

Abstract
Bipolar disorder (BD) is a progressive psychiatric disorder characterized by recurrent changes of mood, and is associated with cognitive decline. There is evidence of excitotoxicity, neuroinflammation, upregulated arachidonic acid (AA) cascade signaling and brain atrophy in BD patients. These observations suggest that BD pathology may be associated with apoptosis as well as with disturbed synaptic function. To test this hypothesis, we measured mRNA and protein levels of the pro-apoptotic (Bax, BAD, Caspase-9 and Caspase-3) and anti-apoptotic factors (BDNF and Bcl-2), and of pre- and post-synaptic markers (synaptophysin and drebrin), in postmortem brain from 10 BD patients and 10 age-matched controls. Consistent with the hypothesis, BD brains showed significant increases in protein and mRNA levels of the pro-apoptotic factors and significant decreases of levels of the anti-apoptotic factors and the synaptic markers, synaptophysin and drebrin. These differences may contribute to brain atrophy and progressive cognitive changes in BD.
Keywords: bipolar disorder, apoptosis, caspase, BDNF, synaptophysin and drebrin
INTRODUCTION
Bipolar disorder (BD) is a prevalent, severe, and highly disabling psychiatric disease characterized by recurrent depressive and manic episodes, and is associated with increased morbidity and mortality due to general medical conditions, such as obesity, diabetes and cardio-disease (1).
BD has multiple risk alleles consistent with a polygenic inheritance (2), but its pathological mechanisms are not agreed on. Studies showing increased brain levels of pro-inflammatory cytokines and increased glutamatergic function suggest roles for excitotoxicity and neuroinflammation in the disease (3–6).
Studies have also reported altered apoptotic factors and their mediated responses in BD. Changes include DNA damage in peripheral blood of BD patients (7), increased pro-apoptotic serum activity in BD patients (8) and mitochondrial dysfunction (9). Additionally, in vivo imaging and postmortem studies have revealed significant brain atrophy in BD patients, with a decrease in cortical thickness (10), as well as reduced numbers and/or sizes of glia and neurons in discrete brain areas (11). These studies implicate the involvement of cell death in the pathophysiology of BD.
Studies have shown that drugs used to treat BD do not induce DNA damage; rather some enhance DNA repair (12). Lithium and valproate inhibit glutamate-induced DNA fragmentation in cerebral cortical neurons (13). These drugs can suppress caspase-3 activity and stimulate B-cell lymphoma-2 (Bcl-2) expression, which render a cell less susceptible to apoptosis (14–16). Chronic administration of lithium at clinically relevant doses has been shown to enhance neurogenesis in rat hippocampus, increasing both the Bcl-2 level and the percent of new cells that display a neuronal phenotype (17, 18).
Upregulated AA signaling has been associated with neuroinflammation, excitotoxicity and apoptosis (19–21). Lithium and carbamazepine when given long-term to rats to produce a therapeutically relevant plasma concentration, downregulate components of the brain arachidonic acid (AA, 20:4n-6) cascade (22), such as Ca2+-dependent AA-selective cytosolic phospholipase A2 (cPLA2) and cyclooxygenase-2 (COX-2). The postmortem BD brain demonstrates increased expression levels of cPLA2 and COX-2 in prefrontal cortex (23). Furthermore, a recent study reported increased markers of excitotoxicity and neuroinflammation in the BD frontal cortex (24).
BD has been associated with cognitive defects (25, 26), and decreased synaptic markers such as synaptophysin and drebrin, also associated with cognitive defects, have been reported in Alzheimer disease brain (27, 28). Taken together, these observations suggest that BD also may be associated with altered pre- and post-synaptic brain markers.
To further clarify the possible involvement of apoptosis and synaptic loss in BD, we measured mRNA and protein levels of apoptotic factors such as Bcl-2, caspase-3/-9, Bcl-2-associated X protein (BAX), Bcl-2-associated death promoter (BAD) and brain derived neurotrophic factor (BDNF), and the protein levels of synaptophysin and drebrin, in the postmortem frontal cortex of BD patients and control subjects, matched for age, postmortem interval (PMI) and pH. We used the frontal cortex because studies have shown structural, metabolic and signaling abnormalities in this particular brain region of bipolar patients (29–34). The current study presents potential alterations in apoptotic factors with synaptic loss in postmortem brain of BD patients. An abstract of part of this work has been published elsewhere (35)
MATERIALS AND METHODS
Postmortem brain samples
This study was approved by the Institutional Review Board of McLean Hospital and the Office of Human Subjects Research (OHSR) of NIH (# 4380). Frozen postmortem human frontal cortex (Brodmann area 9) was provided by the Harvard Brain Tissue Resource Center (McLean Hospital, Belmont, MA) under Public Health Service grant number R24MH068855 (awarded to J.S. Rao.), from 10 BD patients and 10 age-matched controls. Mean age, postmortem interval (PMI) and pH of the frozen brain samples did not differ significantly between the BD and control groups: age (years, control: 43 ± 3.5 vs BD: 49 ± 7.2), PMI (hours, control: 27 ± 1.5 vs BD: 21 ± 3.0) and brain pH (control: 6.6 ± 0.16 vs BD: 6.7 ± 0.09). The pH of the frozen brain samples was measured by the method of Harrison et al. (36). The BD patients had been exposed to various psychotropic medications, as reported in an earlier publication (Table 1) (37).
Table 1.
