

Abstract
Helicobacter pylori encodes a potential virulence factor, agmatine deiminase (HpAgD), which catalyzes the conversion of agmatine to N-carbamoyl putrescine (NCP) and ammonia - agmatine is decarboxylated arginine. Agmatine is an endogenous human cell signaling molecule that triggers the innate immune response in humans. Unlike H. pylori, humans do not encode an AgD; it is hypothesized that inhibition of this enzyme would increase the levels of agmatine, and thereby enhance the innate immune response. Taken together, these facts suggest that HpAgD is a potential drug target. Herein we describe the optimized expression, isolation, and purification of HpAgD (10–30 mg/L media). The initial kinetic characterization of this enzyme has also been performed. Additionally, the crystal structure of wild-type HpAgD has been determined at 2.1 Å resolution. This structure provides a molecular basis for the preferential deimination of agmatine, and identifies Asp198 as a key residue responsible for agmatine recognition, which has been confirmed experimentally. Information gathered from these studies led to the development and characterization of a novel class of haloacetamidine based HpAgD inactivators. These compounds are the most potent AgD inhibitors ever described.
1. Introduction
Helicobacter pylori are gram negative bacteria that colonize the gastrointestinal tract, and infection by this pathogen can lead to gastritis, peptic ulcer diseases, and even cancer [1, 2]. Although ~50% of the worldwide population is infected by this bacterium [3], most are asymptomatic, and only a subset (~15–20%) will present with the aforementioned disease pathologies [2]. Nevertheless, the high incidence of H. pylori infection, coupled with the potential for debilitating effects, often necessitates therapeutic intervention. Current therapeutic approaches include: (i) the proton pump inhibitor1 (PPI) triple therapy (i.e. a PPI plus amoxicillin and clarithromycin), (ii) the bismuth-based triple/quadruple therapy (i.e. bismuth, metronidazole, tetracycline, and a PPI), and (iii) the sequential therapy, which includes a PPI and amoxicillin for the first 5 days, followed by a triple therapy of PPI, clarithromycin, and tinidazole for the remaining 5 days [4]. While generally successful, there are a number of problems associated with these therapies, including: antibiotic resistance, adverse side effects (e.g., allergic reactions), and cost – the latter issue is especially true in developing countries [4–6]. Thus, novel therapeutic approaches targeting this pernicious bacterium are needed.
H. pylori strains encode a number of pathogenecity factors that contribute to disease onset and progression, including: urease, flagella, vaculating cytotoxin A (VacA), and the cytotoxin associated gene (cag) pathogenicity island [2]. Urease contributes to acid resistance and flagella allow for bacterial movement. VacA is a pore-forming toxin that breaks down the gastric epithelium, leading to gastric ulceration and necrosis. Finally, the cag pathogenecity island, which encodes a Type 4 secretion system, is associated with increased inflammation, accumulation of phagocytic cells in the intestines, and release of interleukin (IL)-8 [1, 7]. In addition to these well established pathogenicity factors, H. pylori also encode an Agmatine Deiminase (AgD), which may contribute to disease pathogenesis vide infra.
Agmatine is a naturally occurring molecule that is generated by the decarboxylation of arginine. Although it is unclear whether humans synthesize agmatine directly [8], it is clear that this molecule is present in human tissues [9–11]; likely sources include dietary sources or even beneficial bacteria present in the intestinal flora. In bacteria, the in vivo hydrolysis of agmatine by AgD results in the formation of N-carbamoyl putrescine (NCP) and ammonia (Fig. 1). AgDs are an integral part of the Agmatine Deiminase System (AgDS) in a number of bacterial species (e.g., Pseudomonas aeruginosa, Listeria monocytogenes, Enterococcus faecalis, Yersinia pestis and Streptococcus mutans) [12–14], where they contribute to polyamine biosynthesis. For example, S. mutans possess a 4 gene operon (aguBDAC) that encodes for an agmatine-putrescine antiporter (aguD), an AgD (aguA), a putrescine carbamoyltransferase (aguB) that converts NCP into putrescine and carbamoylphosphate, and a carbamate kinase (aguC) that transfers the phosphate group from carbamoylphosphate to ADP (Fig. 1)[14]. This system, which ultimately converts agmatine into putrescine, ATP, ammonia, and carbon dioxide, is thought to play an important role in acid resistance and competitive survival. It has also been shown that, under specific growth conditions, exogenously added agmatine slows the growth of wild type strains and prevents the growth of strains that lack components of the AgDS [14, 15]. Interestingly, H. pylori, and a number of other human pathogens (e.g., Legionella pneumophila (strain Philadelphia 1), Streptococcus pneumonia, Vibrio parahaemolyticus RIMD 2210633, Lactococcus lactis) encode AgDs, i.e. the enzymes responsible for converting agmatine into NCP, but lack a complete AgDS. Instead, these bacteria encode an alternate agmatine degradation pathway that involves the initial formation of NCP, which is subsequently hydrolyzed to form putrescine, via the actions of NCP amidohydrolase. The essential role of putrescine in polyamine biosynthesis suggests that this alternate agmatine degradation pathway, and the AgDs in particular, plays an important role in the life cycle of these organisms. This pathway may also contribute to acid resistance and competitive survival of the bacteria, similarly to the AgDS described above.
Fig. 1.

Bacterial agmatine degradation pathways. In bacteria, arginine is converted to agmatine via the actions of Arginine Decarboxylases (ADC). Agmatine is then hydrolyzed to form N-carbamoyl putrescine (NCP) by agmatine deiminase (AgD). In bacteria encoding an Agmatine Deiminase System, NCP is then converted into putrescine and carbamoylphosphate by putrescine transcarbamoylase (PTC). Carbamate kinase (CK) then catalyzes the transfer of the phosphate moiety in carbamoylphosphate to ADP, forming ATP, as well as ammonia and CO2. Other pathogenic bacteria encode an alternate non-ATP forming agmatine degradation pathway that involves the conversion of NCP into putrescine, ammonia, and CO2 via the actions of an N-carbamoylputrescine amidohydrolase (CPA).
