

Abstract
Molecular imaging is a new and evolving field that employs a targeted approach to noninvasively assess biologic processes in vivo. By assessing key elements in specific cellular processes prior to irreversible end-organ damage, molecular tools will allow for earlier detection and intervention, improving management and outcomes associated with cardiovascular diseases. The goal of those working to expand this field is not just to provide diagnostic and prognostic information, but rather to guide an individual’s pharmacological, cell-based, or genetic therapeutic regimen. This article will review molecular imaging tools in the context of our current understanding of biological processes of the myocardium, including angiogenesis, ventricular remodeling, inflammation, and apoptosis. The focus will be on radiotracer-based molecular imaging modalities with an emphasis on clinical application. Though this field is still in its infancy and may not be fully ready for widespread use, molecular imaging of myocardial biology has begun to show promise of clinical utility in acute and chronic ischemia, acute myocardial infarction, congestive heart failure, as well as in more global inflammatory and immune-mediated responses in the heart-like myocarditis and allogeneic cardiac transplant rejection. With continued research and development, molecular imaging promises to be an important tool for the optimization of cardiovascular care.
INTRODUCTION
As basic and preclinical research develops a more complex understanding of the cellular and molecular interactions that take place within the cardiovascular system under both physiologic and pathologic conditions, physician-scientists have an opportunity to design more precise and effective medical therapies. Moreover, in the current climate of health care reform where cost-effectiveness and limited resources demand efficient and effective medical evaluation and treatment, molecular imaging may serve as a critical tool that allows evaluation of the efficacy of current treatment strategies for optimized individual-based health care delivery. This personalized health care will require enough knowledge of the human genome and proteome to model molecular interactions from levels of gene expression to the complex milieu and kinetics of protein expression and post-translational modification. To detect and monitor both the disease and the therapeutic intervention in hopes of optimizing care and minimizing the burden of side effects and invasive injuries (as well as containing cost), the design of new tools to image-specific molecular events must take place that allows accurate in vivo assessment and risk stratification of the patient.
Molecular imaging is defined as the application of imaging using biologically targeted markers, and it was born out of the recognition that the high sensitivity of radionuclide imaging techniques could allow for the detection of specific biological processes.1 It is important that the molecular target identified adequately represents the process being studied. Moreover, the target must then lend itself to a readily synthesizable probe(s) that bind with a high degree of specificity. From there, an imaging technology that provides the best combination of sensitivity and both spatial and temporal resolution to identify and localize the probe within the target organ system must be universally available and economically feasible. Molecular imaging approaches are currently being developed for most of today’s expanding range of imaging modalities; including nuclear, magnetic resonance, X-ray computed tomography (CT), optical fluorescence, bioluminescence, and ultrasound.2 Though each modality carries strengths and weaknesses, it is likely that the practical limitations of cost and widespread availability will determine which modalities become adapted for clinical use.
Single photon emission computed tomography (SPECT) and positron emission tomography (PET) are radionuclide imaging techniques that have been used for over three decades. Radiolabeling has the unique advantage of augmenting the signal intensity of miniscule amounts of any substance of interest. For example, PET can detect picomolar and nanomolar concentrations of a molecule of interest.2 Though SPECT offers the advantage of decreased cost and widespread availability, PET offers the advantages of increased sensitivity in conjunction with the improved ability to quantitate as well as repetitively image using tracers with ultra short half-lives. In the past, nuclear imaging modalities have been limited by attenuation artifacts from soft tissue and partial volume effects. More recent systems combining CT imaging with either SPECT or PET have allowed for attenuation correction, leading to improved image quantification, and registration of functional data with anatomical structure.
This article will review several important areas of active cardiovascular molecular imaging research, including angiogenesis, ventricular remodeling, inflammation, and apoptosis, with a focus on radionuclide imaging techniques. Key molecular events or signaling proteins involved in each process were identified through basic and preclinical research. It will be noted that some molecular signals overlap between biological processes, which underscores the significance of basic research in furthering our understanding of the complex setting in which any molecular event takes place.
MYOCARDIAL PROCESSES
Moving from Insult to Injury to the Failing Heart
Moving from the healthy to failing heart, a number of processes are involved. Some of these processes, as mentioned, overlap between different stages of disease progression. Others are present only at certain times. Figure 1 illustrates the various processes as one move from the early insult of atherosclerotic initiation to the failing heart. The biological processes of angiogenesis, cell death, inflammation, and ventricular remodeling are triggered throughout the progression of disease. The specific biomarkers that will be discussed in detail later are highlighted. Early, myocardial disease starts with an insult, the focus for this article will be atherosclerosis and the myocardial consequences of this disease process. Atherosclerosis is a chronic inflammatory disease which leads to the progression of lipid laden vascular plaques as an initial vascular insult, ultimately leading to myocardial ischemic injury.3,4 Monocyte adhesion and transendothelial migration result in activation and differentiation, with amplifying production of cytokines/chemokines, as well as foam cell development when these subendothelial macrophages endocytose oxidized lipid. The progression of atherosclerotic disease can lead to the development of both acute and chronic ischemia in areas of myocardium secondary to poor perfusion through narrowed blood vessels. Subsequent response signals may stimulate angiogenic and arteriogenic responses in order to restore perfusion. Moreover, rupture of cholesterol plaques and subsequent thrombosis can lead to myocardial infarction, tissue injury, and cell death. As myocardial tissue moves from hypoxic stress to injury to subsequent cell death, there is a wide spectrum of dynamic changes that take place including apoptosis and eventually necrosis. Inflammatory reactions are stimulated both locally and globally throughout the heart to remodel the tissue architecture in an attempt to adapt to irreversible tissue damage. Fibrosis and scarring set in and along the way neurohormonal signaling pathways are upregulated in adjustment to the failing heart.
Figure 1.

Schematic spectrum from insult to heart failure using atherosclerotic disease as an example. As the myocardium moves through different phases of the spectrum, various biological processes like angiogenesis, inflammation, cell death, and ventricular remodeling occur. Each process is marked by molecular events that serve as targets for imaging. There is overlap temporally as well as molecularly between the processes underscoring the importance of the pathological circumstances in which an event takes place. The processes highlighted will be discussed in more detail throughout this article.
Angiogenesis
Angiogenesis is defined as the process of sprouting new capillaries from preexisting microvessels.5 There is a great deal of interest in understanding the processes of angiogenesis in order to design therapeutic treatments that allow revascularization through augmentation of the angiogenic response.6–11 This angiogenic process often occurs in association with arteriogenesis, which represents a remodeling of larger, pre-existing vascular channels or collateral vessels feeding the new microvascular network. The goal for any myocardial revascularization strategy would be to initiate angiogenesis and arteriogenesis in manner which improves tissue perfusion.12–14
Angiogenesis and arteriogenesis are stimulated by external processes such as ischemia, hypoxia, inflammation, and shear stress. Hypoxia is a well-established stimulator of angiogenesis.15 Hypoxic conditions such as myocardial ischemia from atherosclerotic disease or acute myocardial infarction result in upregulation of the transcriptional activator hypoxia-inducible factor 1 (HIF-1).16 This upregulation of HIF-1 protein leads to the transcription of a number of hypoxia-inducible genes, including the key angiogenic mediators; vascular endothelial growth factor (VEGF), platlet-derived growth factor (PDGF), TGF-β, and the VEGF receptors, Flt-1 (VEGFR-1) and FLK-1 (VEGFR-2).15,17–20 These angiogenic mediators signal the key players, endothelial cells, smooth muscle cells, blood derived macrophages, and circulating stem cells, which all play distinctive roles in the angiogenic process. There is the careful interaction of these cells with each other as well as within the tissue of extracellular matrix (ECM) proteins. The process itself consists of a series of endothelial cell responses to angiogenic stimulation such as degradation of ECM, budding from parent vessels, proliferation, migration, tube formation, and ultimately maturation and maintenance of the new vessel.21
During angiogenesis, integrins, a family of heterodimeric (αβ) cell-surface receptors that mediate divalent, cation-dependent, cell-cell and cell-matrix adhesion and signaling through tightly regulated interactions with their respective ligands, are upregulated to coordinated cellular responses.22 Endothelial cells make use of integrins to adhere to one another and the ECM to construct and extend new vessels. Peak expression of one integrin in particular, αvβ3 integrin, has been shown to occur 12–24 hours after initiation of angiogenesis with FGF-2.23 Integrins are capable of mediating an array of cellular processes, including cell adhesion, migration, proliferation, differentiation, and survival via a number of signal transduction pathways.24,25 Activation of c-Jun NH2-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK) may lead to endothelial cell-induced remodeling of the ECM in response to mechanical stimuli. Specifically, the endothelial cell integrin, αvβ3, allows cells to interact with the ECM in a way that aids in endothelial cell migration.26 Through outside-in signaling, integrin αvβ3 also plays a critical roll in the survival of cells undergoing angiogenesis.23
Given their significance in angiogenesis, VEGF receptors and integrin αvβ3 represent ideal molecular imaging targets for imaging. Much of the current data in the field stems from studies that make use of these molecules.
APPROACHES TO RADIONUCLIDE IMAGING OF ANGIOGENESIS
VEGF Receptors
VEGF receptors have been targeted for imaging techniques in models of ischemia-induced angiogenesis. Radiolabeled-VEGF121 has been used to affectively identify angiogenesis in a rabbit model of hindlimb ischemia.27 In this study, KDR and Flt-1 receptor expression was increased in the immunohistochemistry analysis of the skeletal muscle, supporting the theoretical hypoxic-driven angiogenic response. Unfortunately, the biodistribution was 20-fold higher levels in critical organs (liver, kidneys) compared with ischemic limb, rasing a practical concern regarding clinical application.
A PET tracer, 64Cu-6DOTA-VEGF121, was recently developed for imaging angiogenesis in a rat model of myocardial infarction (Figure 2).28 Rats underwent ligation of the left coronary artery and subsequent PET imaging at various time points post-myocardial infarction. Co-registration of images was carried out using CT, and the zone of infarct was demonstrated using 18F-FDG uptake. The study demonstrated that 64Cu-6DOTA-VEGF121-specific signal was present in the infarct region and peaked on day 3 consistent with the changing levels of VEGFR expression in the tissue as analyzed by immunofluorescence microscopy. Similar studies have been carried out in a murine model of hindlimb ischemia-induced angiogenesis utilizing 64Cu-6DOTA-VEGF121.29
Figure 2.
