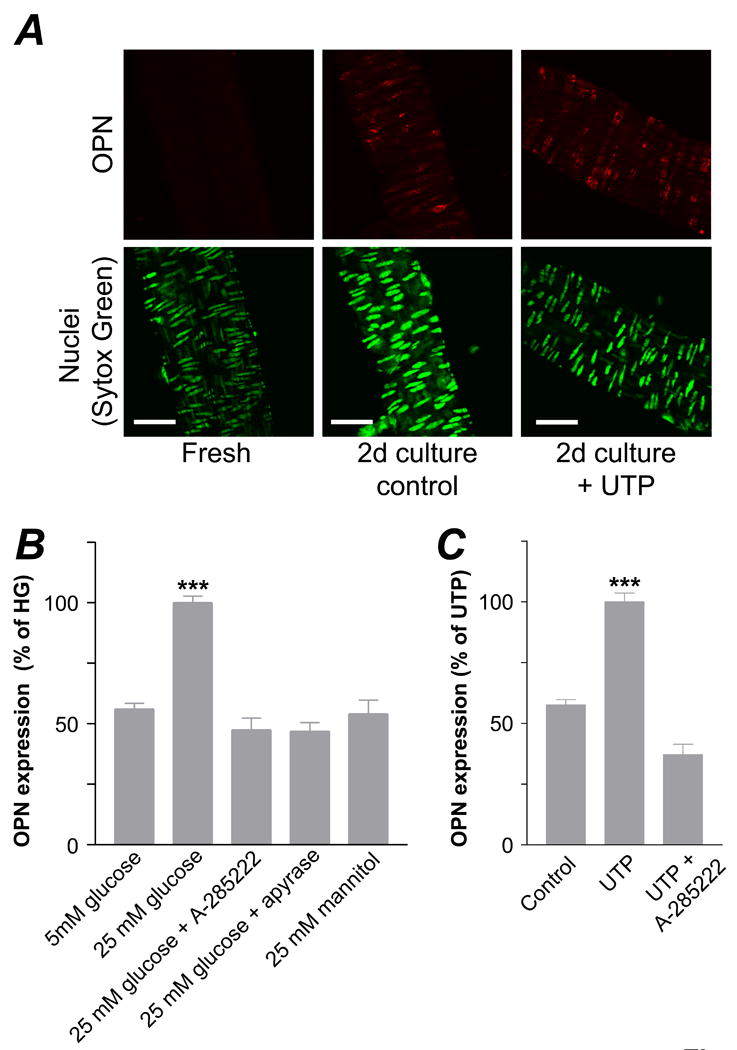
A. Confocal images showing expression of OPN (red, upper panels) in cerebral arteries and nuclear staining with SYTOX Green (lower panels). Arteries were stained immediately after dissection (fresh) or after 2d organ culture in the presence or absence of UTP (10 μmol/L, supplemented daily). Bars=50 μm. B. Summarized confocal data showing OPN expression in cerebral vessels cultured for 2d in 5 mmol/L and 25 mmol/L glucose (90.1 and 450.5 mg/dL respectively) with or without A-285222 (1 μmol/L) or apyrase (0.32 U/mL), or in 25 mmol/L mannitol. Values are normalized to 25 mmol/L glucose (N=13, 26-126 images analyzed in each group, ***P < 0.001 for 25 mmol/L glucose vs. all other bars). C. Summarized confocal data showing OPN expression in cerebral arteries cultured for 2d with or without UTP (10 μmol/L) in the presence or absence of A-285222 (1 μmol/L). Values are normalized to UTP (N=6, 26 to 75 images analyzed in each group, ***P < 0.001 for UTP vs. all other bars).
Absence of NFATc3 prevents UTP-induced increases in OPN expresion
Cerebral arteries and aortas from NFATc3+/+ and NFATc3-/- mice were cultured for 3 days in the presence or absence of UTP. In vessels from NFATc3+/+ mice, a significant increase in OPN expression upon UTP stimulation was observed, whereas no changes were found in vessels from NFATc3-/- animals (Figures 4A and B).
Figure 4. Involvement of NFATc3 in the regulation of OPN expression.

