

Abstract
In this study, two alternatively spliced forms of the mouse death-associated protein kinase (DAPK) have been identified and their roles in apoptosis examined. The mouse DAPK-α sequence is 95% identical to the previously described human DAPK, and it has a kinase domain and calmodulin-binding region closely related to the 130–150 kDa myosin light chain kinases. A 12-residue extension of the carboxyl terminus of DAPK-β distinguishes it from the human and mouse DAPK-α. DAPK phosphorylates at least one substrate in vitro and in vivo, the myosin II regulatory light chain. This phosphorylation occurs preferentially at Ser-19 and is stimulated by calcium and calmodulin. The mRNA encoding DAPK is widely distributed and detected in mouse embryos and most adult tissues, although the expression of the encoded 160-kDa DAPK protein is more restricted. Overexpression of DAPK-α, the mouse homolog of human DAPK has a negligible effect on tumor necrosis factor (TNF)-induced apoptosis. Overexpression of DAPK-β has a strong cytoprotective effect on TNF-treated cells. Biochemical analysis of TNF-treated cell lines expressing mouse DAPK-β suggests that the cytoprotective effect of DAPK is mediated through both intrinsic and extrinsic apoptotic signaling pathways and results in the inhibition of cytochrome c release from the mitochondria as well as inhibition of caspase-3 and caspase-9 activity. These results suggest that the mouse DAPK-β is a negative regulator of TNF-induced apoptosis.
Apoptosis is a carefully regulated cellular event with important roles in a number of processes that occur during development and contribute to tissue homeostasis. Dysregulation of apoptosis can result in cancer, autoimmune diseases, and neurodegenerative disorders. A large number of signaling molecules involved in regulating the commitment and progression of apoptosis have been identified, and their complex interactions are being investigated. There is now also considerable evidence that many protein kinases have roles in apoptosis and may help regulate the signaling pathways that ultimately determine the critical balance in the choice between life and death (1, 2).
Myosin II motor activities have been implicated in the general regulation of morphological changes that occur during the execution phase of apoptosis (3). In smooth and nonmuscle cells, the phosphorylation of myosin II by myosin light chain kinase (MLCK)1 is a key event leading to the activation of myosin II motor activities and the production of forces for contraction, migration, adhesion, and cytokinesis (4). It is now known that other protein kinases in addition to the conventional calcium/calmodulin (Ca2+/CaM)-dependent MLCKs can phosphorylate myosin II regulatory light chain (RLC). These kinases include p21-activated kinase (PAK), rho-activated kinase (RHOK), and death-associated protein kinase (DAPK) (5–7). Thus, multiple signaling pathways converge at the myosin regulatory light chain, and it is likely that each of these pathways modulates myosin motor activities to generate forces necessary for the formation or disassembly of signaling complexes and their intracellular trafficking. A recent study has shown that myosin II motor activities activated by the conventional Ca2+/CaM-dependent MLCK has an important role in regulating the translocation of at least one death receptor, TNFR-1, to the plasma membrane (8), suggesting an additional role in regulation of the apoptotic response in cells.
DAPK is a Ca2+/CaM-dependent Ser/Thr protein kinase that was identified as a positive mediator of interferon-γ-induced apoptosis in HeLa cells (9, 10). Several other kinases related to DAPK have also been identified, and all have strong sequence homology that is restricted to the kinase domain of DAPK (11). This kinase family includes ZIP/DLK, DAPK-related apoptosis-inducing kinases 1 and 2, DAPK2, and dystrophin-related protein-1 (12–16). These kinases have been shown in vitro to phosphorylate RLC isolated from skeletal muscle (10, 13, 15, 16) or smooth muscle myosin (17), but to date no in vivo substrates have been identified. The significance of RLC phosphorylation by DAPKs is unknown, although DAPK, dystrophin-related protein-1, and ZIP/DLK are or can become associated with the actomyosin cytoskeleton (10, 16, 18). Similar to other apoptotic regulators, ectopic overexpression of the DAPK family members induces morphological and biochemical changes associated with apoptosis, and this family of protein kinases is considered to be positive regulators of apoptosis. Although the signaling pathway through which members of the DAPK family promote apoptotic cell death is not understood, it has been shown that DAPK acts upstream of p53 to regulate p53 activity in a p19ARF-dependent manner (19).
This study describes the cloning and characterization of two alternatively spliced mouse DAPKs. Mouse DAPK-α and DAPK-β are highly related to the previously described proapoptotic human DAPK. However, ectopic overexpression of the murine DAPK-α or DAPK-β does not promote apoptosis as previously shown for the human DAPK (9, 10). In addition, in TNF-treated cells, overexpression of DAPK-β is cytoprotective and suppresses caspase-3 and -9 activity and mitochondrial cytochrome c release. Together these studies show that DAPK can protect cells from apoptosis.
MATERIALS AND METHODS
Cloning and Expression of Murine DAPKs
A cDNA probe encoding the kinase domain of the mouse 130-kDa MLCK (20) was used in a low stringency screen of a mouse AT2 cardiac myocyte gt11 cDNA library, generously provided by the Indiana University School of Medicine. Positive isolates were subcloned and sequenced. One cDNA identified from this library screen had significant homology to the kinase domain of MLCK and was extended by subsequent screens to yield a 4.9-kb cDNA encoding the full-length DAPK-α. Sequencing of several other positive cDNAs obtained from subsequent library screens identified a cDNA encoding a DAPK-β that was distinguished at the 3′ end by a putative alternative splice that would result in extending the carboxyl-terminal coding region of DAPK by 12 residues.
Antibodies
Two monoclonal anti-human DAPK (BD/Transduction Laboratories, clone 17, and Sigma, clone 55) were used at dilutions of 1:250 and 1:10,000, respectively, and gave similar results. Omniprobe polyclonal and monoclonal antibodies (Santa Cruz Biotechnology) and Xpress tag antibody (Invitrogen), all of which recognize the Xpress epitope tag (Invitrogen), were used at a dilution of 1:1,000 and 1:2,500, respectively. A polyclonal antibody to purified myosin II regulatory light chains was generated and characterized in this laboratory. The cytochrome c antibody is from PharMingen (San Diego, CA), and the poly(A)DP-ribose polymerase (PARP) antibody (Santa Cruz Biotechnology) recognizes both full-length and caspase-cleaved PARP.
MDCK and HeLa Cell Lines Expressing Wild-type and Mutant DAPK
MDCK or HeLa cell lines expressing either DAPK-α or DAPK-β under the control of a tetracycline-inducible transactivator were constructed by the transfection of a pCDNA4/TO plasmid containing DAPKs into MDCK or HeLa cells already expressing tetracycline-VP16 transactivator (21, 22). Stable zeocin resistant cell lines were selected and characterized for tetracycline-regulated expression of DAPKs. A similar strategy was used to generate MDCK cell lines expressing mutant DAPK-α (K42A) and DAPK-β (K42A). The exogenously expressed mouse wild-type and mutant DAPKs have an amino-terminal Xpress epitope tag that is recognized by both the Omniprobe and Xpress tag antibodies. Parental cells and cell lines expressing DAPKs were maintained routinely in Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) fetal calf serum, 2 mM glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. To regulate expression of DAPK in the MDCK and HeLa cell lines, Doxycycline (Dox) is increased from 0 (repression) to 2 (maximal induction) μg/ml. Under maximal induction conditions, stable and approximately equal expression levels of the DAPKs were achieved in each of the cell lines. For each experiment, controls included MDCK and HeLa parental cells expressing only the tetracycline transactivator and cells under maximal repression (0 μg/ml Dox for MDCK and HeLa cells). In all cases the repressed cells gave similar results as the parental cell line. Overexpression of DAPKs did not have a deleterious effect on growth, doubling time, or morphology. Western blotting and immunofluorescence were used routinely to monitor the expression of the exogenous DAPKs to ensure that the cell lines were expressing each DAPK at equal levels. Cells were maintained at subconfluent levels (~30–50% density) during analysis. MDCK cells are sensitive to TNF and do not require inhibition of protein synthesis with cyclohexamide (8). A biochemical apoptotic response is apparent in MDCK cells within 8 h of exposure to TNF; however, morphological changes including apoptotic blebbing are not readily apparent until between 24 and 48 h of treatment. HeLa cells were treated with 10 ng/ml TNF in the presence of 10 μg/ml cyclohexamide to induce apoptosis, which becomes morphologically apparent within 2–3 h.
Western Blotting
Western blotting was performed as described previously (23). Equivalent amounts of total cellular protein or immunoprecipitates were fractionated by electrophoresis through an SDS-polyacrylamide gel and transferred to nitrocellulose. Immunoreactive proteins on Western blots were visualized using the Supersignal West Dura or West Pico detection systems (Pierce) according to manufacturer directions. Cell extracts were prepared from cells or tissues by homogenization in a lysis buffer containing 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS, 0.15 M NaCl, 10 mM sodium phosphate, pH 7.2, 2 mM EDTA, 50 mM sodium fluoride, 0.2 mM sodium vanadate, 20 μg/ml leupeptin, 40 μg/ml aprotinin, 60 μg/ml N-tosyl-L-phenylalanyl chloromethyl ketone, 60 μg/ml Nα-p-tosyl-L-lysine-chloromethyl ketone, and 100 μg/ml phenylmethylsulfonyl fluoride. After SDS-PAGE and transfer to nitrocellulose, Western blotting was performed using the appropriate anti-DAPK or Xpress antibodies.
