

Abstract
G-protein–coupled receptors (GPCRs) are the largest family of transmembrane signaling proteins in the human genome. Events in the GPCR signaling cascade have been well characterized, but the receptor composition and its membrane distribution are still generally unknown. Although there is evidence that some members of the GPCR superfamily exist as constitutive dimers or higher oligomers, interpretation of the results has been disputed, and recent studies indicate that monomeric GPCRs may also be functional. Because there is controversy within the field, to address the issue we have used total internal reflection fluorescence microscopy (TIRFM) in living cells to visualize thousands of individual molecules of a model GPCR, the M1 muscarinic acetylcholine receptor. By tracking the position of individual receptors over time, their mobility, clustering, and dimerization kinetics could be directly determined with a resolution of ~30 ms and ~20 nm. In isolated CHO cells, receptors are randomly distributed over the plasma membrane. At any given time, ~30% of the receptor molecules exist as dimers, and we found no evidence for higher oligomers. Two-color TIRFM established the dynamic nature of dimer formation with M1 receptors undergoing interconversion between monomers and dimers on the timescale of seconds.
Keywords: acetylcholine receptor, dimerization, G-protein–coupled receptor, receptor clustering, receptor mobility
Muscarinic acetylcholine receptors are members of the major subfamily A of G-protein–coupled receptors (GPCRs). The M1 subtype is selectively distributed in the forebrain and is considered to play an important role in cognition and memory (1, 2). A critical question is how these receptors bind acetylcholine and produce downstream signaling via heterotrimeric G proteins. In particular, a major issue is whether muscarinic receptors exist as obligate dimers or higher oligomers. Initial events in the GPCR signaling cascade can be monitored by measuring radioligand binding to membrane-bound receptors and associated heterotrimeric G proteins and by measuring early biochemical or electrophysiological signals. However, the composition of receptor complexes is not generally known at the level of individual molecular species. Within the GPCR field there is considerable controversy regarding the molecular state of receptors at the surface of living cells. There is incontrovertible evidence that some members of the small subfamily C of GPCRs are constitutive dimers (reviewed in refs. 3 and 4) and substantial evidence that certain members of the largest subfamily A may exist as dimers (4 –6) or even as higher oligomers (6, 7). There is also evidence that GPCR heterodimers may exist (reviewed in ref. 8). However, the principal experimental approach used for many in vivo studies has been resonance energy transfer, and interpretation of its data has led to controversy (9 –12). Adding to the debate, some recent studies have indicated that the monomeric state of some GPCRs can act as a functional unit (13 –16). Using bulk methods, it has been difficult to unambiguously determine the oligomeric state and mobility of molecules within living cells. However, single-molecule approaches can give direct information about how molecules move and interact within the cellular context. Total internal reflection fluorescence microscopy (TIRFM) is a wide-field imaging method that enables hundreds or thousands of individual molecules to be visualized within living cells at sufficiently high spatial and temporal resolution that they can be observed and tracked over a period of several seconds. Using this approach, we have been able to observe the transient formation and disruption of M1 receptor dimers in real time.
Results
To study individual muscarinic receptors, it was necessary to label them specifically with a fluorescent species having suitable biophysical characteristics in terms of absorption wavelength and resistance to photobleaching. Two fluorescent muscarinic antagonists were synthesized (SI Text, Figs. S1–S3, and Tables S1 and S2), and one of these, Cy3B–telenzepine (17) (Fig. 1A), was found to have a very high affinity for M1 receptors with a K d of 35 pM (−logK d 10.46 ± 0.06) at 37 °C and very slow dissociation kinetics (t 1/2 7 ± 2 h at 37 °C and 23–110 h at 23 °C), determined by measuring the time-dependent binding of a fast-associating muscarinic radioligand, [3H]-N-methylscopolamine, in the presence and absence of Cy3B–telenzepine (SI Text, Fig. S4, and Table S3). A second ligand, Alexa488–telenzepine (Table S3 and SI Text), had a lower affinity for the M1 receptor (K d 0.5 nM at 23 °C, −logK d 9.32 ± 0.07) but still had very slow dissociation kinetics (t 1/2 3.6 ± 0.6 h). The time dependence and equilibrium binding of both fluorescent antagonists were compatible with the simple model in which the receptor can be occupied by either antagonist or radioligand (SI Text) and not by models with more complex stoichiometries.