A & B. Summarized data from confocal immunofluorescence experiments showing increased OPN expression upon UTP stimulation for 3 d in intact cerebral arteries and aortas from NFATc3+/+ mice, but no effect in arteries from NFATc3-/- mice. Aortas were divided in halves and cultured either with or without UTP (100 μmol/L, N=3, 45-57 images analyzed in each group, ***P < 0.001 vs. all other bars). For the cerebral arteries, half of the arteries from each mouse were cultured with UTP and the other half without (10 μmol/L, N=3, 15-21 images in each group, ***P < 0.001 vs. all other bars). C. VSMCs and intact aortas were incubated for 3 hours in 5 mmol/L glucose or 25 mmol/L glucose with or without 1 μmol/L A-285222. NFATc3 enrichment by ChIP was performed and associated DNA complexes analyzed by quantitative PCR (in triplicates). Primers specific to NFAT binding site 3 of the OPN promoter were used. Aortas from 3-4 mice were used for each IP reaction, experiments were performed 3-5 times (for VSMCs and aortas, respectively), ***P < 0.001. D. OPN mRNA expression in fresh and cultured aortas (2 days in 5 mmol/L or 25 mmol/L glucose containing media). Results are shown as ratio of OPN expression to cyclophilin B, normalized to 5 mmol/L glucose. N=2, 10, 18 for fresh, 5 mmol/L and 25 mmol/L glucose respectively, in triplicate, *P < 0.05; **P < 0.01 and ***P < 0.001.
NFAT binds to elements in the OPN promoter region
A search of the mouse OPN promoter region revealed four potential NFAT binding sites (Supplemental Figure II). Two of these were located in the proximal promoter region (sites 3 and 4), whereas the other two in the distal region with only nine bases separating them (sites 1 and 2). Nuclear extracts from VSMCs and T-cells and a labeled NFAT consensus probe were used in EMSA experiments resulting in the formation of two complexes (Supplemental Figure II, left gels). The slower migrating complex was competed for by the addition of a 500-fold excess of unlabelled consensus oligo-nucleotide, or by an oligo containing sites 1 and 2, or site 3; but not site 4. This complex was not affected by a mutated NFAT binding site oligo. When site 3 was used as a probe (Supplemental Figure II, right panel), the slower migrating complex was competed for by the NFAT consensus site, by sites 1-2 and by site 3. Inclusion of a control antibody (anti-actin) did not disturb the binding of any of the complexes, while inclusion of an NFATc3 antibody largely reduced the formation of the slower migrating complex. Antibodies directed against NFATc4, NFATc1 or NFATc2 did not affect the binding of any of the complexes (Supplemental Figure III).
To address whether NFATc3 binds to the OPN promoter in vivo, we used a quantitative ChIP assay. VSMCs from aorta explants were incubated for 3 hours in low glucose (5 mmol/L) medium or in high glucose (25 mmol/L) with or without A-285222 (1 μmol/L), followed by formaldehyde fixation, imunoprecipitation of NFATc3 and real-time PCR analysis of immunoprecipitated DNA and input DNA using primers designed to flank the NFAT site 3 described above. As shown in Figure 4C (black bars), high glucose leads to enhanced NFATc3 binding to site 3, and this is prevented by inhibition of NFAT with A-285222. A similar pattern of response was obtained when experiments were repeated using intact aortas (Fig. 3C, grey bars). Further, we show that culture of intact arteries in high glucose for 2 days, leads to significantly increased OPN mRNA when compared to arteries cultured in low glucose (Figure 3D). Culture per se effectively increased OPN mRNA expression, which was low but still detected in freshly dissected arteries (Figure 3D).
In vivo inhibition of NFAT signaling prevents diabetes-induced OPN expression
Previous studies have shown that OPN expression is enhanced in arteries of diabetic patients and STZ-treated rats. In mice, increased OPN expression has been reported in arteries from db/db mice 11. Here we provide evidence for increased OPN expression in STZ treated mice. Eight weeks after the first STZ injection, subvalvular aortic sections were imaged using confocal immunofluorescence. Expression was significantly increased in the media of diabetic animals and correlated with blood glucose levels (r = 0.639, p<0.001, Supplemental Figure V). OPN expression in valve leaflets was lower than in the media, but still exhibited a positive correlation to blood glucose (r = 0.655, p<0.001, not shown). Plasma levels of OPN were significantly increased in diabetic animals and correlated to blood glucose (r = 0.588, p<0.005, Supplemental Figure V).