Northern Blotting and RNase Protection
RNA was prepared from mouse tissues and cell lines using the Totally RNA kit (Ambion). For Northern blotting, 20 μg of total RNA/lane was fractionated on a 1.2% agarose gel. The RNA was transferred to Brightstar Plus positively charged nylon membrane (Ambion) using a vacuum blotter (Bio-Rad), UV cross-linked using a Stratalinker (Stratagene), and prehybridized for 60 min at 65 °C. A 550-bp 32P-labeled antisense riboprobe (cRNA) corresponding to bp 3370–3920 was transcribed from DAPK cDNA using the Maxiscript kit (Ambion). The probe was added to the blot in prehybridization buffer at a concentration of 1 × 106 cpm/ml buffer and hybridized overnight at 65 °C. Final wash conditions were 15 mM sodium citrate, pH 7.0, 0.15 M NaCl (0.1× SSC), and 0.1% SDS at 65 °C for 10 min. The blot was rinsed in 2× SSC and exposed to X-OMAT AR film with an intensifying screen for 1 week.
RNase protection was performed using the reagents and protocols from the RPA III kit (Ambion). A 507-bp 32P-labeled probe was prepared using the Maxiscript kit (Ambion) from a StuI-linearized template from DAPK-β. The probe generated contains an additional 43 nucleotides from the pGEM polylinker. A β-actin probe was used as a positive control. The hybridization of the cRNA probe and total RNA was performed at 42 °C overnight followed by a 30-min RNase digestion (2.5 units/ml RNase A and 100 units/ml RNase T1) and ethanol precipitation. Protected fragments were separated on a 6% acrylamide/7 M urea gel and exposed to film with an intensifying screen at −70 °C for 3 days.
Immunoprecipitation and Kinase Activity Measurements
Transiently transfected COS-1 cells expressing Xpress-tagged DAPK were washed with PBS, and lysates were prepared in lysis buffer (20 mM MOPS, pH 7, 1% Nonidet P-40, 10% glycerol, 0.5 mM EGTA, 50 mM MgCl2, 0.3 M NaCl, 6 μg/ml Nα-p-tosyl-L-lysine-chloromethyl ketone and N-tosyl-L-phenylalanyl chloromethyl ketone, 10 μg/ml (p-amidinophenyl)methanesulfonyl fluoride, 20 μg/ml leupeptin, 40 μg/ml aprotinin, and 1 mM Pefabloc SC). The lysate was clarified by centrifugation, and the supernatant was precleared using protein A-Sepharose. DAPK was immunoprecipitated by the addition of protein A beads precomplexed with rabbit anti-mouse IgG and monoclonal omniprobe antibody. Immune complexes bound to protein A-Sepharose 4B were washed twice with lysis buffer and then twice with kinase assay buffer (50 mM MOPS, pH 7, 10 mM magnesium acetate, and 1 mM dithiothreitol). Protein A-Sepharose beads containing the immunopurified DAPK were resuspended in kinase assay buffer, and equivalent volumes were used for in vitro RLC phosphorylation reactions.
Assays to determine the specific activity of the recombinant murine 160-kDa DAPKs were performed as described previously (24). The amount of DAPK immunoprecipitated per assay was estimated by ligand blotting with biotin-conjugated calmodulin using purified 150-kDa bovine tracheal MLCK as a standard. RLCs were phosphorylated in 50-μl kinase assay buffer (50 mM MOPS, pH 7, 10 mM MgAc, 0.6 mM CaCl2, 1 mM dithiothreitol, 1.2 μM CaM, and 22.5 μM chicken gizzard RLC) containing [γ-32P]ATP (200 cpm/pmol) diluted in 1 mm ATP for 15 min. To determine whether both forms of DAPK undergo autophosphorylation, the immunoprecipitated DAPK was analyzed following SDS-PAGE gel and autoradiography after completion of the kinase assay. To determine whether DAPK phosphorylates myosin regulatory light chains at the activating sites (Ser-19 and Thr-18) and RLC associated with myosin II, kinase assays were performed using either 1 μg of purified mutant recombinant regulatory light chain or 10 μg of myosin II, and the results were analyzed following SDS-PAGE and autoradiography. The myosin II was purified from partially fractionated human platelets, and Western blotting using isoform-specific myosin antibodies confirmed that both nonmuscle myosin IIA and IIB were present.
Glycerol Gel Analysis of Myosin II RLC Phosphorylation
The phosphorylation of RLCs was determined as described (25, 26). Briefly, the cellular proteins were precipitated with 10% trichloroacetic acid; the pellets were washed with acetone and dissolved in 8 M urea, 20 mM Tris, 23 mM glycine, and 10 mM dithiothreitol. Western blotting with an anti-myosin II RLC antibody was used to identify unphosphorylated, monophosphorylated, and diphosphorylated forms of RLC after fractionation through a 10% glycerol-polyacrylamide gel and transfer to nitrocellulose. The relative abundance of each RLC band was determined by scanning densitometry. The scan data were used to calculate the myosin II RLC phosphorylation index as described previously (8).
Apoptotic Analysis
For transient expression analysis, Cos or HeLa cells were seeded at 1 × 105 cells/30-mm dish, transfected with vectors as indicated for expression of DAPKs, and an empty vector (pCDNA4TO, mock) together with a vector encoding β-galactosidase. Transfections were carried out using Fugene 6 (Roche, Indianapolis, IN) according to manufacturer protocol. At 24 h after transfection, HeLa cells were treated with TNF (10 ng/ml) and cyclohexamide (10 μg/ml) for 3 h and then fixed and stained for β-galactosidase expression. Control transfections (−TNF), were incubated for 48 h before analysis. The percentage of apoptotic cells were determined by scoring the number of transfected (LacZ+) cells having apoptotic morphology with condensed cytoplasm and several apparent plasma membrane blebs. At least 100 LacZ-positive blue cells were counted in each well, and each experiment was independently repeated eight times. To examine the expression levels of DAPK and LacZ proteins, cells treated in parallel were lysed in SDS lysis buffer (1% SDS and 50 mM Tris, pH 7.4) and examined by Western blotting to detect DAPK or β-galactosidase expression.
Quantification of apoptotic cell death in conditionally regulatable HeLa or MDCK cell lines expressing DAPKs was performed by seeding the cells at 5 × 104 cells/well in 6-well tissue culture dishes and culturing the cells in the presence (+) or absence (−) of Dox for 24 h to induce stable levels of the exogenous DAPKs. At 24 h post-seeding, TNF (10 ng/ml) or vehicle (Me2SO, final concentration <0.01%) was added (t = 0). At the indicated times, viable attached cells were identified using trypan blue exclusion and counted. Cell viability is expressed as the percentage of the surviving TNF-treated cells compared with the surviving control cells not treated with TNF. Caspase-8, caspase-3, and caspase-9 activity were determined after extraction of the cells with CHAPS lysis buffer (0.1% CHAPS, 100 mM NaCl, 100 μM EDTA, 10 mM dithiothreitol, and 50 mM HEPES, pH 7.4). After centrifugation, equal amounts of total cellular proteins were incubated at 37 °C in assay buffer (CHAPS lysis buffer plus 10% glycerol), and the assay was initiated by the addition of either 200 μM Ac-IETD-pNA (caspase-8), 200 μM Ac-DEVD-pNA (caspase-3), or 200 μM Ac-LEHD-pNA (caspase-9) (Calbiochem, La Jolla, CA). The change in absorbance at 405 nm with time was monitored by spectrophotometry and converted to caspase activity (pmol/min/mg of total protein). Pure p-nitroaniline (pNA, Calbiochem) was used for calibration of the standard A405 curve. For every cell sample, the background was determined by adding the caspase-specific inhibitors, Ac-IETD-CHO (caspase-8), Ac-DEVD-CHO (caspase-3), or Ac-LEHD-CHO (caspase-9) (Calbiochem) as negative control. DNA fragmentation was analyzed by enzymatic detachment of the adherent cells, which were pooled with floating cells and then fixed in 5% acetic acid/95% ethanol at −20 °C and stained with 50 μg/ml propidium iodide (Sigma). Cells were analyzed with a Becton-Dickinson (Mountain View, CA) FACStar plus, and the data were computed with CellQuest. At least 10,000 cells were counted for each condition.
