

Abstract
Background
To gain insight into the molecular diversity of antimicrobial peptides and proteins in the oyster Crassostrea gigas, we characterized and compared the sequence polymorphism of the antimicrobial peptides (AMPs), Cg-Defensins (Cg-Defs) and Cg-Proline Rich peptide (Cg-Prp), and of the bactericidal permeability increasing protein, Cg-BPI. For that, we analyzed genomic and transcript sequences obtained by specific PCR amplification and in silico searches.
Results
High diversification among the three antimicrobial effectors was evidenced by this polymorphism survey. On the basis of sequence phylogenies, each AMP aggregates into clearly defined groups of variants and is the product of a multigenic family displaying a variety of gene structures. In contrast, Cg-bpi forms a single group and is encoded by a single gene copy. Moreover, we identified for both AMPs several genetic mechanisms of diversification such as recombination, parallel mutations leading to phylogenetic homoplasy and indel events. In addition, the non synonymous to synonymous substitutions ratio by codon (dN/dS) revealed several negatively and positively selected sites for both AMPs, suggesting that directional selection pressures have shaped their sequence variations.
Conclusions
This study shows for the first time in a mollusc that antimicrobial peptides and proteins have been subject to distinct patterns of diversification and we evidence the existence of different evolutionary routes leading to such sequence variability.
Background
The Pacific oyster, Crassostrea gigas, is a filter feeder bivalve mollusc which is continually exposed to microorganisms naturally present in the marine environment. Thus, the capacity to control the microflora and overcome infections is essential for the oyster survival. The hemocytes, immunocompetent cells, play a central role in innate antimicrobial response. Indeed, in molluscs, hemocytes infiltrate injured tissues, phagocyte microorganisms, produce antimicrobial peptides (AMPs) and release factors such as lectins and reactive oxygen species (ROS) [1-3].
In C. gigas, several antimicrobial effectors have been recently characterized. An AMP, member of the defensin family named Cg-Defm, was identified from the oyster mantle [4]. Then, two additional defensins named Cg-Defhs (Cg-Defh1 and Cg-Defh2)have been characterized from hemocytes [5]. Both Cg-defm and Cg-defhs appear to be continuously expressed in different tissues. More recently, a new peptide, Cg-Prp, which belongs to the proline-rich AMP family, has been found in hemocytes [6]. Whereas Cg-Prp displays alone a weak in vitro antimicrobial activity, it revealed synergistic activity with Cg-Defm against both Gram positive and negative bacteria [6]. Additionally, a member of the bactericidal/permeability-increasing protein (BPI) family has been characterized [7]. Cg-bpi expression is induced in hemocytes after oyster bacterial challenge and constitutive in various tissue epithelia of unchallenged oysters. Similar to vertebrate BPIs, the oyster BPI binds LPS, displays bactericidal activity against Gram negative bacteria and increases the permeability of the bacterial cytoplasmic membrane [7].
High levels of sequence diversity were reported to be a characteristic of several antimicrobial effectors pertaining to the innate immunity system of invertebrates [8]. The functional significance of this diversity has been addressed in some cases. Multiple variants of lectins from the horseshoe crab Carcinoscorpius rotundicauda are able to recognize and differentiate bacteria and fungi [9]. The AMP drosomycin from Drosophila melanogaster presents six isoforms with different antifungal activities [10]. Thus, diversification of antimicrobial effectors by accumulation of multiple variations around an originally unique form may provide significant means of acquiring microbial target specificity [11], concerned in the evolutionary arms race between pathogens and their hosts.
In this work, we have investigated the diversity of three oyster antimicrobial effectors and shown the existence of a high diversity in the transcript and genomic sequences of three antimicrobials, two peptides (Cg-prp and Cg-defs) and one protein (Cg-bpi). We also evidence distinct phylogenies between them and propose the existence of a combination of different genetic mechanisms from which sequence variability arises.
Results
C. gigas antimicrobial peptides and BPI protein show distinct phylogeny conformations
To characterize the diversity of all the genes, i.e. the two antimicrobial peptides Cg-defs and Cg-prp, the antimicrobial protein Cg-bpi and the non immune gene Cg-actin, we performed an exhaustive sequencing of cloned PCR products and collected sequences in the Sigenae database http://www.sigenae.org/. Finally, we got a total of 256 cDNA sequences and 60 gDNA sequences that we aligned and used to build phylogenies (Table 1). Alignments are available as Popset in GenBank; cDNA sequences of Cg-prp, Cg-defs, Cg-bpi and Cg-actin: FJ669353-FJ669402, FJ669323-FJ669352, FJ669296-FJ669322, FJ669287-FJ669295 respectively and gDNA sequences of Cg-prp and Cg-defs: FJ669252-FJ669286 and FJ669403-FJ669423 respectively. We obtained for both AMPs, Cg-prp and Cg-defs, clearly structured phylogenies where sequences clustered in distinct groups, whereas Cg-bpi and Cg-actin sequences formed star-like phylogenies instead (Figure 1). On this basis, we identified three distinct and more or less distant groups of Cg-defs (Figure 1a, group I, II and III), each one composed by multiple variants of Cg-defh1, Cg-defh2 and Cg-defm, respectively.
Table 1.
Number of nucleotide sequences and general polymorphism values of antimicrobial effectors and non immune gene
| Cg-prp | Cg-defm | Cg-defhs | Cg-bpi | Cg-actin | |
|---|---|---|---|---|---|
| PCR gDNA | 35 | 11 | 14 | 0 | 0 |
| PCR cDNA | 57 | 40 | 43 | 36 | 40 |
| in silico | 12 | 2 | 15 | 1 | 10 |
| total | 104 | 53 | 72 | 37 | 50 |
| different* | 50 | 18 | 12 | 28 | 9 |
| Length (bp) | 177/183 | 195/219 | 195 | 1431 | 1128 |
| SNP ratio 3 | 4.06 | 12.9 | 5.2 | 16.8 | 66.3 |
| Hd | 0.983 | 0.857 | 0.454 | 0.987 | 0.093 |
| Pi | 0.044 | 0.009 | 0.022 | 0.009 | 0.002 |
*N° of sequences with a minimum of 1 nucleotide difference founded at least two times (PCR and/or in silico searches)
SNP ratio: ratio of the number of polymorphic sites to the length of CDS i.e. reflect the density of polymorphic sites
Hd: Proportion of different sequences
Pi: nucleotide diversity
Figure 1.