Characteristics of control and bipolar disorder subjects
| Group | Age, (yr) | Sex | PMI, (h) | Cause of death | Medications |
|---|---|---|---|---|---|
| Control | 32 | F | 29 | Cardiopulmonary attack | Antibiotics |
| Control | 46 | M | 30 | Cardiopulmonary attack | Insulin |
| Control | 54 | M | 24 | Cardiopulmonary attack | Insulin |
| Control | 36 | M | 21 | Electrocution | Vitamins |
| Control | 41 | M | 30 | Cardiopulmonary attack | None |
| Control | 49 | M | 27 | Cardiopulmonary attack | Vitamins |
| Control | 35 | M | 20 | Cardiac arrest | Not available |
| Control | 35 | M | 26 | unknown | Not available |
| Control | 45 | M | 24 | unknown | Not available |
| Control | 25 | M | 15 | Myocardial Infarction | Not available |
| BD | 29 | M | 20 | Suicide | Paxil |
| BD | 74 | M | 7 | Pneumonia | Neurontin |
| BD | 51 | F | 35 | Ischemic heart disease | Ambien |
| BD | 47 | F | 16 | Major system failure | Lithium carbonate |
| BD | 40 | M | 30 | Suicide | Risperidone |
| BD | 75 | M | 20 | Myocardial infarction | Prozac, Avandia |
| BD | 90 | F | 19 | Ventricular tachycardia | Lithium carbonate, |
| BD | 27 | M | 20 | Suicide | Lithium carbonate |
| BD | 25 | F | 11 | Suicide | Not available |
| BD | 35 | M | 42 | Suicide | Lithium |
PMI, postmortem interval; F, Female; M, Male
Preparation of cytosolic and membrane extracts
Cytosolic and membrane extracts were prepared from postmortem frontal cortex of BD and control subjects as previously described (38). Briefly, frontal cortex tissue was homogenized in a buffer containing 20 mM Tris-HCl (pH 7.4), 2 mM EGTA, 5 mM EDTA, 1.5 mM pepstatin, 2 mM leupeptin, 0.5 mM phenylmethylsulfonyl fluoride, 0.2 U/ml aprotinin and 2 mM dithiothreitol. The homogenate was centrifuged at 100,000g for 60 min at 4°C. The resulting supernatant was the cytosolic fraction, and the pellet was resuspended in the homogenizing buffer containing 0.2% (w/v) Triton X-100. The suspension was kept at 4°C for 60 min with occasional stirring and then centrifuged at 100,000g for 60 min at 4°C. The resulting supernatant was the membrane fraction. Protein concentrations of the membrane and cytosolic fractions were determined with Bio-Rad protein Reagent (Bio-Rad, Hercules, CA).
Western blot analysis
Protein (50 μg) from the cytosolic and membrane extracts was separated on 4–20% SDS-polyacrylamide gels (PAGE) (Bio-Rad). Following electrophoresis, the proteins were transferred to a nitrocellulose membrane. Protein blots were incubated overnight in Tris-Buffered-Saline buffer, containing 5% nonfat dried milk and 0.1% Tween-20, with specific primary antibodies for BAD, Bcl-2, Bax, Caspase-3 (1:1000 dilution), Caspase-9 (1:500) (Cell Signaling, Beverly, MA), Drebrin and Synaptophysin (1:10000) (Abcam, Cambridge, MA). Protein blots were incubated with appropriate HRP-conjugated secondary antibodies (Bio-Rad) and visualized using a chemiluminescence reaction (Amersham, Piscataway, NJ) on X-ray film (Kodak, Rochester, NY). Optical densities of immunoblot bands were measured using Alpha Innotech Software (Alpha Innotech, San Leandro, CA) and were normalized to β–actin (Sigma, St. Louis, MO) to correct for unequal loading. All experiments were carried out twice with 10 controls and 10 BD postmortem brain samples. Values were expressed as percent of control.
Total RNA isolation and real time RT-PCR
Total RNA was isolated from the frontal cortex using an RNeasy mini kit for brain and lipid tissue (Qiagen, Valencia, CA). Complementary DNA (cDNA) was prepared from total RNA using a high capacity cDNA Archive kit (Applied Biosystems, Foster City, CA). RNA integrity number (RIN) was measured using Bioanalyzer (Agilent 2100 Bioanalyzer, Santa Clara, CA). RIN values were 6.9 ± 0.4 and 7.1 ± 0.5 (Mean ± SEM) for control and BD samples, respectively. mRNA levels (Bcl-2, Bax, BAD, Caspase-3, Caspase-9, BDNF, Drebrin and Synaptophysin) were measured by quantitative RT-PCR, using an ABI PRISM 7000 sequence detection system (Applied Biosystems). Specific primers and probes for Bcl-2, Bax, BAD, Caspase-3, Caspase-9, BDNF, Drebrin and Synaptophysin were purchased from TaqManR gene expression assays (Applied Biosystems), and consisted of a 20X mix of unlabeled PCR primers and Taqman minor groove binder probe (FAM (6-carboxy-fluorescein) dye-labeled). The fold-change in gene expression was determined by the ΔΔCT method (39). Data are expressed as the relative level of the target gene in the postmortem BD patients normalized to the endogenous control (β-globulin) and relative to the control (calibrator), as previously described (40). All experiments were carried out twice in triplicates, and the data were expressed relative to controls.
BDNF protein levels
BDNF protein levels were measured in brain cytosolic extracts using an ELISA kit according to the manufacturer’s instructions (Chemicon International, Temecula, CA, USA). Values are expressed in pmol/mg protein.
Immunohistochemical analysis
Frozen tissue was sectioned as described earlier (41). Caspase-3 immunohistochemistry with paraffin-embedded postmortem brain sections was performed using SignalStain Cleaved Caspase-3 (Asp175) IHC detection Kit according to the manufacturer’s instructions (Cell Signaling). Counterstaining was performed with methyl green (Vector Laboratories, Inc. Burlingame, CA).