Cytochrome c and NF-κB Analysis
Hela or MDCK cells were scraped into PBS supplemented with a protease inhibitor mixture (20 μg/ml leupeptin, 40 μg/ml aprotinin, 60 μg/ml N-tosyl-L-phenylalanyl chloromethyl ketone, 60 μg/ml Nα-p-tosyl-L-lysine-chloromethyl ketone, 100 μg/ml phenylmethylsulfonyl fluoride, and 100 μg/ml (p-amidinophenyl)methanesulfonyl fluoride) and lysed by passage through a 23-gauge needle 10 times. Cytosol (devoid of mitochondria) and pellet (including mitochondria and nuclei) fractions were separated by centrifugation at 14,000 rpm for 30 min. Western blotting of both fractions was used to determine the relative amounts of cytochrome c and NF-κB. Each analysis was repeated at least three times, and the relative levels of expression were quantified by densitometry.
RESULTS
Cloning and Expression of Two Forms of Mouse DAPK
A low stringency screen of a mouse AT2 cardiomyocyte cDNA library using the kinase domain of the mouse 130-kDa MLCK identified two cDNA clones that represent the murine homologs of human DAPK. The two mouse DAPK clones represent alternative splice forms, differing only at their carboxyl termini. A sequence alignment of the mouse and human DAPKs (Fig. 1A) shows that the murine DAPKs are ~95% identical to the previously described human DAPK (9). The murine DAPK-β appears to be an alternatively spliced DAPK that has a unique carboxyl terminus that extends DAPK-α by 12 residues. All the structural features between the mouse and human DAPKs are highly conserved including the kinase, calmodulin binding, ankyrin repeats, P-loops, and death domain (Fig. 1B). Within the kinase domain (residues 13–263) there are two nonconserved residues, and throughout the remainder of the molecule there are ~25 nonconserved changes in the sequence (Fig. 1A). The significance of these findings, if any, is not known. The kinase domain of DAPK is ~40–50% identical to the 130–150-kDa conventional MLCK, and importantly, residues involved in binding and phosphorylation of myosin II RLC and activation by Ca2+/CaM are highly conserved (24).
FIG. 1. Alignment of the sequences of the mouse and human DAPKs.

A, the alignment of the deduced amino acid sequence of the mouse and human DAPK (9). The symbols below the alignment refer to residues that are similar (.) or identical (*). The arrows denote the kinase domain (residues 13–263, solid arrow brackets), calmodulin binding (residues 288–320, dotted arrow brackets), ankyrin repeats (373–637, dashed arrow brackets), and death domain (residues 1301–1391, brackets). The 12 residues corresponding to the DAPK-β tail are shown in bold letters extending the carboxyl terminus of DAPK-α from 1431 to 1443 residues. B, a schematic representation of the motifs predicted from the deduced sequence of DAPK-α and DAPK-β. Numbers indicate the beginning and end of each domain. CaM, calmodulin binding; Ank, ankyrin repeats.
The full-length cDNAs of DAPK-α and DAPK-β or two mutants with an alanine substitution at the ATP-reactive lysine within the kinase domain, K42A (αK42A, βK42A), which is predicted to inactivate kinase activity, were cloned into pcDNA3His such that the predicted translation start site at the amino terminus was fused in frame to the Xpress/hexahistidine tag, and the plasmids were transfected into Cos cells for expression. Fig. 2A shows the results of a Western blot to detect the wild-type recombinant DAPKs using either a DAPK-specific antibody or the Omniprobe monoclonal antibody (Santa Cruz Biotechnology) to detect the Xpress-tagged DAPK. Blots reacted with the anti-DAPK antibody revealed that the molecular mass of the mouse DAPKs is indistinguishable from the 160-kDa endogenous DAPK present in Cos cells. A smaller, proteolytic breakdown product of ~100 kDa is also detected with this antibody. There is also no apparent size difference between DAPK-α and DAPK-β isoforms (~160 and 161 kDa, respectively) when they are separated on a 5% SDS-PAGE gel. Identical results were obtained for the mutant cDNAs (data not shown).
FIG. 2. Western blotting and ribonuclease protection to detect DAPK of mouse embryonic and adult tissues.

A, both panels are Western blots detecting expression of endogenous and recombinant DAPK-7α and DAPK-β in Cos cells. In the left panel, the recombinant DAPK detected in Cos cells transiently expressing DAPK-α or DAPK-β co-migrates with the endogenous DAPK detected in mock-transfected cells. In the right panel, an antibody to the Xpress tag detects the transiently expressed recombinant DAPKs. B, an autoradiogram of the ribonuclease protection analysis. The antisense cRNA probe is homologous to bp 4506–5009 of DAPK-β and contains 43 bp of vector sequence, making the total probe length 546 bp. Protection of a 507-and 445-bp probe fragment confirms that mRNAs corresponding to DAPK-β and DAPK-α, respectively, are present in adult liver and throughout development.
Ribonuclease Protection Analysis Suggests That an Alternatively Spliced Form of DAPK Is Widely Expressed
Ribonuclease protection assays were used to confirm the presence of mRNAs for both isoforms of DAPK. DAPK-β has a deletion of 485 bp that occurs immediately prior to the UGA stop codon. The splicing results in a deletion of 485 nucleotides of mRNA present in the 3′-untranslated region of DAPK-α and the extension of the open reading frame for an additional 12 residues to generate DAPK-β. The DAPK-β cDNA was linearized at a StuI site located at bp 4506; the resulting template allowed us to generate a cRNA probe that distinguishes the DAPK-α and DAPK-β isoforms. The results of the ribonuclease protection analysis are shown in Fig. 2B. Two protected fragments of 507 and 445 bp were detected, corresponding to the predicted sizes of the 32P-labeled cRNA probe protected by the mRNAs encoding DAPK-β and DAPK-α, respectively. Both of these protected fragments appear in adult liver, whole embryos at days 10, 15, and 19, and embryonic heart (at days 12 and 15), showing that DAPK-β is expressed during early development as well as in adult liver tissue.
Expression of DAPK in Cells and Tissues
Northern blotting with a probe corresponding to bp 3370–3920 (residues 1033–1215) of the DAPK cDNA identified a predominant mRNA of ~6.0 kb in mouse embryos obtained at days 10, 15, and 19 and many adult tissues tested as well as one cell line (Fig. 3A). The relative levels of the 6.0-kb mRNA were highest in the embryo tissues and in adult bladder, uterus, vas deferens, liver, kidney, and 3T3 mouse fibroblasts. Western blotting with a monoclonal anti-DAPK antibody (Transduction Laboratories) revealed the presence of a 160-kDa band that is detectable in several adult tissues including bladder, uterus, vas deferens, lung, liver, and kidney (Fig. 3B). The 100-kDa protein is a proteolytic breakdown product of DAPK. Comparison of the Northern and Western blotting revealed that for several tissues a 6.0-kb mRNA is present, but no DAPK protein is detectable. These tissues generally had lower relative levels of the 6.0-kb mRNA, suggesting that either DAPK is expressed in skeletal muscle, testes, stomach, colon, and ileum but at levels below the detectable limit of the antibody or expression is post-transcriptionally regulated.
FIG. 3. Northern and Western blotting to detect expression of DAPK.

Northern (A) and western (B) blotting to detect DAPK mRNA and protein in mouse embryonic and adult tissues are shown. For the Northern blots, 20-μg samples of total RNA isolated from the indicated mouse tissues were separated on a 1.2% agarose gel and probed with a cRNA probe corresponding to bp 3370–3920 of the mouse DAPKs. A 6-kb mRNA is detected. For Western blotting, a monoclonal antibody against DAPK (Transduction Laboratories) was used to detect the 160-kDa protein corresponding to DAPK. The smaller 100-kDa protein may be a proteolytic breakdown product of DAPK.
DAPK Phosphorylates Myosin II RLC in Vitro and in Vivo
The kinase activity of the human DAPK has been determined in vitro using commercially available myosin II RLC purified from rabbit skeletal muscle as a substrate (10). However, the kinase domain of DAPK is most similar to the kinase domain of the conventional Ca2+/CaM-dependent 130–150-kDa MLCKs (also referred to as smooth muscle MLCK) and less similar to the kinase domain of the skeletal muscle MLCKs. Because the skeletal and smooth MLCKs have different substrate preferences, in vitro phosphorylation assays were performed in the presence of each type of RLC to determine whether DAPK will phosphorylate both skeletal and smooth/nonmuscle myosin RLCs or smooth/nonmuscle RLC preferentially. Immunopurified DAPK-α was incubated with either skeletal or smooth muscle RLCs in the presence and absence of Ca2+/CaM for 30 min at 30 °C and analyzed by SDS-PAGE (Fig. 4A). These results demonstrate that RLCs purified from smooth muscle myosin are better substrates for DAPK in vitro than skeletal muscle RLC and that DAPK phosphorylation of RLC is Ca2+/CaM-dependent. Fig. 4B demonstrates that DAPK can phosphorylate recombinant RLCs as well as RLC associated with intact nonmuscle myosin II and that this phosphorylation depends on the presence of Ca2+/CaM. In these assays, immunoprecipitated DAPK was incubated with purified RLCs or intact myosin purified from human platelets in the presence or absence (+EGTA) of Ca2+/CaM, and the 32P incorporated into RLCs was detected following SDS-PAGE and autoradiography. To determine whether DAPK, similar to MLCK, preferentially phosphorylates Ser-19 of the RLC, in vitro kinase assays using recombinant wild-type RLCs or RLCs with mutations in key phosphorylatable residues were performed (5). The expressed recombinant RLCs are derived from human umbilical vein endothelial cells and represent smooth/nonmuscle myosin-type RLCs (6, 27). Phosphorylation of RLCs by immunoprecipitated DAPK occurred on mutant RLCs where either Thr-18 or Ser-19 were replaced with Ala (T18A, S19A). Alteration of Thr-18 had a minimal effect on the level of phosphorylation, whereas alteration of S19A reduced the level of phosphorylation 3-fold, suggesting that Ser-19 is the preferred site of phosphorylation. Replacement of both Ser-19 and Thr-18 with Ala (T18S19AA) ablated RLC phosphorylation by DAPK. Substitution of Arg-16 with Ala (R16A) reduced the ability of DAPK to phosphorylate RLC by ~6-fold, demonstrating that similar to the conventional MLCKs, DAPK requires the Arg-16 residue for maximal phosphorylation of RLC by DAPK. Together these results show that similar to the conventional MLCKs, Ser-19 is the primary RLC residue phosphorylated by DAPK and that phosphorylation of Thr-18 is also possible.