We then tested whether inhibition of NFAT signaling in vivo had any impact on OPN expression in aorta. For this, diabetic and control mice were treated with A-285222 or vehicle, administered i.p. once a day for the duration of the experiment. STZ-treated mice developed diabetes during week 2, whereas blood glucose remained unchanged in control mice (Figure 5A). After 4 weeks, OPN expression was significantly increased in aortas from diabetic animals when compared to normoglycemic mice (Figure 5B). Inhibition of NFAT signaling with A-285222 significantly reduced OPN expression in aorta. To test the specific involvement of NFATc3 isoform in the regulation of OPN expression in vivo, similar experiments were performed in NFATc3+/+ and NFATc3-/- mice. As shown in Fig. 5D, OPN expression was significantly increased in the aorta of STZ-treated NFATc3+/+ mice, whereas the effect was blunted in STZ-treated NFATc3-/- mice, despite the fact that they developed hyperglycemia to the same extent as NFATc3+/+ mice (Fig. 5C).
Figure 5. Lack of NFATc3 protein or in vivo treatment with A-285222 prevents OPN induction.
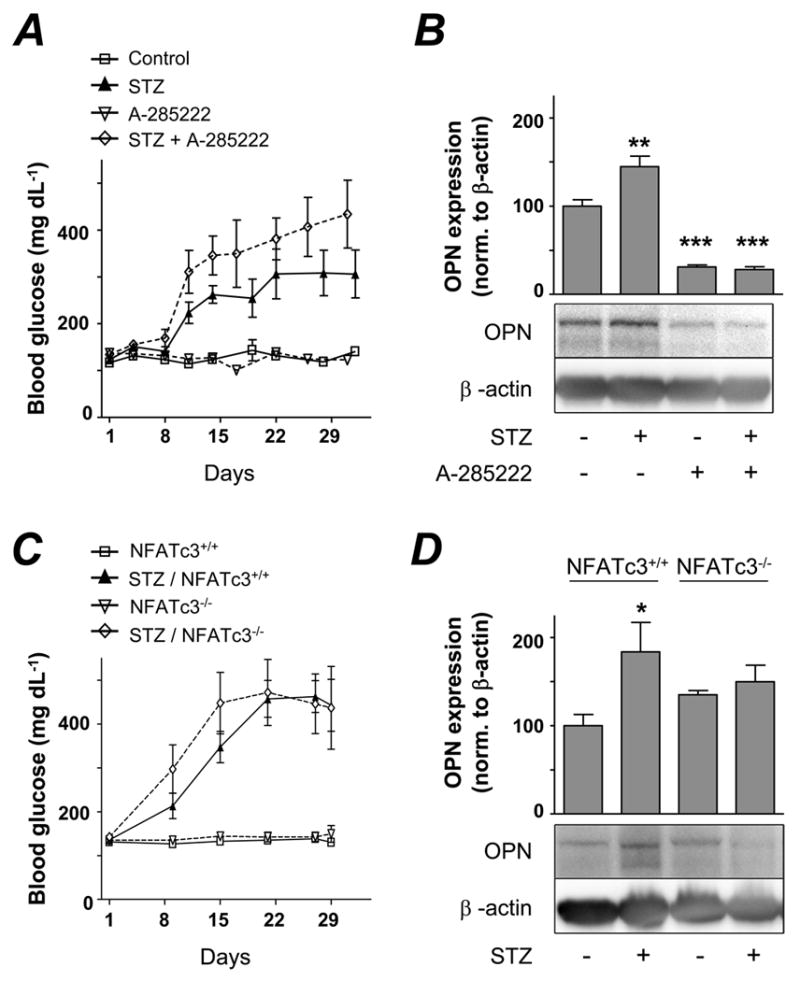
A. Blood glucose levels in BalB/c mice treated with STZ or vehicle. Mice received i.p. injections once a day of A-285222 or equal volumes of saline (control). B. Summarized data and representative immunoblot from western blot experiments showing OPN expression normalized to β-actin in the aorta of mice treated as in A (N=7-8 in each group). **P < 0.01 and ***P < 0.001 vs. untreated control. C. Blood glucose levels in NFATc3+/+ and NFATc3-/- mice treated with STZ or vehicle. D. Summarized data and representative immunoblot from western blot experiments showing OPN expression normalized to β-actin in the aorta of mice treated as in C (N=6-8 in each group).
Already during week 2, diabetic mice treated with A-285222 (0.29 mg/kg body weight) exhibited more pronounced hyperglycemia and weight loss than diabetic mice receiving saline (9.6% vs. 2.2% weight loss at day 16). In an attempt to prevent further weight loss, we adjusted the dose of A-285222 to 0.15 mg/kg body weight from day 16 until the end of the experiment. This lower concentration was better tolerated, since the rate of weight loss decreased. Strong evidence has been provided for the involvement of NFAT the regulation of insulin transcription in β-cells 26; therefore we asked whether the differences in blood glucose levels could be due to NFAT-inhibition on pancreatic islets. Reduced insulin production was observed in 6 out of 8 of the STZ + A-285222 treated mice, as shown by their lower insulinogenic index when compared to STZ-treated mice; plasma glucagon levels were not affected by A-285222 treatment (Supplemental Figure VI). Treatment with A-285222 did not affect glycemia or body weight in non-diabetic animals (3.2% vs. 5.5% weight loss for A-285222 vs. control).