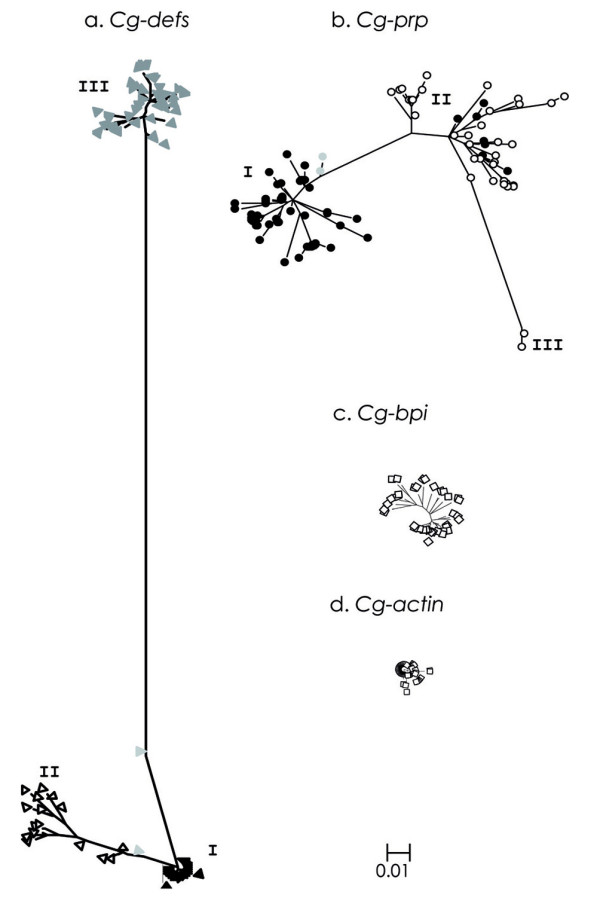
Phylogenies from CDS of three antimicrobial effectors and non immune gene from C. gigas. a. Cg-defs: I. Cg-defh1 (black triangles); II. Cg-defh2 (white triangles) and III. Cg-defm (dark gray triangles). Recombinant sequences between Cg-defh1 and Cg-defh2 are indicated (light gray triangles). b. Cg-prp: I. Cg-prp short forms (black circles), II. Cg-prp short and long forms, and III. Cg-prp long forms (white circles). Cg-prp recombinant sequences are indicated (gray circles). c. Cg-bpi (white rhombus) and d. Cg-actin (white squares). Construction of cladograms were performed with PHYML v2.4.4 using the models GTR+Γ for Cg-defs, HKY85+Γ for Cg-prp, TN93+I for Cg-bpi and F84+Γ for Cg-actin determined by the Akaike information criterion. For accurate comparison between the four genes, cladograms were drawn in the same scale.
Furthermore, we uncovered two lengths of transcripts for Cg-defm, one corresponding to the coding sequence (CDS) of 195 bp previously described [4] and a longer one identified here for the first time with a CDS of 219 bp. Similarly, we identified two lengths of transcripts for Cg-prp, the one already described with a CDS of 183 bp [6] and a novel shorter one with an indel of 6 nucleotides in the C-terminal region. Likewise to Cg-defs, Cg-prp sequences are distributed into three different groups (Figure 1b). Group I is only composed of short sequences, group II of long and short sequences, while group III contained only two long and highly divergent sequences. In addition, whenever we got genomic sequences, they are evenly distributed in every group. In contrast to both AMPs, only one group of sequences and homogeneous sequence lengths were obtained for Cg-bpi (Figure 1c) and Cg-actin (Figure 1d).
The four genes were compared considering the total nucleotide diversity (Pi) from all identified sequences for each gene, their haplotype diversity (Hd) and the mean number of Single Nucleotide Polymorphism (SNPs) in the coding region. Comparison between these values gave evidence of different constraint forces acting on each gene (Table 1). Besides, the average density of SNPs identified for Cg-actin was in agreement with SNP values estimated for the genome of C. gigas of one SNP every 60 bp, also characterised by direct sequencing of PCR products of several genes [12].
Additionally, the comparison of mean nucleotide diversity (Pi) and the mean nucleotide divergence between groups of each AMP showed that Cg-prp presents more diverse but less divergent groups compared to Cg-defs. Cg-prp presents Pi values of 0.023, 0.035 and 0.006 within group I, II and III, respectively, whereas Cg-defs groups present lower Pi values of 0.001, 0.02 and 0.009 within group I, II and III, respectively. Interestingly, Cg-defh1 (group I) appears to be highly conserved as compared to Cg-defh2 (group II). Cg-bpi and Cg-actin, which both yield an unstructured phylogeny, present a Pi of 0.009 and 0.002 respectively.
Besides, values of mean nucleotide divergences between groups of AMPs show a higher divergence of Cg-defs groups compared to Cg-prp ones. Cg-defs present mean divergence values of 0.058 between group I and II; 0.214 between group I and III and 0.223 between group II and III, while Cg-prp present lower values of 0.063 between group I and II; 0.101 between group I and III and 0.081 between group II and III. This phylogenetic analysis indicates that Cg-defm is definitively distinct from Cg-defhs. We then separate Cg-defm and Cg-defhs for further polymorphism analysis.
Antimicrobial peptides are encoded by multigene families with different gene structures
The evidence of different groups revealed by AMP phylogenies and the number of genomic sequence variants found in one single individual strongly suggest the existence of multiple gene copies. We have therefore estimated the gene copy number of the four molecules analysed in this study. The gene copy number estimation on 3 individuals revealed that Cg-prp and Cg-defs are both represented by a high number of copies with mean values of 13 and 48 copies, respectively. It must be noticed that the primers used for this quantification did not allow the distinction between the different defensins. Thus, this value represents the sum of Cg-defhs and Cg-defm copies. In contrast, Cg-bpi is found as a single copy gene. Cg-actin is represented by a higher copy number (mean value of 168 copies), and as for Cg-defs and Cg-prp, its number of copies varies among the three oysters analysed (Table 2).
Table 2.
Estimation of gene copy number of antimicrobial effectors and non immune gene by real time PCR
| gene | Sample | Primer efficiency (E) | Calculated copy number | Estimated copy number | ||
|---|---|---|---|---|---|---|
| Mean1 | SD | Mean2 | SD | |||
| Cg-bpi | pDNA | 2.01 | ± 0.006 | |||
| gDNA | 2.06 | ± 0.060 | 0.95 | ± 0.10 | 1 | |
| Cg-prp | pDNA | 1.99 | ± 0.008 | |||
| gDNA | 2.03 | ± 0.040 | 12.59 | ± 0.99 | 11 - 13 | |
| Cg-defs | pDNA | 2.02 | ± 0.004 | |||
| gDNA | 2.03 | ± 0.010 | 47.46 | ± 6.4 3 | 41 - 53 | |
| Cg-actin | pDNA | 1.99 | ± 0.030 | |||
| gDNA | 2.07 | ± 0.040 | 168.04 | ± 10. 4 | 156 - 176 | |
gDNA: genomic DNA from C. gigas
pDNA: plasmid DNA used as standard
1 Mean of primer efficiency (E = 10-slope) from three technical replicates
2 Mean of three biological replicates
SD: standard deviation
Because both Cg-defs and Cg-prp AMPs are present as multigene families, we also characterized their gene structure by exhaustive gDNA sequencing from one individual. The genomic organization of Cg-prp and Cg-defs revealed two different structures for Cg-prp and one single structure for Cg-defs (Figure 2). The two structures of Cg-prp correspond either to genes with or without an intron, both arrangements coding for the long and short peptide variants. All Cg-prp genes containing the intron have the same exonic organization. Exon 1 contains the sequence encoding the signal peptide and half of the residues of the anionic domain (103 bp) while exon 2 encodes the end of the anionic domain and the cationic domain region (177-183 bp) (figure 2a). Nevertheless, we identified several indel events between introns of long and short variants. In addition, intron sequences of short Cg-prp variants showed length variations ranging from 571 to 588 bp and multiple SNPs. In contrast to Cg-prp, we found a single genomic structure for Cg-defs. Exon 1 contains the sequence encoding part of the signal peptide (43 bp) and exon 2 encodes the remaining part of the signal peptide region and the mature peptide region (149 bp), except for the last two residues encoded by exon 3 (5 bp) (figure 2b). However, no homology was found between intron sequences of Cgdefm and Cg-defhs, confirming thus the very ancient divergence time between the two lineages of these genes. Moreover, introns between Cg-defm and Cg-defhs differ by length variations. Cg-defs and Cg-prp AMP genes contain canonical GT/AG splicing recognition sequence located at the end of each intron.