FIG. 4. In vitro and in vivo phosphorylation of myosin II RLCs by DAPK.

A–E, results of in vitro kinase assays. RLC was incubated in the presence of Ca2+ and calmodulin (+Ca2+) or EGTA (−Ca2+), [32P]ATP, and immunoprecipitated Xpress-tagged DAPK for 15 min and analyzed as described under “Materials and Methods.” The reactions were followed either by SDS-PAGE and autoradiography or by counting 32P incorporation into RLCs. A, an autoradiogram showing the relative levels of phosphorylation of RLCs isolated from smooth (sm) or skeletal (sk) muscle by immunoprecipitated DAPK-α after transient expression in HeLa cells. B and C, in vitro kinase assays utilizing purified recombinant wild-type or mutant RLCs or purified myosin II followed by SDS-PAGE and autoradiography. All incubations were in the presence of either Ca2+/CaM or EGTA, which is used to chelate calcium. D, kinetic analysis showing rates of phosphorylation of myosin RLC by immunoprecipitated DAPK-α, DAPK-β, DAPK-α(K42A) and DAPK-β(K42A) determined in the presence of Ca2+ and calmodulin (+Ca2+) or EGTA (−Ca2+) for the indicated times. The line denoted −Ca2+ Controls; MDCK parental represents all the control (+EGTA) reactions that had nearly identical levels of 32P incorporation into RLC, and for simplicity a single line is shown on the graph. The panel at the top is a representative Western blot showing that the relative levels of immunoprecipitated DAPK used in the assay were approximately equal. DAPK was immunoprecipitated and detected using the anti-Xpress antibody. E, an autoradiogram showing the relative levels of autophosphorylation (upper) and RLC phosphorylation (lower) by DAPK-α, mutant DAPK-α(K42A), DAPK-β, and mutant DAPK-β(K42A) immunoprecipitated from MDCK cell lines. Control reactions were X-press antibody immunoprecipitates from lysates of the parental MDCK cells (Mock-IP). After the kinase assays, the DAPKs and RLCs were analyzed by SDS-PAGE and autoradiography. F, myosin RLC phosphorylation levels in MDCK cell lines that overexpress DAPK-α, DAPK-α(K42A), DAPK-β, or DAPK-β(K42A) that have either been treated with (+) or without (−) TNF (10 ng/ml) for 1 h. The results shown are representative of at least three independent experiments.
To determine the specific activity of DAPK, the kinetics of phosphorylation of DAPK were analyzed in an in vitro assay. Fig. 4D summarizes the relative rates of phosphorylation of RLCs by wild-type or mutant DAPK-α and DAPK-β immunoprecipitated with X-press antibody in the presence or absence of Ca2+/CaM. This experiment shows that the relative rates of RLC phosphorylation by the wild-type DAPK-α and DAPK-β are nearly identical. Unexpectedly, both mutant DAPK forms have some residual kinase activity in the presence of Ca2+/CaM. For clarity, a single line (−Ca2+ Controls; MDCK parental) is shown to represent the nearly identical rates of phosphorylation that were obtained in the absence of Ca2+/CaM for the wild-type or mutant DAPKs and a mock immunoprecipitation from parental MDCK cells. A Western blot (Fig. 4D, upper) of the immunoprecipitates revealed that approximately equal amounts of each DAPK were present in the assay. The rate of phosphorylation of RLC by DAPK was estimated by quantifying the relative amounts of DAPK in the in vitro kinase assay as described under “Materials and Methods.” The specific activity of the wild-type DAPKs was estimated to be 0.053 pmol/min/ng. Comparison of this activity to the rate determined for MLCKs (36–39 pmol/min/ng) suggests that DAPK has an ~700-fold lower RLC phosphorylation activity compared with the conventional 130–150 kDa MLCKs (25, 26). The residual catalytic activity of the K42A mutants was estimated to be ~20% of the activity of the wild-type DAPKs.
Fig. 4E shows the autoradiographic results obtained following gel electrophoresis of the in vitro kinase assays shown in Fig. 4D. Control reactions representing X-press tag immunoprecipitates from the parental MDCK cells (Mock-IP) were also included. These autoradiographs show that RLC phosphorylation by the wild-type DAPKs is stimulated by the presence of Ca2+/CaM and that in the absence of Ca2+/CaM, activity is attenuated significantly but not completely abolished. Because the human DAPK has been shown to undergo autophosphorylation, we examined the autoradiographs to determine whether this was true for the mouse DAPKs. The appearance of the 160-kDa bands on these gels suggests that the wild-type DAPKs undergo autophosphorylation that is stimulated by Ca2+/CaM. This analysis also revealed that both DAPK-α(K42A) and DAPK-β(K42A), are capable of residual RLC phosphorylation and autophosphorylation.
To learn whether DAPK can phosphorylate RLC in vivo, MDCK cell lines expressing wild-type and mutant DAPKs were treated with TNF, and the levels of phosphorylation of endogenous RLCs were quantified by densitometry following urea-glycerol PAGE and Western blotting with an antibody to RLC (Fig. 4F). The results of this analysis show that there is no statistical difference between the in vivo RLC phosphorylation levels obtained for MDCK cell lines that overexpress either DAPK-α or DAPK-β in the absence of TNF stimulation. In these cell lines, TNF stimulates a 3–4-fold increase in RLC phosphorylation from an average of 0.42 mol of Pi/mol of RLC to 1.7 mol of Pi/mol of RLC. Consistent with their residual kinase activity, cells overexpressing the mutant kinase-defective (K42A) DAPKs have reduced amounts of TNF-stimulated RLC phosphorylation. In comparison, treatment of the parental MDCK cells with TNF stimulates only a modest increase in RLC phosphorylation from 0.46 mol of Pi/mol of RLC to 0.72 mol of Pi/mol of RLC. These experiments confirm that DAPK can phosphorylate nonmuscle myosin II RLCs in vivo. Together the results shown in Fig. 4 reveal that the biochemical properties of DAPK-α and DAPK-β are indistinguishable and that these kinases can phosphorylate Thr-18 and Ser-19 in the myosin RLC both in vitro and in vivo. In comparison to the conventional Ca2+/CaM-dependent MLCKs, the apparent rate of RLC phosphorylation in vitro by DAPK is significantly lower than the one determined previously for the conventional ML-CKs, and there is an incomplete reliance on Ca2+/CaM for activity.
Transient Overexpression of DAPK-β Protects Cells from TNF-induced Apoptosis
Ectopic overexpression of some apoptosis regulatory proteins is known to induce apoptosis (28–33). In addition, previous studies have shown that overexpression of the human DAPK promotes apoptosis in the absence of any apoptotic stimuli (10). To learn whether this is true for the mouse DAPKs, HeLa cells were co-transfected with either DAPK-α or DAPK-β and β-galactosidase or β-galactosidase and empty pcDNA3His vector. At 24 h the cells were then treated with TNF (10 ng/ml) in the presence of cyclohexamide (10 μg/ml) for 3 h prior to fixation and staining to detect transfected cells expressing β-galactosidase. Cells were scored as apoptotic when they exhibited condensed cytoplasm and several visible membrane blebs. The average ratio of apoptotic cells expressing β-galactosidase to total transfected β-galactosidase-positive cells was calculated from eight independent transient expression assays. The Western blot in Fig. 5 shows that the transient expression level of either DAPK-α or DAPK-β is similar, as are the relative levels of expression of β-galactosidase, suggesting that the differences in apoptosis are not the result of dramatic differences in co-transfection or expression. The results obtained from transient expression analysis in HeLa cells were similar to those obtained with the conditionally regulatable cell lines (see below). HeLa cells transiently expressing either DAPK-α or DAPK-β had a statistically significant lower level of apoptotic β-galactosidase-positive cells (53 ± 1.9% and 40 ± 1.1%, respectively) compared with the level found for control cells (60 ± 1.8%). In addition, in the absence of TNF, the very low level of apoptosis in the DAPK-transfected cells after 48 h of expression was comparable with that found for the LacZ control transfected cells under the same conditions (≤1 ± 0.1%). Together these results show that transient overexpression of either form of the mouse DAPK in HeLa cells suppresses the appearance of TNF-induced apoptotic morphology. In addition, in the absence of TNF, neither DAPK-α nor DAPK-β enhanced apoptosis. Finally, although both forms of mouse DAPK are cytoprotective, DAPK-β has a more potent antiapoptotic function.