In addition to its potential importance to H. pylori growth and survival, H. pylori AgD (HpAgD) may also contribute to disease pathogenesis through its ability to degrade agmatine. This is the case because agmatine seems to play important, albeit incompletely defined, roles in human cell signaling pathways [16]. For example, agmatine is known to bind to the α2-adrenergic and imidazoline receptors, possess neuromodulatory activity [9, 10], trigger nitric oxide production [16, 17], modulate the innate immune response [18], and decrease blood glucose in type 2 diabetic rats [19]. Agmatine can also down regulate the synthesis of other polyamines. For example, increased levels of agmatine can result in decreased levels of ornithine decarboxylase, a key enzyme in putrescine synthesis; this decrease leads to antiproliferative effects on a number of colonic, hepatic, and neuronal tumor cells [20]. Interestingly, H. pylori are known to increase the expression of ornithine decarboxylase in macrophages and, as a consequence, increase the levels of spermine, a polyamine that is known to inhibit the effector function of macrophages. This activity has been suggested as a bacterial defense response to avoid being killed by the innate immune response [21].
In humans, it has also been shown that agmatine stimulates the release of gastrin [22], which in turn, increases the release of protons into the gut. Additionally, agmatine levels are markedly decreased in tumors of colonic origin [23]. These findings are relevant to H. pylori infections because they suggest that HpAgD may contribute to tumor cell proliferation and acid resistance by decreasing the endogenous levels of agmatine. In total, the above described actions of agmatine in humans suggest that, by decreasing the in vivo concentration of agmatine, HpAgD may, in addition to other virulence factors, modulate human cell signaling, thereby providing a mechanism to avoid (or control) detection by the human immune system. A similar role has been suggested for H. pylori arginase: decreased levels of arginine, due to bacterial arginase activity, attenuate the innate immune response [24]. If this hypothesis is correct, AgD inhibitors should, in addition to their potential direct effects on bacterial growth, provide a mechanism to increase the concentration of endogenous agmatine and thereby alter the virulence associated with infection by these pathogenic bacteria. The presence of AgD homologues in other pathogenic bacteria suggests that AgD inhibitors may show broad utility.
H. pylori AgD (HpAgD) is a 330 amino acid, 38 kDa protein that is annotated in both genomic and structural databases (PDB ID 2CMU) as a putative peptidyl arginine deiminase (PAD). PADs are members of the guanidinium modifying family of enzymes, a family that includes enzymes that hydrolyze the guanidinium groups of arginine, methylarginine, peptidyl-arginine, and agmatine to form the ureido containing derivatives [25]. In PAD4, and other guanidinium modifying enzymes, there are 4 key catalytic residues: Asp350, His471, Asp473 and Cys645 (human PAD4 numbering) [26]. Structure based alignments of HpAgD and PAD4 indicate that Asp84, His202, Asp204, and Cys324 are the corresponding residues in HpAgD. Based on this sequence conservation and insights gained from mechanistic studies of PAD4 [27], Asp84 and Asp204 likely play critical roles in binding and orienting the substrate guanidinium for nucleophilic attack by the side chain thiolate of Cys324 (Fig. 2). His202 would then be expected to act as a general acid, donating a proton to the departing ammonia either before or after collapse of the tetrahedral intermediate. His202 is also thought to act as a general base, activating a water molecule for nucleophilic attack, thus leading to hydrolysis of the thiouronium intermediate generated in the previous step.
Fig. 2.

Proposed catalytic mechanism: Cys324 is the active site nucleophile and His202 acts as a general acid, donating a proton to the departing amine, leading to the formation of an S-alkyl thiouronium intermediate. His 202 activates a water molecule for nucleophilic attack on the thiouronium intermediate. This leads to the formation of a second tetrahedral intermediate that collapses to eliminate the Cys thiolate and generate NCP.
While HpAgD shows homology to the PADs, and other guanidinium modifying enzymes, we demonstrate here for the first time that this enzyme is actually an AgD, as originally suggested by bioinformatic analyses [13, 28]. We further demonstrate that this is a hydrolytic reaction and that NCP and ammonia are produced in equimolar amounts. Site directed mutagenesis experiments additionally confirm the key catalytic role of Cys324. The structure of HpAgD, which has been determined to 2.1 Å resolution, also provides a molecular basis for the preferential deimination of agmatine by this enzyme, and identifies Asp198 as a key residue responsible for agmatine recognition, which has been confirmed by site directed mutagenesis. Finally, we describe the design, synthesis, and characterization of two haloacetamidine-based inactivators of HpAgD, i.e. N-(4-aminobutyl)-2-fluoro-ethanimidamide (ABFA) and N-(4-aminobutyl)-2-chloro-ethanimidamide (ABCA). These compounds, which inactivate HpAgD by modifying Cys324, are the most potent AgD inhibitors described to date.
2. Materials and methods
2.1. Chemicals and general experimental methods
Agmatine sulfate, L-arginine, streptomycin, and N-α-Benzoyl-L-arginine ethyl ester (BAEE) were acquired from Sigma Aldrich (St. Louis, MO). N-α-Benzoyl-L-arginine amide (BAA) was obtained from Acros (Hampton, NH). Guanidine-HCl was purchased from EMD Bioscience (Darmstadt, Germany). Deuterium oxide was obtained from Cambridge Isotope Laboratories (Andover, MA). N-α-Benzoyl-L-arginine methyl ester (BAME) was purchased from MP Biomedicals (Irvine, CA). Histone H4 was isolated and purified as previously described [29]. The Ac-H4-5 peptide (Ac-SGRGK-COO−) was synthesized using the Fmoc-strategy, purified by reverse phase HPLC, and its structure confirmed by MALDI-MS.
Electrospray ionization mass spectra were acquired using a Waters Micromass quadrapole-time-of-flight (Q-TOF) tandem mass spectrometer in the positive ion mode. Matrix assisted laser desorption/ionization (MALDI) mass spectra were acquired with a Bruker Ultraflex II MALDI-TOF/TOF mass spectrometer. DNA sequencing was performed at the Synthesis and Sequencing Facility at Johns Hopkins University School of Medicine.
2.2. Construction of an expression vector for HpAgD
An expression vector encoding HpAgD (PDB ID 2CMU) was obtained from SGX Pharmaceuticals, Inc.. Using this vector as the template for PCR reactions, the HpAgD gene was cloned into a modified pET28 vector using the following primers: 5′-AAAAAACATATGAAAAGAATGTTAGCGGAGTTTG-3′ and 5′-AAAAAACTCGAGTCAATAAAGTTGCATCGTTACACAATG-3′. The forward primer contains an NdeI restriction site (underlined) followed by 22 bases that correspond to the 5′-coding region of the HpAgD gene. The reverse primer contains an XhoI restriction site (underlined) followed by 27 bases that correspond to the 3′-coding region of HpAgD. The resulting pET28 HpAgD construct was sequenced to ensure that no mutations were incorporated during the PCR amplification. Although this construct was expressed and purified by immobilized metal ion affinity chromatography (IMAC), little to no activity was apparent. This lack of activity was also found when HpAgD was expressed using the original vector. However, active enzyme could be purified in both high yield and high purity using conventional methods (vide infra). Because IMAC was not required for enzyme purification, the N-terminal 6x-His tag was removed by subcloning the HpAgD gene into the pET21 vector, thereby generating the p21HpAgD expression construct.