Fig. 1.

TIRFM recording of a single CHO cell, transfected with M1 muscarinic receptors and labeled with the fluorescent antagonist, Cy3B–telenzepine. (A) Structural formula of Cy3B–telenzepine. (B) A single video frame (33-ms exposure) at the start of a recording. The insert shows a 5 × 5-μm2 expanded region containing 15 objects, 4 having intensity consistent with two fluorophores (attributed to receptor dimers) and 11 with intensity similar to a single fluorophore. A pseudocolor look-up table is shown inset. The first 100 frames of the recording are shown in Movie S1. (C) A total of 2,659 tracks of moving M1 receptors identified from the entire recording of the cell shown in B. The insert shows nine trajectories that illustrate the random nature of the diffusive process. Movie S2 shows the identification of tracks by the tracking algorithm. (D) Plot of average mean square displacement (MSD) versus the time interval (δt) for the trajectories shown in C. The plot is linear (r 2 = 0.99), over 1 μm in length, with a 5-s timescale, which is consistent with receptor movement following a random walk, and shows no evidence for anomalous diffusive behavior. (E) Histogram of the intensity of 911 objects identified in the single video frame shown in B. The shape of the distribution is due to the presence of two populations, attributed to receptor dimers (red arrowhead, intensity ~120 counts/pixel) and monomers (black arrowhead, ~60 counts/pixel). The spread of the data arises from a combination of photon noise and intensity variation between spots that are located at different regions within the specimen.
Visualization of Single M1 Muscarinic Receptors.
The high affinity of the Cy3B–telenzepine ligand allows stoichiometric labeling of M1 receptors on CHO cells, with up to 97% being labeled by 0.1–1 nM concentrations (see SI Text). The slow off-rate means that any unbound or nonspecifically bound ligand can be washed away with no substantial loss of receptor labeling over a period of hours. The level of receptor expression used in the present study is in the range found for the expression of M1 receptors in rat cerebral cortex [~1 pmol receptor per mg membrane protein (18)].
When the basal plasma membrane of live cells was viewed using laser-based TIRFM (SI Text), numerous moving fluorescent spots were observed, and their positions and intensities were recorded using an electron multiplying (EMCCD) video camera system at a rate of either 25 or 33 frames·s−1. To confirm specific labeling of M1 receptors by the Cy3B–telenzepine ligand, cells were pretreated with 10−5 M atropine (a potent muscarinic antagonist). We found just a small number of stationary spots adhering to the glass coverslip surface (nonspecific binding), but cell labeling was completely blocked and no moving fluorescent spots were observed at the plasma membrane. Figure 1B shows a single video frame taken from a 48-s recording of the basolateral cell surface of a living CHO cell where the transfected M1 receptors were labeled with 1 nM Cy3B–telenzepine. A large number of fluorescent spots with intensities comparable to those of single molecules adsorbed to a glass slide were observed. A region of interest in Fig. 1B is highlighted and enlarged to illustrate the pixelated appearance of the image of the individual receptors. The first 3 s of this recording is shown in Movie S1. At the laser excitation intensity used in this recording, fluorescent spots photobleached with a half time of ~5 s. If, after photobleaching, the laser was turned off for several minutes, unbleached labeled receptors diffused to the basal surface of the cell from regions that had not been exposed to the evanescent field produced by the TIRF illumination. This fluorescence recovery procedure could be repeated a number of times to obtain multiple recordings from the same cell.
Fluorescent molecules were identified and tracked using digital image analysis (19). An example of a recording is in Fig. 1C and Movie S2. The mean lateral diffusion coefficient of the Cy3B–telenzepine-labeled receptors at 23 °C was 0.089 ± 0.019 μm2·s−1 (mean ± SD for 35,208 tracks from 23 cells, range 0.06–0.13 μm2·s−1 for different cells). A similar diffusion coefficient was found for Alexa488-labeled M1 receptors [0.079 ± 0.006 μm2·s−1 (mean ± SD for 15,944 tracks from nine cells)]. At 37 °C, the labeled receptors moved faster, with a mean lateral diffusion coefficient of 0.16 ± 0.04 μm2·s−1 (3,540 tracks from eight cells). The higher diffusion rate at 37 °C made it more difficult to track receptors at the expression levels found in most cells examined (range ~0.5–2 receptors·μm−2).