Figure 2.
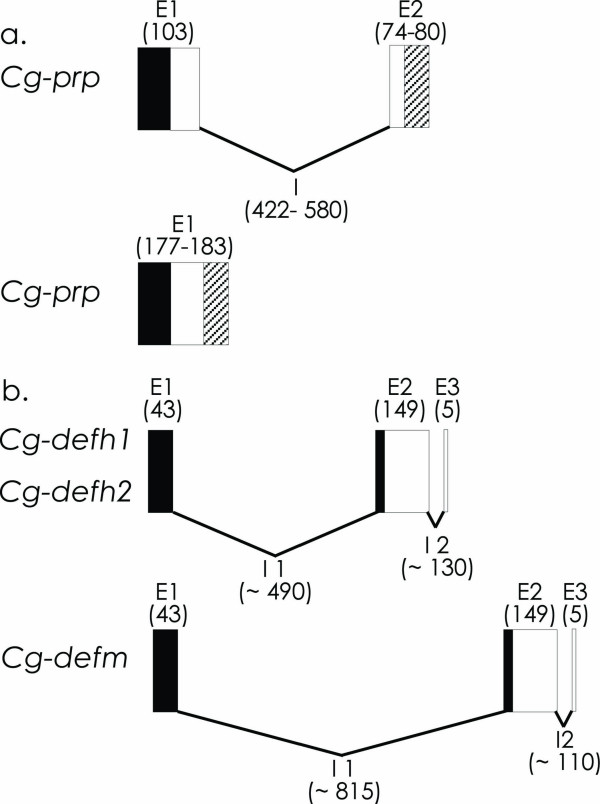
Gene organization of Cg-defs and Cg-prp from C. gigas. a. Cg-prp genomic structures. Black boxes indicate signal peptide region; white boxes indicate anionic domain region and striped boxes indicate cationic domain region. b. Cg-defhs and Cg-defm gene structures. Black boxes indicate signal peptide region and white boxes indicate mature peptide region. Numbers indicate the length of exons and introns (bases pairs). Note the gene size is not to scale.
Antimicrobial peptides and protein variants are produced by several mechanisms of diversification
The cDNA and gDNA sequence diversity and the variable gene copy number observed between C. gigas families of antimicrobials prompted us to investigate the genetic processes that participated to the diversification. Thus, we analyzed the repartition of synonymous and non synonymous substitutions in each group from AMP phylogenies (figure 3). Each group of Cg-defs presents a specific distribution of substitutions all along the CDS (figure 3a). Thus, Cg-defh1 is the most overall conserved antimicrobial peptide, with only a few numbers of substitutions. Cg-defh2, in contrast, presents a high number of synonymous substitutions and Cg-defm presents a profile which differs from hemocyte defensins with higher number of nonsynonymous substitutions. Cg-prp shows a different profile compared to Cg-defs in terms of the distribution of substitutions. Indeed, the substitutions localized in the signal and anionic peptide region are shared by groups, while substitutions in the cationic peptide region are specific of each group (figure 3b). Altogether, these results show the existence of different constraint levels between and within the groups of AMPs (Cg-defs and Cg-prp).
Figure 3.

Cladograms Cg-defs and Cg-prp and schematic representation of aligned CDS with synonymous and nonsynonymous substitutions. a. Cg-defs. b. Cg-prp. Identical sequences are grouped and the number is indicated at the right of each branch of cladograms and sequence symbols are the same as in figure 1. Nonsynonymous (blue boxes) and synonymous substitutions (yellow boxes) are localized over schematic alignment. Different groups of sequences (I, II and III) and obvious recombinants (R) are boxed. Indel events are represented by dark lines (Cg-prp) and striped box (Cg-defs) and circles indicate parallel homoplasic mutations. SP: signal peptide; MP: Mature peptide; AD: Anionic Domain; CD: Cationic Domain. For accurate comparison between AMPs, cladograms were drawn in the same scale.
Sequence alignments gave also evidence of several genetic mechanisms of diversification such as recombination and parallel mutations leading to phylogenetic homoplasy. Clues of recombination were detected by the visual inspection of alignments and by the construction of cladograms using different domains of each antimicrobial (data not shown) that lead to inconsistent clustering of sequences: several sequences of Cg-defhs, Cg-prp and Cg-bpi moving from one clade to another according to the domain used, suggesting past recombination events between members of different groups. We then calculated the minimum number of recombination events (Rm) for all genes. Cg-defhs, Cg-prp and Cg-bpi showed Rm values of 5, 6 and 10, respectively. In contrast, we did not find any evidence of recombination for Cg-defm and Cg-actin. Because of the large difference in size between our genes and since the probability of recombination is proportional to the length of each sequence, we calculated a ratio of Rm/CDS length for a correct comparison between AMPs and Cg-bpi protein. The ratios of Rm/CDS length for Cg-defhs, Cg-prp and Cg-bpi were 0.030, 0.027 and 7 × 10-3 respectively, which gave evidence of a higher probability in AMPs to present recombination between alleles compared to Cg-bpi.
In addition to the increase in the gene copy number described above, other processes that participate in AMP diversification are indel events which produce length variations in the CDS. Long variants of Cg-defm are obtained by an indel of two nucleotides at the 3'end (position 191) which changes the open reading frame and generates a longer molecule of 24 nucleotides, coding for a peptide of 73 amino acids with eight extra amino acids compared to the original form. For Cg-prp, the two lengths are generated by an indel of six nucleotides near the 3'end (position 171), corresponding to a repetition of the precedent nucleotides. These forms code for peptides of 59 and 62 amino acids.
Antimicrobial peptides and protein show a great number of variants and AMP diversity is shaped by directional selection pressure
From all sequences analyzed in this study, we identified 43 variants of Cg-Prp, 24 variants of Cg-BPI, 9 variants of Cg-Defm, 10 variants of Cg-Defhs and only 5 variants of Cg-Actin. Thus, we then analyzed if the sequence diversity has been shaped by directional selection pressures. The ratios of non synonymous to synonymous substitutions (dN/dS) for each codon of the four molecules indicate several sites under negative and positive selection for both AMPs (Cg-Defs and Cg-Prp), but not for Cg-BPI and Cg-Actin (Figure 4). To obtain dN/dS ratios, we first excluded all obvious recombinant sequences. Then, to assess the significance of the findings, we carried out a likelihood ratio test (LRT) and considered only codons showing significantly negative and positive selection (p < 0.05). Ten and 11 residues showing significant negative selection were identified in Cg-Defs and Cg-Prp respectively. For both AMPs, we detected a lower number of positively selected sites (3 and 6 residues in Cg-Defs and Cg-Prp respectively).