Statistical analysis
Data are expressed as mean ± SEM. Statistical significance of means was calculated using a two-tailed unpaired t-test. Pearson correlations were made between age, post-mortem interval and pH of the frontal cortex, and mRNA levels of tested genes in post-mortem brain from controls and BD patients combined. When three groups were compared (e.g. controls, all BD subjects and the subgroups of BD treated with lithium or controls, all BD subjects and the subgroup of BD that died by suicides), statistical significance was determined using a Bonferroni’s multiple comparison test. Statistical significance was set at p < 0.05.
RESULTS
Increased protein and mRNA levels of BAD in frontal cortex from BD patients
Mean protein level of pro-apoptotic factor BAD was increased significantly, by 55% (p < 0.05), in BD compared with control frontal cortex (Figure 1a). Further, the mean mRNA level of BAD was significantly increased by 2.16 fold (p < 0.01) in BD compared with control brain (Figure 1b).
Figure 1.

Mean BAD protein (a) (with representative immunoblot) in control (n = 10) and BD frontal cortex (n = 10). Data are ratios of optical densities of BAD protein to β-actin, expressed as percent of control. mRNA level of bad (b) in postmortem control (n = 10) and BD (n = 10) frontal cortex, measured using real time RT-PCR. Data are mRNA level of bad in the BD patients normalized to the endogenous control (β-globulin) and relative to control level (calibrator), using the ΔΔCT method. Mean ± SEM, *p < 0.05, **p < 0.01.
Decreased protein and mRNA levels of Bcl-2 in frontal cortex from BD patients
Compared with control brain, there were significant decreases in mean protein and mRNA levels of anti-apoptotic factor Bcl-2 (Figure 2a and b) by 32% (p < 0.01) and 0.57 fold (p < 0.01), respectively, in the BD brain.
Figure 2.

Representative immunoblot of Bcl-2 (a) and Bax protein level (c) in frontal cortex of controls (n = 10) and BD patients (n = 10). Data are ratios of optical density of Bcl-2 and Bax to β-actin, expressed as percent of control. mRNA level of bcl-2 (b) and bax (d) in postmortem control (n = 10) and BD (n = 10) frontal cortex, measured using real time RT-PCR. Bar graph (e) is Bax to Bcl-2 ratio in frontal cortex of controls and BD patients. Mean ± SEM, *p < 0.05, **p < 0.01.
Increased protein and mRNA levels of Bax and increased ratio of Bax/Bcl-2 in frontal cortex from BD patients
As illustrated in Figure 2, compared with control, protein and mRNA levels of pro-apoptotic factor Bax were elevated by 57% (p < 0.05) and 2.9 fold (p < 0.01), respectively, in BD frontal cortex (Figure 2c and d). The ratio of Bax to Bcl-2 was significantly increased in BD frontal cortex compared with control (Figure 2e) (p < 0.01).
Increased protein and mRNA levels of Caspase-9 and -3 in frontal cortex from BD patients
Mean protein levels of initiator Caspase-9 and effector Caspase-3 were significantly elevated by 66% and 91%, respectively, in BD brains relative to controls (Figures 3a and 3c; p < 0.05). Mean mRNA levels of Caspase-9 and -3 were also significantly elevated by 4.8 and 5.8 fold, respectively, in BD brains compared with controls (Figures 3b and 3d; p < 0.001). Increased active Caspase-3 was observed in BD compared with control brain by immunohistochemistry (Figure 3e).
Figure 3.

Representative immunoblot of Caspase-9 (a) and -3 (c) protein levels in frontal cortex of controls (n = 10) and BD patients (n = 10). Data are ratios of optical density of Caspases to β-actin, expressed as percent of control. mRNA levels of Caspase-9 (b) and -3 (d) in postmortem control (n = 10) and BD (n = 10) frontal cortex, measured using real time RT-PCR. Mean ± SEM, *p < 0.05, ***p < 0.001. (e) Representative histology of control and BD frontal cortex. Active Caspase-3 was detected by kit as described in materials and methods. Scale bar, 100 μm (lower) and 20 μm (upper). Red stains indicated by arrows represent the expression of active Caspase-3 in BD frontal cortex. Counterstaining was performed using methyl green.
Decreased protein and mRNA levels of BDNF in frontal cortex from BD patients
Compared with control brain, there were significant decreases in the mean protein (p < 0.001) and mRNA (p < 0.01) levels of BDNF (Figure 4a and 4b) in the BD brain.
Figure 4.

Protein level of BDNF (a) was determined by ELISA. mRNA levels of BDNF (b) in postmortem control (n = 10) and BD (n = 10) frontal cortex, measured using real time RT-PCR. Mean ± SEM, **p < 0.01, *** < 0.001.
Decreased protein levels of synaptophysin and drebrin in frontal cortex from BD patients
Mean protein levels of Synaptophysin (Figure 4a) (38%, p < 0.05) and Drebrin (Figure 4c) (40%, p < 0.01) and mean mRNA levels of Synaptophysin and Drebrin (Figure 4b and 4d) were significantly decreased in BD compared with control brain (p < 0.01).
Pearson correlations with brain variables
Pearson correlations between variables (age, PMI and pH) and the mRNA levels from across all 20 brain samples (control and BD patients combined) are not statistically significant (Table 2). Using Bonferroni’s multiple comparison tests between controls, all BD subjects and the subgroup of BD with lithium or controls, all BD subjects and the subgroup of BD with suicide did not show significant change in tested marker levels (mRNA and protein) between all BD subjects and the subgroup of BD with lithium or all BD subjects and the subgroup of BD died with suicide.
Table 2.