Discussion
In this study we demonstrate for the first time that changes in blood glucose levels are readily detected by NFATc3 in arterial smooth muscle in vivo, and that the calcineurin/NFATc3 signaling pathway regulates the expression of OPN, which is a key player in the development of vascular disease. Our major findings are: 1) changes in extracellular glucose activate NFATc3 signaling in vivo, 2) NFATc3 has the ability to bind to at least one NFAT binding site (site 3) in the mouse promoter region of OPN, 3) NFATc3 activation leads to increased OPN mRNA and protein expression in intact arteries, 4) glucose-induced OPN expression is dependent on the release of extracellular nucleotides acting on P2Y membrane receptors, 5) glucose-induced OPN expression is prevented by pharmacological inhibition of calcineurin/NFAT signaling and 6) STZ-induced diabetes increases OPN expression in the ascending and thoracic aorta, vascular segments particularly prone to atherosclerosis in diabetic patients, and this is prevented by in vivo treatment with the NFAT inhibitor A-285222 or by lack of NFATc3 protein in arteries from NFATc3-/- mice. These results suggest a role for NFATc3 as a metabolic sensor in the vascular wall of potential relevance for vascular dysfunction in diabetes.
Our previous ex vivo studies show that raising extracellular glucose elicits a time- and dose-dependent increase in NFATc3 nuclear accumulation with a maximum response obtained at 360 mg/dL glucose (20 mmol/L) and after 20 min of exposure 18. The data presented here demonstrates a similar activation pattern in vivo, with significant NFATc3 nuclear accumulation at an average blood glucose level of 316.5 mg/dL (17.6 mmol/L) 18 min post IP-GTT. Based on published data describing NFATc3 nuclear export rates 27, a transient NFATc3 response can be predicted after an IP-GTT, with decreased NFATc3 nuclear accumulation once blood glucose is normalized, may be allowing the system to sense new changes in glycemia and leading to a new wave of transcriptional activity. This could be important in the clinical situation because transient hyperglycemic peaks may be sufficient for NFAT activation. Another interesting observation is that basal levels of NFATc3 nuclear accumulation measured in arteries from control mice (Figure 1A and B), which are perfusion fixed under pressurized conditions, are higher than the levels measured in previous ex vivo experiments in arteries fixed under non-pressurized conditions. This seems in agreement with evidence showing that intraluminal pressure is a stimulus for NFATc3 nuclear accumulation 28.
The regulation of OPN transcription has been suggested to require the concerted action of various transcription factors in VSMCs. The NFAT-binding site 3 identified here is located within the proximal region where both AP-1 and USF have been shown to bind. Although we did not test for possible synergisms between NFAT and AP-1 or USF, it is possible that all three proteins interact to overcome stereological constraints and enable stable assembly within this limited DNA region. Interestingly, in experiments using luciferase-reporter plasmids containing different lengths of the 5′ region of the rat OPN promoter, basal activity of the OPN promoter is greatly reduced when the regions harboring the NFAT site 3 investigated here are deleted 14, 29. On the other hand, the promoter regions identified to mediate glucose-(-112/-62 and -83/-45) and UTP-(-76/+1) induced transcription are adjacent to, but do not overlap with NFAT site 3 29, 30. Possible explanations to this inconsistency could be the incomplete sequence homology between species (mouse vs. rat). Nevertheless, the fact that pharmacological or genetic inhibition of NFAT signaling prevents glucose- and UTP-mediated OPN protein production in intact arteries, together with the ChIP results suggest that NFAT may bind to the OPN promoter under these conditions.