Distribution and Diffusion of M1 Receptors on the Cell Surface.
To characterize the motion of receptors at the membrane, we calculated their mean squared displacement (MSD) over a range of time intervals (δt). The linear relationship between MSD and δt (Fig. 1D) shows that receptor diffusion is consistent with a classical random walk. There was no evidence of restricted or anomalous diffusion over the timescales explored in this study (50 ms–5 s). To test whether receptors are clustered or corralled at the plasma membrane, we examined the distribution of tracks over the duration of the video records and of fluorescent spots observed at the very start of the recording. The distribution of molecules was examined either by counting within 3 × 3-μm2 grids (or quadrants) overlaid on images of the cell or by performing a nearest-neighbor statistical analysis of the objects. We found no evidence for clustering of the receptors at any region of the plasma membrane of isolated cells. As expected, agonists showed no detectable effects when applied to cells where the M1 receptors had been prelabeled to high occupancy by Cy3B–telenzepine.
M1 Receptors Exist as Monomers and Dimers.
A histogram (Fig. 1E) of the intensities of fluorescent spots observed at the beginning of the video record (Fig. 1B) shows an asymmetric distribution skewed to higher intensities than that expected for a single fluorophore. The origin of the asymmetry is evident when the intensities of individual tracks are plotted as a function of time (Fig. 2). Some tracks have intensities that can be categorized as “level 1” [corresponding to the intensity of a single fluorophore, as measured from the intensity of nonspecifically bound ligand molecules attached to the glass slide near the cell being observed (described in more detail in SI Text)], others as “level 2” (corresponding to the intensity of two fluorophores), and a third type of track switches from intensity 2 to 1 (termed 2 → 1) before a track terminates. As described earlier, the binding studies show that one molecule of Cy3B–telenzepine binds to one M1 receptor, so the first two types of track are compatible with the presence of monomers and dimers, respectively. The third type of track is characteristic of two-step photobleaching whereby two molecules of Cy3B–telenzepine are bound to a dimer, and they sequentially photobleach. However, tracks of type 2 → 1 are also compatible with dissociation of the dimer into two monomers because the tracking algorithm would follow only one of the two tracks that are generated upon dimer disruption. The distribution of fluorescence intensities for individual spots is compatible with the presence of monomers (mean ~60 counts/pixel) and dimers (mean ~120 counts/pixel) indicated by the black and red arrowheads in Fig. 1E.
Fig. 2.

Representative examples of some of the different intensity changes shown by individual tracks of Cy3B-labeled M1 receptors. (A) Intensity trajectory corresponding to a single molecule (upper trace), compared to the background intensity of a neighboring region containing no fluorescently labeled receptors (lower trace). (B) Intensity trajectory of a single moving object with an intensity corresponding to two molecules of Cy3B–telenzepine, i.e., a dimer. (C) Intensity trajectory of a single moving object with an intensity corresponding to two Cy3B fluorophores (level 2), which changes level abruptly to that of a single fluorophore (level 1), which is due to either photobleaching or dissociation of the dimer into two monomers. (D) Intensity trajectory that changes abruptly from level 1 to level 2 (i.e., 1 → 2). This might arise if the initially tracked monomer forms a dimer with another Cy3B-labeled receptor. (E) Intensity trajectory that exhibits abrupt changes from level 1 → 2 → 1. (monomer → dimer → monomer). (F) A rare and more complex intensity track that switches from 2 → 1 → 2 (dimer → monomer → dimer). This track could be interpreted as the transient dissociation of a dimer. This behavior was confirmed by dual-color imaging (see Fig. 4G: record 3). Automatic assignment of intensity changes is described in the SI Text and Fig. S5. The signal fluctuations observed in each record are mainly due to photon noise whereas the differences in average intensity level attributed to one or two fluorophores found for the different objects are caused by spatial heterogeneity in the laser illumination and object position within the depth of the evanescent field.