Probabilities and Pearson correlation r squared between brain mRNA levels and subject age, postmortem interval and brain pH.
| N=20 | BDNF | BCl-2 | Caspase-3 | Caspase-9 | BAD | BAX | Drebrin | Synaptophysin | |
|---|---|---|---|---|---|---|---|---|---|
| Age, (yr) | P | 0.08 | 0.07 | 0.89 | 0.26 | 0.58 | 0.12 | 0.56 | 0.911 |
| r2 | 0.15 | 0.16 | 0.00 | 0.06 | 0.01 | 0.12 | 0.01 | 0.00 | |
| PMI, (hr) | P | 0.22 | 0.12 | 0.83 | 0.81 | 0.45 | 0.12 | 0.67 | 0.74 |
| r2 | 0.01 | 0.00 | 0.09 | 0.00 | 0.03 | 0.12 | 0.05 | 0.00 | |
| pH | P | 0.70 | 0.83 | 0.27 | 0.27 | 0.48 | 0.79 | 0.55 | 0.55 |
| r2 | 0.08 | 0.12 | 0.00 | 0.03 | 0.12 | 0.00 | 0.00 | 0.00 |
PMI, postmortem interval
DISCUSSION
The present study demonstrates statistically significant decreases in protein and mRNA levels of anti-apoptotic factors (Bcl-2, BDNF) and of synaptic markers (synaptophysin and drebrin), and significant increases in pro-apoptotic factors (Bax, BAD, active Caspase- 3 and -9) in postmortem prefrontal cortex from BD compared with control subjects.
Recent brain imaging studies have revealed that the volumes of the hippocampus, amygdala, and frontal cortex are decreased in BD patients (42–44), and that numbers and sizes of glia and neurons are reduced in discrete brain areas (45, 46). Several studies have also demonstrated mitochondrial dysfunction and increased pro-apoptotic activity in serum of BD patients (8, 9). Although studies implicate the association of apoptosis in the pathophysiology of BD, a few studies have investigated apoptosis directly in the postmortem brain of BD patients.
Members of the Bcl-2 family play important roles in the regulation of apoptosis. The representative member of this family is Bcl-2, an inner mitochondrial membrane protein with anti-apoptotic activity (47). The Bcl-2 homologue, Bax, a monomeric cytosolic protein, displays a pro-apoptotic function. Bax can homodimerize and trigger the activation of terminal caspase by altering mitochondrial function, which results in the release of apoptosis-promoting factors into the cytoplasm. The ratio between Bax/Bcl-2 appears to be essential in deciding the life or death of a cell (48). In our current study, we showed an increased ratio of Bax/Bcl-2 and increased Caspase-3 and -9 active protein and mRNA levels. These results suggest that there might be an aberration in the apoptotic pathway of the BD brain.
We recently reported significant increases of mRNA and protein levels of calcium-dependent phospholipase A2 (cPLA2), which releases arachidonic acid (AA) from membrane phospholipids, in postmortem brain of BD (23). AA can bind 14-3-3ζ protein, which has important roles in preventing apoptosis by retaining the pro-apoptotic protein BAD, and by reducing the binding of 14-3-3ζ to phosphorylated BAD (49). Release of 14-3-3ζ from BAD allows dephosphorylation of BAD and allows BAD to move from the cytoplasm to the mitochondria, where it can displace Bax, leading to apoptosis (50). In this study, we observed increased BAD protein and mRNA levels in postmortem brain of BD. Increased cPLA2 expression may induce AA release which can promote early steps in the apoptotic pathway, through the dissociation of 14-3-3ζ from phosphorylated BAD.
Serretti et al. (51) used linkage and association methods to identify genes that are involved in BD, which included the BDNF gene. BDNF is a primary neurotrophic factor, and plays important roles in cell survival, cell plasticity, and in the growth and differentiation of new neurons and synapses (52). Animal models that demonstrated upregulated AA signaling and bipolar-like behaviors have been reported to have downregulated brain BDNF expression (53, 54). Furthermore, several drugs approved for treating BD show a neuroprotective effect ascribed to increased BDNF expression (55). In our study, we demonstrated decreased BDNF mRNA and protein levels in the frontal cortex of BD. These data indicate that decreased BDNF may be part of the pathophysiology of BD.
Mood stabilizers utilized in BD, when given long-term to rats, downregulate brain expression of cPLA2 or COX-2, which are key enzymes of AA metabolism (56). Furthermore, cPLA2 and COX-2 are increased in prefrontal cortex of the postmortem BD brain (23). Thus increased cPLA2 would release more AA, which may interrupt the anti-apoptotic action of 14-3-3ζ in the brain. Consistent with these findings, a recent study showed that the inhibition of cPLA2-mediated AA release reduced apoptosis in astrocytes (57). Additionally, mood stabilizers are reported to suppress caspase-3 activity, stimulate Bcl-2 and BDNF expression and enhance neurogenesis in rat hippocampus (14–16). Our findings suggest that deregulation of apoptosis may be involved in BD.
Altered pro- and anti-apoptotic factors may cause changes in neuronal markers. Reports have demonstrated loss of synaptic integrity, associated with decreased expression of the postsynaptic marker drebrin and presynaptic marker synaptophsyin, in the Alzheimer disease brain (58, 59). We observed a significant decrease in protein levels of synaptophysin and drebrin in BD brain compared with control. These decreases may be responsible for cognitive deficits that have been reported in BD patients.
The limitation of the present study is non-availability of medical diagnosis, and lack of information on whether the patients were in the manic or depressive phase at the time of death. However, since several BD patients died by suicide, they may have been in the depressed phase of their illness. Also, the BD patients had been exposed to various drugs not experienced by the control subjects, which may have confounded the results. Therefore, our findings may be related to differences in drug exposure, rather than the BD trait. However, no statistical differences were found in all genes studied in the present study when the BD subjects were compared with the subgroup of BD subjects that were treated with lithium (data not shown). Also, no statistical significance was found when the BD subjects were compared to the BD subjects that died by suicide. However, future studies should examine apoptotic and synaptic markers in brains of patients with schizophrenia using roughly comparable drug exposure as a control, or with unipolar major depression, or with Alzheimer disease to test for disease specificity.