2.3. Purification of HpAgD
The methodology to purify HpAgD is outlined in detail in the Supplemental Data section. Briefly, the p21HpAgD expression construct, transformed into E. coli BL21(DE3) cells, was used for protein expression. Cultures were grown in a baffled flask (37 °C, 250 rpm) to an OD600 of 1.0. Expression of HpAgD was then induced by the addition of IPTG (0.3 mM final) and the cultures were then incubated overnight (25 °C, 250 rpm). Cells were harvested by centrifugation (5000 × g, 10 min, 4 °C), resuspended, and lysed. Streptomycin was then added to clarified cell extracts to afford the precipitation of contaminating DNA and enhance enzyme binding to a Q Sepharose strong anion-exchange column. Protein recovered from this step was further purified using a combination of heparin affinity and size exclusion chromatography. This methodology afforded active HpAgD in high yield (10–30 mg/L of E. coli cell culture) with greater than 95% purity (Table S1; Fig. S1). Enzyme prepared in this fashion was stable for several months at −80 °C.
2.4. Kinetic assay
A previously established discontinuous assay that monitors the production of ureido-containing compounds was adapted for use as an HpAgD activity assay [30, 31]. The steady state kinetic parameters for HpAgD were determined by pre-incubating varying amounts of agmatine in Assay Buffer (50 mM HEPES, pH 8.0, 60 μL final volume) at 37 °C. After 10 min the reactions were initiated by the addition of enzyme. Under these conditions, enzymatic activity was linear with respect to both time and enzyme concentration. Standard errors were typically less than 20%. Initial rates were fit, by a non-linear least squares method, to equation 1 using GraFit (version 5.0.11) [32].
| (1) |
2.5. Substrate specificity
The enzymatic activity of HpAgD was evaluated against several guanidinium containing compounds, including: L-arginine, streptomycin, BAA, guanidine, BAEE, BAME, the Ac-H4-5 peptide, histone H4, and agmatine. All substrates, with the exception of the Ac-H4-5 peptide and histone H4, were tested at a final concentration of 10 mM. The Ac-H4-5 peptide and histone H4 were evaluated at final concentrations of 5 mM and 125 μM, respectively. Enzyme assays were performed as described above, except that the reactions were incubated for 2–3 h to obtain estimates of the reaction rate.
2.6. Metal inhibition
The effects of Ni2+, Zn2+, and Co2+ on HpAgD activity were determined by pre-incubating HpAgD with varying concentrations of each metal ion for 15 min prior to initiating the reaction with agmatine (5.0 mM final). After an additional 15 min, the reactions were quenched and NCP production was quantified using the methodology outlined above. Initial rates were fit to equation 2 using GraFit (version 5.0.11) [32], where [I] is the concentration of inhibitor (in this case the metal ion) and IC50 is the concentration of metal ion that yields half-maximal activity.
| (2) |
2.7. 18Oxygen incorporation studies
Oxygen incorporation experiments were conducted in Assay Buffer made up in either 16O- or 18O- labeled water – the mole percent of 18O-label was 70% final. Agmatine (5.0 mM final) and HpAgD (2.8 μM final) were added and the reaction mixture was incubated at 37 °C for 3 h. The reaction mixtures were desalted using a C18 ZipTip (Milipore), and the deiminated products were analyzed by ESI mass spectrometry.
2.8. Ammonia production
Time course experiments that monitor the production of ammonia and NCP by HpAgD were performed in parallel. For the ammonia production assay, agmatine (10.0 mM) in Assay Buffer was pre-incubated at 37 °C for 10 min prior to the addition of HpAgD (2.8 μM final). Aliquots (60 μL) of the reaction mixture were removed and quenched with iodoacetamide (50 mM, 180 μL) at 0, 2, 4, 6, 10, and 15 min. NCP production was measured using the assay described above, and ammonia concentrations were measured fluorometrically [33, 34].
2.9. Synthesis of N-(4-aminobutyl)-2-chloro-ethanimidamide (ABCA)
To a flask containing dry methanol (8 mL) was added N-1-Boc-1,4-diaminobutane hydrochloride (Bachem, 75.0 mg, 0.33 mmol), ethyl-2-chloroacetimidate (104.3 mg, 0.66 mmol), and cesium carbonate (162.5 mg, 0.50 mmol). Ethyl-2-chloroacetimidate was prepared according to previously established methods [35, 36]. The reaction mixture was stirred overnight at rt before being concentrated under reduced pressure. The concentrated sample was loaded onto a column of silica gel and eluted with 40% methanol in ether to afford N-1-Boc-ABCA as a clear residue. Boc-deprotection was accomplished by treating the residue with neat TFA (5 mL) at 0 °C. After 30 min, the mixture was allowed to warm to rt and stir for an additional 30 min. The TFA was then removed, and the remaining residue was dissolved in water (10 ml). This solution was extracted with ether (3 mL) and the aqueous portion was lyophilized to afford the title compound (90 mg, 95% yield) as a clear residue. 1H NMR (D2O, 300.107 MHz): δ 4.25 (s, 2H), 3.22 (m, 2H), 2.87 (m, 2H), 1.58 (m, 4H). 13C NMR (D2O, 75.47 MHz): δ 62.98, 42.15, 39.22, 39.06, 24.21, 23.82. HR-ESI-MS (C6H15N3Cl+): calculated 164.0954, observed 164.0948.
2.10. Synthesis of N-(4-aminobutyl)-2-fluoro-ethanimidamide (ABFA)
ABFA was prepared using the methodology outlined above for ABCA, except that ethyl-2-fluoroacetimidate (93.5 mg, 0.66 mmol) was used in place of ethyl-2-chloroacetimidate. Ethyl-2-fluoroacetimidate was prepared according to previously established methods [35, 36]. The title compound (72 mg, 84% yield) was obtained as a clear residue. 1H NMR (D2O, 300.107 MHz): δ 5.11 (d, JH-F = 45 Hz, 2H), 3.24 (m, 2H), 2.87 (m, 2H), 1.57 (m, 4H). 13C NMR (D2O, 75.47 MHz): δ 162.73 (d, JC-F = 11.1 Hz), 77.75 (d, JC-F = 177.5 Hz), 41.56, 39.04, 24.20, 23.93. HR-ESI-MS (C6H15N3F+): calculated 148.1250, observed 148.1255.