Because the conditions used in these experiments were such that ~97% of the receptors were labeled, it is important to note that, if the M1 receptors were constitutive dimers, the percentage of objects of intensity 2 at the start of the recording would be close to 100% but would progressively fall to zero as photobleaching occurred. We found that the starting percentage of dual-labeled receptors (level 2) never approached 100% (typically 10–15%) and showed no strong time-dependent change during the recording period (SI Text). These findings are incompatible with the idea that M1 receptors form constitutive dimers. Furthermore, we did not observe objects at the plasma membrane with an intensity corresponding to three or more fluorophores or objects showing three or more photobleaching steps. Our data provide no evidence for the presence of receptor trimers or higher oligomers as suggested by atomic force microscopy observations of rhodopsin in retinal preparations (7). However, estimates of the steady-state fraction of M1 dimers from these data are not precise because both photobleaching and the nature of the video tracking method lead to an underestimation.
Evidence for Reversible Dimer Formation.
Visual inspection of individual track intensities from a single cell allowed assignment to three categories (1, 2, and 2 → 1), giving 82%, 7%, and 3%, respectively, of 1346 identified tracks. The remainder were more complex or could not be unambiguously assigned. To make this procedure more rigorous, an automated procedure was developed to assign intensity trajectories to specific categories, and this also allowed more complex intensity changes to be categorized for very large data sets (SI Textand Fig. S5). Using this procedure, the percentage of tracks in different categories for the same cell described above (i.e., 1, 2, and 2 → 1) was 81%, 9%, and 3%, respectively. The major population, that of level 1, showed no evidence of two-step photobleaching, which means that level 1 cannot correspond to a dually labeled receptor dimer nor level 2 to a receptor tetramer. We found that more complex intensity trajectories accounted for the remainder, and these categories were dominated by 1 → 2, 1 → 2 → 1, and 2 → 1 → 2 trajectories (3%, 3%, and 0.5%, respectively) (Fig. 2). These changes in intensity are compatible with reversible dimer formation, which can be tested specifically by inspecting the individual frames of original video data (Fig. 3). In many cases, as illustrated here, we found that the more complex intensity trajectories arose from two objects of intensity 1, coalescing to form a single spot of intensity 2, which then diffused for some time before the objects separated (i.e., the original spots returned to intensity 1). However, optical diffraction means that, if two point sources of light are separated by less than 300 nm (the Rayleigh limit), the images coalesce and appear as a single, bright spot. This can make interpretation of track intensity fluctuations ambiguous because it is not clear if molecules have associated or simply passed close by.
Fig. 3.

Illustration of two molecules of Cy3B-labeled M1 receptors reversibly forming a dimer. One hundred sequential frames (30-ms exposure time/frame) of two molecules of Cy3B-labeled M1 receptors diffusing on the surface of a CHO cell. These molecules are initially monomers (about the first 21 frames), form a dimer (single red spot) for ∼40 frames (∼1 s), and then dissociate into monomers, possibly for the rest of the recording. There may be a transient reformation of short lifetime dimers for about three frames in the latter part of the recording, but it is not possible to exclude this being a chance overlap of the images of two molecules that are not interacting but diffusing independently close to each other. (Lower) A color look-up table. Two-color imaging (see text and Fig. 4) was used to study the details of M1 receptor dimerization.
Two-Color TIRFM Measurements of M1 Receptor Dynamics.
To address the issue of whether M1 receptors undergo reversible dimer formation, we devised a dual-color imaging technique in which receptors were labeled in a 1:1 ratio with Cy3B–telenzepine and Alexa488–telenzepine (SI Text). The known affinities of the two ligands, derived from bulk studies, enabled us to control the labeling ratio. We used alternating dual-color TIRFM (SI Text) to visualize the movements of receptors. Excitation was switched between two lasers (488 and 566 nm) so the specimen was illuminated with blue and then green light on alternate, sequential video frames. A dual bandpass emission filter allowed either the green or red fluorescence signals to be separated from the scattered laser excitation light so the monochrome video images represented the intensity of green and red fluorescence. Video recordings of up to 50 s were stored, and the image stack could later be displayed using appropriate color look-up tables (Fig. 4 A–F, SI Text, Movies S3 and S4, and Table S4).
Fig. 4.