In summary, postmortem frontal cortex from BD patients compared with control cortex showed significantly decreased anti-apoptotic factor (Bcl-2 and BDNF) protein and mRNA levels, and reduced protein levels of synaptic markers (synaptophysin and drebrin), but increased protein and mRNA expression of pro-apoptotic factors (Bax, BAD and caspase-9/-3). These alterations may enhance apoptosis in the frontal cortex of BD patients. Apoptosis and synaptic loss may occur in the presence of neuroinflammation and excitotoxicity in the BD brain, and may be triggered or interact with these process (24). These multiple pathological processes may be the basis of disease progression, evidenced by reports of progressive mood disturbance, brain atrophy and cognitive decline. Therapeutic strategies aimed at downregulating apoptotic processes and neuronal degeneration might be effective in slowing the progression of BD. Mood stabilizers may help to do this (56, 60).
Figure 5.

Representative immunoblot of Synaptophysin (a) and Drebrin (c) protein levels in frontal cortex of controls (n = 10) and BD patients (n = 10). Data are ratios of optical density of synaptophysin and drebrin to β-actin, expressed as percent of control. mRNA levels of Synaptophysin (b) and Drebrin (d) in postmortem control (n = 10) and BD (n = 10) frontal cortex, measured using real time RT-PCR. Mean ± SEM, *p < 0.05, **p < 0.01, *** < 0.001.
Figure 6.
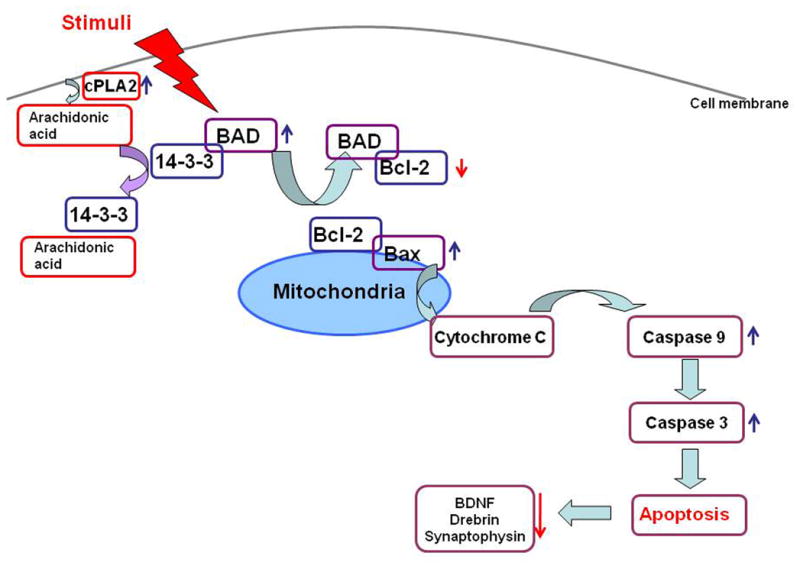
Schematic diagram of apoptotic pathway in brain of bipolar disorder patients. Various stimuli can initiate apoptosis cascade, including diseases. Once triggered, pro-apoptotic factor BAD is dissociated from the complex with 14-3-3ζ, and replace anti-apoptotic factor Bcl-2, leading to release cytochrome C from mitochondria, which in turn activates Capases-9 and Caspase-3. Apoptotic change may result in loss of synaptic integrity with decreased expression of synaptic markers, Synaptophysin and Drebrin.
Acknowledgments
We thank the Harvard Brain Bank, Boston, MA for providing the postmortem brain samples under PHS grant number R24MH068855. This research was entirely supported by the Intramural Research Programs of the National Institute on Aging and the National Institute of Environmental Health Sciences, National Institutes of Health Bethesda, MD 20892. We thank the NIH Fellows Editorial Board and Dr. Eugene Streicher for proofreading the manuscripts.