FIG. 5. Overexpression of DAPK does not promote apoptosis, and DAPK-β is cytoprotective in HeLa cells.
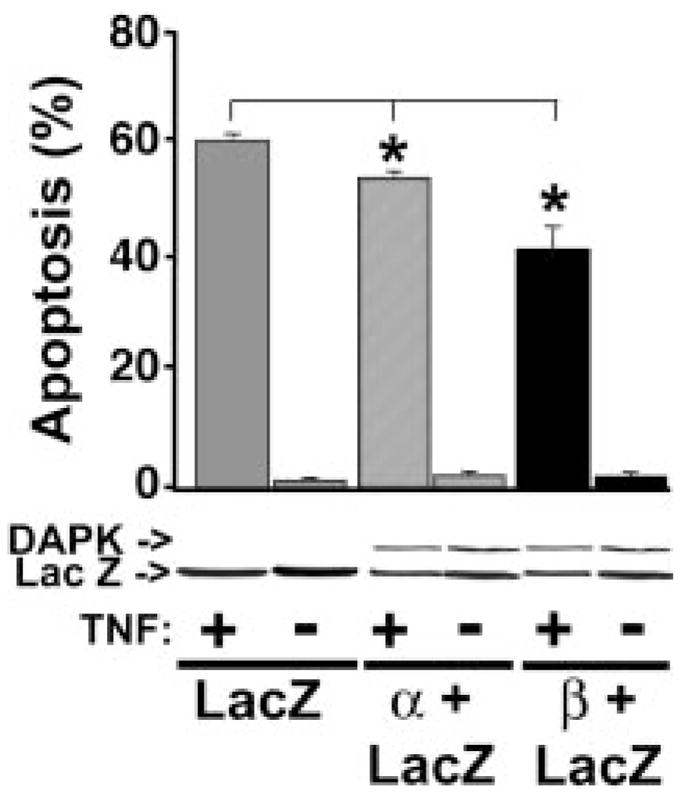
HeLa cells transiently expressing β-galactosidase (LacZ) or β-galactosidase and DAPK-α or DAPK-β were treated with 10 ng/ml TNF and 10 μg/ml cyclohexamide or vehicle (−TNF) to induce apoptosis. After 3 h of TNF or 48 h of vehicle, the cells were fixed and stained to detect β-galactosidase activity. The number of β-galactosidase-positive cells that had membrane blebs consistent with apoptosis were counted to determine the fractional amount of apoptosis. The results are representative of eight independent experiments. The lower panel is a representative Western blot to show that the expression levels of Xpress-tagged DAPK-α, DAPK-β, and LacZ were approximately equal. The results shown represent the mean ± S.E. from at least three independent experiments. Significance (*) indicates that p ≤ 0.01.
Mouse DAPK Regulation of TNF-induced Apoptosis
To aid us in determining whether the cytoprotective effects of DAPK observed in TNF-treated HeLa cells transiently expressing mouse DAPK were the result of antagonizing an apoptotic or necrotic TNF-induced cell death and to examine the mechanism by which mouse DAPK interferes with TNF signaling, we generated tetracycline-inducible HeLa and MDCK cell lines expressing both wild-type and kinase-defective forms of DAPK. For all the following experiments the controls that were performed included both the MDCK or HeLa parental cell lines as well as the DAPK cell lines in the repressed state (−Dox) in the presence and absence of TNF. Analysis of the cell lines in the tetracycline-inducible configuration occurred after a 24-h period of treatment in the presence of Dox prior to the addition of TNF or vehicle. For clarity in some figures the data obtained for the repressed state of the DAPK (−Dox) cell line is not included, because these control experiments had results that were indistinguishable from those obtained with the parental cell lines. This finding suggests that clonal variants with defects in apoptotic signaling have not been selected during generation of these cell lines. Although MDCK cells are sensitive to the apoptotic effects of TNF and do not require cyclohexamide, the progression of apoptosis is slower, requiring ~24 h of TNF treatment for biochemical evidence of apoptosis to become evident (8). In contrast, HeLa cells required the addition of 10 μg/ml cyclohexamide to promote the apoptotic effects of TNF and morphological changes such as nuclear condensation and blebbing consistent with apoptosis become visibly apparent within 2–3 h.
Fig. 6, A and D, shows the relative expression levels of DAPKs in the MDCK and HeLa cell lines in both the uninduced (−Dox) and induced (+Dox) configurations. These Western blots show that the induced levels of all of the DAPKs are approximately equal and that in the absence of Dox, the expression of the exogenous kinases is tightly regulated with only a marginal amount of leakiness occurring in the absence of Dox. The differences in the relative levels of TNF-induced apoptosis in the MDCK cell lines are shown in Fig. 6B. For these experiments parental MDCK cells and the four cell lines were pretreated with Dox for 24 h to induce (+Dox) equal levels of expression of the exogenous DAPKs. Parallel control cultures were treated identically but DAPK expression was not induced (−Dox). The cells were then either treated with 10 ng/ml TNF or vehicle for 60 h, a dose previously determined (8) to induce a maximal apoptotic response in parental MDCK cells. Apoptotic levels were determined by counting the number of viable, trypan blue-excluding cells and compared with the number of viable cells in the untreated controls. The results of these experiments summarized in Fig. 6B show that DAPK-α has a modest but statistically significant cytoprotective effect and reduces apoptosis from 65 ± 1.2% to 56 ± 1.6%. In contrast, overexpression of DAPK-β in MDCK cells resulted in a marked reduction of TNF-induced apoptosis to 29 ± 4.4%. Expression of either kinase-defective (K42A) DAPK completely reversed the cytoprotective effect of the wild-type DAPKs and increased apoptosis to 73 ± 2.7% for DAPK-α(K42A) and 92 ± 2.8% for DAPK-β(K42A). It is unclear as to why the overexpression of DAPK-β(K42A) promotes TNF-induced apoptosis to a level greater than that found for DAPK-α. It is unlikely this effect is caused by differences in expression levels of the exogenous DAPKs, because under our experimental conditions relatively equal and stable levels of expression were obtained. However, it is possible that the kinase-defective DAPK-β(K42A) is a more potent dominant negative than DAPK-α(K42A) and can compete more effectively against the cytoprotective effects of the endogenous DAPKs expressed in MDCK cells.
FIG. 6. Characterization of MDCK and HeLa cell lines expressing wild-type and mutant DAPK-α and DAPK-β.

Conditional MDCK (A, B, and C) or HeLa (D, E, and F) cell lines expressing DAPK-α, DAPK-α(K42A), DAPKβ, or DAPK-β(K42A) were established as described under “Materials and Methods.” For each experiment, the appropriate amount of Dox was added to the cell cultures, and 24 h later either 10 ng/ml TNF or vehicle was added (t = 0) to initiate the experiments. Western blotting using the Omniprobe antibody demonstrates the relative levels of DAPK expression in MDCK (A) or HeLa (D) cell lines after culture in the presence of doxycycline (Dox) for 24 h. All experiments were conducted using conditions that result in these levels of expression. The percentage of apoptosis was measured in the MDCK (B) or HeLa (F) cell lines by determining cell viability at the indicated times using trypan blue exclusion. C, Western blotting shows the temporal appearance of the 85-kDa PARP fragment in response to TNF in MDCK cells. E, TNF-induced apoptosis dose-response curves for HeLa cell lines expressing the indicated DAPKs. The results shown represent the mean ± S.E. from at least three independent experiments. Significance (*) indicates that p ≤ 0.01.
HeLa cell lines expressing DAPKs were examined to determine the sensitivity of these cell lines to TNF. The dose-response curve shown in Fig. 6E revealed that expression of wild-type DAPK-α and DAPK-β reduces the sensitivity of HeLa cells to TNF-induced apoptosis at all concentrations of TNF between 2.5 ng/ml up to 50 ng/ml. In contrast, overexpression of both K42A DAPK mutants increases the sensitivity of HeLa cells to TNF-induced apoptosis. A concentration of TNF of 10 ng/ml induced maximal levels of apoptosis in the parental HeLa cell and all four of the DAPK cell lines in the repressed configuration (−Dox) (data not shown), and this concentration was used for the experiments shown in Figs. 6F, 7, and 8. Fig. 6F compares the levels of apoptosis in the four DAPK-expressing HeLa cells to those in the parental HeLa cells in the presence (+Dox) or absence (−Dox) of exogenous DAPK. These results paralleled those from the MDCK cell lines, and together these studies suggest that overexpression of DAPK-β suppresses TNF-induced apoptosis, with DAPK-α having a negligible cytoprotective effect (Fig. 6F). Experiments conducted in the absence of TNF showed that there was no significant increase in the very low level of apoptosis (<1% of the cells) normally observed in HeLa parental cells in response to induction for up to 48 h of expression of any of the DAPKs, confirming that overexpression of mouse DAPK is not proapoptotic.