To date, it is not clear what the exact role of individual NFAT isoforms is in the vasculature. Although all NFAT isoforms are expressed in aortic VSMCs (data not shown), the luciferase experiments using NFAT-luc/NFATc3+/+, NFAT-luc/NFATc3+/- and NFAT-luc/NFATc3-/- mice demonstrate that NFATc3 is responsible for the increased NFAT-dependent transcriptional activity measured in diabetic animals. EMSA results suggest that probably only NFATc3 is able to bind to the identified NFAT-sites in the OPN promoter. ChIP results confirm that this isoform binds to the OPN promoter in vivo. The experiments using NFATc3-/- mice further support a role for this isoform in the regulation of OPN expression. Recent studies have shown that activation of NFAT, and more specifically NFATc3, promotes VSMC proliferation and IL-6 production in human resistance arteries 23, drives the expression of the inflammatory protein vascular cell adhesion molecule (VCAM)-1 31 and plays a role during pulmonary hypertension 32. Also, NFATc3 down-regulates the expression of voltage-gated K+ channels (Kv2.1) and the β1 subunit of large conductance, Ca2+-activated K+ channels, mediating angiotensin II-induced hypertension 33. Taken together, these events all contribute to arterial dysfunction and if combined with enhanced OPN expression may intensify the severity of vascular disease.
Several important findings emerged from the in vivo treatment of mice with the NFAT blocker A-285222. Interestingly, the arteries of mice treated with A-285222 exhibit dramatically reduced OPN expression despite glucose levels even higher than in non-treated diabetic mice. Also, treatment of normoglycemic mice with A-285222 resulted in OPN expression below basal levels, supporting the idea of a constitutively active NFAT signaling pathway under control conditions. This is the first in vivo study in mice using A-285222. The only pharmacokinetic or pharmacodynamic data available is from a study by Birsan et al 34 in which cynomolgus monkeys were treated with A-285222 orally twice a day for several days. Already after one day, a marked inhibition of NFAT signaling, as assessed by reduced T-cell cytokine production was observed. They also showed that treatment with A-285222 was possible over long periods of time and that no neurotoxicity develops if administered at 5-7.5 mg/kg body weight. In our hands, a much lower dose was required for the clear vascular effects reported here (0.15-0.29 mg/kg body weight). However, the undesired effects observed on β-cell function points out the need of further adjustments of the drug dose and/or a more local delivery of the compound.
The concept of the calcineurin/NFAT signaling pathway as a sensor in the vascular wall of potential relevance for vascular dysfunction in diabetes is novel. Despite all the evidence linking hyperglycemia to increased risk of microvascular and macrovascular disease 35, 36, the molecular mechanisms underlying vascular damage are not completely understood. Most studies focus on the generation of oxidative stress as the pathogenic mechanism linking hyperglycemia to endothelial dysfunction, an initial step in atherosclerosis 37. In endothelial cells, hyperglycemia promotes the production of reactive oxygen species (ROS), which in turn, activates four seemingly independent biochemical pathways: activation of the aldose reductase, protein kinase C and hexosamine pathways, and increase of advanced glycation end products 38. Activation of these pathways triggers the production of atherogenic circulating adhesion molecules and inflammatory cytokines. The identification of an additional pathway in this study, able to sense hyperglycemia in both large and resistance arteries adds a new layer of complexity to the process of diabetic angiopathy and reveals a potential therapeutic target and biomarker.
Supplementary Material
Acknowledgments
We thank Bodil Israelsson, Ingrid Söderberg and Irena Ljungcrantz for skillful technical assistance; Dr. Paul Mc Guire for assistance in the experiments involving STZ-treated NFAT-luc mice and Dr. Olga Kotova for reading the manuscript.
Sources of support: Grants from the Crafoord, Albert Påhlsson, Lars Hierta Memorial, Åke Wiberg, Knut & Alice Wallenberg and Swedish Heart and Lung Foundations. Also from the Royal Physiographic Society in Lund, Malmö Hospital Research Funds, Swedish Research Council (#2006-5287), Vascular Wall Programme and Lund University Diabetes Centre. NIH HL081682, AHA SDG to Brian R. Wamhoff.
Footnotes
Disclosures: None.