Figure 4.

Alignments of amino acids sequences of putative isoforms for Cg-defs and Cg-prp from C. gigas. a. Cg-Defm (FJ669323-FJ669336) and Cg-defhs (FJ669341- FJ669352). b. Cg-Prp (FJ669354- FJ669402). Highlighted sites showed evidence of positive (in black) or negative (in gray) selection (p < 0.05). Dots indicate identical residues and gaps (-) were introduced to obtain maximum sequence identity. The different domains are indicated in white boxes at the bottom of alignments.
In Cg-Defs, the signal peptide is well conserved for each class of defensin. Besides, the cleavage site for signal peptidase and the position of the cysteines is completely conserved in all variants (Figure 4a). We identified a high number of amino acid substitutions between Cg-Defs and several substitutions occurring in the mature peptide which change the physicochemical properties of the encoded amino acid. Thus, the polarity and/or charge of amino acids are modified in sites 28 (Gly/Arg), 29 (Asn/Asp), 31 (Leu/Tyr), 32 (Lys/Glu), 35 (Asn/Ser/Arg) 38 (Lys/Gln/Arg), 41 (Ser/Gly), and 60 (Asp/Gly). In contrast, substitutions occurring in the signal peptide do not modify the physico-chemical properties of the encoded amino acid. Interestingly, long forms of Cg-defm present two extra positive charged amino acids in position 67 and 70 (Lys) and one extra negative charged amino acid in position 68 (Glu).
In Cg-Prp, the signal peptide is the most variable region and the cleavage site is completely conserved among all variants, but the putative chymotrypsin cleavage site between the anionic and cationic domain presents an amino acid variation in position 43 (Tyr/Iso) (Figure 4b). Although this mutation is found in both long and short variants, the chymotrypsin cleavage site remained unchanged. Amino acid substitutions are identified in both signal and mature peptide region and they mainly concern the polarity of residues, like sites 7 (Tyr/Ile/Cys), 8 (Tyr/Phe), 14 (Ser/Gly), 26 (Leu/Ser), 45 (Ile/Tyr), 47 (Ser/Pro) and 57 (Pro/Gln). We also found complex sites with four and six different amino acid substitutions per site in which polarity and charge are modified, like sites 20 (His/Phe/Lys/Tyr/Asp/Gln) and 21 (Leu/Arg/Ile/Gln).
Discussion
Our results showed the existence of high and variable sequence diversity of C. gigas antimicrobial peptides (two AMPs,Cg-prp and Cg-defs) and protein (Cg-bpi), produced by a combination of different genetic mechanisms. Phylogenetic trees constructed with CDS from transcripts and genomic sequences showed a distinct phylogeny shape for each gene. The antimicrobial protein Cg-bpi expressed in hemocytes presents higher number of variants compared to non immune gene Cg-actin, but it is less diverse than AMPs. In contrast to both AMPs, Cg-bpi variants fall into one single group encoded by one single gene copy, being in accordance with the one copy present in human [13]. Thus, the sequence diversity observed for Cg-bpi results from allelic polymorphism only, with occasional recombination between alleles at a lower frequency when compared to AMPs. Though sequences obtained by PCR approach come from relatively limited sample size (N = 144) but with a large spectrum of oyster origin and challenge conditions, the sequence sampling saturation curve (plot of the number of different haplotypes to the total number of sequences performed) have a sigmoid shape indicating that sequencing effort was sufficient in our population to approach the plateau of saturation for each gene (data not shown). Furthermore, we have complemented the sample size by adding all the C. gigas ESTs available in databases. These ESTs come from more than 20 cDNA libraries (different tissues, experimental conditions and developmental stages) retrieved in the last decade. The combination of these two distinct sources of sequences is probably not exhaustive but covers a large enough range of variants to give a good insight of the overall polymorphism of the genes.
Cg-prp, which is expressed only in hemocytes, presents the highest number of transcript variants that are distributed into three groups. This partitioned phylogeny results in part from group specific mutations affecting only the cationic region. These key mutations in the potential active domain have segregated into two major lineages that are further diversified on other sites. There, high polymorphism in signal peptide and anionic region induces high sequence diversity within each group. Interestingly, we observed two very divergent sequences forming a third distinct group. Such a long branch in the phylogeny leading to few sequences may be indicative of an ongoing or recent process of neo-functionalization or subfunctionalization. These kinds of phenomena are often invoked in the course of evolution of duplicate genes [14], here supported by the finding of multiple copies for Cg-prp. In addition to this, sequence polymorphism of Cg-prp is produced by recombination and convergent mutations leading to phylogenetic homoplasy. Besides, Cg-prp is encoded by a lower number of copies than Cg-defs but shows higher number of different transcripts, indicating that allelic polymorphism associated with higher recombination frequency is important for this antimicrobial effector. Noticeably, intronless genes were found for Cg-Prp as previously described in other organisms where it was demonstrated to accelerate mRNA maturation process [15]. On the opposite, intron presence was also described to have positive effect on transcript stability [16]. Nevertheless, the role of different genomic structures and its correlation with the antimicrobial function remains unclear.
Cg-defs expressed in hemocytes and mantle tissue exhibit the highest divergence between groups but a lower diversity within each one. Moreover, each Cg-defs group displays different conservation level, showing that Cg-defh1 is less diversified than Cg-defh2, Cg-defm and even Cg-actin. Since we collected the different Cg-defs in the same conditions, we can rule out a technical bias inducing a highly redundant sequencing of the same molecule Cg-defh1 and leading to a skewed appreciation of the relative variability of each group. Thus, two major hypotheses could explain this extremely low level of variability: (i) Cg-defh1 appeared recently and is not yet diversified albeit frequent in the genome we explored, or (ii) Cg-defh1 could be an older variant but have undergone strong selective pressure. There is no apparent reason why Cg-defh1 could be more under selective constraints than the other Cg-defs. So the polymorphism reduction it displays is more likely due to a recent selective sweep than to purifying selection acting in a long term manner. We can postulate that this selective sweep could be consecutive to infections by pathogens that Cg-defh1 was particularly effective to combat. In addition, the fact that Cg-actin also appears more polymorphic at nucleotide level than Cg-defh1 is also in favour of a selective sweep on Cg-defh1, since it indicates that the polymorphism reduction concerns both the synonymous and the non-synonymous sites while purifying selection would affect mainly non-synonymous mutations. However, further studies are needed to clarify this point.
Cg-defs also present recombination parallel homoplasic events and a multigene family as observed for Cg-prp. Nevertheless, the absence of recombination between Cg-defm and Cgdefhs, together with the lack of homology between their introns, indicates that these two lineages are separated for a longer time than Cg-prp. This could be associated with the tissue specific expression of Cg-defm [4] and Cg-defhs [5] probably acquired after a duplication event followed by sub-functionalization. The tissue specific expression has already been described for several AMPs like mice α-defensins, which might be linked to differences in innate immune functions [17]. Likewise, sequence diversification of AMPs by gene duplication has been reported for vertebrates and invertebrates [18,19].