Abbreviations
- AA
arachidonic acid
- BD
bipolar disorder
- BDNF
brain derived neurotrophic factor
- Bcl-2
B-cell lymphoma-2
- Bax
Bcl-2-associated X protein
- BAD
Bcl-2-associated death promoter
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kupfer DJ. The increasing medical burden in bipolar disorder. Jama. 2005 May 25;293(20):2528–2530. doi: 10.1001/jama.293.20.2528. [DOI] [PubMed] [Google Scholar]
- 2.Baum AE, Akula N, Cabanero M, Cardona I, Corona W, Klemens B, et al. A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Molecular psychiatry. 2008 Feb;13(2):197–207. doi: 10.1038/sj.mp.4002012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim YK, Jung HG, Myint AM, Kim H, Park SH. Imbalance between pro-inflammatory and anti-inflammatory cytokines in bipolar disorder. J Affect Disord. 2007 Dec;104(1–3):91–95. doi: 10.1016/j.jad.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 4.Mueller HT, Meador-Woodruff JH. NR3A NMDA receptor subunit mRNA expression in schizophrenia, depression and bipolar disorder. Schizophrenia research. 2004 Dec 1;71(2–3):361–370. doi: 10.1016/j.schres.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 5.Mundo E, Tharmalingham S, Neves-Pereira M, Dalton EJ, Macciardi F, Parikh SV, et al. Evidence that the N-methyl-D-aspartate subunit 1 receptor gene (GRIN1) confers susceptibility to bipolar disorder. Molecular psychiatry. 2003 Feb;8(2):241–245. doi: 10.1038/sj.mp.4001218. [DOI] [PubMed] [Google Scholar]
- 6.O’Brien SM, Scully P, Scott LV, Dinan TG. Cytokine profiles in bipolar affective disorder: focus on acutely ill patients. Journal of affective disorders. 2006 Feb;90(2–3):263–267. doi: 10.1016/j.jad.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 7.Andreazza AC, Frey BN, Erdtmann B, Salvador M, Rombaldi F, Santin A, et al. DNA damage in bipolar disorder. Psychiatry research. 2007 Sep 30;153(1):27–32. doi: 10.1016/j.psychres.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 8.Politi P, Brondino N, Emanuele E. Increased proapoptotic serum activity in patients with chronic mood disorders. Archives of medical research. 2008 Feb;39(2):242–245. doi: 10.1016/j.arcmed.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 9.Shao L, Martin MV, Watson SJ, Schatzberg A, Akil H, Myers RM, et al. Mitochondrial involvement in psychiatric disorders. Ann Med. 2008;40(4):281–295. doi: 10.1080/07853890801923753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lyoo IK, Sung YH, Dager SR, Friedman SD, Lee JY, Kim SJ, et al. Regional cerebral cortical thinning in bipolar disorder. Bipolar disorders. 2006 Feb;8(1):65–74. doi: 10.1111/j.1399-5618.2006.00284.x. [DOI] [PubMed] [Google Scholar]
- 11.Rajkowska G. Depression: what we can learn from postmortem studies. Neuroscientist. 2003 Aug;9(4):273–284. doi: 10.1177/1073858403252773. [DOI] [PubMed] [Google Scholar]
- 12.Gasiorowski K, Brokos B. DNA repair of hydrogen peroxide-induced damage in human lymphocytes in the presence of four antimutagens. A study with alkaline single cell gel electrophoresis (comet assay) Cellular & molecular biology letters. 2001;6(4):897–911. [PubMed] [Google Scholar]
- 13.Shao L, Young LT, Wang JF. Chronic treatment with mood stabilizers lithium and valproate prevents excitotoxicity by inhibiting oxidative stress in rat cerebral cortical cells. Biological psychiatry. 2005 Dec 1;58(11):879–884. doi: 10.1016/j.biopsych.2005.04.052. [DOI] [PubMed] [Google Scholar]
- 14.Chuang DM. Neuroprotective and neurotrophic actions of the mood stabilizer lithium: can it be used to treat neurodegenerative diseases? Critical reviews in neurobiology. 2004;16(1–2):83–90. doi: 10.1615/critrevneurobiol.v16.i12.90. [DOI] [PubMed] [Google Scholar]
- 15.Mora A, Gonzalez-Polo RA, Fuentes JM, Soler G, Centeno F. Different mechanisms of protection against apoptosis by valproate and Li+ European journal of biochemistry/FEBS. 1999 Dec;266(3):886–891. doi: 10.1046/j.1432-1327.1999.00919.x. [DOI] [PubMed] [Google Scholar]
- 16.Mora A, Sabio G, Risco AM, Cuenda A, Alonso JC, Soler G, et al. Lithium blocks the PKB and GSK3 dephosphorylation induced by ceramide through protein phosphatase-2A. Cellular signalling. 2002 Jun;14(6):557–562. doi: 10.1016/s0898-6568(01)00282-0. [DOI] [PubMed] [Google Scholar]
- 17.Chen G, Rajkowska G, Du F, Seraji-Bozorgzad N, Manji HK. Enhancement of hippocampal neurogenesis by lithium. Journal of neurochemistry. 2000 Oct;75(4):1729–1734. doi: 10.1046/j.1471-4159.2000.0751729.x. [DOI] [PubMed] [Google Scholar]
- 18.Chen RW, Chuang DM. Long term lithium treatment suppresses p53 and Bax expression but increases Bcl-2 expression. A prominent role in neuroprotection against excitotoxicity. The Journal of biological chemistry. 1999 Mar 5;274(10):6039–6042. doi: 10.1074/jbc.274.10.6039. [DOI] [PubMed] [Google Scholar]
- 19.Chang YC, Kim HW, Rapoport SI, Rao JS. Chronic NMDA administration increases neuroinflammatory markers in rat frontal cortex: cross-talk between excitotoxicity and neuroinflammation. Neurochem Res. 2008 Nov;33(11):2318–2323. doi: 10.1007/s11064-008-9731-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee HJ, Rao JS, Chang L, Rapoport SI, Bazinet RP. Chronic N-methyl-D-aspartate administration increases the turnover of arachidonic acid within brain phospholipids of the unanesthetized rat. J Lipid Res. 2008 Jan;49(1):162–168. doi: 10.1194/jlr.M700406-JLR200. [DOI] [PubMed] [Google Scholar]