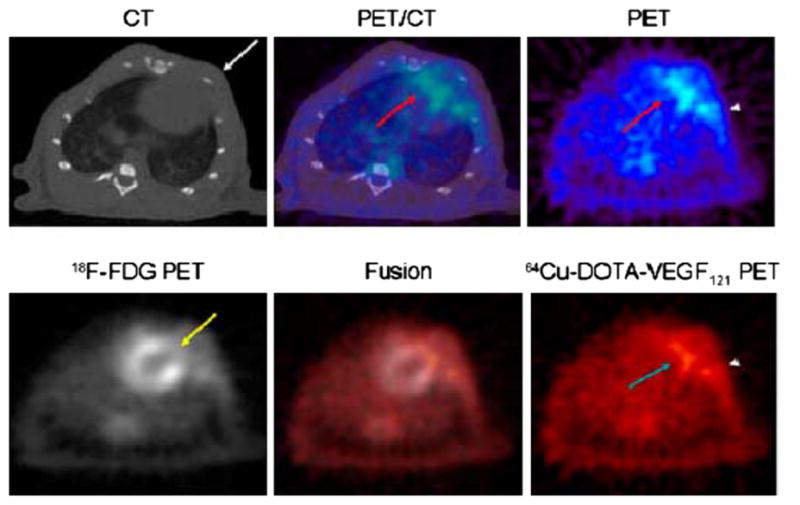
Myocardial 64Cu-DOTA-VEGF121 and 18F-FDG PET-CT imaging after MI. Upper images demonstrate co-registered images of microCT (left), PET (right), and fused PET/CT image (center) in a representative animal after myocardial infarction. The 64Cu-DOTA-VEGF121 signal is detected by PET in the anterolateral myocardium (PET and fused images, red arrow). Intercostal muscle layer is designated on microCT image with a white arrow. There is some increased uptake in area of surgical wound (PET image, arrowhead). Lower images demonstrate 64Cu-DOTA-VEGF121 (left), 18F-FDG (right), and 64Cu-DOTAVEGF121/18F-FDG fused image (middle). 18 F-FDG scan shows that coronary artery ligation resulted in development of a scar by lack of 18F-FDG uptake (yellow arrow) and that uptake of 64Cu-DOTA-VEGF121 occurs in the region of that scar (turquoise arrow). Fusion of both scans results in complementation of 18F-FDG and 64 Cu-DOTA-VEGF121 signals. Again, increased uptake in area of surgical wound is designated with an arrowhead (Reprinted with permission from Rodriguez-Porcel et al.28).
Given mixed clinical findings regarding clinical studies attempting therapeutic angiogenesis, there is a need for developing a reporter system in tandem with therapeutic delivery to assess treatment efficiency.6–11 Along those lines, a cardiac-specific reporter has been developed as a gene expression system for use in rats with microPET imaging.30 Briefly, the system involves adenovirus delivery of mutated thymidine kinase under the control of a cytomegalovirus promoter-driving expression in myocardial cells. The reporter probe is 18F-FHBG which crosses the myocardial membrane and gets phosphorylated by the thymidine kinase. Phosphorylation traps the 18F-FHGB in the myocardium for subsequent microPET imaging. Early studies revealed that the localized site of the mutated thymidine kinase, HSV1-sr39tk, corresponded closely with that defined by postmortem autoradiography, histology, and immunohistochemistry.31 Other studies have demonstrated the feasibility of utilizing a similar reporter system in pigs using a clinical PET scanner.32 The reporter system was then linked to VEGF to assess feasibility of developing an approach that links therapy and imaging.33 Early experiments with rat embryonic cardiomyocytes revealed a strong correlation that both the mutated thymidine kinase and VEGF were expressed in the same cells. Further studies involved injection of the VEGF/thymidine kinase reporter system in models of ischemia. Using microPET, cardiac transgene expression was assessed and the in vivo imaging correlated well with ex vivo tissue studies for gamma counting, thymidine kinase activity, and VEGF levels. There appeared to be increased capillaries and small blood vessels in the VEGF-treated myocardium, however, there was no improvement in perfusion assessed by nitrogen-13 ammonia imaging or metabolism assessed with 18F-FDG imaging. Though more preclinical work is needed, these studies suggest that a reporter system can be developed to help visualize the effectiveness of delivering VEGF gene therapies for stimulation of angiogenesis.
Integrin αvβ3
Imaging angiogenic vessels through targeting of αvβ3 integrin was first proposed through a series of magnetic resonance imaging studies using a monoclonal antibody to integrin αvβ3 tagged with a paramagnetic contrast agent.34 The studies were complicated by poor clearance of the tracer from the blood pool. Later studies made use of a number of αvβ3 antagonists that were radiolabeled.35,36
An 111In-labeled quinolone (111In-RP748) revealed high affinity and selectivity for αvβ3 integrin in adhesion assays.37 Subsequent studies using a cy3-labeled homolog of 111In-RP748, demonstrated preferential binding to activated αvβ3 integrins on endothelial cells in culture, with localization to cell-cell contact points.38 The first imaging of ischemia-induced myocardial angiogenesis using 111In-RP748 was carried out in rat and canine models of infarction (Figure 3).39 111In-RP748 demonstrated favorable kinetics for in vivo SPECT imaging ischemia-induced angiogenesis of the heart. Relative 111In-RP748 activity was markedly increased in the infarcted region acutely and persisted for at least 3 weeks post-reperfusion.39,40 Targeted imaging with 111In-RP748 has demonstrated integrin αvβ3 activation early postinfarction, suggesting a role for this technique in early detection of angiogenesis as well as for detection of chronic ischemia.40 The recent imaging of αvβ3 integrin by the PET imaging tracer, 18F-Galakto-RGD, in a 35-year-old patient with a transmural myocardial infarction 2 weeks prior demonstrates the feasibility of detecting angiogenesis in the myocardium in humans (Figure 4).41
Figure 3.
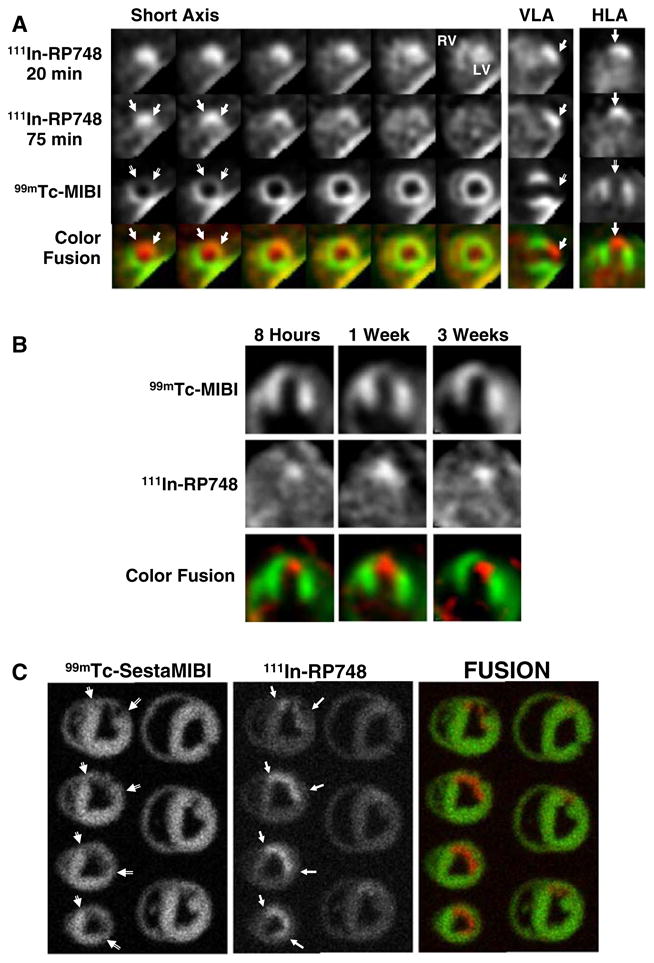
In vivo and ex vivo 111In-RP748 and 99mTc-sestamibi (99mTc-MIBI) images from dogs with chronic infarction. Serial in vivo 111In-RP748 SPECT short-axis, vertical long axis (VLA), and horizontal long axis (HLA) images in a dog 3 wks post-LAD infarction at 20 minutes, and 75 minutes postinjection in standard format (Figure 2A). 111In-RP748 SPECT images were registered with 99mTc-MIBI perfusion images (third row). The 75 minutes 111In-RP748 SPECT images were colored red and fused with MIBI images (green) to better demonstrate localization of 111In-RP748 activity within the heart (color fusion, bottom row). Right ventricular (RV) and left ventricular (LV) blood pool activity are seen at 20 minutes. Filled arrows indicate region of increased 111In-RP748 uptake in anterior wall. This corresponds to the anteroapical 99mTc-sestamibi perfusion defect (open arrow). Sequential 99mTc-sestamibi (top row) and 111In-RP748 in vivo SPECT HLA images at 90 minutes postinjection (middle row) from a dog at 8 hours (Acute), and 1 and 3 weeks post-LAD infarction (Figure 2B). Increased myocardial 111In-RP748 uptake is seen in anteroapical wall at all three time points, although appears to be maximal at 1 week postinfarction. Color fusion 99mTc-MIBI (green) and 111In-RP748 (red) images (bottom row) demonstrate 111In-RP748 uptake within 99mTc-MIBI perfusion defect. Ex vivo 99mTc-sestamibi (left) and 111In-RP748 (center) images of myocardial slices from a dog 3 weeks post-LAD occlusion, with color fusion image on right (Figure 2C). Short-axis slices are oriented with anterior wall on top, RV on left. Open arrows indicate anterior location of nontransmural perfusion defect region, and filled arrows indicate corresponds area of increased 111In-RP748 uptake (Reprinted with permission from Meoli et al.39).
Figure 4.

Cine-MRI (CMR) with delayed enhancement (arrows) extending from the anterior wall to the apical region in the four-chamber (A) and two-chamber (D) view. (B, E) Identically reproduced location and geometry with severely reduced myocardial blood flow using 13N-ammonia, corresponding to the regions of delayed enhancement by CMR (arrows). (C, F) Focal 18F-RGD signal co-localized to the infarcted area. This signal may reflect angiogenesis within the healing area (arrows). (G, I) Polar map (I: 3D) of myocardial blood flow assessed by 13N-ammonia indicating severely reduced flow in the distal LAD-perfused region. (H, J) Co-localized 18F-RGD signal corresponding to the regions of severely reduced 13N-ammonia flow signal, reflecting the extent the of αvβ3 expression within the infarcted area (Reprinted with permission from Makowski et al.41).