It is remarkable that gene duplication, as well as strong selective pressure (either positive or negative), did not affect identically the genes that we have studied; the antimicrobial peptides,Cg-defs and Cg-prp, have been a target for both, while antimicrobial protein Cg-bpi was not. This may reflect a difference in the way these two types of molecules participate in the immune response.
Concerning the sites under selection, the sites subject to purification found for both AMPs correspond to residues under strong functional constraints. In Cg-Defs, seven of eight Cys residues are under negative selection and Cys residues are known to be essential for the antimicrobial function of these peptides [20]. In Cg-Prp, the conserved Pro-Arg motif has been involved in antimicrobial activity of this class of AMPs [21] and the conserved C-terminal Gly is expected to be eliminated, obtaining a C-terminally amidated peptide which would increase its cationic properties [6].
In addition to these constrained sites, sites subject to diversifying selection of the AMPs could also have a role in antimicrobial function. In Cg-Defs, positively selected sites concern the modification of the charge and/or polarity of certain amino acids, and these changes could probably affect the affinity of the peptides to bacterial membranes. This finding is in accordance with several sites in human beta-defensin 1, in which amino acid charge is directly involved in the antimicrobial activity [22]. In Cg-Prp, from all positively selected sites, we identified a site in the active domain where a change of a Pro toward a Gln occurred. Because the activity of proline-rich AMPs [23] relies of course on proline residues, the functional characterization of these variants in terms of efficiency could help in understanding their role in the antimicrobial response. The functional significance of other positively and negatively sites along the coding region of Cg-defs and Cg-prp remains unknown and needs further investigation. Nevertheless, we propose, following several authors [24,25] that strong selective pressures on AMPs are directed toward the acquisition of novel microbial target specificities.
Evidence for directional selection driving evolution has been addressed in several cases [26-28] and it is consistent with the general hypothesis of co-evolution or "arms race" [29]. In this hypothesis, the pathogen evolves continuously to escape from the immune response of host and, consequently, the immune system of host evolves to improve barriers against pathogens. Thus, such interactions might provide rapid evolution in genes concerned in host-pathogen interactions. The emergence of Cg-defh1, a form of Cg-defhs that seems to have largely spread out in the oysters we analyzed, could be a good illustration of this.
Besides the sites under directional pressures, both AMPs show different lengths of mature peptide which could modify their antimicrobial activities. In one hand, Cg-Defm long variants present a longer C-terminal tail with eight extra amino acid residues that could influence the affinity to the bacterial membranes. Indeed, different C-terminal tails of alpha-defensins from rat are already described and are predicted to be exposed to the surface where they might confer a different antimicrobial spectrum [30]. On the other hand, the two lengths of Cg-Prpare produced by an indel of an Arg-Pro motif in the cationic domain, which is described to be involved in the antimicrobial activity of proline rich AMPs [23]. All these differences between antimicrobial effectors could be related to specific antimicrobial functions and result from co-evolution between host and pathogen species.
Conclusions
All these data support the hypothesis that oysters, continuously exposed to an environment rich in microorganisms, have developed specific strategies of innate immunity to increase the antimicrobial defense upon infection through effector diversification. In addition, considering the synergistic activities already reported between the two AMPs, Cg-Prp and Cg-Defm, [6] and between several isoforms of the same AMP in other species [31,32], the observed diversity could also improve the antimicrobial response through synergic activities. The next step of our work is now to establish the functional significance of this diversity.
Methods
Animals
Hemocytes were sampled from C. gigas adult oysters under 24 experimental conditions (total of 144 oysters): oysters of three geographic origins (6 oysters per condition; Atlantic coast-La Tremblade, Normandie-Bay des Veys and Mediterranean Sea-Thau lagoon) were exposed to four kinds of bacterial challenges and hemolymph were sampled and pooled at two post challenge times. The bacterial challenges performed by oyster immersion were (1) alive non virulent Micrococcus luteus and Vibrio tasmaniensis (2.5 × 108 bacteria/L for each strain), (2) alive virulent V. splendidus (5 × 108 bacteria/L), (3) mix of heat killed virulent V. splendidus and V. aestuarianus (2.5 × 108 bacteria/L for each strain) and (4) unchallenged oysters. For each condition, hemolymph was collected at 12 and 24 hours after challenge from the pericardial cavity through the adductor muscle. After hemolymph collection, hemocytes were isolated by centrifugation to discard plasma (700 g for 10 min. at 4°C) for further RNA extraction. For RNA extraction of mantle tissue, oysters from the Mediterranean Sea (Thau lagoon) were harvested by dissection. All experimental infections were performed according to the Ifremer animal care guideline and policy.
For the genomic DNA extractions (gDNA), hemocytes were recovered individually as described above from three oysters used for gene quantification and from one oyster used for gene structure determination originating from the Mediterranean Sea (Thau lagoon).
RNA extraction and cDNA synthesis
Total RNA was extracted from C. gigas hemocytes and mantle tissues using Trizol reagent according to manufacturer instructions (Invitrogen™). RNA was then treated with DNAse I (Invitrogen™), 15 min at room temperature and inactivated by heat, 10 min at 65°C. A second precipitation was performed with sodium acetate 0.3 M to eliminated DNAse and degraded DNA. Then, quantification and quality of total RNA were determined using a NanoDrop spectrophotometer (NanoDrop Technologies) and agarose gel electrophoresis, respectively. Following heat denaturation of 1 μg of total RNA (65°C for 5 min), first strand synthesis was carried out using 50 ng oligo-(dT)12-18, 1 mM dNTPs, 1 unit RnaseOUT (Invitrogen™) and 200 units M-MLV reverse transcriptase in reverse transcriptase buffer (Invitrogen™) following the manufacturer protocol.
Genomic DNA extraction
Genomic DNA (gDNA) was isolated from oyster hemocytes individually, by incubation in buffer (100 mM NaCl, 10 mM Tris-HCl pH 8, 25 mM EDTA pH 8, 0.5% SDS and 0.1 mg/ml proteinase K), 4 h at 50°C, followed by phenol/chloroform extraction. gDNA was precipitated by the addition of cold ethanol (-80°C) and treated with 50 μg/ml RNAse (Invitrogen™), 30 min at 37°C. This was followed by a second DNA precipitation and DNA purity and concentration were determined as described above for RNA samples.