FIG. 7. Overexpression of DAPK-β is cytoprotective in TNF-treated HeLa cells.

Flow cytometric analysis of TNF-treated HeLa cell lines expressing DAPK-α or DAPK-β. After 3 h of TNF treatment, cells were dissociated, fixed, and treated with propidium iodide (PI). The DNA content of 10,000 cells was analyzed by flow cytometry as described under “Materials and Methods.” The results shown are representative of three independent experiments. The percentage of apoptotic cells having fragmented sub-G1 DNA(<G1) content in the bracketed peak is indicated for each cell line. Propidium iodide fluorescence is in arbitrary units.
FIG. 8. Caspase activity and cytochrome c release in TNF-treated HeLa cell lines expressing wild-type and mutant DAPK.

For each experiment, at 24 h post-induction 10 ng/ml TNF and cyclohexamide (10 μg/ml) was added to initiate the experiment. Caspase-8 (A), caspase-3 (B), or caspase-9 (C) activity was determined directly in cell extracts by quantitating the cleavage of IETD-pNA (caspase-8), LEHD-pNA (caspase-9), or DEVD-pNA (caspase-3). The results shown represent the mean ± S.E. from at least three independent experiments. Significance (*) indicates that p ≤ 0.01. D, the release of cytochrome c from mitochondria was determined in the presence and absence of TNF by Western blotting of the post-mitochondrial supernatant from the indicated HeLa cell lines expressing DAPKs. The panel shown is representative of three independent analyses.
Previous studies have shown that MDCK cells undergo a maximal apoptotic response to 10 ng/ml TNF in the absence of cyclohexamide (8). In the present study we used Western blotting to examine the cleavage of PARP, which is an endogenous substrate for caspase-3 (Fig. 6C) (33). The temporal appearance of the 89-kDa PARP fragment in MDCK cells expressing DAPK-α or the kinase-defective DAPK-α(K42A) in MDCK is similar to the parental MDCK cells and becomes detectable within 24 h. Consistent with the levels of apoptosis determined for these cell lines (Fig. 6B), overexpression of DAPK-β results in a significant delay in the appearance of the 89-kDa PARP fragment, from 24 to greater than 72 h. In contrast, MDCK cells expressing DAPK-β(K42A) appear to have a slightly higher level of the 89-kDa PARP proteolytic fragment at 24 h, a result consistent with the enhanced apoptotic rate observed for this cell line (Fig. 6B). The appearance of the 89-kDa fragment from PARP indicates that all the MDCK cell lines were undergoing a TNF-induced caspase-mediated apoptotic death rather than a necrotic death. Together, these results confirm that DAPK-β antagonizes TNF-induced apoptotic death and that in MDCK cells its activities are upstream of caspase-3 or another effector caspase that is able to cleave PARP. To confirm further that the HeLa cell lines were undergoing an apoptotic death induced by treatment with TNF, the cleavage of DNA into small fragments was examined by flow cytometry after treatment with TNF and cyclohexamide for 3 h (Fig. 7). The results of this analysis showed that overexpression of DAPK-β significantly decreased the appearance of HeLa cells having fragmented sub-G1 (<G1) DNA from 36 to 15%, whereas expression of DAPK-α had little or no effect on the number of cells with sub-G1 fragmented DNA and confirms that these HeLa cell lines are undergoing apoptotic cell death induced by TNF.
To examine where in the TNF apoptotic signaling pathway DAPK-α and DAPK-β exert their cytoprotective effects, the activities of caspase-8, caspase-3, and caspase-9 were determined in MDCK and HeLa cell lines expressing DAPK using colorimetric peptide substrates that are reported to be relatively specific (caspase-3 and caspase-8) or moderately specific (caspase-9) for these caspases. Fig. 8 shows the results of these kinetic analyses in HeLa cells. This analysis revealed that overexpression of either wild-type or kinase-defective mutant DAPKs had no effect on the level of caspase-8 activity, the apical initiator caspase in the extrinsic death receptor signaling pathway (Fig. 8A). This result suggested that DAPK activity is important at a more distal point in the TNF signaling pathway. In contrast, expression of DAPK-β had significant effects on the activities of two effector caspases, caspase-3 and caspase-9, in these HeLa cell lines (Fig. 8, B and C). Consistent with its strong cytoprotective effects, expression of DAPK-β suppressed the levels of both caspase-3 and caspase-9 activities to result in a 3- and 5-fold decrease, respectively. These experiments also showed that the kinase activity of DAPK-β was essential to antagonize both caspase-3 and caspase-9 activities as overexpression of the kinase-defective DAPK-β(K42A) reverses the antagonism and, interestingly, seems to stimulate caspase activity between 2- and 5-fold.
To further refine the signaling pathway being regulated by DAPK expression, the release of cytochrome c from mitochondria was examined by Western blotting of fractionated HeLa cell lines in the presence or absence of TNF. The Western blot shown in Fig. 8D shows that TNF-induced release of cytochrome c to a post-mitochondrial supernatant prepared from fractionated HeLa cells is inhibited by the expression of DAPK-α or DAPK-β. Consistent with our other results, the overexpression of the kinase-defective DAPK-α(K42A) reverses the inhibition of release, and the DAPK-β(K42A) form results in an increased release that seems higher than the level of cytochrome c release from the parental HeLa cell line. Together these results suggest that the antiapoptotic effect of DAPK-β occurs after activation of caspase-8 and before mitochondrial release of cytochrome c, which leads to activation of effector caspases such as those having caspase-3- and caspase-9-like activities. Our data characterizing the effect of overexpression of the mouse DAPK-α on TNF-induced apoptosis is ambiguous. In HeLa cells, a small but statistically significant decrease in the level of apoptosis was found after either conditional or transient expression, suggesting that the expression of DAPK-α has a small cytoprotective effect in cells. This was supported by the relatively small decrease in cytochrome c release from the mitochondria. In contrast, we were unable to detect a significant decrease in caspase activity, which may reflect the sensitivity of these activity assays. Overall these results suggest that DAPK-α has a negligible if any cytoprotective effect in cells. It is also clear that overexpression of the mouse DAPK-α does not promote apoptosis either in stimulated or unstimulated cells under transient or conditionally regulated expression conditions.
To determine whether the antiapoptotic effects of DAPK in TNF-treated cells are being potentiated by TNF signaling through the NF-κB pathway, we used Western blotting and densitometry to compare the relative amounts of nuclear NF-κB in the HeLa cell lines to the parental HeLa cells. These experiments were performed in the presence or absence of TNF and cyclohexamide (Fig. 8). The results of this analysis revealed that the levels of NF-κB in the nuclear fraction are not altered significantly in response to DAPK expression. This experiment suggests that DAPK-β mediates its cytoprotective effects by a mechanism that does not involve increased NF-κB stimulation in response to TNF.
DISCUSSION
Two murine homologs of the 160-kDa human DAPK have been cloned and characterized. The murine DAPKs are represented by two alternatively spliced forms designated DAPK-α and DAPK-β. Both of these Ser/Thr protein kinases are ~95% identical to the previously described human DAPK. The mouse DAPK-α form corresponds to the previously described human DAPK (9), and DAPK-β, a previously unknown form of DAPK, is distinguished by the presence of an additional 12 residues that extend its carboxyl terminus.
Our studies show that overexpression of both mouse DAPK forms results in a distinctly different apoptotic outcome than the one described for the human DAPK, because neither form of the mouse kinase stimulates apoptosis (9). In addition, the ectopic overexpression of the murine DAPK-β has a strong cytoprotective effect in TNF-treated cells, suggesting that this kinase can antagonize TNF-induced apoptotic signaling. We also show that the cytoprotective effect of the mouse DAPK-β depends on its kinase activity, becaus diminishing kinase activity by the mutation of a single lysine residue (K42A) within the kinase domain reverses the cytoprotective effects of this kinase in HeLa and MDCK cells and stimulated apoptosis.
The reason for the disparity in the apoptotic outcome resulting from overexpression of the mouse DAPK-α compared with the human DAPK, which lacks the 12 residues present at the carboxyl-terminus of the mouse DAPK-β, is not clear. Possible reasons include differences in the levels of exogenous expression of the human and mouse DAPKs, different experimental conditions, or cell-specific differences in the interaction of DAPK and other apoptosis regulatory proteins. In support of the latter, a yeast two-hybrid interaction screen has identified a novel protein that binds the ankyrin repeats of DAPK and exogenous expression of this protein, called DIP (DAPK-interacting protein) promotes TNF-induced apoptosis.2 Further investigation of DAPK as an apoptosis regulator will be required to clarify the role of this kinase as a pro- or antiapoptotic modulator.