A new biodegradable positron emitting nanoprobe targeted at αvβ3 integrin has been designed for noninvasive imaging of angiogenesis with a PET-based system (Figure 5).42 The nanoprobe has a core-shell architecture that allows radiolabeling with radiohalogens, which are linked to the core and to protect them from dehalogenation. The terminal ends of the outer shell are covalently linked to cyclic-RGD peptides to confer specificity to αvβ3 integrin. This nanoprobe revealed enhanced binding of αvβ3 integrin using in vitro and cellular assays. The nanoprobe demonstrated favorable biodistribution when compared with untargeted probe in rodents with a slight increase in uptake in phagocytic rich organs like the spleen, liver, and kidneys. Using a 76Br-labeled derivative of the nanoprobe, the investigators targeted αvβ3 integrin in a murine model of hindlimb ischemia. In vivo PET imaging revealed specificity of the probe to the ischemic limb, using the nonischemic limb and a version of the probe lacking the cyclic-RGD peptides for controls. Ex vivo imaging and histological analysis allowed for quantification of the radioactivity leading to further association of the probe to areas of increased αvβ3 integrin expression and new vessel formation. The application of nanotechnology to angiogenesis imaging is an exciting new area of research; however, further studies will be required to assess the feasibility applying this system to the clinical arena.
Figure 5.

Noninvasive PET/CT images of angiogenesis induced by hindlimb ischemia in a murine model. Molecular models of the dendritic nanoprobes structures are shown in the top right inset in each set of images. Nontargeted dendritic nanoprobe (left set of images). Uptake of αvβ3-targeted dendritic nanoprobes (right set of images) was higher in ischemic hindlimb (right limbs) as compared with control hindlimb (left limbs) (Reprinted with permission from Almutairi et al.42).
INFLAMMATION
Myocardial inflammation is a broad term that characterizes a number of interactions myocardial cells, the ECM, vascular cells, and immigrant cells like lymphocytes, neutrophils, and macrophages. The ultimate goal of the inflammatory reaction appears to be removal of damaging or harmful substances and subsequent healing. As such, it is understandable that inflammation plays an important role in many cardiovascular processes including myocardial infarction, reperfusion injury, angiogenesis, apoptosis, cardiac allograft rejection, and myocarditis.
Initial attempts at imaging inflammatory processes involved radiolabeling leukocytes with 99mTc or 111In. These techniques require removal of blood and in vitro labeling and generated a number of concerns, including nonspecific activation of the labeled cells that interfered with specificity of targeting.43,44 18F-labeled deoxyglucose (FDG) PET imaging takes advantage of increased metabolic activity of inflammatory cells, but changes in glucose uptake can be associated with other tissues and disease processes including tumors, again leading to nonspecificity.45–47 Thus, there is a tremendous amount of interest in developing more specific noninvasive imaging techniques to detect inflammation in myocardium.
APPROACHES TO RADIONUCLIDE IMAGING OF INFLAMMATION
Antimyosin Antibodies
Injury to myocytes in the setting of inflammation leads to the disruption of cellular membranes and the release of myosin heavy chain. Early attempts to visualize myosin in inflammatory reactions involving myocyte damage during myocardial infarction utilized 111In-labeled antimyosin antibodies.48 99mTc-labeled monoclonal antibody fragments have been used to quantitate the degree of myosin exposure in patients in the setting of acute myocardial infarction and correlate it with necrosis.49 Inflammation associated with myocarditis were also carried out using 111In-antimyosin antibodies.50,51 Though these initial studies showed promise, the background antibody binding to necrotic debris in the cell was high, yielding a very low specificity (25%–50%).52
Antitenascin-C Antibody
A monoclonal antibody against tenascin-C, an ECM protein involved in wound healing and inflammation, has been identified as another potential imaging agent. In rodent models of myocarditis, 111In-labeled antitenascin-C localizes to the sites of myocardial inflammation.53 Using a dual isotope SPECT approach with 111In-antitenascin-C and 99mTc-sestamibi, the antibody localized to the injured wall by in vivo imaging. 111In-antitenascin-C monoclonal antibody fragments have been used in a dual isotope SPECT imaging approach with 99mTc-MIBI to study in vivo expression of tenascin-C in a rodent model of myocardial infarction (Figure 6)54 In this study 111In-antitenascin-C activity was associated with the 99mTc-MIBI perfusion defect, supporting this agent as a possible marker for inflammation associated with myocardial infarction.
Figure 6.
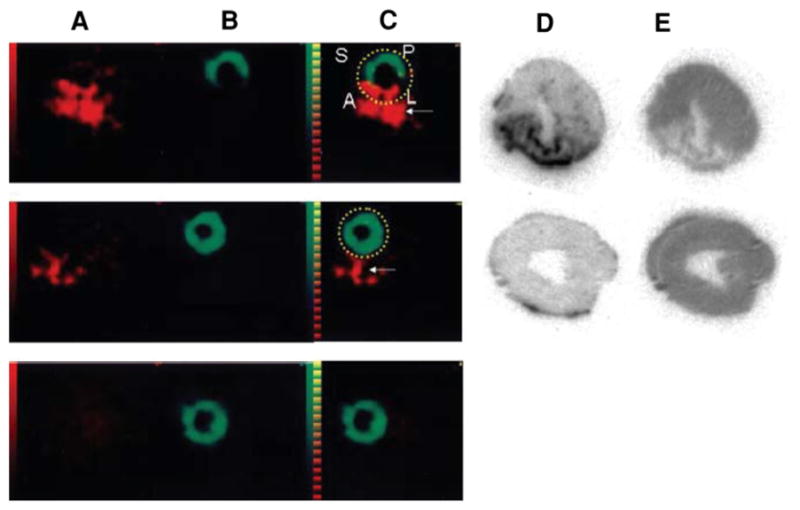
Comparison of SPECT imaging between 111In-anti-TNC-Fab and 99mTc-MIBI. Transverse dual isotope SPECT images (A–C) and autoradiographies of the same rats (D, E). The uptake of 111In-anti-TNC-Fab (red in A, C, D) and 99mTc-MIBI (green in B, C, E) in acute MI heart (upper panels), in sham-operated heart (middle panels), and in normal rat heart (lower panels). A indicates anterior left ventricular wall; L, lateral left ventricular wall; P, posterior left ventricular wall; and S, septal wall. Red color indicates the uptake of 111In-anti-TNC-Fab and green color, the uptake of 99mTc-MIBI. Yellow broken lines circle myocardium. White arrows indicate sutured incision of the left intercostal space just below the myocardium (Reprinted with permission from Odaka et al.54).
LTB4 Receptor
LTB4 is a lipid mediator synthesized from arachidonic acid and secreted by neutrophils, macrophages, and endothelial cells as a potent chemotactic agent.55,56 The LTB4 receptor can be found on neutrophils and signaling through this receptor stimulates endothelial adhesion and superoxide production. A radiolabeled LTB4 receptor antagonist, 99mTc-RP517, was developed for in vivo imaging of inflammation.57,58 99mTc-RP517 localized to sites of inflammation induced by S. aureas and E. coli infection, and chemical (phorbolester)-induced bowel inflammation.
When prepared with human peripheral whole blood in vitro, fluorinated RP517 localized to neutrophils by fluorescence activated cell sorter (FACs) analysis.59 99mTc-RP517 was injected into open chest dogs before occlusion and reperfusion in a canine model of postischemic myocardial inflammation. There was an inverse relationship between radiotracer uptake and occlusion flow, suggesting localization of the imaging agent to the site of ischemic inflammation (Figure 7). Ex vivo segment analysis revealed that 99mTc-RP517 correlated with the neutrophil enzyme, myeloperoxidase. One concern regarding the application of 99mTc-RP517 is the lipophilic nature of the molecule, resulting in high hepatobiliary clearance and thus large amounts of gastrointestinal uptake. To overcome this, alternative constructs of the LTB4 antagonist are currently being evaluated.60
Figure 7.
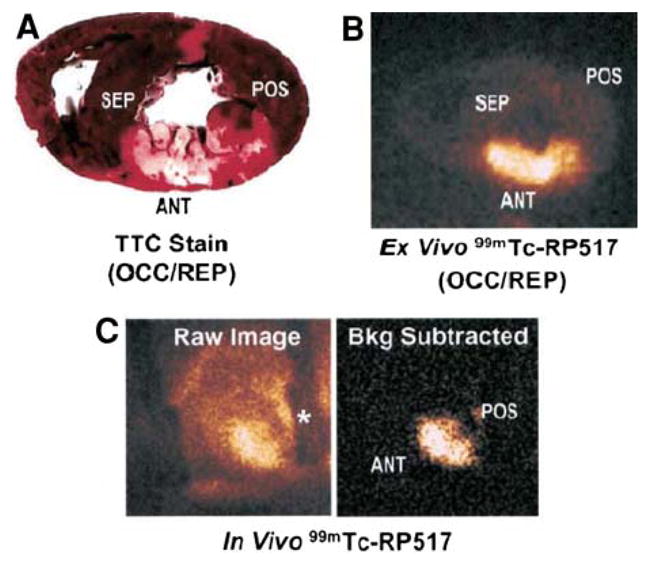
Imaging ischemic inflammation with the LTB4 receptor antagonist, RP517. TTC-stained heart slice (A) and ex vivo 99mTc-RP517 image (B) of the same heart slice. (C) Raw (left) and background subtracted (right) in vivo 99mTc-RP517 images acquired from a dog 60 minutes after reperfusion. Background subtraction was performed to eliminate the surgically related tracer uptake in the field of view. The shadow on the raw image denoted by an asterisk is the metal rib spreader. Note that focal 99mTc-RP517 uptake was readily observed in the inflamed anteroseptal region of the heart on both ex vivo and in vivo images. Tracer uptake was negligible in the normal, posterior wall. (Reprinted with permission from Riou et al.59).
CELL DEATH
Apoptosis is the physiological process of programmed cell death, whereby organisms selectively target cells to be eliminated when they are no longer needed. The cardiovascular pathologies of cardiomyopathy, heart failure, myocarditis, and myocardial infarction are associated with increased levels of apoptosis, particularly in the myocyte. There is a subset of cell death that occurs as an outcome of these pathological processes considered to be outside of programmed cellular mechanisms termed necrosis. A recent study evaluating a role of apoptosis and necrosis in the setting of acute myocardial infarction revealed a potential therapeutic role for cyclosporine.61 The intervention is hypothesized to minimize peri-infarct, reperfusion-related cell death that takes place in the setting of revascularization. It is estimated that 30% of cardiomyocytes in the injured myocardium become apoptotic as a result of ischemia reperfusion injury, and animal models of acute infarction demonstrate that up to 50% of the final size of the infarct can be related to lethal reperfusion injury.62,63 Other animal studies demonstrate that inhibition of apoptosis with caspase-inhibitors is cardioprotective.64–66 There is also data that suggests early apoptosis may be the pathological substrate leading from ischemia to necrosis.67 An ability to assess cell death anywhere along the spectrum of apoptosis to necrosis would allow investigators to fine tune a therapeutic regimen and optimize clinical outcome.