- 21.Rao JS, Ertley RN, Rapoport SI, Bazinet RP, Lee HJ. Chronic NMDA administration to rats up-regulates frontal cortex cytosolic phospholipase A2 and its transcription factor, activator protein-2. J Neurochem. 2007 Sep;102(6):1918–1927. doi: 10.1111/j.1471-4159.2007.04648.x. [DOI] [PubMed] [Google Scholar]
- 22.Rapoport SI, Bosetti F. Do lithium and anticonvulsants target the brain arachidonic acid cascade in bipolar disorder? Arch Gen Psychiatry. 2002 Jul;59(7):592–596. doi: 10.1001/archpsyc.59.7.592. [DOI] [PubMed] [Google Scholar]
- 23.Rao JS, Kim HW, Rapoport SI. Society for Neuroscience. San Diego: 2007. Up-regulated arachidonic acid cascade enzymes and their transcription factors in post-mortem frontal cortex from bipolar disorder patients; p. 707.705/Z704. [Google Scholar]
- 24.Rao JS, Harry GJ, Rapoport SI, Kim HW. Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Mol Psychiatry. 2009 Jun 2; doi: 10.1038/mp.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McIntosh AM, Moorhead TW, McKirdy J, Hall J, Sussmann JE, Stanfield AC, et al. Prefrontal gyral folding and its cognitive correlates in bipolar disorder and schizophrenia. Acta Psychiatr Scand. 2009 Mar;119(3):192–198. doi: 10.1111/j.1600-0447.2008.01286.x. [DOI] [PubMed] [Google Scholar]
- 26.Sachs G, Schaffer M, Winklbaur B. Cognitive deficits in bipolar disorder. Neuropsychiatr. 2007;21(2):93–101. [PubMed] [Google Scholar]
- 27.Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol. 1997 Aug;56(8):933–944. doi: 10.1097/00005072-199708000-00011. [DOI] [PubMed] [Google Scholar]
- 28.Harigaya Y, Shoji M, Shirao T, Hirai S. Disappearance of actin-binding protein, drebrin, from hippocampal synapses in Alzheimer’s disease. J Neurosci Res. 1996 Jan 1;43(1):87–92. doi: 10.1002/jnr.490430111. [DOI] [PubMed] [Google Scholar]
- 29.Rubinsztein JS, Fletcher PC, Rogers RD, Ho LW, Aigbirhio FI, Paykel ES, et al. Decision-making in mania: a PET study. Brain. 2001 Dec;124(Pt 12):2550–2563. doi: 10.1093/brain/124.12.2550. [DOI] [PubMed] [Google Scholar]
- 30.Suhara T, Nakayama K, Inoue O, Fukuda H, Shimizu M, Mori A, et al. D1 dopamine receptor binding in mood disorders measured by positron emission tomography. Psychopharmacology. 1992;106(1):14–18. doi: 10.1007/BF02253582. [DOI] [PubMed] [Google Scholar]
- 31.Rajkowska G. Cell pathology in bipolar disorder. Bipolar disorders. 2002 Apr;4(2):105–116. doi: 10.1034/j.1399-5618.2002.01149.x. [DOI] [PubMed] [Google Scholar]
- 32.Lyoo IK, Kim MJ, Stoll AL, Demopulos CM, Parow AM, Dager SR, et al. Frontal lobe gray matter density decreases in bipolar I disorder. Biological psychiatry. 2004 Mar 15;55(6):648–651. doi: 10.1016/j.biopsych.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 33.Buchsbaum MS. Brain imaging in the search for biological markers in affective disorder. The Journal of clinical psychiatry. 1986 Oct;47( Suppl):7–12. [PubMed] [Google Scholar]
- 34.Buchsbaum MS, Wu J, DeLisi LE, Holcomb H, Kessler R, Johnson J, et al. Frontal cortex and basal ganglia metabolic rates assessed by positron emission tomography with [18F]2-deoxyglucose in affective illness. Journal of affective disorders. 1986 Mar–Apr;10(2):137–152. doi: 10.1016/0165-0327(86)90036-4. [DOI] [PubMed] [Google Scholar]
- 35.Kim HW, Rapoport SI, Rao JS. Society for Neuroscience. Washington DC: 2008. Increased expression of apoptotic factors in postmortem brain from bipolar disorder patietnts; p. 414.413. [Google Scholar]
- 36.Harrison PJ, Heath PR, Eastwood SL, Burnet PW, McDonald B, Pearson RC. The relative importance of premortem acidosis and postmortem interval for human brain gene expression studies: selective mRNA vulnerability and comparison with their encoded proteins. Neurosci Lett. 1995 Nov 24;200(3):151–154. doi: 10.1016/0304-3940(95)12102-a. [DOI] [PubMed] [Google Scholar]
- 37.Rao JS, Rapoport SI, Kim HW. Decreased GRK3 but not GRK2 expression in frontal cortex from bipolar disorder patients. Int J Neuropsychopharmacol. 2009 Apr;29:1–10. doi: 10.1017/S146114570900025X. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Dwivedi Y, Rizavi HS, Rao JS, Pandey GN. Modifications in the phosphoinositide signaling pathway by adrenal glucocorticoids in rat brain: focus on phosphoinositide-specific phospholipase C and inositol 1,4,5-trisphosphate. J Pharmacol Exp Ther. 2000 Oct;295(1):244–254. [PubMed] [Google Scholar]
- 39.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods (San Diego, Calif. 2001 Dec;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 40.Rao JS, Ertley RN, Lee HJ, Rapoport SI, Bazinet RP. Chronic fluoxetine upregulates activity, protein and mRNA levels of cytosolic phospholipase A2 in rat frontal cortex. Pharmacogenomics J. 2006 Nov–Dec;6(6):413–420. doi: 10.1038/sj.tpj.6500391. [DOI] [PubMed] [Google Scholar]