The characterization of the mouse DAPKs has shown that there is a strong distinction in the apoptotic outcomes of TNF-treated cells that overexpress mouse DAPK-α compared with mouse DAPK-β. This distinction is evidenced by the strong cytoprotective effect that the expression of DAPK-β confers on TNF-treated cells compared with the negligible cytoprotective effect that results from overexpression of DAPK-α. Because the only difference between DAPK-α and DAPK-β is the additional 12 residues present at the carboxyl terminus, these results suggest that the “tail” region strongly regulates the cytoprotective activities of DAPK. Previous studies examining this region of the human DAPK support the importance of this region, because deletion of the human DAPK carboxyl-terminal tail enhances the ability of mutated human DAPK to promote apoptosis (34, 35). Similarly, exogenous expression of the human death domain (residues 1320–1371) or the carboxyl-terminal tail (residues 1415–1431) was shown to partially rescue cells from TNF-induced apoptosis (34). These results have led to the proposal that the carboxyl-terminal tail may be involved in negative regulation of the proapoptotic functions of human DAPK (34) and by analogy are likely to regulate the antiapoptotic functions of the mouse DAPK. Therefore, in addition to the requirement for catalytic activity, which is regulated by Ca2+/CaM, critical regulation of the antiapoptotic activities of the mouse DAPK seem to be imposed by its carboxyl-terminal tail. It has already been noted that this carboxyl-terminal tail region of DAPK is rich in serine, threonine, and tyrosine residues, which suggests that it may be a potential target for regulation by additional signaling pathways.
Ribonuclease protection analysis verified the presence of mRNA encoding DAPK-β, and extensive Northern and Western blotting analysis revealed that the mouse DAPK is ubiquitously expressed and detectable both during early development and in adult tissues. Ribonuclease protection analysis also suggests that mRNAs encoding both forms of DAPK are present during development and in adult liver tissue at approximately equal levels. However, it is clear from comparing the Northern and Western blotting results that there is at least some discordance between these two detection methods. For example, many tissues having detectable mRNA have very low or undetectable levels of DAPK protein, suggesting that expression of DAPK is post-transcriptionally regulated, although it is also possible that the level of DAPK expression in some tissues is below the limits of detection by the antibody. In addition, given the clear difference in the cytoprotective effects of the mouse DAPK-α and DAPK-β, it will be critical to determine which forms are expressed in cells and tissues before a complete understanding of the role of DAPK in apoptosis regulation can be defined.
Our biochemical characterization of the mouse DAPKs revealed that these protein kinases preferentially phosphorylate smooth/nonmuscle RLCs in vitro and have little or no activity toward RLCs purified from skeletal muscle. Surprisingly, we found that mutation of an important lysine residue within the kinase domain to alanine (K42A) significantly reduces but does not completely ablate DAPK activity. We have also established that in vitro DAPK can phosphorylate RLCs associated with myosin and have identified the in vitro phosphorylation site in the RLC. These results show that DAPK is a member of the MLCK family that preferentially phosphorylates smooth/non-muscle-type RLCs at the activating site, Ser-19, and to a lesser extent at Thr-18. Finally, in vivo studies examining changes in RLC phosphorylation demonstrate that TNF induces a significant increase in the diphosphorylation of RLCs in MDCK cells overexpressing a wild-type but not a kinase-defective DAPK. This finding suggests that at least one in vivo function for DAPK relates to the activation of myosin II motor activities during TNF-induced apoptosis. However, additional studies will be required to determine the importance and physiological role that myosin motor activities have during apoptosis. One potential role that already has been suggested is the regulation of the morphological changes associated with membrane blebbing in apoptotic cells (3). Our previous studies have suggested that modulation of myosin RLC phosphorylation by the over-expression of the conventional Ca2+/CaM-dependent MLCK has an important role in regulating TNF-induced apoptosis by controlling the amount of TNFR-1 (TNF receptor 1) on the plasma membrane (8). Our previous studies have shown that increased RLC phosphorylation caused by everexpression of the conventional MLCK potentiates TNF-induced apoptosis rather than antagonizing it as seen in these studies overexpressing DAPK. The reason for this apparent contradiction is not yet known, but it may be possible that DAPK and MLCK have distinct intracellular distributions in cells, associate with different signaling proteins, or may have additional unknown substrates, and further studies will be required to resolve these issues.
The delay in TNF-induced cleavage of PARP in MDCK cell lines overexpressing DAPK-β suggests that the cytoprotective effects of DAPK are exerted prior to the activation of executioner caspases such as caspase-3, which cleave PARP during the late stages of apoptosis. Overexpression of DAPK-β strongly antagonizes the activities of caspase-3 and caspase-9 or caspases with similar activities that can cleave the substrates used in this study. This result refines the placement of DAPK in the apoptotic signaling cascade to a point prior to or during the activation of these effector caspases. Suppression of cytochrome c release from the mitochondria of cells stimulated with TNF, by expression of both forms of DAPK, also depends on kinase activity and can be reversed by expression of the kinase-defective forms of DAPK. Because cytochrome c release is upstream of the activation of both of the effector caspases examined, these results suggest that the apoptotic regulation by DAPK may work by signaling upstream of mitochondrial alterations that lead to the release of cytochrome c and formation of the apoptosome. The release of cytochrome c from mitochondria is a critical event in the intrinsic apoptotic pathway but can also occur in the death receptor-dependent extrinsic apoptotic pathway through the caspase-8-dependent cleavage and activation of Bid, a proapoptotic Bcl-2 family member (36–40). The latter pathway is thought to be an amplification loop in the extrinsic pathway that converges at the activation of caspase-3 (41). Recently, a potential link between DAPK regulation of apoptosis and the p19 (ARF)-MDM-p53 pathway was identified (19). In this study, expression of human DAPK was found to activate expression of p53 via a p19ARF-dependent mechanism. This finding together with the demonstration that p53 promotes apoptosis through mitochondrial release of cytochrome c and caspase activation (42, 43) suggest that DAPK regulates apoptosis at a signaling point that is upstream of the intrinsic mitochondrial pathway. Despite the disparity in apoptotic outcome identified for the mouse DAPKs in these studies, our finding that the expression of mouse DAPK-β abrogates caspase-3 and caspase-9 activity as well as suppresses mitochondrial release of cytochrome are also consistent with positioning DAPK regulation at a point that is upstream of the intrinsic pathway and suggests that this form of DAPK can antagonize p53 promotion of apoptotic cell death. Finally, the lack of any significant change in NF-κB movement to the nucleus in the HeLa DAPK (Fig. 9) cell lines suggests that the cytoprotective effects of DAPK-β are not the result of an enhancement of TNF-induced activation of NF-κB.
FIG. 9. Nuclear levels of NF-κB do not change in response to DAPK expression.
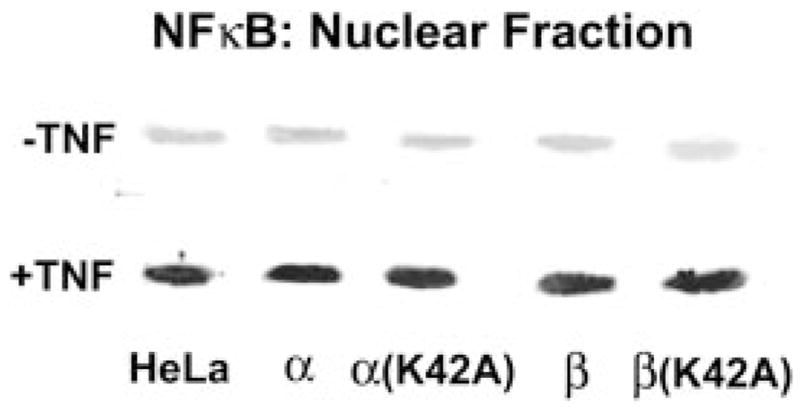
Nuclear levels of NF-κB were determined in the presence and absence of TNF by Western blotting of a nuclear fraction from the indicated HeLa cell lines expressing DAPKs. The panel shown is representative of three independent analyses.
In summary, we have established that in vivo DAPK can phosphorylate RLCs associated with myosin II in a Ca2+/CaM-dependent manner, making DAPK a member of the MLCK family. Characterization of the mouse homologs of the previously described human DAPK revealed that the mouse DAPKs do not promote apoptosis. Paradoxically, the overexpression of DAPK-β confers a potent cytoprotective effect that antagonizes TNF-stimulated apoptotic death. This finding suggests the unique carboxyl-terminal 12 residues present in DAPK-β have an important role in regulating the cytoprotective activity of DAPK. Our data also show that the DAPK cytoprotective activity impacts the apoptotic signaling pathway at a point upstream of the mitochondrial release of cytochrome c and the subsequent activation of caspase-3 and caspase-9.