5' and 3' RACE, molecular cloning and sequencing
Rapid amplification of cDNA ends (RACE) were performed from hemocytes and mantle tissue RNA to obtain both 5'UTR and 3'UTR of Cg-defh1 and Cg-defh2 and the 5'UTR of Cg-defm. We designed three specific antisense primers from Cg-defh cDNA partial coding sequences (CDS) [GenBank DQ400101 and GenBank DQ400102] for the 5' RACE, one specific sense primer for the 3' RACE and three antisense primers for the 5' RACE from Cg-defm cDNA CDS [GenBank AM050547]. Primers are listed in table 1. The procedure was performed according to the manufacturer 5'/3' RACE protocol (Roche, Mannheim, Germany). Briefly, 5' RACE for Cg-defhs was carried out using 1 μg of total RNA, 1.6 μM of Cg-defh5' sp1 antisense primer, 1 mM dNTPs, 1 unit/μl RnaseOUT (Invitrogen™) and 200 units/μl M-MLV reverse transcriptase in reverse transcriptase buffer (Invitrogen™). The cDNA was then treated with two units of RNaseH for 20 min at 37°C. ssDNA product was purified with High Pure PCR Product purification kit (Roche), and a poly A end was added with 1 μL of terminal transferase (80 U/μL) (Promega). The 5'UTR for Cg-defhs was obtained after two PCR reactions, the first reaction with the antisense primer Cg-defh5'sp2 and the oligo (dT) anchor primer (Roche) and the second one with the antisense primer Cg-defh5'sp3 and the race (dT) anchor primer (Roche). 5'RACE for Cg-defm was performed with the three antisense primers as described above. 3' RACE for Cg-defhs was performed in the same conditions as 5' RACE but using the oligo (dT) anchor primer (Roche) for first strand synthesis. Only one round of PCR was performed using the specific sense primer Cg-defh3 'sp1 and race (dT) anchor primer (Roche) to obtain 3'UTR. Each RACE-PCR products was gel purified using a QIAQuick® gel extraction kit (Qiagen, Germany) and cloned using TOPO TA (Invitrogen™) with the pCR 2.1 TOPO® vector. Plasmids were transformed into TOP10 E. coli cells, and plasmids were purified using Wizard™ SV miniprep DNA purification kit (Promega). Plasmid sequencing was performed using Big Dye™ Terminator sequencing kit on a DNA sequencer model ABI Prism 3130XL (Applied Biosystems).
PCR amplification and sequencing from cDNA and gDNA
PCR amplifications from cDNA were performed using specific primers designed in the 5'UTR and in 3'UTR from sequences of Cg-prp [GenBank BQ426670], Cg-bpi [GenBank AY165040], Cg-defm [GenBank AM050547] and Cg-actin [GenBank EW779066]. Specific primers for Cg-defhs and sense primer for Cg-defm were designed in the 5'UTR and 3'UTR from sequences retrieved through RACE assays described above. The sequences of each specific pair of primers are listed in Table 3. Cg-prp, Cg-defhs, Cg-bpi and Cg-actin were amplified from cDNA synthesised from a pool sample of hemocyte RNAs and Cg-defm was amplified from cDNA synthesised from a pool sample of mantle tissue RNAs. PCR reactions were carried out with GOTaq polymerase according to manufacturer instructions (Promega), using 1 μl of synthesized cDNA under the following conditions: 10 min at 95°C, then 35 cycles at 95°C for 30 s, 57°C for 1 min, 72°C for 2 min and a final elongation step at 72°C for 7 min. PCR products were cloned and sequenced as described above.
Table 3.
List of primers
| Sense primers (5'3') | Antisense primers (5'3') | |||
|---|---|---|---|---|
| gene | PCR amplification from cDNA and gDNA | |||
| Cg-prp | Cg-prp2F | CACCATGTTCTCTCGGAGGA | Cg-prp3R | TCTCTTCATCAAAACAAAGTCG |
| Cg-defm | Cg-defm5F | CCACTTTCTGGTTTGCTGAG | Cg-defU4R | TCTTGGTCAGATTCAGWCTGG |
| Cg-defh | Cg-defh4F | CTACCAGTTGTTCATACAGAG | Cg-defU4R | TCTTGGTCAGATTCAGWCTGG |
| Cg-bpi | Cg-bpi1F | CTACCAGTTGTTCATACAGAG | Cg-bpi1R | GGATTTAATATATCCGTCTTCTG |
| Cg-actin | Cg-actin1F | CTTCACAATGGGAGATGAAGA | Cg-actin1R | GTAAACTCCTATCACAGCCAC |
| gene copy number estimation | ||||
| Cg-prp | Cg-prpUq1F | CCACCATGTTCTCTCGGAGG | Cg-prpUq1R | CTTGTAGAACGGGCTAGCAC |
| Cg-defs | Cg-defUq1F | CTGCAGACATGGCTTTTGCTG | Cg-defUq1R | GTACATCTTGACCAGAGCGTG |
| Cg-bpi | Cg-bpiq2F | AGATAGAAATAGGAATGGACGG | Cg-bpiq2R | GTTATAGATCCACGCTGCTCC |
| Cg-actin | Cg-actq2F | CAGCTATGTAGGAGACGAG | Cg-actq3R | CACGGAGTTCATTGTAGAAGG |
| 5' and ' RACE | ||||
| Cg-defh | Cg-defh3'sp1 | GCTGGATTTGGGTGTCCG | race (dT) anchor | GACCACGCGTATCGATGTCGAC |
| Cg-defh | Cg-defh5' sp1 | CACAGTAGCCCGCTCTACA | ||
| Cg-defh | oligo (dT) anchor | GACTCGAGTCGACATCGATTTTTTTTTTTTTTTTV | Cg-defh5' sp2 | CGGACACCCAAATCCAGC |
| Cg-defh | race (dT) anchor | GACCACGCGTATCGATGTCGAC | Cg-defh5' sp3 | CCATYTCTGCAGAAACCATC |
| Cg-defm | Cg-defm5' sp1 | CATCTTAACCAGAGCGTGGC | ||
| Cg-defm | oligo (dT) anchor | GACTCGAGTCGACATCGATTTTTTTTTTTTTTTTV | Cg-defm5' sp2 | GGACTTGCAGTGATTGTTGC |
| Cg-defm | race (dT) anchor | GACCACGCGTATCGATGTCGAC | Cg-defm5' sp3 | CATCAGAAGGACAGCTAGTG |
Genomic sequences were obtained by PCR amplification on gDNA from one individual sample with the same specific pair of primers used to amplify cDNA (Table 1), in a total volume of 25 μl. Template (100 ng of gDNA) was amplified under the following conditions: 10 min at 95°C, followed by 35 cycles at 95°C for 1 min, 57°C for 1 min, 72°C for 3 min and a final elongation step at 72°C for 10 min. PCR products were cloned and sequenced as described above.
In silico approach
Sequences previously identified for Cg-prp, Cg-defm, Cg-defh1, Cg-defh2, Cg-bpi and Cg-actin were used for the search of homologous sequences among the 29,745 unique sequences contained in the Sigenae C. gigas EST database http://www.sigenae.org/[33,34].
From all the identified sequences, we kept only sequences with the complete CDS. In addition, to avoid scoring PCR or sequencing artefacts, we have only considered sequences shared between PCR and in silico approaches or those observed at least twice in the same approach.
Gene copy number estimation
For each gene, specific pairs of primers were designed from a conserved region on one exon selected by comparison of all the sequences compiled in this study (Table 3). As standard of gene quantification, we used equimolar amounts of pCR 2.1 TOPO ® vector plasmid DNA (pDNA) containing the inserts of interest which were pooled and diluted in 40 ng of herring sperm DNA. Efficiency of amplification was calculated by serial dilutions ranging from 103 to 109 copies per reaction which were tested in duplicate with each primer pair. qPCR assays were then performed in duplicate with four serial dilutions of gDNA (between 5 to 40 ng per reaction) from 3 individuals separately, and we used the concentration of 20 ng to estimated the gene number. qPCR was carried out on the LightCycler 480 System (Roche). The reaction mixture consisted of LightCycler 480 master mix (1×) and 0.5 μM of each primer and was submitted to: 10 min of denaturation at 95°C, 40 cycles of 10s of denaturation at 95°C, 20s of annealing at 60°C and 20s of extension at 72°C, and fluorescence detection. After an initial 10 s denaturation step at 95°C, a melting curve was obtained from a start temperature of 65°C to a final temperature of 95°C, with an increase of 0.06°C/s. The data were analyzed using LightCycler 480 software version 1.5.0.39 and the 2nd derivative max algorithm.