Two-color TIRFM of labeled M1 receptors moving on a single CHO cell. (A) A 33-ms frame of Alexa488–telenzepine-labeled M1 receptors (green). (B) Image of the same cell recorded 33 ms later with the Cy3B–telenzepine-labeled receptors (red). Scale bar (5 μm) in B applies to A–F. (C) Superposition of images in A and B. Yellow spots indicate candidate dimers or chance colocalization. (D) A total of 825 trajectories of Alexa488–telenzepine-labeled M1 receptors identified during the 44-s recording. (E) A total of 970 trajectories of Cy3B–telenzepine-labeled M1 receptors. (F) A total of 241 trajectories of M1 dimers labeled with both green and red fluorophores. (G) Different tracks showing dimer formation and dissociation. The x and y coordinates are shown for five examples in the upper and lower sections of each numbered plot. Coincidence of the tracks in both the x and y dimensions for a period >660 ms was taken as evidence of dimer formation (separation <160 nm). The green and red objects are not viewed simultaneously but 33 ms apart so the objects may move during that time. The five examples shown demonstrate different behaviors: (1) trajectory of a two-color dimer formation' (2) trajectory of a two-color dimer dissociation; (3) trajectory of a two-color dimer that dissociates after ∼1.5 s and then reforms a dimer; (4, 5) formation and dissociation of two two-color dimers. (Inset) A histogram of the lifetimes of 84 M1 receptor dimers taken from trajectories similar to those in examples 4 and 5 and collected in 0.5-s bins. The solid line is a monoexponential fit for a mean lifetime of 0.5 s.
The initial densities of green and red fluorescently labeled receptors were each ~0.9 μm−2, reflecting the 1:1 labeling stoichiometry. At this receptor density, individual receptors appear as diffraction-limited spots of light. When two adjacent frames representing green and red fluorescence intensities are overlaid (Fig. 4C), regions where both green and red channels are of high intensity give rise to a yellow color. [Yellow spots can indicate the presence of dimers or the chance presence of two monomers within the same diffraction-limited (~300 nm) distance in the membrane.] Furthermore, overlap between individual spots might be imperfect if the time delay (33 ms) between acquisition of the image pair had allowed a receptor dimer (labeled with one green and one red fluorophore) to diffuse by more than 300 nm. Although the root mean square displacement is <x> = 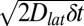 ≈ 80 nm for δt = 33 ms, the random nature of diffusion means that larger and smaller excursions in position will occur between adjacent frames by chance.
≈ 80 nm for δt = 33 ms, the random nature of diffusion means that larger and smaller excursions in position will occur between adjacent frames by chance.
By tracking green and red spots separately (12,485 red and 9,469 green tracks from 9 cells, giving D lat = 0.097 μm2·s−1; Table S4), a statistical analysis of the coincidence of tracks could be made. Given the diffusion coefficient of the receptors and assuming that they diffuse independently, the probability of the molecular trajectories overlapping (i.e., centroids of the spots remaining with 300 nm of each other) by chance for more than 10 consecutive frames is exceedingly low (<<1%). We used this as a criterion to distinguish overlap of the spots by chance from true association (dimerization) of the receptors. Analysis of coincidence of the two-color channels showed that 2089 tracks coincided (i.e., ~20% of the total observations) for part of their trajectory. As an independent check, we offset the green and red video frames of the cell shown in Fig. 4 (by shifting the coordinates of the red frames by 500 nm) and found that no tracks then overlapped. We found that dual-labeled tracks diffused more slowly (0.065 μm2·s−1 at 23 °C) than the estimate obtained for monomers, which is consistent with the increased radius of a dimer compared to a monomer (20). We found no evidence for anomalous diffusion of dimers, which might be expected if dimers were specifically anchored to a larger intracellular complex or localized to specific membrane domains or the cytoskeleton.
Closer inspection of the green and red coincident tracks revealed three types of behavior: (i) those that started as dimers and separated into monomers; (ii) those that started as monomers and formed dimers; and (iii) those that started as monomers, formed dimers, and then separated into monomers. The behavior of the last group is equivalent to that shown in Fig. 3 for the singly Cy3B–telenzepine-labeled receptors. Trajectories of examples of these three groups of tracks are shown in Fig. 4G. The x-coordinates of green and red tracks are shown in the upper sections of records 1–5 in Fig. 4G, and the y-coordinates in the lower sections. Coincidence of the green and red tracks, i.e., the presence of a dimer, is manifest by the overlap of green and red tracks in both the x- and y-coordinates. Although the tracks that show two-color coincidence represent ~20% of the total tracks (Table S4), these tracks are dimers for only ~50% of the time (e.g., tracks in Fig. 4G), and thus the estimate of the percentage of two-color dimers, at any one time, is ~10%.