Acknowledgments
We thank Paul Herring and Loren Field at the Indiana University School of Medicine for helpful comments and reagents.
Footnotes
The abbreviations used are: MLCK, myosin light chain kinase; CaM, calmodulin; RLC, regulatory light chain; DAPK, death-associated protein kinase; TNF, tumor necrosis factor; kb, kilobase(s); PARP, poly(A)DP-ribose polymerase; Dox, doxycycline; PAGE, polyacrylamide gel electrophoresis; bp, base pair(s); MOPS, 4-morpholinepropanesulfonic acid; pNA, p-nitroaniline; NF-κB, nuclear factor κB.
Y. Jin and P. J. Gallagher, manuscript in preparation.
This work was supported by American Heart Association Grant GIA 95009230 (to P. J. G.) and National Institutes of Health Grants RO1HL54118 (to P. J. G.) and RO1HL45788 (to R. B. W).
References
- 1.Anderson P. Microbiol Mol Biol Rev. 1997;61:33–46. doi: 10.1128/mmbr.61.1.33-46.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Utz PJ, Anderson P. Cell Death Differ. 2000;7:589, 602. doi: 10.1038/sj.cdd.4400696. [DOI] [PubMed] [Google Scholar]
- 3.Mills JC, Stone NL, Erhardt J, Pittman RN. J Cell Biol. 1998;140:627, 636. doi: 10.1083/jcb.140.3.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kamm KE, Stull JT. J Biol Chem. 2001;276:4527–4530. doi: 10.1074/jbc.R000028200. [DOI] [PubMed] [Google Scholar]
- 5.Chew TL, Masaracchia RA, Goeckeler ZM, Wysolmerski RB. J Muscle Res Cell Motil. 1998;19:839–854. doi: 10.1023/a:1005417926585. [DOI] [PubMed] [Google Scholar]
- 6.Goeckeler ZM, Wysolmerski RB. J Cell Biol. 1995;130:613, 627. doi: 10.1083/jcb.130.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Eyk JE, Arrell DK, Foster DB, Strauss JD, Heinonen TY, Furmaniak-Kazmierczak E, Cote GP, Mak AS. J Biol Chem. 1998;273:23433–23439. doi: 10.1074/jbc.273.36.23433. [DOI] [PubMed] [Google Scholar]
- 8.Jin Y, Atkinson SJ, Marrs JA, Gallagher PJ. J Biol Chem. 2001;276:30342–30349. doi: 10.1074/jbc.M102404200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deiss LP, Feinstein E, Berissi H, Cohen O, Kimchi A. Genes Dev. 1995;9:15, 30. doi: 10.1101/gad.9.1.15. [DOI] [PubMed] [Google Scholar]
- 10.Cohen O, Feinstein E, Kimchi A. EMBO J. 1997;16:998, 1008. doi: 10.1093/emboj/16.5.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kogel D, Prehn JH, Scheidtmann KH. Bioessays. 2001;23:352, 358. doi: 10.1002/bies.1050. [DOI] [PubMed] [Google Scholar]
- 12.Kawai T, Matsumoto M, Takeda K, Sanjo H, Akira S. Mol Cell Biol. 1998;18:1642–1651. doi: 10.1128/mcb.18.3.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kogel D, Plottner O, Landsberg G, Christian S, Scheidtmann KH. Oncogene. 1998;17:2645, 2654. doi: 10.1038/sj.onc.1202204. [DOI] [PubMed] [Google Scholar]
- 14.Sanjo H, Kawai T, Akira S. J Biol Chem. 1998;273:29066–29071. doi: 10.1074/jbc.273.44.29066. [DOI] [PubMed] [Google Scholar]
- 15.Kawai T, Nomura F, Hoshino K, Copeland NG, Gilbert DJ, Jenkins NA, Akira S. Oncogene. 1999;18:3471, 3480. doi: 10.1038/sj.onc.1202701. [DOI] [PubMed] [Google Scholar]
- 16.Inbal B, Shani G, Cohen O, Kissil JL, Kimchi A. Mol Cell Biol. 2000;20:1044–1054. doi: 10.1128/mcb.20.3.1044-1054.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murata-Hori M, Suizu F, Iwasaki T, Kikuchi A, Hosoya H. FEBS Lett. 1999;451:81, 84. doi: 10.1016/s0014-5793(99)00550-5. [DOI] [PubMed] [Google Scholar]
- 18.Page G, Kogel D, Rangnekar V, Scheidtmann KH. Oncogene. 1999;18:7265, 7273. doi: 10.1038/sj.onc.1203170. [DOI] [PubMed] [Google Scholar]
- 19.Raveh T, Droguett G, Horwitz MS, DePinho RA, Kimchi A. Nat Cell Biol. 2001;3:1–7. doi: 10.1038/35050500. [DOI] [PubMed] [Google Scholar]
- 20.Herring BP, Dixon S, Gallagher PJ. Am J Physiol. 2000;279:C1656–C1664. doi: 10.1152/ajpcell.2000.279.5.C1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barth AI, Pollack AL, Altschuler Y, Mostov KE, Nelson WJ. J Cell Biol. 1997;136:693, 706. doi: 10.1083/jcb.136.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaush CR, Hard WL, Smith TF. Proc Soc Exp Biol Med. 1966;122:931–935. doi: 10.3181/00379727-122-31293. [DOI] [PubMed] [Google Scholar]
- 23.Gallagher PJ, Jin Y, Killough G, Blue EK, Lindner V. Am J Physiol. 2000;279:C1078–C1087. doi: 10.1152/ajpcell.2000.279.4.C1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gallagher PJ, Herring BP, Trafny A, Sowadski J, Stull JT. J Biol Chem. 1993;268:26578–26582. [PMC free article] [PubMed] [Google Scholar]
- 25.Gallagher PJ, Herring BP, Griffin SA, Stull JT. J Biol Chem. 1991;266:23936–23944. [PMC free article] [PubMed] [Google Scholar]
- 26.Gallagher PJ, Herring BP, Stull JT. J Muscle Res Cell Motil. 1997;18:1–16. doi: 10.1023/a:1018616814417. [DOI] [PubMed] [Google Scholar]
- 27.Bresnick AR. Curr Opin Cell Biol. 1999;11:26–33. doi: 10.1016/s0955-0674(99)80004-0. [DOI] [PubMed] [Google Scholar]
- 28.Boldin MP, Mett IL, Varfolomeev EE, Chumakov I, Shemer-Avni Y, Camonis JH, Wallach D. J Biol Chem. 1995;270:387–391. doi: 10.1074/jbc.270.1.387. [DOI] [PubMed] [Google Scholar]
- 29.Hsu H, Xiong J, Goeddel DV. Cell. 1995;81:495, 504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 30.Stanger BZ, Leder P, Lee TH, Kim E, Seed B. Cell. 1995;81:513, 523. doi: 10.1016/0092-8674(95)90072-1. [DOI] [PubMed] [Google Scholar]
- 31.Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. Cell. 1995;81:505, 512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 32.Haupt Y, Rowan S, Shaulian E, Vousden KH, Oren M. Genes Dev. 1995;9:2170, 2183. doi: 10.1101/gad.9.17.2170. [DOI] [PubMed] [Google Scholar]
- 33.Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Cell. 1995;81:801, 809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 34.Raveh T, Berissi H, Eisenstein M, Spivak T, Kimchi A. Proc Natl Acad Sci U S A. 2000;97:1572–1577. doi: 10.1073/pnas.020519497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohen O, Inbal B, Kissil JL, Raveh T, Berissi H, Spivak-Kroizaman T, Feinstein E, Kimchi A. J Cell Biol. 1999;146:141, 148. doi: 10.1083/jcb.146.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reed JC. Cell. 1997;91:559, 562. doi: 10.1016/s0092-8674(00)80442-0. [DOI] [PubMed] [Google Scholar]
- 37.Li H, Zhu H, Xu CJ, Yuan J. Cell. 1998;94:491, 501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 38.Gross A, Yin XM, Wang K, Wei MC, Jockel J, Milliman C, Erdjument-Bromage H, Tempst P, Korsmeyer SJ. J Biol Chem. 1999;274:1156–1163. doi: 10.1074/jbc.274.2.1156. [DOI] [PubMed] [Google Scholar]
- 39.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Cell. 1998;94:481, 490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 40.Green DR, Reed JC. Science. 1998;281:1309, 1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 41.Bossy-Wetzel E, Green DR. J Biol Chem. 1999;274:17484–17490. doi: 10.1074/jbc.274.25.17484. [DOI] [PubMed] [Google Scholar]
- 42.Marchenko ND, Zaika A, Moll UM. J Biol Chem. 2000;275:16202–16212. doi: 10.1074/jbc.275.21.16202. [DOI] [PubMed] [Google Scholar]
- 43.Schuler M, Bossy-Wetzel E, Goldstein JC, Fitzgerald P, Green DR. J Biol Chem. 2000;275:7337–7342. doi: 10.1074/jbc.275.10.7337. [DOI] [PubMed] [Google Scholar]