2.11. Determination of IC50 values
The effects of ABFA and ABCA on HpAgD activity were determined by pre-incubating HpAgD with varying concentrations of each compound in Assay Buffer for 15 min prior to the addition of agmatine (0.5 mM final). After 15 min, the reactions were flash frozen in liquid nitrogen and the results analyzed using the methodology outlined above for NCP quantification. Initial rates were fit to equation 2 using GraFit (version 5.0.11) [32].
2.12. Inactivation kinetics of ABFA and ABCA
Varying concentrations of ABFA and ABCA were pre-incubated with agmatine (0.5 mM) in Assay Buffer at 37 °C. After 10 min, the reactions were initiated by the addition of HpAgD. Aliquots (60 μL) of the reactions were removed and flash frozen in liquid nitrogen at various time points. NCP levels were quantified using the methodology outlined above. The non-linear progress curves were fit to equation 3, using GraFit (version 5.0.11) [32], where vi is the initial velocity, kobs.app is the apparent pseudo first order rate constant for inactivation, t is time, and [NCP] is the concentration of NCP produced during the reaction.
| (3) |
To obtain kobs, the apparent pseudo first order rate constant (kobs.app) was multiplied by the transformation (1+[S]/Km). Because saturation was observed in the plots of kobs versus [I], the data were fit to equation 4 using GraFit (version 5.0.11) [32]. In this equation, kinact corresponds to the maximal rate of inactivation and KI is the concentration of I that yields half-maximal inactivation.
| (4) |
2.13. Substrate protection experiments
To evaluate the ability of agmatine to protect HpAgD against inactivation by ABFA and ABCA, progress curves were generated for reactions, both in the presence and in the absence of inactivator (ABFA: 500 μM or ABCA: 175 μM), at two different concentrations of agmatine (0.5 or 1.0 mM). The Assay Buffer was incubated at 37 °C for 10 min before HpAgD was added to initiate the reaction. Aliquots (60 μL) of the reaction were removed, flash frozen, and analyzed at 0, 2, 4, 6, 10, 12 and 14 min. The progress curves were fit to equation 3 as described above.
2.14. Irreversibility of inactivation
To demonstrate that ABFA and ABCA irreversibly inactivate HpAgD, a solution of HpAgD (2.8 μM) was treated with an excess of each inhibitor (1.0 mM final) at 37 °C. After 1 h, the reactions were dialyzed against 50 mM HEPES pH 8.0, 1.0 mM EDTA, and 10% glycerol for 20 h. The residual activity was measured using our standard assay for NCP production.
2.15. Site-directed mutagenesis
The C324A and D198A HpAgD mutants were generated using the Quik Change Site Directed Mutagenesis Kit (Stratagene) using pET21-HpAgD as the template. The forward and reverse primers for the C324A mutant are 5′-CAGCATGGAAGCTTGCATGCTGTAACGATGCAACTTTATTG-3′ and 5′-CAATAAAGTTGCATCGTTACAGCATGCAAGCTTCCATGCTG-3′, respectively. The forward and reverse primers for the D198A mutant are 5′-CTATTTAAAGGGCGATGCTACGGATAGCCATACCG-3′ and 5′-CGGTATGGCTATCCGTAGCATCGCCCTTTAAATAG-3′, respectively. Both mutated genes were sequenced in their entirety to ensure that only the desired mutation had been incorporated into the HpAgD expression construct. The mutant enzymes were expressed and purified as described above for the wild-type enzyme.
2.16. Tryptic digest
To identify the site of inactivator modification, HpAgD (100 μg, 26 μM) was incubated at 37 °C with ABFA (132 μM, 5 eq.), ABCA (132 μM, 5 eq.), or no inactivator in Assay Buffer for 30 min. A tryptic digest was then performed using Seq. Grade Modified Trypsin (Promega) in a final ratio of 1:20 (enz:sub) and incubated overnight at 37 °C. The digested sample was prepared for MALDI-TOF MS and MALDI-TOF MS/MS using a C-18 ZipTip.
2.17. Crystallization of HpAgD
Crystals of HpAgD were grown from 15% PEG 4K, 10 mM sodium citrate pH 5.6, and 7% isopropanol by vapor diffusion, hanging-drop method. Crystals were transferred to a cryoprotectant solution, mother liquor plus 15% ethylene glycol, and then flash frozen in a cryo stream.
2.18. X-ray diffraction data measurement and processing
X-ray diffraction experiments were carried out at the SER-CAT ID beamline at APS, Argonne National Laboratory. Data were collected on a single crystal of HpAgD with approximate dimensions of 0.60 × 0.30 × 0.25 mm. The data were indexed and processed with HKL2000 [37]. Data collection and processing statistics are shown in Table 1.
Table 1.
Crystallographic Data and Refinement Statistics
| X-Ray Source | APS SER-CAT 22ID | |
|---|---|---|
| PDB ID | 3HVM | |
| Wavelength (Å) | 1.00000 | |
| Space group | P3221 | |
| Unit cell dimensions: | a (Å) | 52.08 |
| c (Å) | 246.40 | |
| Resolution Range (Å) (highest shell) | 50.0-2.10 (2.18-2.10) | |
| Mosaicity (degree) | 0.47 | |
| Completeness (%),(highest shell) | 76.0(59.8) | |
| Total linear R-merge | 0.043 | |
| Reflections Observed | 65011 | |
| I over average sigma(I) | 16.5 | |
| Redundancy, (highest shell) | 3.6 (3.2) | |
| Rmsd bonds (Å) | 0.016 | |
| Rmsd angles (°) | 1.73 | |
| Ramachandran Statistics | ||
| Residues in most favored regions (%) | 265 (88.6) | |
| Residues in additionally allowed regions (%) | 32 (10.7) | |
| Residues in generaously allowed regions (%) | 1 (0.3) | |
| Residues in disallowed regions (%) | 1 (0.3) | |
| R Refmac5 | 0.200 | |
| Rfree Refmac5 | 0.259 | |
2.19. Structure determination and refinement
The structure of HpAgD (PDB code 2CMU), was used as the search model in the molecular replacement method carried out with Phaser [38] from CCP4 [39]. Model rebuilding and crystallographic refinements were conducted using TURBO graphics software [40] and the Refmac5 [41] component of CCP4 [39] respectively.