Gene copy number was calculated by absolute quantification. Standard curves for gDNA and pDNA of each gene were constructed from the mean Ct values using linear regression, from which slope and correlation coefficients were calculated. PCR efficiency was determined by plotting the threshold cycle (Ct) as a function of Log10 concentration of the template used. Using an estimation of the C. gigas genome size of about 823 Mb, the number of molecules represented in 20 ng of gDNA was then calculated. Quantification was finally achieved by plotting the measured threshold cycle (Ct) on the standard curve obtained with the serial dilutions of pDNA. pDNA and gDNA can be used similarly as reference molecules if their PCR efficiencies and correlation coefficients are identical [35].
Sequence data analysis
The multiple alignments were generated using the MAFFT alignment program (http://align.bmr.kyushu-u.ac.jp/mafft/online/server/, version 6) [36]. Prediction of signal peptide was performed with the SignalP program (http://www.cbs.dtu.dk/services/SignalP/, version 3.0) [37]. Construction of phylogenetic trees was performed with PHYML v2.4.4 [38] using the models with the best Akaike information criterion (Phymltest function from the APE package) [39].
On the basis of the uncovered sequence phylogenies, we defined clearly distinct groups of sequences that were used in further polymorphism analyses, whereas sequences with ambiguous positions were not considered. Measure of sequence diversity as the mean nucleotide diversity (Pi) (intra-group) and the mean nucleotide divergence (inter-groups) values were calculated using the Maximum Composite Likelihood method included in the Molecular Evolutionary Genetics Analysis program (http://www.megasoftware.net/ version 4.0) [40]. Polymorphism values and minimum numbers of recombination events (Rm) were calculated with DNAsp program (http://www.ub.es/dnasp/ version 4.20.2) [41]. The ratio of the rate of non-synonymous substitutions (dN) to the rate of synonymous substitutions (dS) for each codon was calculated with Selecton web server http://selecton.tau.ac.il/index.html[42], based on M8 evolutionary model which allows for positive selection. Statistical significance of results (p < 0.05) was assessed using the likelihood ratio test (LRT) which compares the log likelihood of M8 model to the log likelihood of M8a alternative model, that allows for negative and neutral selection.
Authors' contributions
JdL, EB and YG participated in the conception and design of the study and in its coordination. PS produced the genetic data and carried out the analyses. ED provided advice and support for the analysis and helped design the sequence phylogenies. PS and JdL wrote the manuscript with contributions from all the other authors. All authors read and approved the final manuscript.
Contributor Information
Paulina Schmitt, Email: Paulina.Schmitt@ifremer.fr.
Yannick Gueguen, Email: Yannick.Gueguen@ifremer.fr.
Erick Desmarais, Email: desmarais@univ-montp2.fr.
Evelyne Bachère, Email: Evelyne.Bachere@ifremer.fr.
Julien de Lorgeril, Email: Julien.De.Lorgeril@ifremer.fr.
Acknowledgements
We are very grateful to D. Destoumieux-Garzón for helpful discussion and M. Leroy for technical assistance. This work was supported in part by the Ifremer, the CNRS and the University of Montpellier II. Data used in this work were partly produced through molecular genetic analysis technical facilities of the IFR119 "Montpellier Environnement Biodiversité" and the "plateau qPCR de la plateforme Montpellier GenomiX". P. Schmitt was supported by a doctoral funding from Program Becas Chile-CONICYT.
References
- Terahara K, Takahashi KG, Nakamura A, Osada M, Yoda M, Hiroi T, Hirasawa M, Mori K. Differences in integrin-dependent phagocytosis among three hemocyte subpopulations of the Pacific oyster " Crassostrea gigas". Dev Comp Immunol. 2006;30(8):667–683. doi: 10.1016/j.dci.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Mitta G, Vandenbulcke F, Roch P. Original involvement of antimicrobial peptides in mussel innate immunity. FEBS Lett. 2000;486(3):185–190. doi: 10.1016/S0014-5793(00)02192-X. [DOI] [PubMed] [Google Scholar]
- Bugge DM, Hegaret H, Wikfors GH, Allam B. Oxidative burst in hard clam (Mercenaria mercenaria) haemocytes. Fish Shellfish Immunol. 2007;23(1):188–196. doi: 10.1016/j.fsi.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Gueguen Y, Herpin A, Aumelas A, Garnier J, Fievet J, Escoubas JM, Bulet P, Gonzalez M, Lelong C, Favrel P. Characterization of a defensin from the oyster Crassostrea gigas. Recombinant production, folding, solution structure, antimicrobial activities, and gene expression. J Biol Chem. 2006;281(1):313–323. doi: 10.1074/jbc.M510850200. [DOI] [PubMed] [Google Scholar]
- Gonzalez M, Gueguen Y, Desserre G, de Lorgeril J, Romestand B, Bachere E. Molecular characterization of two isoforms of defensin from hemocytes of the oyster Crassostrea gigas. Dev Comp Immunol. 2007;31(4):332–339. doi: 10.1016/j.dci.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Gueguen Y, Bernard R, Julie F, Paulina S, Delphine DG, Franck V, Philippe B, Evelyne B. Oyster hemocytes express a proline-rich peptide displaying synergistic antimicrobial activity with a defensin. Mol Immunol. 2008;46(4):516–22. doi: 10.1016/j.molimm.2008.07.021. [DOI] [PubMed] [Google Scholar]
- Gonzalez M, Gueguen Y, Destoumieux-Garzon D, Romestand B, Fievet J, Pugniere M, Roquet F, Escoubas JM, Vandenbulcke F, Levy O. Evidence of a bactericidal permeability increasing protein in an invertebrate, the Crassostrea gigas Cg- BPI. Proc Natl Acad Sci USA. 2007;104(45):17759–17764. doi: 10.1073/pnas.0702281104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulenburg H, Boehnisch C, Michiels NK. How do invertebrates generate a highly specific innate immune response? Mol Immunol. 2007;44(13):3338–3344. doi: 10.1016/j.molimm.2007.02.019. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Ng PM, Wang L, Ho B, Ding JL. Diversity in lectins enables immune recognition and differentiation of wide spectrum of pathogens. Int Immunol. 2006;18(12):1671–1680. doi: 10.1093/intimm/dxl101. [DOI] [PubMed] [Google Scholar]
- Yang WY, Wen SY, Huang YD, Ye MQ, Deng XJ, Han D, Xia QY, Cao Y. Functional divergence of six isoforms of antifungal peptide Drosomycin in Drosophila melanogaster. Gene. 2006;379:26–32. doi: 10.1016/j.gene.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Litman GW, Cannon JP, Rast JP. New insights into alternative mechanisms of immune receptor diversification. Adv Immunol. 2005;87:209–236. doi: 10.1016/S0065-2776(05)87006-3. full_text. [DOI] [PubMed] [Google Scholar]