References
- 1.Ohmori R, Momiyama Y, Taniguchi H, Takahashi R, Kusuhara M, Nakamura H, Ohsuzu F. Plasma osteopontin levels are associated with the presence and extent of coronary artery disease. Atherosclerosis. 2003;170:333–337. doi: 10.1016/s0021-9150(03)00298-3. [DOI] [PubMed] [Google Scholar]
- 2.Kato R, Momiyama Y, Ohmori R, Tanaka N, Taniguchi H, Arakawa K, Kusuhara M, Nakamura H, Ohsuzu F. High plasma levels of osteopontin in patients with restenosis after percutaneous coronary intervention. Arterioscler Thromb Vasc Biol. 2006;26:e1–2. doi: 10.1161/01.ATV.0000194157.26665.e6. [DOI] [PubMed] [Google Scholar]
- 3.Golledge J, Muller J, Shephard N, Clancy P, Smallwood L, Moran C, Dear AE, Palmer LJ, Norman PE. Association between osteopontin and human abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2007;27:655–660. doi: 10.1161/01.ATV.0000255560.49503.4e. [DOI] [PubMed] [Google Scholar]
- 4.Scatena M, Liaw L, Giachelli CM. Osteopontin: A Multifunctional Molecule Regulating Chronic Inflammation and Vascular Disease. Arterioscler Thromb Vasc Biol. 2007;27:2302–2309. doi: 10.1161/ATVBAHA.107.144824. [DOI] [PubMed] [Google Scholar]
- 5.Strom A, Franzen A, Wangnerud C, Knutsson AK, Heinegard D, Hultgardh-Nilsson A. Altered vascular remodeling in osteopontin-deficient atherosclerotic mice. J Vasc Res. 2004;41:314–322. doi: 10.1159/000079205. [DOI] [PubMed] [Google Scholar]
- 6.Matsui Y, Rittling SR, Okamoto H, Inobe M, Jia N, Shimizu T, Akino M, Sugawara T, Morimoto J, Kimura C, Kon S, Denhardt D, Kitabatake A, Uede T. Osteopontin Deficiency Attenuates Atherosclerosis in Female Apolipoprotein E-Deficient Mice. Arterioscler Thromb Vasc Biol. 2003;23:1029–1034. doi: 10.1161/01.ATV.0000074878.29805.D0. [DOI] [PubMed] [Google Scholar]
- 7.Chiba S, Okamoto H, Kon S, Kimura C, Murakami M, Inobe M, Matsui Y, Sugawara T, Shimizu T, Uede T, Kitabatake A. Development of atherosclerosis in osteopontin transgenic mice. Heart Vessels. 2002;16:111–117. doi: 10.1007/s003800200005. [DOI] [PubMed] [Google Scholar]
- 8.Yamaguchi H, Igarashi M, Hirata A, Tsuchiya H, Sugiyama K, Morita Y, Jimbu Y, Ohnuma H, Daimon M, Tominaga M, Kato T. Progression of diabetic nephropathy enhances the plasma osteopontin level in type 2 diabetic patients. Endocr J. 2004;51:499–504. doi: 10.1507/endocrj.51.499. [DOI] [PubMed] [Google Scholar]
- 9.Kase S, Yokoi M, Saito W, Furudate N, Ohgami K, Kitamura M, Kitaichi N, Yoshida K, Kase M, Ohno S, Uede T. Increased osteopontin levels in the vitreous of patients with diabetic retinopathy. Ophthalmic Res. 2007;39:143–147. doi: 10.1159/000102936. [DOI] [PubMed] [Google Scholar]
- 10.Takemoto M, Yokote K, Nishimura M, Shigematsu T, Hasegawa T, Kon S, Uede T, Matsumoto T, Saito Y, Mori S. Enhanced Expression of Osteopontin in Human Diabetic Artery and Analysis of Its Functional Role in Accelerated Atherogenesis. Arterioscler Thromb Vasc Biol. 2000;20:624–628. doi: 10.1161/01.atv.20.3.624. [DOI] [PubMed] [Google Scholar]
- 11.Martin AS, Du P, Dikalova A, Lassegue B, Aleman M, Gongora MC, Brown K, Joseph G, Harrison DG, Taylor WR, Jo H, Griendling KK. Reactive oxygen species-selective regulation of aortic inflammatory gene expression in Type 2 diabetes. Am J Physiol Heart Circ Physiol. 2007;292:H2073–2082. doi: 10.1152/ajpheart.00943.2006. [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Louden C, Ohlstein EH, Stadel JM, Gu JL, Yue TL. Osteopontin expression in platelet-derived growth factor-stimulated vascular smooth muscle cells and carotid artery after balloon angioplasty. Arterioscler Thromb Vasc Biol. 1996;16:1365–1372. doi: 10.1161/01.atv.16.11.1365. [DOI] [PubMed] [Google Scholar]