3. Results and discussion
3.1. Purification of HpAgD
An expression vector encoding HpAgD (PDB ID 2CMU) was obtained from SGX Pharmaceuticals. In addition to the HpAgD open reading frame, this construct encodes two additional N-terminal residues and four additional C-terminal residues that are linked, in frame, to a sequence encoding a C-terminal 6x-His-tag. Although this construct has previously been used to express and purify recombinant HpAgD in E. coli (PDB ID: 2CMU) the enzyme is inactive (data not shown). Due to the proximity of the C-terminus to the active site cysteine (Cys324), we postulated that the lack of activity may be a result of the C-terminal His-tag and/or the additional C-terminal residues. In order to mitigate these effects, the superfluous residues were deleted and the His tag was moved to the N-terminus by sub-cloning the HpAgD gene into a modified pET28 vector. While this new construct, i.e. pET28HpAgD, was used to purify the enzyme by IMAC, the enzyme also possessed essentially no activity (data not shown). By contrast, conventional purification methods yielded active enzyme in both high yield and high purity. Upon recognizing that the His-tag was unnecessary for enzyme purification, the HpAgD gene was subcloned into the pET21 vector to eliminate this tag, thereby enabling the expression and purification of the untagged wild-type protein, also in both high yield (10–30 mg/L of E. coli cell culture) and purity (Table S1 and Fig. S1). Consistent with previous studies with Arabidopsis thaliana AgD [42] and Pseudomonas aeruginosa AgD [28], HpAgD also purifies as a dimer by size exclusion chromatography (not shown).
3.2. Substrate specificity studies
Given the fact that HpAgD is annotated as a PAD, we initially examined whether this enzyme could deiminate a number of protein and peptide substrates, including, histone H4, a peptide based on the N-terminus of histone H4 (i.e., the Ac-H4-5 peptide), as well as a number of benzoylated arginine derivatives (i.e., BAA, BAEE, BAME), all of which are known PAD substrates [30]. However, none of these compounds served as a substrate for the enzyme. Given this result, a variety of guanidinium containing small molecules were then tested for their ability to act as HpAgD substrates. These potential substrates included: guanidine, L-arginine, asymmetrically dimethylated arginine (ADMA), streptomycin, and agmatine. A comparison of the kcat/Km values obtained for these various potential substrates revealed that HpAgD modifies agmatine at rates that are >104-fold faster than the rates observed for all other substrates tested (Table 2). Thus, this enzyme, like other AgDs [42, 43], demonstrates an exquisite preference for agmatine. Consistent with the fact that agmatine is the preferred substrate, is our finding that the Km for agmatine is in the low μM range (Table 3). It is noteworthy that this value is nearly identical to that obtained for the E. faecalis orthologue [44]. Somewhat surprisingly the kcat is quite slow (~5 min−1) relative to both the E. faecalis enzyme and other deiminases. Although the reasons for this slow turnover are not known, the specificity constant, i.e., kcat/Km (1.47 × 105 min−1M−1), is quite close to that obtained for both P. aeruginosa AgD (4.2 × 105 min−1M−1) [28] and PAD4 (3.0 × 105 min−1M−1) [30], indicating that HpAgD is a functional enzyme with reasonable kinetic parameters. Perhaps the unique role of the enzyme in bacterial physiology is the explanation for the relatively low kcat.
Table 2.
kcat/Km values for potential guanidinium containing substrates of Wild Type and Mutant HpAgD
| Substrate | Wild Type (min−1 M−1) | C324A (min−1 M−1) | D198A (min−1 M−1) |
|---|---|---|---|
| H4d,f | ≤ 8.55×10−6 | NDa | ≤ 2.58×10−7 |
| L-Arginineb,f | ≤ 1.03 | NDa | ≤ 8.96 |
| Streptomycinb,e | ≤ 2.26 | NDa | ≤ 1.98 |
| Ac-H4-5c,e | ≤ 0.23 | NDa | ≤ 1.41 |
| BAAb,e | ≤ 4.32 | NDa | ≤ 7.71 |
| Guanidineb,e | ≤ 4.87 | NDa | ≤ 4.07 |
| BAEEb,f | ≤ 5.08 | NDa | ≤ 8.30 |
| BAMEb,f | ≤ 6.23 | NDa | ≤ 13.1 |
| ADMAb,f | ≤ 17.2 | NDa | ≤ 1.46 |
| Agmatine | 147 ×103 | ≤ 19.2 | ≤ 1.7×103 |
ND=Not Determined;
10 mM Substrate;
5 mM Substrate;
125 μM Substrate;
2 h incubation;
3 h incubation.
Table 3.
Steady-State Kinetic Parameters for Agmatine with Wild Type and Mutant HpAgD
| Enzyme | Substrate | kcat (min−1) | Km (μM) | kcat/Km (min−1M−1) |
|---|---|---|---|---|
| Wild Type | Agmatine | 4.90 ± 0.29 | 33 ± 8.8 | 147 ×103 |
| C324Aa | Agmatinea | NDb | NDb | ≤ 19.2 |
| D198Aa | Agmatine | 0.28 ± 0.01 | 170 ± 79.8 | 1.7×103 |
10 mM agmatine,
ND=Not Determined.
3.3. Product specificity of HpAgD
To confirm that the major products of the reaction are ammonia and NCP, the production of these compounds was measured as a function of time. These studies were performed for two reasons: (i) our assay for NCP production actually detects the presence of ureido groups, and (ii) other guanidinium modifying enzyme family members can catalyze amidino group transfer reactions [13]. Thus it was conceivable that the first tetrahedral intermediate could break down to form putrescine and a thiouronium intermediate that could ultimately be hydrolyzed to form urea. As depicted in Fig. 3, the results indicate that ammonia and NCP are produced in approximately equimolar amounts, thereby confirming that HpAgD catalyzes the production of agmatine to form NCP and ammonia. To confirm that HpAgD is a hydrolase, HpAgD catalyzed reactions were performed in 18O-labeled water. Control reactions in 16O-labeled water were also performed. The observed masses (i.e., 132 and 134 Da) are consistent with those expected for the production of NCP (i.e., 132 and 134 Da) in 16O- and 18O-labeled, respectively. The 2 unit mass shift observed for the reaction performed in 18O-labeled water also confirms that HpAgD catalyzes the hydrolytic deimination of agmatine to form NCP.
Fig. 3.