Depending on the initiating signals, there are two major pathways for cell death; intrinsic and extrinsic.68 The intrinsic pathway is generated from within the cell through DNA damage, mitochondrial signals, and oncogene activation, leading to activation of caspase enzymes. The extrinsic pathway is initiated through extracellular signals that target cell membrane receptors like Fas, a death receptor. The culmination of this event through either pathway is the activation of a key effector, caspase-3.69 Soon after the activation of caspase-3, the energy-dependent asymmetric distribution of phospholipids that enables the definition of various subregions within the lipid bilayer of cell membranes is lost, leading to increased phosphatidyl serine (PS) on the outer cell membrane.70 The exposure of PS on the surface of the cell makes it a target for binding the protein, annexin V.70–72
As myocardial ischemia or infarction persists, cells move from early apoptotic signals to complete necrosis. Breakdown of mitochondrial respiration and loss membrane potential lead to the accumulation of calcium in the mitochondria of infarcted or severely injured myocardium.73,74 With loss of membrane potential cellular structures also begin to dissipate. Positively charged histones and other organelle proteins are exposed from the protection of their membrane barriers. These changes in early necrotic tissue have been utilized for imaging techniques that seek to identify early necrosis in acute myocardial infarctions and are discussed in more detail below.
APPROACHES TO RADIONUCLIDE IMAGING IN APOPTOSIS AND NECROSIS
Annexin V
99mTc-labeled annexin A5 was utilized for imaging the distribution of cells expressing PS noninvasively. Radiolabeling involved derivatization of annexin A5 with hydrazinonicotinamine (HYNIC), which binds to reduced 99mTc.75 The initial studies were carried out in mice with fulminant hepatic apoptosis through the injection of an anti-Fas antibody, which initiates an apoptotic cascade, particularly in hepatocytes.76
99mTc-labeled annexin V was subsequently utilized in humans to detect in vivo cell death in patients presenting with myocardial infarction (Figure 8).77 Patients presenting with their first myocardial infarction within 6 hours of symptom-onset underwent standard revascularization with percutaneous intervention. Within 2 hours of revascularization, SPECT imaging was performed utilizing 99mTc-labeled annexin V. This was followed by perfusion imaging 6–8 weeks after discharge using 99mTc-sestamibi. Regional retention of 99mTc-labeled annexin V correlated with the perfusion defect identified 6–8 weeks after discharge, providing a proof of concept that annexin-V imaging can be utilized for noninvasive detection of myocardial cell death.
Figure 8.
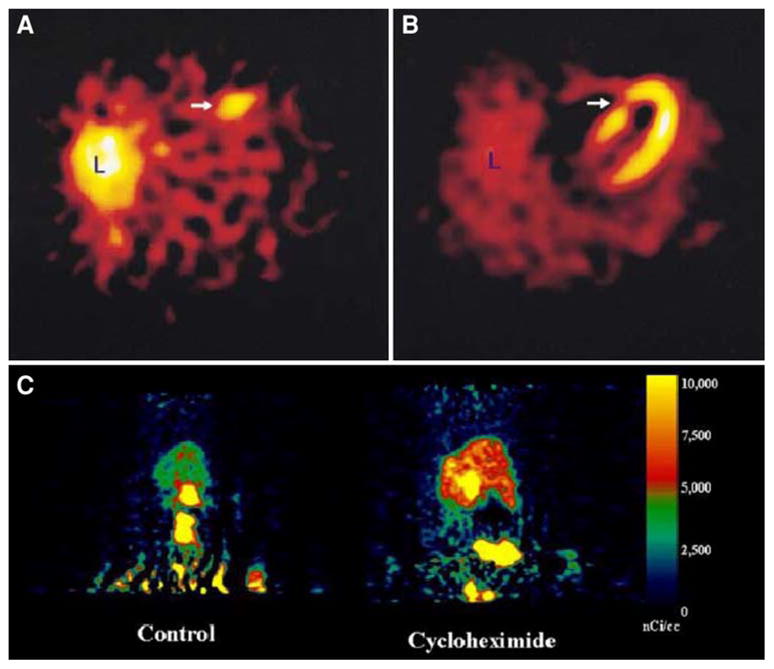
In vivo imaging of apoptosis. (A, B) (upper panels): Transverse tomographic images of acute anteroseptal infarction in a patient. (A) Arrow shows increased 99mTc-labeled annexin-V uptake in the anteroseptal region 22 hours after reperfusion. (B) Perfusion scintigraphy with sestamibi 6–8 weeks after discharge shows an irreversible perfusion defect which coincides with the area of increased 99mTc-labeled annexin-V uptake (arrow). (Reprinted with permission from Hofstra et al.77). (C) (Lower panel): Whole-body MicroPET images of caspase-3 specific inhibitor, 18F-WC-II-89, distribution in a control rat (left) and cycloheximide-treated rat (right). Images were summed from 10 to 60 minutes after intravenous injection of approximately 150 μCi of 18F-WC-II-89 (Reprinted with permission from Zhou et al.83).
Heart transplant rejection is characterized by perivascular and interstitial mononuclear inflammatory infiltrates associated with myocyte apoptosis and necrosis.78 In a study of 18 patients undergoing apoptotic imaging within 1 year of cardiac transplantation, annexin-V retention correlated with the severity of rejection, suggesting a potential role for annexin V imaging as a surrogate for detection of allograft rejection in place of serial biopsies in patients following heart transplantation though more clinical studies are needed.79
Myocarditis is another pathological condition where apoptosis is known to occur.80 In a rat model of auto-immune myocarditis, 99mTc-labeled annexin V retention corresponded to histological TUNEL staining for areas of myocardial apoptosis. Interestingly, 99mTc-labeled annexin V positive areas could be differentiated from areas of inflammation identified by 14C-labeled deoxyglucose. This suggests that one could potentially differentiate between inflammation and active apoptosis with dual isotope molecular imaging. To date, no studies have attempted to employ this technique in conjunction with FDG-PET in human cases of myocarditis. Along similar line, a recent study was carried out in rat model of cardiac ischemia-reperfusion in which dual isotope imaging using 99mTc-labeled annexin V and 111In-labeled antimyosin antibodies to map a temporal relationship between membrane PS disruption and myosin exposure.81 It suggested that PS exposure occurs very early within 20 minutes of ischemia; however, membrane disruption to the point of myosin exposure occurred 3–4 hours out from initial injury. More studies like this will be needed in order to fully understand the relationship of molecular events not only just with regard to the pathologic process, but also with respect to how the different biological processes influence each other.
Caspase Inhibitors
Because PS can be exposed on the surface of cells in physiologic conditions other than apoptosis, there is interest in developing more specific apoptosis tracers. Recently, caspase-3 inhibitors have been synthesized and labeled with 18F as potential PET tracers for in vivo imaging of apoptosis (Figure 8).82,83 These caspase-3 targeted tracers have shown favorable biodistribution and clearance. MicroPET imaging in a murine model of hepatic apoptosis has shown specificity of the tracer to the liver; however, further analysis in cardiovascular models will be necessary to determine feasibility of utilizing this new class of tracers for cardiac applications in human.
Pyrophosphate and Glucarate
The in vivo noninvasive detection of myocardial infarction will allow for early diagnosis and treatment in patients when electrocardiographic changes may not be evident or when biomarkers may not distinguish between ischemic injury associated with acute plaque rupture vs unstable angina and demand related ischemia. In the former condition, the early detection of plaque rupture may be treatable with mechanical or pharmacological revascularization, preventing irreversible loss of large areas of myocardium. In addition to visualizing apoptosis, several studies have demonstrated that certain agents allow for the visualization of ongoing myocardial necrosis as a mechanism of identifying acute infarction potentially even in the presence of prior myocardial infarction. 99mTc-labeled pyrophosphate has been shown to bind to areas of necrosis and is thought to bind exposed mitochondrial calcium.73,74 99mTc-pyrophosphate has a moderate degree of sensitivity for acute infarction depending on the presence of Q wave infarction or a non-ST elevation infarction.84 The specificity for acute myocardial infarction is considered to be between 60% and 80%. However, 99mTc-pyrophosphate has not gained widespread clinical use because of its limitation in the detection of early infarction. In addition, there may be persistent positivity beyond the period of acute injury.
99mTc-glucarate imaging provides an alternative to 99mTc-pyrophosphate imaging for the detection of acute infarction.74 99mTc-glucarate enters the necrotic cells by passive diffusion following breakdown of the sarcolemma, and binds to exposed histones in the nucleus of the myocytes. Canine models for ischemia and infarction reveal a high affinity of 99mTc-glucarate for necrotic tissue over ischemic but viable myocardium.85 Moreover, establishment of partial reperfusion did not inhibit the ability to detect acute infarct in canines (Figure 9).86 In a rabbit model of infarction, 99mTc-glucarate did not accumulate in areas of ischemia and could be imaged in areas of infarction as early as 10 minutes post-reperfusion and within 30–60 minutes in nonreperfused zones.87 Initial data in patients revealed that 99mTc-glucarate is able to noninvasively diagnose myocardial infarction in patients presenting with chest pain with a sensitivity that is dependent on the onset of symptoms, specifically within the first 9 hours of symptom-onset.88 99mTc-glucarate also demonstrates rapid blood clearance and good target to background signal and is currently under active investigation as a tool to detect early infarction in a number of clinical trials.
Figure 9.

In vivo serial gamma camera 99mTc-glucarate scintigraphic images of a single heart, acquired at 1, 15, 30, 60, 120, and 240 minutes after tracer injection. A hot spot is clearly visualized involving the lateral wall at 30 minutes, and persists to 240 minutes (arrow). The schematic diagram at the far right shows the location of the right ventricular cavity (RV), septal wall (S), left ventricular cavity (LV), right ventricular free wall (RVW), and left ventricular posterolateral wall (PLW) (Reprinted with permission from Johnson et al.86).