- 13.Giachelli CM, Bae N, Almeida M, Denhardt DT, Alpers CE, Schwartz SM. Osteopontin is elevated during neointima formation in rat arteries and is a novel component of human atherosclerotic plaques. J Clin Invest. 1993;92:1686–1696. doi: 10.1172/JCI116755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Renault MA, Jalvy S, Belloc I, Pasquet S, Sena S, Olive M, Desgranges C, Gadeau AP. AP-1 Is Involved in UTP-Induced Osteopontin Expression in Arterial Smooth Muscle Cells. Circ Res. 2003;93:674–681. doi: 10.1161/01.RES.0000094747.05021.62. [DOI] [PubMed] [Google Scholar]
- 15.Renault MA, Jalvy S, Potier M, Belloc I, Genot E, Dekker LV, Desgranges C, Gadeau AP. UTP Induces Osteopontin Expression through a Coordinate Action of NF{kappa}B, Activator Protein-1, and Upstream Stimulatory Factor in Arterial Smooth Muscle Cells. J Biol Chem. 2005;280:2708–2713. doi: 10.1074/jbc.M411786200. [DOI] [PubMed] [Google Scholar]
- 16.Gomez MF, Stevenson AS, Bonev AD, Hill-Eubanks DC, Nelson MT. Opposing actions of inositol 1,4,5-trisphosphate and ryanodine receptors on nuclear factor of activated T-cells regulation in smooth muscle. J Biol Chem. 2002;277:37756–37764. doi: 10.1074/jbc.M203596200. [DOI] [PubMed] [Google Scholar]
- 17.Liu Z, Dronadula N, Rao GN. A Novel Role for Nuclear Factor of Activated T Cells in Receptor Tyrosine Kinase and G Protein-coupled Receptor Agonist-induced Vascular Smooth Muscle Cell Motility. J Biol Chem. 2004;279:41218–41226. doi: 10.1074/jbc.M406917200. [DOI] [PubMed] [Google Scholar]
- 18.Nilsson J, Nilsson LM, Chen YW, Molkentin JD, Erlinge D, Gomez MF. High Glucose Activates Nuclear Factor of Activated T Cells in Native Vascular Smooth Muscle. Arterioscler Thromb Vasc Biol. 2006;26:794–800. doi: 10.1161/01.ATV.0000209513.00765.13. [DOI] [PubMed] [Google Scholar]
- 19.Kawamura H, Yokote K, Asaumi S, Kobayashi K, Fujimoto M, Maezawa Y, Saito Y, Mori S. High glucose-induced upregulation of osteopontin is mediated via Rho/Rho kinase pathway in cultured rat aortic smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:276–281. doi: 10.1161/01.ATV.0000112012.33770.2a. [DOI] [PubMed] [Google Scholar]
- 20.Nilsson LM, Nilsson-Ohman J, Zetterqvist AV, Gomez MF. Nuclear factor of activated T-cells transcription factors in the vasculature: the good guys or the bad guys? Curr Opin Lipidol. 2008;19:483–490. doi: 10.1097/MOL.0b013e32830dd545. [DOI] [PubMed] [Google Scholar]
- 21.Lipskaia L, Pourci ML, Delomenie C, Combettes L, Goudouneche D, Paul JL, Capiod T, Lompre AM. Phosphatidylinositol 3-Kinase and Calcium-Activated Transcription Pathways Are Required for VLDL-Induced Smooth Muscle Cell Proliferation. Circ Res. 2003;92:1115–1122. doi: 10.1161/01.RES.0000074880.25540.D0. [DOI] [PubMed] [Google Scholar]
- 22.Liu Z, Zhang C, Dronadula N, Li Q, Rao GN. Blockade of Nuclear Factor of Activated T Cells Activation Signaling Suppresses Balloon Injury-induced Neointima Formation in a Rat Carotid Artery Model. J Biol Chem. 2005;280:14700–14708. doi: 10.1074/jbc.M500322200. [DOI] [PubMed] [Google Scholar]
- 23.Nilsson LM, Sun ZW, Nilsson J, Nordstrom I, Chen YW, Molkentin JD, Wide-Swensson D, Hellstrand P, Lydrup ML, Gomez MF. Novel blocker of NFAT activation inhibits IL-6 production in human myometrial arteries and reduces vascular smooth muscle cell proliferation. Am J Physiol Cell Physiol. 2007;292:C1167–1178. doi: 10.1152/ajpcell.00590.2005. [DOI] [PubMed] [Google Scholar]
- 24.McDonald OG, Wamhoff BR, Hoofnagle MH, Owens GK. Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo. J Clin Invest. 2006;116:36–48. doi: 10.1172/JCI26505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wamhoff BR, Bowles DK, McDonald OG, Sinha S, Somlyo AP, Somlyo AV, Owens GK. L-type voltage-gated Ca2+ channels modulate expression of smooth muscle differentiation marker genes via a rho kinase/myocardin/SRF-dependent mechanism. Circ Res. 2004;95:406–414. doi: 10.1161/01.RES.0000138582.36921.9e. [DOI] [PubMed] [Google Scholar]