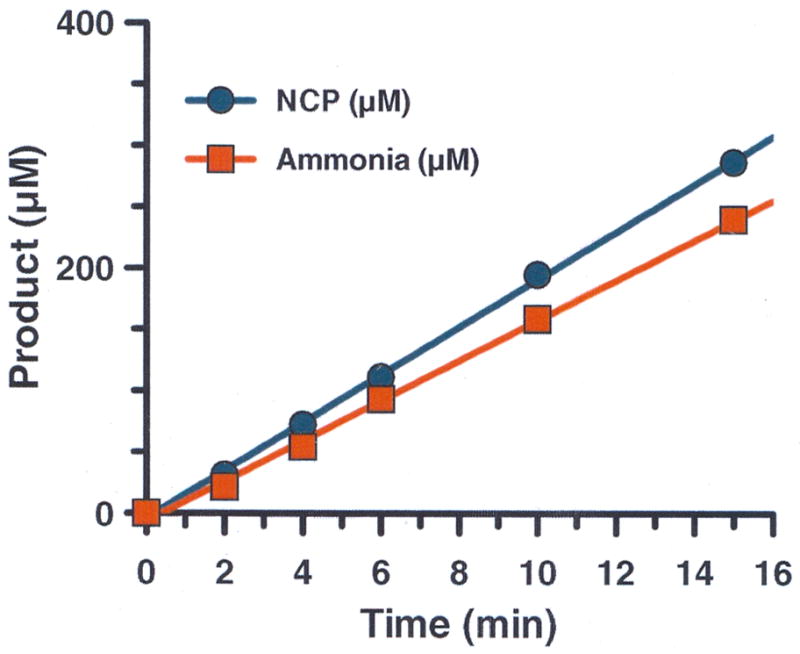
The reaction catalyzed by HpAgD leads to the production of NCP and ammonia in equimolar amounts.
3.4. Metal inhibition studies
Our inability to purify active HpAgD via IMAC prompted us to investigate the inhibitory effects of metal ions that are typically used for IMAC (i.e. Ni2+, Zn2+, and Co2+). IC50 values were therefore determined for the aforementioned metal ions. The results of these studies indicate that each of the metal ions inhibit HpAgD with varying degrees of potency – the IC50 values for Ni2+, Zn2+, and Co2+ are 100 ± 3.9, 30 ± 3.0, and 13 ± 5.5 μM, respectively. Interestingly, AgDs from A. thaliana and Chorella virus are readily purified by IMAC [42, 43], suggesting that the active site of HpAgD is uniquely susceptible to metal ion inhibiton. While the mechanism by which these metal ions inhibit HpAgD is not known, it is possible that these cations inactivate the enzyme by catalyzing the oxidation of the active site cysteine or, alternatively, inhibit HpAgD activity by coordinating with the active site cysteine and histidine. There is some precedence for the latter possibility as Stone et al have shown that zinc coordinates with the corresponding residues in P. aueruginosa DDAH [45]. Note that exhaustive treatments with EDTA could not reactivate the enzyme (data not shown).
3.5. Structure and mutagenesis of HpAgD
To aid our understanding of HpAgD catalysis and substrate recognition, the structure of the enzyme was determined to 2.1 Å resolution. The overall structure of HpAgD consists of five repeated ββαβ subdomains that are arranged in space to form a pseudo-five-fold symmetric structure that has the appearance of an α/β propeller. The active site is at the ‘boss’, or hub, of the propeller in a deep pocket that contains key active site residues (Fig. 4A).
Fig. 4.


Structure of HpAgD. (A) The overall structure of HpAgD. The catalytic side chains, i.e. Asp84, His202, Asp204, and Cys324, are shown. (B) Superposition of the structures of HpAgD (in blue), PAD4 (in green) and EfAgD (in yellow). Calcium ions, represented by black spheres, and sulfates are from the PAD4 structure. The position of the active site is marked by BAA (in red).
The structure of His-tagged selenomethionine-containing HpAgD has previously been determined to 2.5 Å resolution (PDB ID: 2CMU). Although this enzyme was purified by IMAC, and presumably represents a catalytically inactive form of the enzyme, the overall structure of the enzyme is very similar to that reported here. The rmsd of Cα positions between the two structures is 0.26 Å; the deviations over 1 Å are only for loop 262–266 with the largest deviation being 1.3 Å. We have recalculated electron density maps for the unpublished structure (PDB ID: 2CMU), using structure factors deposited in the PDB. Although extra electron density adjacent to the active site Cys is apparent in omit maps comparing our structure to the 2CMU structure, the maps are inconclusive. A water molecule placed next to the active site cysteine is at a distance of 2.5 Å from the β-sulfur atom of the active site Cys. This distance is too short for a non-bonded oxygen-sulfur interaction. As a Ni-column was used for the purification of this enzyme, it is possible that the extra electron density corresponds to a nickel ion coordinated to the active site cysteine, however, the environment of the sulfur ligand does not appear to be consistent with this hypothesis. The surrounding residues are Asp199 at 3.14 Å and His204 at 3.17 Å from the ligand. These distances are significantly greater than the expected distances for oxygen and nitrogen coordinating to nickel ion (~2.2A), but correspond very well to interactions with a water molecule. Additionally, the electron density peak for the sulfur of cysteine is higher than the peak for the hypothetical nickel ion. These findings suggest that there may be at most partial occupancy for nickel. The observed distance is not consistent with the alternative hypothesis of sulfur oxidation. However a combination of these two hypotheses with partial occupancy would explain the observed lack of activity.
The structure of the enzyme is also quite similar to that observed for other members of the guanidinium modifying family of enzymes, including both PAD4 and the AgD from E. faecalis (EfAgD). No major differences are observed in the superpositions of HpAgD with PAD4 and EfAgD (Fig. 4B) with an overall rmsd of 2.66 Å and 1.59 Å respectively. The large displacement value for the difference between HpAgD and PAD4 is due to the movement of flexible loops.
To identify residues that are important for catalysis, a structure based alignment of PAD4 and HpAgD was generated using the Matchmaker function in UCSF Chimera [46]. The results of these computational studies showed that Asp84, His202, Asp204, and Cys324 are analogous to the key catalytic residues in PAD4 (i.e. Asp350, His471, Asp473, and Cys645) [47]. A superposition of these residues in HpAgD and PAD4 (rmsd = 0.145 Å), using Lsqkab [48] in CCP4 [39], demonstrates that they are positioned appropriately to catalyze agmatine deimination (Fig. 5A). One major difference in these two active sites is the presence of a well positioned, and highly ordered, water molecule that is roughly equidistant between His202 and Cys324 and occupies the space otherwise occupied by the guanidinium carbon of the substrate. Although the existence of this water molecule could be an artifact of crystallization at pH 5.6, its presence suggests the potential formation of a water mediated shared ion pair that could facilitate proton transfer from the Cys324 thiol to the imidazole moiety of His202 (Fig. 5A); thereby generating the catalytically competent protonation states of these two residues. The presence of a water mediated shared ion pair would also be expected to prevent non-specific oxidation of the active site cysteine. While PAD4 is also thought to possess a shared ion pair [26], additional studies are necessary to validate a role for this water molecule in HpAgD catalysis, and determine whether proton transfer occurs prior to or concomitant with substrate binding.