VENTRICULAR REMODELING
Ventricular remodeling is a complex biological process that involve inflammation, angiogenesis, repair, and healing with specific biochemical and structural alterations in the myocardial infarct and peri-infarct regions as well as remote regions.89,90 The process is felt to be one of adaptation to form a scar that allows a degree of mechanical stability. The remodeling process involves several key cell types and structural elements, including myocardial cells, endothelial cells, inflammatory cells, and the ECM. Early in the first weeks after a myocardial infarction, an innate immune response initiates a complex process of wound healing in the necrotic tissue. This process evolves into a more chronic remodeling process that can involve hypertrophy, chamber dilation and, depending on the success of healing or lack thereof, heart failure.90
Matrix metalloproteinases (MMPs) are a family of zinc-containing enzymes that play a key role in ventricular remodeling by degradation of the ECM.91,92 MMPs are tightly regulated on several levels via transcriptional, post-transcriptional, and post-translational mechanisms. With regard to ventricular remodeling, MMPs appear to play an integral role in infarct expansion and left ventricular dilation. Gene deletion of MMPs has been demonstrated to have some cardioprotective effects from ventricular dilation and rupture postinfarct.93 Pharmacologic inhibition of MMPs has also been shown to decrease left ventricular dilation in infarct models.94–96
Factor XIII has been shown to be crucial in organizing the new matrix of the scar by involvement with ECM turnover and regulation of inflammatory cascades.97,98 Mice with decreased levels of factor XIII demonstrate increased ventricular dilation and postinfarct rupture. Patients with infarct rupture were demonstrated to have lower levels of factor XIII in their myocardium. Factor XIII is activated by thrombin and often decreased in the setting of acute myocardial infarction in part because of therapeutic inhibition of thrombin. It has been hypothesized that supplementing Factor XIII activity may have a beneficial role in post infarct remodeling.
A critical system that is locally activated during ventricular remodeling and contributes to the progression to heart failure is the renin-angiotensin system.99,100 As healing and remodeling are unsuccessful in the failing heart, there is increased expression of prorenin, renin, and angiotensin-converting enzyme (ACE). Activation of this system through signaling pathways mediated by the angiotensin II type I receptor (AT1) leads to myocyte hypertrophy, interstitial and perivascular collagen deposition, and myocyte apoptosis.99 Inhibition of this pathway has been demonstrated to reverse the functional abnormalities associated with this negative remodeling.
RADIOTRACER-BASED IMAGING OF LEFT VENTRICULAR REMODELING
MMPs
By radiolabeling molecules that target MMPs like pharmacological inhibitors that specifically bind to the catalytic domain, MMP activation postinfarct can be visualized in vivo (Figure 10).101 Initial studies involved nonimaging techniques with 111In-labeled broad-spectrum MMP inhibitor (RP782), a molecule that selectively targets activated MMPs. This MMP targeted agent demonstrated a favorable biodistribution in a murine model of myocardial infarction. One week after myocardial infarction, 111In-RP782 localized primarily within the infarct region, although a lesser increase in retention was seen in the remote noninfarcted regions of the heart, consistent with global MMP activation and remodeling.
Figure 10.

Imaging of MMP activity postinfarction. Hybrid Micro-SPECT/CT reconstructed short-axis images were acquired without x-ray contrast (A) in control sham-operated mouse (left) and selected mice at 1 week (middle) and 3 weeks (right) after MI, after injection of 201Tl (top row, green) and 99mTc-RP805 (middle row, red). A black-and-white (B&W) and multicolor fusion image is shown on bottom. Control heart demonstrates normal myocardial perfusion and no focal 99mTc-RP805 uptake within the heart, although some uptake is seen in chest wall at the thoracotomy site (dashed arrows). All post-MI mice have a large anterolateral 201Tl perfusion defect (yellow arrows) and focal uptake of 99mTc-RP805 in defect area. A dashed circle is drawn around the heart to demonstrate localization of 99mTc-RP805, the MMP radiotracer, within the infarcted area of the heart. Some activity is also seen in the peri-infarct border zone. Additional micro-SPECT/CT images were acquired by use of a higher-resolution SPECT detector after the administration of x-ray contrast, at 1 week (B) and 3 weeks (C) after MI. The contrast agent permitted better definition of the LV myocardium, which is highlighted by white dotted line. Representative short-axis (SA), horizontal long-axis (HLA), and vertical long-axis (VLA) images are shown. Focal uptake of 99mTc-RP805 is seen within the central infarct and peri-infarct regions, which again corresponds to 201Tl perfusion defect (Reprinted with permission Su et al.101).
Further imaging studies were carried out utilizing 99mTc-labeled analog of RP782 (99mTc-RP805) and hybrid SPECT/CT imaging with a dual isotope protocol involving 99mTc-RP805 imaging and adjunctive 201Tl-perfusion imaging. The dual isotope imaging studies revealed MMP activation within the perfusion defect region. This suggests that MMP activation is taking place primarily within the sites of injury and supports the concept that molecules that target MMP activation might be utilized to evaluate ventricular remodeling. An area of future investigation would be to address whether pharmacologic interventions known to favorably affect remodeling (i.e., angiotensin converting enzyme (ACE) inhibitors) might influence MMP levels, allowing prognostication of an individual’s response to therapy.
Factor XIII
111In-DOTA-FXIII is a radiolabeled glutaminase Factor XIII substrate analog that Factor XIII cross-links to ECM proteins (Figure 11).98 111In-DOTA-FXIII has been shown to accumulate in areas of increased factor XIII activity, using microSPECT/CT imaging in a murine model of myocardial infarction. Mice treated with factor XIII intravenously exhibited increased factor XIII activity as reflected by 111In-DOTA-FXIII in the infarct zone. The mice also demonstrated more rapid inflammatory turnover of neutrophils and increased recruitment of macrophages to the site of infarction. There was also increased collagen synthesis and capillary density in the factor XIII-treated animals, suggesting improved healing postinfarction. The 111In-labeled peptide substrate was demonstrated to be decreased in myocardial infarcts of animals treated with the direct thrombin inhibitor, dalteparin. Moreover, dalteparin treatment increased the risk of infarct rupture. These experimental studies suggest there may be a role to image for Factor XIII activity in an individual with myocardial infarction and to therapeutically supplement activity where indicated to assist with positive ventricular remodeling, although further study is needed.
Figure 11.

In vivo molecular imaging of transglutaminase factor XIII (FXIII) activity predicts survival and evolution of heart failure. (A–I) Longitudinal imaging study (MRI day 2, SPECT-CT day 3, second MRI day 21); on day 2 (A, D, G), late enhancement MRI showed similar infarct size in all groups. FXIII-treatment led to higher SPECT signal (H, K). In dalteparin-treated mice, the SPECT signal was lower (E, K). Serial MRI showed attenuated left ventricular (LV) dilation in FXIII-treated mice (L). Due to reduced survival in dalteparin (DP)-treated mice, the second MRI on day 21 was not acquired (F). * P < .05, ** P < .001 (Reprinted with permission from Nahrendorf et al.98).
ACE Inhibitors and AT1 Antagonists
A number of ACE inhibitors and AT1 antagonists have been radiolabeled for molecular imaging techniques.100 In a study of explanted hearts from patients with ischemic cardiomyopathy, 18F-fluorobenzoyllisinopril was used to assess ACE levels in infarcted myocardium and fibrosed tissue.102 The study demonstrated that the radiolabeled ACE inhibitor bound with some degree of specificity to areas adjacent to the infarct. Other studies using AT1 antagonists have demonstrated a differential between ACE activity and AT1 levels.103 In an ovine model of heart failure, ACE activity was primarily in the vascular endothelium while AT1 was upregulated in the myofibroblasts of the infarct region. In a murine model of acute myocardial infarction, a 99mTc-labeled AT1 receptor peptide analog was developed and demonstrated specificity to the myofibroblasts that localized to the infarct region in the weeks following the infarction. These early studies suggest the changes in the renin-angiotensin system that take place within an infarction may be utilized to identify those at risk for developing significant heart failure after myocardial infarction. Much more work is needed to assess the feasibility of these agents for imaging of postinfarction remodeling in clinical trials.
Integrin αvβ3
Because collagen deposition and fibrosis in the failing heart appear to be mediated by myofibroblasts, markers that indicate increased myofibroblast recruitment and activity are of interest to the field.104 Myofibroblasts demonstrate an upregulation of angiotensin receptors as mentioned above; however, they also demonstrate an upregulation of integrin moieties as well.103,105 Taking advantage of this molecular event, a recent study used the 99mTc-labeled cy5.5-RGD peptide analog, CRIP, to image upregulated αvβ3 integrins in a murine model of myocardial infarction.106 Utilizing CT for registration, in vivo microSPECT analysis confirmed localization of the CRIP to the infarct and border zones. Fluorescence imaging and histological analysis of explanted hearts revealed that CRIP co-localized to areas of myofibroblasts in the infarct region, although earlier studies suggest specific binding to endothelial cells and smooth muscle cells of newly formed microvascular networks. This issue needs further studies for clarification. Additional studies suggest that treating animals with either captopril or a combination of captopril and losartan altered the uptake of the CRIP-αvβ3 integrin signal. This preliminary study suggests that critical changes in the ventricular myocardium and vasculature postmyocardial infarction can not only be imaged using a molecular approach, but also that a response to remodeling therapy may also be assessed.
CONCLUSIONS
Molecular imaging represents an in vivo, targeted approach to noninvasively assess biological processes within the myocardium associated with atherosclerosis and subsequent ischemic injury. The goal of molecular imaging is to develop an approach for studying disease process as well as efficacy of a therapeutic treatment. The relatively high sensitivity of radiotracer-based imaging systems such as SPECT and PET have been of great use in the practical application of molecular imaging techniques. Though much of the research has focused on animal models of disease, studies have demonstrated the feasibility of targeted imaging approaches in the evaluation of angiogenesis, inflammation, apoptosis, and ventricular remodeling. Studies in humans are actively being pursued in several of these areas. By combining nuclear and CT imaging modalities, issues of attenuation artifact or partial volume effect are being overcome. Dual isotope protocols for monitoring physiological parameters (metabolism or perfusion) with targeted molecular probes show promise in the areas of myocardial infarction and angiogenesis. Combining gene therapy with PET reporter constructs is an active area of research with regard to the optimization of therapeutic angiogenesis. The role for apoptotic imaging in understanding reperfusion injury and the effects of therapeutic interventions also have potential clinical value. Imaging the activation of MMPs, active factor XIII, or the levels of renin-angiotensin system activation during ventricular remodeling may guide therapeutic regimens that could help positively influence outcomes postinfarction. In conclusion, targeted, radiotracer-based molecular imaging is rapidly becoming feasible and will likely play a more important role in the evaluation and management of cardiovascular disease as the field moves from the preclinical to clinical arena.