From an analysis of the lifetimes of the 84 individual dimers in the third group (Fig. 4G: records 4 and 5 and inset), it was possible to estimate the dissociation rate of the dimers as ~1.3·s−1 (or a lifetime of ~0.5 s at 23 °C). Analysis of the categories of tracks shown in Fig. 4G: records 1 and 2 gave estimated dimer lifetimes of ~0.5 s (1,243 tracks) and ~0.8 s (372 tracks), respectively.
For some tracks, we could also detect transient dissociation–dimerization events where the two-color dimers separated for short periods of time (Fig. 4G, record 3). Finally, we also observed that 5.7% and 4.6% of the green and red tracks did not overlap with the other color but instead exhibited two-step photobleaching compatible with the presence of homodimers of Alexa488–telenzepine- or Cy3B–telenzepine-labeled receptors (1,079 tracks, ~10% of the total observations; Table S4), which is equivalent to what is observed in the one-color data (Fig. 2C). Thus the total percentage of single-color and two-color dimers observed is ~20% (equivalent to ~30% of the total receptor molecules present as dimers).
Discussion
In this study, M1 muscarinic receptors labeled with fluorescent telenzepine analogs were observed as individual, mobile, fluorescent spots that were evenly distributed on the cell surface. The high affinity and very slow off-rates of the telenzepine analogs enabled us to observe ~97% of all receptors present at the plasma membrane. Individual receptors were tracked with high spatial resolution using digital image analysis, and their lateral diffusion, on the timescale of seconds, was found to be consistent with the predictions of a random walk model and in good agreement with estimates from bulk studies of other GPCRs (21, 22). We found no evidence for restricted or anomalous diffusion of M1 receptors, unlike reports for gold-tagged μ-opioid receptors (23, 24).
The simplest interpretation of our single-color imaging experiments is that each muscarinic receptor binds a single fluorescently tagged telenzepine molecule. This means that the intensities of the optical signals report the oligomeric state of receptor complexes. The pattern of intensity changes for individual spots of light tracked over time indicates that the receptors exist in a dynamic equilibrium between monomeric and dimeric states and that higher oligomeric states are completely absent. This finding was confirmed by dual-color labeling experiments in which transient formation and disruption of dual-labeled species were directly observed. Although this is the simplest interpretation of the data, we cannot exclude the possibility that muscarinic receptors exhibit “half of the sites binding.” Each fluorophore would then report the position of a dimer, and instead the data would suggest dimer-to-tetramer transitions. It has been suggested from bulk studies of muscarinic receptor subtypes that they may be oligomers (25) (M2 receptors), covalently linked dimers (26) (M3 receptors), or predominantly constitutive dimers (27) (M1–M3 receptors). However, we know of no evidence that M1 receptors show “half of the sites binding” stoichiometry (see SI Text for further discussion) or reports showing that they form heterodimers with other endogenous GPCRs in CHO cells. So, our single- and dual-color imaging data are most easily explained by the presence of a steady-state mixture of M1 receptor monomers and dimers that associate and dissociate on the timescale of seconds.
In cell biology terms, M1 receptors are behaving as a poised system. The steady-state proportion of dimers in vivo will depend on the level of receptor expression, on any heterogeneity in their distribution within the membrane, and possibly on the cellular milieu. The pharmacological nature of any bound ligand, whether it be an agonist, neutral antagonist, or inverse agonist, might also bring about changes in the proportion of dimers. This could well be important if the dimers behave differently from monomers, for example, in their functional properties or in their ability to be delivered to the cell surface or internalized. If our findings of reversible dimer formation transfer to other members of family A of GPCRs, it is likely that the affinities of the receptors for each other (e.g., in homo- or heterodimers) and hence dimer lifetimes could well be different from values reported here. Transient receptor dimerization, as also suggested by recent fluorescence recovery after photobleaching studies (28), could have unanticipated physiological consequences as well as providing a molecular explanation for apparently conflicting data in the literature. Using the single-color and two-color imaging approaches described here, it is now practicable to follow and analyze quantitatively very large numbers of individual molecules of GPCRs (or other cell-surface proteins) in living cells with high spatial and temporal resolution.