- Sauvage C, Bierne N, Lapegue S, Boudry P. Single Nucleotide polymorphisms and their relationship to codon usage bias in the Pacific oyster Crassostrea gigas. Gene. 2007;406(1-2):13–22. doi: 10.1016/j.gene.2007.05.011. [DOI] [PubMed] [Google Scholar]
- Linzmeier RM, Ganz T. Copy number polymorphisms are not a common feature of innate immune genes. Genomics. 2006;88(1):122–126. doi: 10.1016/j.ygeno.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290(5494):1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- Wang HF, Feng L, Niu DK. Relationship between mRNA stability and intron presence. Biochem Biophys Res Commun. 2007;354(1):203–208. doi: 10.1016/j.bbrc.2006.12.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu DK. Protecting exons from deleterious R-loops: a potential advantage of having introns. Biol Direct. 2007;2:11. doi: 10.1186/1745-6150-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson J, Putsep K, Chu H, Kays RJ, Bevins CL, Andersson M. Regional variations in Paneth cell antimicrobial peptide expression along the mouse intestinal tract. BMC Immunol. 2008;9:37. doi: 10.1186/1471-2172-9-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas P, El Amri C. The dermaseptin superfamily: A gene-based combinatorial library of antimicrobial peptides. Biochim Biophys Acta. 2008;1788(8):1537–50. doi: 10.1016/j.bbamem.2008.09.006. [DOI] [PubMed] [Google Scholar]
- Pujol N, Zugasti O, Wong D, Couillault C, Kurz CL, Schulenburg H, Ewbank JJ. Anti-fungal innate immunity in C. elegans is enhanced by evolutionary diversification of antimicrobial peptides. PLoS Pathog. 2008;4(7):e1000105. doi: 10.1371/journal.ppat.1000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjicharalambous C, Sheynis T, Jelinek R, Shanahan MT, Ouellette AJ, Gizeli E. Mechanisms of alpha-defensin bactericidal action: comparative membrane disruption by Cryptdin-4 and its disulfide-null analogue. Biochemistry. 2008;47(47):12626–12634. doi: 10.1021/bi800335e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markossian KA, Zamyatnin AA, Kurganov BI. Antibacterial proline-rich oligopeptides and their target proteins. Biochemistry (Mosc) 2004;69(10):1082–1091. doi: 10.1023/B:BIRY.0000046881.29486.51. [DOI] [PubMed] [Google Scholar]
- Pazgier M, Prahl A, Hoover DM, Lubkowski J. Studies of the biological properties of human beta-defensin 1. J Biol Chem. 2007;282(3):1819–1829. doi: 10.1074/jbc.M607210200. [DOI] [PubMed] [Google Scholar]
- Otvos L Jr. The short proline-rich antibacterial peptide family. Cell Mol Life Sci. 2002;59(7):1138–1150. doi: 10.1007/s00018-002-8493-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padhi A, Verghese B. Molecular diversity and evolution of myticin-C antimicrobial peptide variants in the Mediterranean mussel, Mytilus galloprovincialis. Peptides. 2008;29(7):1094–1101. doi: 10.1016/j.peptides.2008.03.007. [DOI] [PubMed] [Google Scholar]
- Hollox EJ, Armour JA. Directional and balancing selection in human betadefensins. BMC Evol Biol. 2008;8:113. doi: 10.1186/1471-2148-8-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzaro BP. Natural selection on the Drosophila antimicrobial immune system. Curr Opin Microbiol. 2008;11(3):284–289. doi: 10.1016/j.mib.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohuet A, Krishnakumar S, Simard F, Morlais I, Koutsos A, Fontenille D, Mindrinos M, Kafatos FC. SNP discovery and molecular evolution in Anopheles gambiae, with special emphasis on innate immune system. BMC Genomics. 2008;9:227. doi: 10.1186/1471-2164-9-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger E, Grunau C, Pierce RJ, Hirai H, Gourbal B, Galinier R, Emans R, Cesari IM, Cosseau C, Mitta G. Controlled Chaos of Polymorphic Mucins in a Metazoan Parasite (Schistosoma mansoni) Interacting with Its Invertebrate Host (Biomphalaria glabrata) PLoS Negl Trop Dis. 2008;2(11):e330. doi: 10.1371/journal.pntd.0000330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Bielawski JP. Statistical methods for detecting molecular adaptation. Trends Ecol Evol. 2000;15(12):496–503. doi: 10.1016/S0169-5347(00)01994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil A, Hughes AL, Zhang G. Rapid evolution and diversification of mammalian alpha-defensins as revealed by comparative analysis of rodent and primate genes. Physiol Genomics. 2004;20(1):1–11. doi: 10.1152/physiolgenomics.00150.2004. [DOI] [PubMed] [Google Scholar]
- Lauth X, Babon JJ, Stannard JA, Singh S, Nizet V, Carlberg JM, Ostland VE, Pennington MW, Norton RS, Westerman ME. Bass hepcidin synthesis, solution structure, antimicrobial activities and synergism, and in vivo hepatic response to bacterial infections. J Biol Chem. 2005;280(10):9272–9282. doi: 10.1074/jbc.M411154200. [DOI] [PubMed] [Google Scholar]
- Mangoni ML, Epand RF, Rosenfeld Y, Peleg A, Barra D, Epand RM, Shai Y. Lipopolysaccharide, a key molecule involved in the synergism between temporins in inhibiting bacterial growth and in endotoxin neutralization. J Biol Chem. 2008;283(34):22907–22917. doi: 10.1074/jbc.M800495200. [DOI] [PubMed] [Google Scholar]
- Fleury E, Huvet A, Lelong C, de Lorgeril J, Boulo V, Gueguen Y, Bachere E, Tanguy A, Moraga D, Fabioux C. Generation and analysis of a 29,745 unique Expressed Sequence Tags from the Pacific oyster (Crassostrea gigas) assembled into a publicly accessible database: the GigasDatabase. BMC Genomics. 2009;10:341. doi: 10.1186/1471-2164-10-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigenae C. gigas EST database. http://public-contigbrowser.sigenae.org:9090/Crassostrea_gigas/index.html
- Toyota A, Akiyama H, Sugimura M, Watanabe T, Kikuchi H, Kanamori H, Hino A, Esaka M, Maitani T. Quantification of genetically modified soybeans using a combination of a capillary-type real-time PCR system and a plasmid reference standard. Biosci Biotechnol Biochem. 2006;70(4):821–827. doi: 10.1271/bbb.70.821. [DOI] [PubMed] [Google Scholar]
- MAFFT alignment program. http://align.bmr.kyushu-u.ac.jp/mafft/online/server/
- SignalP program. http://www.cbs.dtu.dk/services/SignalP/
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52(5):696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics. 2004;20(2):289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- Molecular Evolutionary Genetics Analysis program. http://www.megasoftware.net/
- Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19(18):2496–2497. doi: 10.1093/bioinformatics/btg359. http://www.ub.es/dnasp/ DNAsp program. [DOI] [PubMed] [Google Scholar]
- Selecton web server. http://selecton.tau.ac.il/index.html