References
- 1.Pichler A, Piwnica-Worms D. Overview of cardiovascular molecular imaging. In: Gropler RJ, Glover DK, Sinusas AJ, Tegtmeyer H, editors. Cardiovascular molecular imaging. New York: Informa Healthcare USA, Inc; 2007. pp. 1–8. [Google Scholar]
- 2.Sinusas AJ, Bengel F, Nahrendorf M, Epstein FH, Wu JC, Villanueva FS, et al. Multimodality cardiovascular molecular imaging, Part I. Circ Cardiovasc Imaging. 2008;1:244–56. doi: 10.1161/CIRCIMAGING.108.824359. [DOI] [PubMed] [Google Scholar]
- 3.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–95. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 4.Weber C, Zernecke A, Libby P. The multifaceted contributions of leukocyte subsets to atherosclerosis: Lessons from mouse models. Nat Rev Immunol. 2008;8:802–15. doi: 10.1038/nri2415. [DOI] [PubMed] [Google Scholar]
- 5.Fam NP, Verma S, Kutryk M, Stewart DJ. Clinician guide to angiogenesis. Circulation. 2003;108:2613–8. doi: 10.1161/01.CIR.0000102939.04279.75. [DOI] [PubMed] [Google Scholar]
- 6.Giordano FJ, Ping P, McKirnan MD, Nozaki S, DeMaria AN, Dillmann WH, et al. Intracoronary gene transfer of fibroblast growth factor-5 increases blood flow and contractile function in an ischemic region of the heart. Nat Med. 1996;2:534–9. doi: 10.1038/nm0596-534. [DOI] [PubMed] [Google Scholar]
- 7.Simons M, Annex BH, Laham RJ, Kleiman N, Henry T, Dauerman H, et al. Pharmacological treatment of coronary artery disease with recombinant fibroblast growth factor-2: Double-blind, randomized, controlled clinical trial. Circulation. 2002;105:788–93. doi: 10.1161/hc0802.104407. [DOI] [PubMed] [Google Scholar]
- 8.Grines CL, Watkins MW, Helmer G, Penny W, Brinker J, Marmur JD, et al. Angiogenic Gene Therapy (AGENT) trial in patients with stable angina pectoris. Circulation. 2002;105:1291–7. doi: 10.1161/hc1102.105595. [DOI] [PubMed] [Google Scholar]
- 9.Henry TD, Annex BH, McKendall GR, Azrin MA, Lopez JJ, Giordano FJ, et al. The VIVA trial: Vascular endothelial growth factor in Ischemia for Vascular Angiogenesis. Circulation. 2003;107:1359–65. doi: 10.1161/01.cir.0000061911.47710.8a. [DOI] [PubMed] [Google Scholar]
- 10.Stewart DJ, Hilton JD, Arnold JM, Gregoire J, Rivard A, Archer SL, et al. Angiogenic gene therapy in patients with nonrevascularizable ischemic heart disease: A phase 2 randomized, controlled trial of AdVEGF(121) (AdVEGF121) versus maximum medical treatment. Gene Ther. 2006;13:1503–11. doi: 10.1038/sj.gt.3302802. [DOI] [PubMed] [Google Scholar]
- 11.Kastrup J, Jorgensen E, Ruck A, Tagil K, Glogar D, Ruzyllo W, et al. Direct intramyocardial plasmid vascular endothelial growth factor-A165 gene therapy in patients with stable severe angina pectoris A randomized double-blind placebo-controlled study: The Euroinject One trial. J Am Coll Cardiol. 2005;45:982–8. doi: 10.1016/j.jacc.2004.12.068. [DOI] [PubMed] [Google Scholar]
- 12.Cai W, Chen X. Multimodality molecular imaging of tumor angiogenesis. J Nucl Med. 2008;49(Suppl 2):113S–28S. doi: 10.2967/jnumed.107.045922. [DOI] [PubMed] [Google Scholar]
- 13.Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358:2039–49. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sasayama S, Fujita M. Recent insights into coronary collateral circulation. Circulation. 1992;85:1197–204. doi: 10.1161/01.cir.85.3.1197. [DOI] [PubMed] [Google Scholar]
- 15.Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–5. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- 16.Lee SH, Wolf PL, Escudero R, Deutsch R, Jamieson SW, Thistlethwaite PA. Early expression of angiogenesis factors in acute myocardial ischemia and infarction. N Engl J Med. 2000;342:626–33. doi: 10.1056/NEJM200003023420904. [DOI] [PubMed] [Google Scholar]
- 17.Brogi E, Schatteman G, Wu T, Kim EA, Varticovski L, Keyt B, et al. Hypoxia-induced paracrine regulation of vascular endothelial growth factor receptor expression. J Clin Invest. 1996;97:469–76. doi: 10.1172/JCI118437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Banai S, Jaklitsch MT, Shou M, Lazarous DF, Scheinowitz M, Biro S, et al. Angiogenic-induced enhancement of collateral blood flow to ischemic myocardium by vascular endothelial growth factor in dogs. Circulation. 1994;89:2183–9. doi: 10.1161/01.cir.89.5.2183. [DOI] [PubMed] [Google Scholar]
- 19.Li J, Brown LF, Hibberd MG, Grossman JD, Morgan JP, Simons M. VEGF, flk-1, and flt-1 expression in a rat myocardial infarction model of angiogenesis. Am J Physiol. 1996;270:H1803–11. doi: 10.1152/ajpheart.1996.270.5.H1803. [DOI] [PubMed] [Google Scholar]
- 20.Hu CJ, Iyer S, Sataur A, Covello KL, Chodosh LA, Simon MC. Differential regulation of the transcriptional activities of hypoxia-inducible factor 1 alpha (HIF-1alpha) and HIF-2alpha in stem cells. Mol Cell Biol. 2006;26:3514–26. doi: 10.1128/MCB.26.9.3514-3526.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dufraine J, Funahashi Y, Kitajewski J. Notch signaling regulates tumor angiogenesis by diverse mechanisms. Oncogene. 2008;27:5132–7. doi: 10.1038/onc.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiong JP, Stehle T, Diefenbach B, Zhang R, Dunker R, Scott DL, et al. Crystal structure of the extracellular segment of integrin alpha Vbeta3. Science. 2001;294:339–45. doi: 10.1126/science.1064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brooks PC, Montgomery AM, Rosenfeld M, Reisfeld RA, Hu T, Klier G, et al. Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell. 1994;79:1157–64. doi: 10.1016/0092-8674(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 24.Schwartz MA, Schaller MD, Ginsberg MH. Integrins: Emerging paradigms of signal transduction. Annu Rev Cell Dev Biol. 1995;11:549–99. doi: 10.1146/annurev.cb.11.110195.003001. [DOI] [PubMed] [Google Scholar]
- 25.Hynes RO. Integrins: Bidirectional, allosteric signaling machines. Cell. 2002;110:673–87. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 26.Clyman RI, Mauray F, Kramer RH. Beta 1 and beta 3 integrins have different roles in the adhesion and migration of vascular smooth muscle cells on extracellular matrix. Exp Cell Res. 1992;200:272–84. doi: 10.1016/0014-4827(92)90173-6. [DOI] [PubMed] [Google Scholar]
- 27.Lu E, Wagner WR, Schellenberger U, Abraham JA, Klibanov AL, Woulfe SR, et al. Targeted in vivo labeling of receptors for vascular endothelial growth factor: Approach to identification of ischemic tissue. Circulation. 2003;108:97–103. doi: 10.1161/01.CIR.0000079100.38176.83. [DOI] [PubMed] [Google Scholar]
- 28.Rodriguez-Porcel M, Cai W, Gheysens O, Willmann JK, Chen K, Wang H, et al. Imaging of VEGF receptor in a rat myocardial infarction model using PET. J Nucl Med. 2008;49:667–73. doi: 10.2967/jnumed.107.040576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Willmann JK, Chen K, Wang H, Paulmurugan R, Rollins M, Cai W, et al. Monitoring of the biological response to murine hindlimb ischemia with 64Cu-labeled vascular endothelial growth factor-121 positron emission tomography. Circulation. 2008;117:915–22. doi: 10.1161/CIRCULATIONAHA.107.733220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu JC, Inubushi M, Sundaresan G, Schelbert HR, Gambhir SS. Positron emission tomography imaging of cardiac reporter gene expression in living rats. Circulation. 2002;106:180–3. doi: 10.1161/01.cir.0000023620.59633.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inubushi M, Wu JC, Gambhir SS, Sundaresan G, Satyamurthy N, Namavari M, et al. Positron-emission tomography reporter gene expression imaging in rat myocardium. Circulation. 2003;107:326–32. doi: 10.1161/01.cir.0000044385.60972.ae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bengel FM, Anton M, Richter T, Simoes MV, Haubner R, Henke J, et al. Noninvasive imaging of transgene expression by use of positron emission tomography in a pig model of myocardial gene transfer. Circulation. 2003;108:2127–33. doi: 10.1161/01.CIR.0000091401.26280.A0. [DOI] [PubMed] [Google Scholar]
- 33.Wu JC, Chen IY, Wang Y, Tseng JR, Chhabra A, Salek M, et al. Molecular imaging of the kinetics of vascular endothelial growth factor gene expression in ischemic myocardium. Circulation. 2004;110:685–91. doi: 10.1161/01.CIR.000013815302213.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sipkins DA, Cheresh DA, Kazemi MR, Nevin LM, Bednarski MD, Li KC. Detection of tumor angiogenesis in vivo by alphaVbeta3-targeted magnetic resonance imaging. Nat Med. 1998;4:623–6. doi: 10.1038/nm0598-623. [DOI] [PubMed] [Google Scholar]
- 35.Haubner R, Wester HJ, Weber WA, Mang C, Ziegler SI, Goodman SL, et al. Noninvasive imaging of alpha(v)beta3 integrin expression using 18F-labeled RGD-containing glycopeptide and positron emission tomography. Cancer Res. 2001;61:1781–5. [PubMed] [Google Scholar]