Materials and Methods
Chemical Synthesis.
The synthesis and characterization of Cy3B–telenzepine and Alexa488–telenzepine are described in the SI Text.
Determination of the Association and Dissociation Kinetics of the Fluorescent Ligands.
The radioligand binding assay protocols used and the results are reported in the SI Text.
Labeling of the M1 Receptors on CHO Cells by the Fluorescent Ligands.
The methods for labeling M1 receptors with the fluorescent ligands for single-color or dual-color TIRFM are described in the SI Text.
Detection of Single Molecules of M1 Receptors.
The TIRFM imaging system used is described in detail in the SI Text.
Analysis of the TIRFM Data.
The automatic single particle tracking algorithm used to identify and track individual muscarinic receptors has been described previously (19) (available at www.nimr.mrc.ac.uk/GMimPro). The algorithm used to identify and analyze pairs of moving “red” and “green” particle tracks in the two-color TIRFM experiments is described in the SI Text. The algorithm used to identify stepwise changes in intensity in the single-color imaging experiments also is described in the SI Text.
Supplementary Material
Acknowledgments
The authors acknowledge the Medical Research Council and the Interdisciplinary Research Collaboration in Bionanotechnology for financial support and Ed Hulme, National Institute of Medical Research (London), for helpful discussions.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. B.K.K. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/cgi/content/full/0907915107/DCSupplemental.
References
- 1.Caulfield MP, Birdsall NJM. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev. 1998;50:279–290. [PubMed] [Google Scholar]
- 2.Langmead CJ, Watson J, Reavill C. Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol Ther. 2008;117:232–243. doi: 10.1016/j.pharmthera.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 3.Pin JP, et al. Allosteric functioning of dimeric class C G-protein-coupled receptors. FEBS J. 2005;272:2947–2955. doi: 10.1111/j.1742-4658.2005.04728.x. [DOI] [PubMed] [Google Scholar]
- 4.Milligan G. A day in the life of a G protein-coupled receptor: The contribution to function of G protein-coupled receptor dimerization. Br J Pharmacol. 2008;153(Suppl 1):S216–S229. doi: 10.1038/sj.bjp.0707490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banères JL, Parello J. Structure-based analysis of GPCR function: Evidence for a novel pentameric assembly between the dimeric leukotriene B4 receptor BLT1 and the G-protein. J Mol Biol. 2003;329:815–829. doi: 10.1016/s0022-2836(03)00439-x. [DOI] [PubMed] [Google Scholar]
- 6.Milligan G, Bouvier M. Methods to monitor the quaternary structure of G protein-coupled receptors. FEBS J. 2005;272:2914–2925. doi: 10.1111/j.1742-4658.2005.04731.x. [DOI] [PubMed] [Google Scholar]
- 7.Fotiadis D, et al. Atomic-force microscopy: Rhodopsin dimers in native disc membranes. Nature. 2003;421:127–128. doi: 10.1038/421127a. [DOI] [PubMed] [Google Scholar]
- 8.Milligan G. G protein-coupled receptor hetero-dimerization: Contribution to pharmacology and function. Br J Pharmacol. 2009;158:5–14. doi: 10.1111/j.1476-5381.2009.00169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chabre M, le Maire M. Monomeric G-protein-coupled receptor as a functional unit. Biochemistry. 2005;44:9395–9403. doi: 10.1021/bi050720o. [DOI] [PubMed] [Google Scholar]
- 10.James JR, Oliveira MI, Carmo AM, Iaboni A, Davis SJ. A rigorous experimental framework for detecting protein oligomerization using bioluminescence resonance energy transfer. Nat Methods. 2006;3:1001–1006. doi: 10.1038/nmeth978. [DOI] [PubMed] [Google Scholar]
- 11.Bouvier M, Heveker N, Jockers R, Marullo S, Milligan G. BRET analysis of GPCR oligomerization: Newer does not mean better. Nat Methods. 2007;4:3–4. doi: 10.1038/nmeth0107-3. author reply 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salahpour A, Masri B. Experimental challenge to a ‘rigorous’ BRET analysis of GPCR oligomerization. Nat Methods. 2007;4:599–600. doi: 10.1038/nmeth0807-599. author reply 601. [DOI] [PubMed] [Google Scholar]