- 26.Heit JJ. Calcineurin/NFAT signaling in the beta-cell: From diabetes to new therapeutics. Bioessays. 2007;29:1011–1021. doi: 10.1002/bies.20644. [DOI] [PubMed] [Google Scholar]
- 27.Gomez MF, Bosc LV, Stevenson AS, Wilkerson MK, Hill-Eubanks DC, Nelson MT. Constitutively elevated nuclear export activity opposes Ca2+-dependent NFATc3 nuclear accumulation in vascular smooth muscle: role of JNK2 and Crm-1. J Biol Chem. 2003;278:46847–46853. doi: 10.1074/jbc.M304765200. [DOI] [PubMed] [Google Scholar]
- 28.Gonzalez Bosc LV, Wilkerson MK, Bradley KN, Eckman DM, Hill-Eubanks DC, Nelson MT. Intraluminal Pressure Is a Stimulus for NFATc3 Nuclear Accumulation: ROLE OF CALCIUM, ENDOTHELIUM-DERIVED NITRIC OXIDE, AND cGMP-DEPENDENT PROTEIN KINASE. J Biol Chem. 2004;279:10702–10709. doi: 10.1074/jbc.M312920200. [DOI] [PubMed] [Google Scholar]
- 29.Bidder M, Shao JS, Charlton-Kachigian N, Loewy AP, Semenkovich CF, Towler DA. Osteopontin Transcription in Aortic Vascular Smooth Muscle Cells Is Controlled by Glucose-regulated Upstream Stimulatory Factor and Activator Protein-1 Activities. J Biol Chem. 2002;277:44485–44496. doi: 10.1074/jbc.M206235200. [DOI] [PubMed] [Google Scholar]
- 30.Asaumi S, Takemoto M, Yokote K, Ridall AL, Butler WT, Fujimoto M, Kobayashi K, Kawamura H, Take A, Saito Y, Mori S. Identification and characterization of high glucose and glucosamine responsive element in the rat osteopontin promoter. J Diabetes Complications. 2003;17:34–38. doi: 10.1016/s1056-8727(02)00189-7. [DOI] [PubMed] [Google Scholar]
- 31.Orr AW, Lee MY, Lemmon JA, Yurdagul A, Jr, Gomez MF, Schoppee Bortz PD, Wamhoff BR. Molecular Mechanisms of Collagen Isotype-Specific Modulation of Smooth Muscle Cell Phenotype. Arterioscler Thromb Vasc Biol. 2008 doi: 10.1161/ATVBAHA.108.178749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Frutos S, Spangler R, Alo D, Bosc LV. NFATc3 mediates chronic hypoxia-induced pulmonary arterial remodeling with alpha-actin up-regulation. J Biol Chem. 2007;282:15081–15089. doi: 10.1074/jbc.M702679200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nieves-Cintron M, Amberg GC, Nichols CB, Molkentin JD, Santana LF. Activation of NFATc3 down-regulates the beta1 subunit of large conductance, calcium-activated K+ channels in arterial smooth muscle and contributes to hypertension. J Biol Chem. 2007;282:3231–3240. doi: 10.1074/jbc.M608822200. [DOI] [PubMed] [Google Scholar]
- 34.Birsan T, Dambrin C, Marsh KC, Jacobsen W, Djuric SW, Mollison KW, Christians U, Carter GW, Morris RE. Preliminary in vivo pharmacokinetic and pharmacodynamic evaluation of a novel calcineurin-independent inhibitor of NFAT. Transpl Int. 2004;3:145–150. doi: 10.1007/s00147-003-0676-1. [DOI] [PubMed] [Google Scholar]
- 35.Klein R. Hyperglycemia and microvascular and macrovascular disease in diabetes. Diabetes Care. 1995;18:258–268. doi: 10.2337/diacare.18.2.258. [DOI] [PubMed] [Google Scholar]
- 36.Davidson JA. Treatment of the patient with diabetes: importance of maintaining target HbA(1c) levels. Curr Med Res Opin. 2004;20:1919–1927. doi: 10.1185/030079904X6291. [DOI] [PubMed] [Google Scholar]
- 37.Yamagishi SI, Nakamura K, Matsui T, Ueda SI, Imaizumi T. Role of postprandial hyperglycaemia in cardiovascular disease in diabetes. Int J Clin Pract. 2007;61:83–87. doi: 10.1111/j.1742-1241.2006.01168.x. [DOI] [PubMed] [Google Scholar]
- 38.Brownlee M. The Pathobiology of Diabetic Complications: A Unifying Mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.