- 36.Haubner R, Wester HJ, Burkhart F, Senekowitsch-Schmidtke R, Weber W, Goodman SL, et al. Glycosylated RGD-containing peptides: Tracer for tumor targeting and angiogenesis imaging with improved biokinetics. J Nucl Med. 2001;42:326–36. [PubMed] [Google Scholar]
- 37.Harris TD, Kalogeropoulos S, Nguyen T, Liu S, Bartis J, Ellars C, et al. Design, synthesis, and evaluation of radiolabeled integrin alpha v beta 3 receptor antagonists for tumor imaging and radiotherapy. Cancer Biother Radiopharm. 2003;18:627–41. doi: 10.1089/108497803322287727. [DOI] [PubMed] [Google Scholar]
- 38.Sadeghi M, Krassilnikova S, Zhang J, et al. Imaging of αvβ3 integrin in vascular injury: Does this reflect increased integrin expression or activaton? Circulation. 2008;108:404. [Google Scholar]
- 39.Meoli DF, Sadeghi MM, Krassilnikova S, Bourke BN, Giordano FJ, Dione DP, et al. Noninvasive imaging of myocardial angiogenesis following experimental myocardial infarction. J Clin Invest. 2004;113:1684–91. doi: 10.1172/JCI20352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kalinowski L, Dobrucki LW, Meoli DF, Dione DP, Sadeghi MM, Madri JA, et al. Targeted imaging of hypoxia-induced integrin activation in myocardium early after infarction. J Appl Physiol. 2008;104:1504–12. doi: 10.1152/japplphysiol.00861.2007. [DOI] [PubMed] [Google Scholar]
- 41.Makowski MR, Ebersberger U, Nekolla S, Schwaiger M. In vivo molecular imaging of angiogenesis, targeting alphavbeta3 integrin expression, in a patient after acute myocardial infarction. Eur Heart J. 2008;29:2201. doi: 10.1093/eurheartj/ehn129. [DOI] [PubMed] [Google Scholar]
- 42.Almutairi A, Rossin R, Shokeen M, Hagooly A, Ananth A, Capoccia B, et al. Biodegradable dendritic positron-emitting nanoprobes for the noninvasive imaging of angiogenesis. Proc Natl Acad Sci U S A. 2009;106:685–90. doi: 10.1073/pnas.0811757106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peters AM, Danpure HJ, Osman S, Hawker RJ, Henderson BL, Hodgson HJ, et al. Clinical experience with 99mTc-hexamethylpropylene-amineoxime for labelling leucocytes and imaging inflammation. Lancet. 1986;2:946–9. doi: 10.1016/s0140-6736(86)90601-x. [DOI] [PubMed] [Google Scholar]
- 44.Peters AM, Saverymuttu SH. The value of indium-labelled leucocytes in clinical practice. Blood Rev. 1987;1:65–76. doi: 10.1016/0268-960x(87)90021-x. [DOI] [PubMed] [Google Scholar]
- 45.Lavender JP, Lowe J, Barker JR, Burn JI, Chaudhri MA. Gallium 67 citrate scanning in neoplastic and inflammatory lesions. Br J Radiol. 1971;44:361–6. doi: 10.1259/0007-1285-44-521-361. [DOI] [PubMed] [Google Scholar]
- 46.Mochizuki T, Tsukamoto E, Kuge Y, Kanegae K, Zhao S, Hikosaka K, et al. FDG uptake and glucose transporter subtype expressions in experimental tumor and inflammation models. J Nucl Med. 2001;42:1551–5. [PubMed] [Google Scholar]
- 47.Kubota R, Yamada S, Kubota K, Ishiwata K, Tamahashi N, Ido T. Intratumoral distribution of fluorine-18-fluorodeoxyglucose in vivo: High accumulation in macrophages and granulation tissues studied by microautoradiography. J Nucl Med. 1992;33:1972–80. [PubMed] [Google Scholar]
- 48.Johnson LL, Seldin DW, Becker LC, LaFrance ND, Liberman HA, James C, et al. Antimyosin imaging in acute transmural myocardial infarctions: Results of a multicenter clinical trial. J Am Coll Cardiol. 1989;13:27–35. doi: 10.1016/0735-1097(89)90544-5. [DOI] [PubMed] [Google Scholar]
- 49.Khaw BA, Gold HK, Yasuda T, Leinbach RC, Kanke M, Fallon JT, et al. Scintigraphic quantification of myocardial necrosis in patients after intravenous injection of myosin-specific antibody. Circulation. 1986;74:501–8. doi: 10.1161/01.cir.74.3.501. [DOI] [PubMed] [Google Scholar]
- 50.Yasuda T, Palacios IF, Dec GW, Fallon JT, Gold HK, Leinbach RC, et al. Indium 111-monoclonal antimyosin antibody imaging in the diagnosis of acute myocarditis. Circulation. 1987;76:306–11. doi: 10.1161/01.cir.76.2.306. [DOI] [PubMed] [Google Scholar]
- 51.Dec GW, Palacios I, Yasuda T, Fallon JT, Khaw BA, Strauss HW, et al. Antimyosin antibody cardiac imaging: Its role in the diagnosis of myocarditis. J Am Coll Cardiol. 1990;16:97–104. doi: 10.1016/0735-1097(90)90463-y. [DOI] [PubMed] [Google Scholar]
- 52.Narula J, Khaw BA, Dec GW, Palacios IF, Newell JB, Southern JF, et al. Diagnostic accuracy of antimyosin scintigraphy in suspected myocarditis. J Nucl Cardiol. 1996;3:371–81. doi: 10.1016/s1071-3581(96)90070-7. [DOI] [PubMed] [Google Scholar]
- 53.Sato M, Toyozaki T, Odaka K, Uehara T, Arano Y, Hasegawa H, et al. Detection of experimental autoimmune myocarditis in rats by 111In monoclonal antibody specific for tenascin-C. Circulation. 2002;106:1397–402. doi: 10.1161/01.cir.0000027823.07104.86. [DOI] [PubMed] [Google Scholar]
- 54.Odaka K, Uehara T, Arano Y, Adachi S, Tadokoro H, Yoshida K, et al. Noninvasive detection of cardiac repair after acute myocardial infarction in rats by 111 In Fab fragment of monoclonal antibody specific for tenascin-C. Int Heart J. 2008;49:481–92. doi: 10.1536/ihj.49.481. [DOI] [PubMed] [Google Scholar]
- 55.Ford-Hutchinson AW. Regulation of leukotriene biosynthesis. Cancer Metastasis Rev. 1994;13:257–67. doi: 10.1007/BF00666096. [DOI] [PubMed] [Google Scholar]
- 56.Yokomizo T, Izumi T, Shimizu T. Leukotriene B4: Metabolism and signal transduction. Arch Biochem Biophys. 2001;385:231–41. doi: 10.1006/abbi.2000.2168. [DOI] [PubMed] [Google Scholar]
- 57.Serhan CN, Prescott SM. The scent of a phagocyte: Advances on leukotriene b(4) receptors. J Exp Med. 2000;192:F5–8. doi: 10.1084/jem.192.3.f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brouwers AH, Laverman P, Boerman OC, Oyen WJ, Barrett JA, Harris TD, et al. A 99Tcm-labelled leukotriene B4 receptor antagonist for scintigraphic detection of infection in rabbits. Nucl Med Commun. 2000;21:1043–50. doi: 10.1097/00006231-200011000-00010. [DOI] [PubMed] [Google Scholar]
- 59.Riou LM, Ruiz M, Sullivan GW, Linden J, Leong-Poi H, Lindner JR, et al. Assessment of myocardial inflammation produced by experimental coronary occlusion and reperfusion with 99mTc-RP517, a new leukotriene B4 receptor antagonist that preferentially labels neutrophils in vivo. Circulation. 2002;106:592–8. doi: 10.1161/01.cir.0000023878.04716.6d. [DOI] [PubMed] [Google Scholar]
- 60.van Eerd JE, Oyen WJ, Harris TD, Rennen HJ, Edwards DS, Liu S, et al. A bivalent leukotriene B(4) antagonist for scintigraphic imaging of infectious foci. J Nucl Med. 2003;44:1087–91. [PubMed] [Google Scholar]
- 61.Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359:473–81. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- 62.Fliss H, Gattinger D. Apoptosis in ischemic and reperfused rat myocardium. Circ Res. 1996;79:949–56. doi: 10.1161/01.res.79.5.949. [DOI] [PubMed] [Google Scholar]
- 63.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–35. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 64.Yaoita H, Ogawa K, Maehara K, Maruyama Y. Attenuation of ischemia/reperfusion injury in rats by a caspase inhibitor. Circulation. 1998;97:276–81. doi: 10.1161/01.cir.97.3.276. [DOI] [PubMed] [Google Scholar]
- 65.Dumont EA, Reutelingsperger CP, Smits JF, Daemen MJ, Doevendans PA, Wellens HJ, et al. Real-time imaging of apoptotic cell-membrane changes at the single-cell level in the beating murine heart. Nat Med. 2001;7:1352–5. doi: 10.1038/nm1201-1352. [DOI] [PubMed] [Google Scholar]
- 66.Hayakawa Y, Chandra M, Miao W, Shirani J, Brown JH, Dorn GW, et al. Inhibition of cardiac myocyte apoptosis improves cardiac function and abolishes mortality in the peripartum cardiomyopathy of Galpha(q) transgenic mice. Circulation. 2003;108:3036–41. doi: 10.1161/01.CIR.0000101920.72665.58. [DOI] [PubMed] [Google Scholar]
- 67.Thimister PW, Hofstra L, Liem IH, Boersma HH, Kemerink G, Reutelingsperger CP, et al. In vivo detection of cell death in the area at risk in acute myocardial infarction. J Nucl Med. 2003;44:391–6. [PubMed] [Google Scholar]
- 68.Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004;5:897–907. doi: 10.1038/nrm1496. [DOI] [PubMed] [Google Scholar]
- 69.Tait JF. Imaging of apoptosis. J Nucl Med. 2008;49:1573–6. doi: 10.2967/jnumed.108.052803. [DOI] [PubMed] [Google Scholar]
- 70.Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM, et al. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: Inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182:1545–56. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, van Oers MH. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 1994;84:1415–20. [PubMed] [Google Scholar]
- 72.van Engeland M, Ramaekers FC, Schutte B, Reutelingsperger CP. A novel assay to measure loss of plasma membrane asymmetry during apoptosis of adherent cells in culture. Cytometry. 1996;24:131–9. doi: 10.1002/(SICI)1097-0320(19960601)24:2<131::AID-CYTO5>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 73.Khaw BA. The current role of infarct avid imaging. Semin Nucl Med. 1999;29:259–70. doi: 10.1016/s0001-2998(99)80014-2. [DOI] [PubMed] [Google Scholar]