- 41.Rao JS, Harry GJ, Rapoport SI, Kim HW. Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Molecular Psychiatry. 2009 doi: 10.1038/mp.2009.47. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blumberg HP, Fredericks C, Wang F, Kalmar JH, Spencer L, Papademetris X, et al. Preliminary evidence for persistent abnormalities in amygdala volumes in adolescents and young adults with bipolar disorder. Bipolar Disord. 2005 Dec;7(6):570–576. doi: 10.1111/j.1399-5618.2005.00264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frazier JA, Breeze JL, Makris N, Giuliano AS, Herbert MR, Seidman L, et al. Cortical gray matter differences identified by structural magnetic resonance imaging in pediatric bipolar disorder. Bipolar Disord. 2005 Dec;7(6):555–569. doi: 10.1111/j.1399-5618.2005.00258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haznedar MM, Roversi F, Pallanti S, Baldini-Rossi N, Schnur DB, Licalzi EM, et al. Fronto-thalamo-striatal gray and white matter volumes and anisotropy of their connections in bipolar spectrum illnesses. Biol Psychiatry. 2005 Apr 1;57(7):733–742. doi: 10.1016/j.biopsych.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 45.Brauch RA, Adnan El-Masri M, Parker JC, Jr, El-Mallakh RS. Glial cell number and neuron/glial cell ratios in postmortem brains of bipolar individuals. Journal of affective disorders. 2006 Mar;91(1):87–90. doi: 10.1016/j.jad.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 46.Rajkowska G, Halaris A, Selemon LD. Reductions in neuronal and glial density characterize the dorsolateral prefrontal cortex in bipolar disorder. Biological psychiatry. 2001 May 1;49(9):741–752. doi: 10.1016/s0006-3223(01)01080-0. [DOI] [PubMed] [Google Scholar]
- 47.Kirkin V, Joos S, Zornig M. The role of Bcl-2 family members in tumorigenesis. Biochim Biophys Acta. 2004 Mar 1;1644(2–3):229–249. doi: 10.1016/j.bbamcr.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 48.Cvejic D, Selemetjev S, Savin S, Paunovic I, Petrovic I, Tatic S. Apoptosis and proliferation related molecules (Bcl-2, Bax, p53, PCNA) in papillary microcarcinoma versus papillary carcinoma of the thyroid. Pathology. 2008 Aug;40( 5):475–480. doi: 10.1080/00313020802026989. [DOI] [PubMed] [Google Scholar]
- 49.Brock TG. Arachidonic acid binds 14-3-3zeta, releases 14-3-3zeta from phosphorylated BAD and induces aggregation of 14-3-3zeta. Neurochem Res. 2008 May;33(5):801–807. doi: 10.1007/s11064-007-9498-3. [DOI] [PubMed] [Google Scholar]
- 50.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996 Nov 15;87(4):619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 51.Serretti A, Mandelli L. The genetics of bipolar disorder: genome ‘hot regions,’ genes, new potential candidates and future directions. Mol Psychiatry. 2008 Aug;13(8):742–771. doi: 10.1038/mp.2008.29. [DOI] [PubMed] [Google Scholar]
- 52.Chuang DM. The antiapoptotic actions of mood stabilizers: molecular mechanisms and therapeutic potentials. Ann N Y Acad Sci. 2005 Aug;1053:195–204. doi: 10.1196/annals.1344.018. [DOI] [PubMed] [Google Scholar]
- 53.Rao JS, Ertley RN, DeMar JC, Jr, Rapoport SI, Bazinet RP, Lee HJ. Dietary n-3 PUFA deprivation alters expression of enzymes of the arachidonic and docosahexaenoic acid cascades in rat frontal cortex. Mol Psychiatry. 2007 Feb;12(2):151–157. doi: 10.1038/sj.mp.4001887. [DOI] [PubMed] [Google Scholar]
- 54.Rao JS, Ertley RN, Lee HJ, DeMar JC, Jr, Arnold JT, Rapoport SI, et al. n-3 polyunsaturated fatty acid deprivation in rats decreases frontal cortex BDNF via a p38 MAPK-dependent mechanism. Mol Psychiatry. 2007 Jan;12(1):36–46. doi: 10.1038/sj.mp.4001888. [DOI] [PubMed] [Google Scholar]
- 55.Hammonds MD, Shim SS. Effects of 4-week Treatment with Lithium and Olanzapine on Levels of Brain-derived Neurotrophic Factor, B-Cell CLL/Lymphoma 2 and Phosphorylated Cyclic Adenosine Monophosphate Response Element-binding Protein in the Sub-regions of the Hippocampus. Basic Clin Pharmacol Toxicol. 2009 Apr 17; doi: 10.1111/j.1742-7843.2009.00416.x. [DOI] [PubMed] [Google Scholar]
- 56.Rao JS, Lee HJ, Rapoport SI, Bazinet RP. Mode of action of mood stabilizers: is the arachidonic acid cascade a common target? Mol Psychiatry. 2008 Jun;13(6):585–596. doi: 10.1038/mp.2008.31. [DOI] [PubMed] [Google Scholar]
- 57.Gabryel B, Chalimoniuk M, Stolecka A, Waniek K, Langfort J, Malecki A. Inhibition of arachidonic acid release by cytosolic phospholipase A2 is involved in the antiapoptotic effect of FK506 and cyclosporin a on astrocytes exposed to simulated ischemia in vitro. J Pharmacol Sci. 2006 Sep;102(1):77–87. [PubMed] [Google Scholar]
- 58.Hatanpaa K, Isaacs KR, Shirao T, Brady DR, Rapoport SI. Loss of proteins regulating synaptic plasticity in normal aging of the human brain and in Alzheimer disease. J Neuropathol Exp Neurol. 1999 Jun;58(6):637–643. doi: 10.1097/00005072-199906000-00008. [DOI] [PubMed] [Google Scholar]
- 59.Leuba G, Walzer C, Vernay A, Carnal B, Kraftsik R, Piotton F, et al. Postsynaptic density protein PSD-95 expression in Alzheimer’s disease and okadaic acid induced neuritic retraction. Neurobiol Dis. 2008 Jun;30(3):408–419. doi: 10.1016/j.nbd.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 60.Rapoport SI, Basselin M, Kim HW, Rao JS. Bipolar disorder and mechanisms of action of mood stabilizers. Brain Research Review. 2009 doi: 10.1016/j.brainresrev.2009.06.003. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]