- 13.Bayburt TH, Leitz AJ, Xie G, Oprian DD, Sligar SG. Transducin activation by nanoscale lipid bilayers containing one and two rhodopsins. J Biol Chem. 2007;282:14875–14881. doi: 10.1074/jbc.M701433200. [DOI] [PubMed] [Google Scholar]
- 14.Gurevich VV, Gurevich EV. GPCR monomers and oligomers: It takes all kinds. Trends Neurosci. 2008;31:74–81. doi: 10.1016/j.tins.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meyer BH, et al. FRET imaging reveals that functional neurokinin-1 receptors are monomeric and reside in membrane microdomains of live cells. Proc Natl Acad Sci USA. 2006;103:2138–2143. doi: 10.1073/pnas.0507686103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whorton MR, et al. Efficient coupling of transducin to monomeric rhodopsin in a phospholipid bilayer. J Biol Chem. 2008;283:4387–4394. doi: 10.1074/jbc.M703346200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harris A, Cox S, Burns D, Norey C. Miniaturization of fluorescence polarization receptor-binding assays using CyDye-labeled ligands. J Biomol Screen. 2003;8:410–420. doi: 10.1177/1087057103256319. [DOI] [PubMed] [Google Scholar]
- 18.Hammer R, Berrie CP, Birdsall NJ, Burgen AS, Hulme EC. Pirenzepine distinguishes between different subclasses of muscarinic receptors. Nature. 1980;283:90–92. doi: 10.1038/283090a0. [DOI] [PubMed] [Google Scholar]
- 19.Mashanov GI, Molloy JE. Automatic detection of single fluorophores in live cells. Biophys J. 2007;92:2199–2211. doi: 10.1529/biophysj.106.081117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saffman PG, Delbrück M. Brownian motion in biological membranes. Proc Natl Acad Sci USA. 1975;72:3111–3113. doi: 10.1073/pnas.72.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horvat RD, Nelson S, Clay CM, Barisas BG, Roess DA. Intrinsically fluorescent luteinizing hormone receptor demonstrates hormone-driven aggregation. Biochem Biophys Res Commun. 1999;255:382–385. doi: 10.1006/bbrc.1999.0185. [DOI] [PubMed] [Google Scholar]
- 22.Nelson S, et al. Characterization of an intrinsically fluorescent gonadotropin-releasing hormone receptor and effects of ligand binding on receptor lateral diffusion. Endocrinology. 1999;140:950–957. doi: 10.1210/endo.140.2.6518. [DOI] [PubMed] [Google Scholar]
- 23.Daumas F, et al. Confined diffusion without fences of a G-protein-coupled receptor as revealed by single particle tracking. Biophys J. 2003;84:356–366. doi: 10.1016/S0006-3495(03)74856-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki K, Ritchie K, Kajikawa E, Fujiwara T, Kusumi A. Rapid hop diffusion of a G-protein-coupled receptor in the plasma membrane as revealed by single-molecule techniques. Biophys J. 2005;88:3659–3680. doi: 10.1529/biophysj.104.048538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma AWS, Redka DS, Pisterzi LF, Angers S, Wells JW. Recovery of oligomers and cooperativity when monomers of the M2 muscarinic cholinergic receptor are reconstituted into phospholipid vesicles. Biochemistry. 2007;46:7907–7927. doi: 10.1021/bi6026105. [DOI] [PubMed] [Google Scholar]
- 26.Zeng FY, Wess J. Identification and molecular characterization of m3 muscarinic receptor dimers. J Biol Chem. 1999;274:19487–19497. doi: 10.1074/jbc.274.27.19487. [DOI] [PubMed] [Google Scholar]
- 27.Goin JC, Nathanson NM. Quantitative analysis of muscarinic acetylcholine receptor homo- and heterodimerization in live cells: Regulation of receptor down-regulation by heterodimerization. J Biol Chem. 2006;281:5416–5425. doi: 10.1074/jbc.M507476200. [DOI] [PubMed] [Google Scholar]
- 28.Dorsch S, Klotz KN, Engelhardt S, Lohse MJ, Bünemann M. Analysis of receptor oligomerization by FRAP microscopy. Nat Methods. 2009;6:225–230. doi: 10.1038/nmeth.1304. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.