- 74.Flotats A, Carrio I. Non-invasive in vivo imaging of myocardial apoptosis and necrosis. Eur J Nucl Med Mol Imaging. 2003;30:615–30. doi: 10.1007/s00259-003-1136-y. [DOI] [PubMed] [Google Scholar]
- 75.Blankenberg FG, Katsikis PD, Tait JF, Davis RE, Naumovski L, Ohtsuki K, et al. In vivo detection and imaging of phosphatidylserine expression during programmed cell death. Proc Natl Acad Sci USA. 1998;95:6349–54. doi: 10.1073/pnas.95.11.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ogasawara J, Watanabe-Fukunaga R, Adachi M, Matsuzawa A, Kasugai T, Kitamura Y, et al. Lethal effect of the anti-Fas antibody in mice. Nature. 1993;364:806–9. doi: 10.1038/364806a0. [DOI] [PubMed] [Google Scholar]
- 77.Hofstra L, Liem IH, Dumont EA, Boersma HH, van Heerde WL, Doevendans PA, et al. Visualisation of cell death in vivo in patients with acute myocardial infarction. Lancet. 2000;356:209–12. doi: 10.1016/s0140-6736(00)02482-x. [DOI] [PubMed] [Google Scholar]
- 78.Laguens RP, Meckert PM, Martino JS, Perrone S, Favaloro R. Identification of programmed cell death (apoptosis) in situ by means of specific labeling of nuclear DNA fragments in heart biopsy samples during acute rejection episodes. J Heart Lung Transplant. 1996;15:911–8. [PubMed] [Google Scholar]
- 79.Narula J, Acio ER, Narula N, Samuels LE, Fyfe B, Wood D, et al. Annexin-V imaging for noninvasive detection of cardiac allograft rejection. Nat Med. 2001;7:1347–52. doi: 10.1038/nm1201-1347. [DOI] [PubMed] [Google Scholar]
- 80.Tokita N, Hasegawa S, Tsujimura E, Yutani K, Izumi T, Nishimura T. Serial changes in 14C-deoxyglucose and 201Tl uptake in autoimmune myocarditis in rats. J Nucl Med. 2001;42:285–91. [PubMed] [Google Scholar]
- 81.Sarda-Mantel L, Hervatin F, Michel JB, Louedec L, Martet G, Rouzet F, et al. Myocardial uptake of 99mTc-annexin-V and 111In-antimyosin-antibodies after ischemia-reperfusion in rats. Eur J Nucl Med Mol Imaging. 2008;35:158–65. doi: 10.1007/s00259-007-0559-2. [DOI] [PubMed] [Google Scholar]
- 82.Faust A, Wagner S, Law MP, Hermann S, Schnockel U, Keul P, et al. The nonpeptidyl caspase binding radioligand (S)-1-(4-(2-[18F]Fluoroethoxy)-benzyl)-5-[1-(2-methoxymethylpyrrolidinyl)s ulfonyl]isatin ([18F]CbR) as potential positron emission tomography-compatible apoptosis imaging agent. Q J Nucl Med Mol Imaging. 2007;51:67–73. [PubMed] [Google Scholar]
- 83.Zhou D, Chu W, Rothfuss J, Zeng C, Xu J, Jones L, et al. Synthesis, radiolabeling, and in vivo evaluation of an 18F-labeled isatin analog for imaging caspase-3 activation in apoptosis. Bioorg Med Chem Lett. 2006;16:5041–6. doi: 10.1016/j.bmcl.2006.07.045. [DOI] [PubMed] [Google Scholar]
- 84.Corbett JR, Lewis M, Willerson JT, Nicod PH, Huxley RL, Simon T, et al. 99mTc-pyrophosphate imaging in patients with acute myocardial infarction: Comparison of planar imaging with single-photon tomography with and without blood pool overlay. Circulation. 1984;69:1120–8. doi: 10.1161/01.cir.69.6.1120. [DOI] [PubMed] [Google Scholar]
- 85.Orlandi C, Crane PD, Edwards DS, Platts SH, Bernard L, Lazewatsky J, et al. Early scintigraphic detection of experimental myocardial infarction in dogs with technetium-99m-glucaric acid. J Nucl Med. 1991;32:263–8. [PubMed] [Google Scholar]
- 86.Johnson G, III, Okada CC, Hocherman SD, Liu Z, Hart C, Khaw BA, et al. (99m)Tc-glucarate imaging for the early detection of infarct in partially reperfused canine myocardium. Eur J Nucl Med Mol Imaging. 2006;33:319–28. doi: 10.1007/s00259-005-1913-x. [DOI] [PubMed] [Google Scholar]
- 87.Narula J, Petrov A, Pak KY, Lister BC, Khaw BA. Very early noninvasive detection of acute experimental nonreperfused myocardial infarction with 99mTc-labeled glucarate. Circulation. 1997;95:1577–84. doi: 10.1161/01.cir.95.6.1577. [DOI] [PubMed] [Google Scholar]
- 88.Mariani G, Villa G, Rossettin PF, Spallarossa P, Bezante GP, Brunelli C, et al. Detection of acute myocardial infarction by 99mTc-labeled D-glucaric acid imaging in patients with acute chest pain. J Nucl Med. 1999;40:1832–9. [PubMed] [Google Scholar]
- 89.Weber KT. Extracellular matrix remodeling in heart failure: A role for de novo angiotensin II generation. Circulation. 1997;96:4065–82. doi: 10.1161/01.cir.96.11.4065. [DOI] [PubMed] [Google Scholar]
- 90.Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: Pathophysiology and therapy. Circulation. 2000;101:2981–8. doi: 10.1161/01.cir.101.25.2981. [DOI] [PubMed] [Google Scholar]
- 91.Creemers EE, Cleutjens JP, Smits JF, Daemen MJ. Matrix metalloproteinase inhibition after myocardial infarction: A new approach to prevent heart failure? Circ Res. 2001;89:201–10. doi: 10.1161/hh1501.094396. [DOI] [PubMed] [Google Scholar]
- 92.Spinale FG. Matrix metalloproteinases: Regulation and dysregulation in the failing heart. Circ Res. 2002;90:520–30. doi: 10.1161/01.res.0000013290.12884.a3. [DOI] [PubMed] [Google Scholar]
- 93.Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000;106:55–62. doi: 10.1172/JCI8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rohde LE, Ducharme A, Arroyo LH, Aikawa M, Sukhova GH, Lopez-Anaya A, et al. Matrix metalloproteinase inhibition attenuates early left ventricular enlargement after experimental myocardial infarction in mice. Circulation. 1999;99:3063–70. doi: 10.1161/01.cir.99.23.3063. [DOI] [PubMed] [Google Scholar]
- 95.Lindsey ML, Gannon J, Aikawa M, Schoen FJ, Rabkin E, Lopresti-Morrow L, et al. Selective matrix metalloproteinase inhibition reduces left ventricular remodeling but does not inhibit angiogenesis after myocardial infarction. Circulation. 2002;105:753–8. doi: 10.1161/hc0602.103674. [DOI] [PubMed] [Google Scholar]
- 96.Yarbrough WM, Mukherjee R, Escobar GP, Mingoia JT, Sample JA, Hendrick JW, et al. Selective targeting and timing of matrix metalloproteinase inhibition in post-myocardial infarction remodeling. Circulation. 2003;108:1753–9. doi: 10.1161/01.CIR.0000091087.78630.79. [DOI] [PubMed] [Google Scholar]
- 97.Nahrendorf M, Hu K, Frantz S, Jaffer FA, Tung CH, Hiller KH, et al. Factor XIII deficiency causes cardiac rupture, impairs wound healing, and aggravates cardiac remodeling in mice with myocardial infarction. Circulation. 2006;113:1196–202. doi: 10.1161/CIRCULATIONAHA.105.602094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nahrendorf M, Aikawa E, Figueiredo JL, Stangenberg L, van den Borne SW, Blankesteijn WM, et al. Transglutaminase activity in acute infarcts predicts healing outcome and left ventricular remodelling: Implications for FXIII therapy and antithrombin use in myocardial infarction. Eur Heart J. 2008;29:445–54. doi: 10.1093/eurheartj/ehm558. [DOI] [PubMed] [Google Scholar]
- 99.Aras O, Messina SA, Shirani J, Eckelman WC, Dilsizian V. The role and regulation of cardiac angiotensin-converting enzyme for noninvasive molecular imaging in heart failure. Curr Cardiol Rep. 2007;9:150–8. doi: 10.1007/BF02938342. [DOI] [PubMed] [Google Scholar]
- 100.Shirani J, Dilsizian V. Imaging left ventricular remodeling: Targeting the neurohumoral axis. Nat Clin Pract Cardiovasc Med. 2008;5(Suppl 2):S57–62. doi: 10.1038/ncpcardio1244. [DOI] [PubMed] [Google Scholar]
- 101.Su H, Spinale FG, Dobrucki LW, Song J, Hua J, Sweterlitsch S, et al. Noninvasive targeted imaging of matrix metalloproteinase activation in a murine model of postinfarction remodeling. Circulation. 2005;112:3157–67. doi: 10.1161/CIRCULATIONAHA.105.583021. [DOI] [PubMed] [Google Scholar]
- 102.Dilsizian V, Eckelman WC, Loredo ML, Jagoda EM, Shirani J. Evidence for tissue angiotensin-converting enzyme in explanted hearts of ischemic cardiomyopathy using targeted radiotracer technique. J Nucl Med. 2007;48:182–7. [PubMed] [Google Scholar]
- 103.Shirani J, Narula J, Eckelman WC, Narula N, Dilsizian V. Early imaging in heart failure: Exploring novel molecular targets. J Nucl Cardiol. 2007;14:100–10. doi: 10.1016/j.nuclcard.2006.12.318. [DOI] [PubMed] [Google Scholar]
- 104.Cleutjens JP, Blankesteijn WM, Daemen MJ, Smits JF. The infarcted myocardium: Simply dead tissue, or a lively target for therapeutic interventions. Cardiovasc Res. 1999;44:232–41. doi: 10.1016/s0008-6363(99)00212-6. [DOI] [PubMed] [Google Scholar]
- 105.Asano Y, Ihn H, Yamane K, Jinnin M, Mimura Y, Tamaki K. Increased expression of integrin alpha(v)beta3 contributes to the establishment of autocrine TGF-beta signaling in scleroderma fibroblasts. J Immunol. 2005;175:7708–18. doi: 10.4049/jimmunol.175.11.7708. [DOI] [PubMed] [Google Scholar]
- 106.van den Borne SW, Isobe S, Verjans JW, Petrov A, Lovhaug D, Li P, et al. Molecular imaging of interstitial alterations in remodeling myocardium after myocardial infarction. J Am Coll Cardiol. 2008;52:2017–28. doi: 10.1016/j.jacc.2008.07.067. [DOI] [PubMed] [Google Scholar]