Fig. 5.



Superpositions of HpAgD and PAD4. (A) The active site cleft of HpAgD. Residues that line the entrance to the active site are highlighted. (B) Superposition of the active site residues of HpAgD (in atom color) and PAD4 (in blue) highlighting the residues that are involved in substrate recognition. A water molecule from HpAgD is shown in red. BAA, from the PAD4 structure, is shown in blue with functional moieties in atom color. (C) Entrance to the active site with HpAgD represented by its molecular surface (gray). The position of BAA, shown using stick representation, was modeled by superpositioning the PAD4 complex.
Based on the above described structural similarity, it is likely that HpAgD and PAD4 use analogous catalytic mechanisms. Briefly, nucleophilic attack by the active site thiolate (Cys324 in HpAgD) on the guanidinium carbon of agmatine results in the formation of a tetrahedral intermediate. Protonation of the departing amine and collapse of the intermediate results in the formation of a stable thiouronium adduct. This thioronium intermediate can then be hydrolyzed to form NCP and regenerate the enzyme (Fig. 2).
In order to verify that Cys324 is critical for catalysis, this residue was changed to an alanine by site directed mutagenesis. This mutant protein was expressed, isolated, and purified in a manner identical to that of the wild-type enzyme. Activity assays of this mutant enzyme indicated only very limited activity (kcat/Km ≤ 19.2 min−1M−1; Table 3). This level of activity represents a ≥ 8,000-fold reduction in HpAgD activity over the wild-type enzyme – a result that is consistent with both the putative role of this residue in nucleophilic catalysis and with results obtained for other guanidinium modifying enzymes (e.g. PAD4 and ADI) [27, 49]. However, it is interesting to note that the mutation of the corresponding Cys in A. thaliana AgD decreases activity by 6.7-fold [42]; this discrepancy is most likely an artifact of the assay employed in those studies.
3.6. Substrate recognition
The overwhelming preference of HpAgD for agmatine can be explained, at least in part, by comparing the structures of PAD4 and HpAgD. The active site cleft of HpAgD is lined with several residues, including: Trp106, Lys109, Tyr110, Thr199, Asp198, Glu263, and Gln318 (Fig. 5). The presence of these residues results in an active site cleft that is considerably narrower than the corresponding region in PAD4 (Fig. 5), likely preventing the binding of peptide or protein-based substrates. Of these residues, Trp106 appears to play a major role in excluding arginine containing peptide substrates, as the 1.5 Å distance between the carboxamide moiety of BAA and Trp106, in the modeled complex, would preclude the binding of such substrates via steric hindrance (Fig. 5). No effect on agmatine binding would be expected because this substrate lacks the carboxamide group. Note that the side chain of Trp106 cannot be repositioned to an alternate rotamer without creating intramolecular clashes. A similar steric hindrance between Trp106 in HpAgD and the carboxylate of arginine is observed in a superimposition of HpAgD and the PaADI•arginine complex (not shown), thereby providing a mechanism to exclude arginine from the binding pocket of HpAgD.
It is also noteworthy that Trp106 occupies the space normally occupied by an arginine residue that corresponds to Arg374 in PAD4 and Arg243 in P. aeruginosa Arginine Deiminase (PaADI). The absence of this residue is significant because, in PAD4, it forms favorable interactions with the backbone carbonyls immediately surrounding the site of deimination. Similarly, in PaADI, this arginine interacts with the α-carboxyl group of free arginine. The loss of these interactions, combined with the narrower active site, would therefore explain, in part, why arginine and peptidyl-arginine are poor HpAgD substrates.
The presence of Asp198 in HpAgD also likely contributes to the exclusion of both arginine and peptidyl-arginine based substrates. This is the case because overlays of unliganded HpAgD and the PAD4 complex show that the Asp198 side chain is 2.4 Å from the carboxamide group of BAA in the PAD4•BAA complex. A similar arrangement is observed in a superimposition of HpAgD and the PaADI•arginine complex (not shown). Thus, electrostatic repulsions and/or sterics would combine to exclude arginine and peptidyl-arginine containing substrates from the HpAgD active site. Although readily accessible alternate rotamers remove the steric hindrance, it is likely that Asp198 plays an additional role in selecting agmatine over arginine or arginine-containing peptide substrates because, in the overlays, this residue is 3.2 Å from the α-amino group of the arginine. Thus, in addition to excluding arginine, this residue may form favorable electrostatic interactions that promote agmatine binding. Consistent with this notion is the fact that a glutamate residue, i.e. Glu214, occupies a nearly identical position in E. faecalis AgD [44]. To verify this possibility experimentally, an Asp198Ala mutant was constructed, expressed, and purified to homogeneity. The results of kinetic analyses (Table 3) show an 86-fold decrease in kcat/Km, a 17-fold decrease in kcat, and a 5-fold increase in Km, consistent with this residue playing an important role in substrate binding. Although this mutation might be expected to ‘open up’ the substrate binding cleft and remove a residue that would form unfavorable interactions with both arginine and peptidyl-arginine containing substrates, agmatine remained the preferred substrate (Table 2).
3.7. Design and synthesis of AgD inactivators
After confirming that HpAgD is in fact a bona fide AgD, we set out to develop inhibitors for this enzyme. Because HpAgD possesses structural and mechanistic features that are similar to PAD4, and given our previously successful efforts to inhibit PAD4 with haloacetamidine-containing peptidyl-arginine analogs [35, 36], we designed and synthesized haloacetamidine-containing agmatine analogs. The structures of these compounds (Fig. 6) replace the guanidinium moiety of agmatine with either a fluoro- or chloroacetamidine moiety. These compounds, denoted N-(4-aminobutyl)-2-fluoro-ethanimidamide (ABFA) and N-(4-aminobutyl)-2-chloro-ethanimidamide (ABCA), were synthesized over two synthetic steps (reaction of mono-Boc protected putrescine with a 2-haloacetimidate followed by deprotection to reveal the terminal amine) and isolated in reasonable yields. Similarly to our PAD4 inactivators F- and Cl-amidine [35, 36], ABFA and ABCA show excellent stability in aqueous buffer (not shown).
Fig. 6.

(A) Structures of ABXA and X-amidine. (B) Proposed mechanisms of inactivation. Both mechanisms lead to the inactivation of HpAgD through a thioether linkage.
3.8. Inactivation of HpAgD with ABFA and ABCA
To begin to characterize the inhibitory properties of ABFA and ABCA, IC50 values were determined. For these reactions, inactivators were pre-incubated with HpAgD for 15 min prior to the addition of agmatine (0.5 mM) to initiate the deimination reaction. The IC50 values obtained from this experiment (ABFA: 6.8 ± 0.26 μM; ABCA: 0.87 ± 0.03 μM; Table 4) indicate that ABCA is a significantly more potent inactivator of HpAgD than is ABFA. This result is not unexpected given the greater leaving group potential of chloride. To examine the selectivity of these compounds, their ability to inactivate PAD4 was determined. The results of these experiments indicate that ABFA and ABCA are very poor inhibitors of PAD4 – essentially no inhibition was observed at concentrations as high as 1 mM. Our previously described PAD4 inhibitors, denoted F-amidine and Cl-amidine (Fig. 6), were also tested to determine if either of these compounds inhibit HpAgD. The results of these experiments demonstrate that these latter compounds are quite poor HpAgD inhibitors – concentrations as high as 500 μM did not afford HpAgD inhibition. The fact that F-amidine and Cl-amidine are poor HpAgD inhibitors is consistent with the fact that this enzyme shows an exquisite preference for agmatine as a substrate. Additionally, the fact that significant selectivity can be achieved, between the PADs and the Agmatine Deiminases, by simply changing the context of the reactive moiety shows the versatility of the haloacetamidine warhead, and bodes well for the future use of this moiety in generating chemical probes for the diverse members of the guanidinium modifying family of enzymes. Consistent with this notion is the fact that fluorescently-tagged versions of F- and Cl-amidine show very little non-specific modification of proteins in whole proteomes [50].
Table 4.
IC50 Values
| Inhibitor | HpAgDa(μM) | PAD4b (μM) |
|---|---|---|
| ABCA | 0.87 ± 0.03 | > 1000 |
| ABFA | 6.8 ± 0.26 | > 1000 |
| Cl-Amidine | >500 | 5.9 ± 0.3 |
| F-Amidine | > 500 | 21.6 ± 0.3 |
0.5 mM Agmatine,
10 mM BAEE
To confirm that ABFA and ABCA irreversibly inactivate HpAgD, pre-formed HpAgD•inactivator complexes were dialyzed for 20 h, after which point, the residual activity was measured. As depicted in Fig. 7, enzyme activity was not recovered, which is consistent with irreversible enzyme inactivation. Substrate protection experiments were subsequently conducted to demonstrate that enzyme inactivation was due to the modification of an active site residue. For these experiments, product formation was measured as a function of time in both the absence and in the presence of both ABFA and ABCA at two different concentrations of agmatine (i.e., 0.5 and 1.0 mM). In the absence of added inhibitor, the progress curves are linear, as expected. However, in the presence of ABFA and ABCA, the progress curves are nonlinear, a result that is consistent with the irreversible inactivation of HpAgD (Fig. 7). Notably, at higher concentrations of substrate, the rate of enzyme inactivation is impeded, demonstrating that substrate can protect against enzyme inactivation. This finding is consistent with the modification of an active site residue.
Fig. 7.

(A) ABFA and ABCA irreversibly inhibit HpAgD. Both inhibitors were reacted with HpAgD for 1h. The ABFA/ABCA:HpAgD complex was dialyzed at which point the residual activity was measured. (B and C) Substrate protection experiment with ABFA (B) and ABCA (C). The amount of product was measured as a function of time with varying amounts of inhibitor and substrate.
To confirm that inactivation was due to the modification of the active site cysteine (i.e., Cys324), MS and MS/MS analyses were performed. For these experiments, pre-formed pAgD•inactivator complexes were digested with trypsin and the resultant peptides characterized by MALDI-MS. Peptide mass fingerprints were then constructed for both HpAgD treated with ABFA and ABCA (Fig. S2). Sequence coverage of 45% and 33% was obtained for the ABFA and ABCA treated HpAgD, respectively. This coverage compared quite favorably to untreated control samples (sequence coverage = 34%). Notably, a tryptic peptide encompassing Cys324, the active site cysteine, was absent from both the ABFA and ABCA treated samples. However, a new signal consistent with the mass of the covalent adduct was apparent in the MALDI-MS spectra obtained from samples treated with either of these compounds. The observed m/z values obtained from digests of the ABCA and ABFA treated enzyme, 1643.816 and 1643.860, respectively, are quite close to the calculated value, i.e. 1643.815. Fragmentation of this ion, and analysis by MALDI-MS/MS, resulted in the production of a series of y and b ions that are consistent with the modification of Cys324 (Fig. 8 and Fig. S3). In total, these results confirm that ABFA and ABCA inactivate HpAgD by modifying Cys324.
Fig. 8.

Site of ABCA modification determined by MALDI-TOF MS and MS/MS. (A) Unmodified HpAgD after tryptic digest. (B) ABCA modified HpAgD after tryptic digest. (C) Sequence of the predicted site of modification. The b and y ions identified by MALDI-TOF MS/MS are shown in red.
The kinetics of HpAgD inactivation by ABFA and ABCA were subsequently determined. In these experiments, product formation, in the presence of varying concentrations of ABCA and ABFA, was measured as a function of time (Fig. 9). The pseudo first order rate constants of inactivation (i.e., kobs), obtained from the non-linear progress curves, after correction for the substrate concentration, were plotted against inactivator concentration. The resultant plots, which are hyperbolic (Fig. 9C and 9D), are consistent with a two step mechanism of inactivation where the inactivator first binds to the enzyme and then reacts with Cys324 to afford the formation of the covalent adduct. The kinact, KI, and kinact/KI values obtained from analysis of the data in Fig. 9C and 9D are 530 ± 150 μM, 2.1 ± 0.28 min−1, 4000 min−1 M−1 for ABFA and 1200 ± 234 μM, 15 ± 1.9 min−1, and 12,500 min−1 M−1 for ABCA. Interestingly, the kinact/KI values for ABFA and ABCA (4,000 min−1 M−1 and 12,500 min−1 M−1, respectively) are similar to those obtained for the inactivation of PAD4 by F- and Cl-amidine (3,000 min−1 M−1 and 13,000 min−1 M−1, respectively) despite the fact that their kcat values differ by greater than 50-fold. This result is at least partially consistent with the fact that these enzymes have very similar kcat/Km values. Based on the fact that the side chain portion of F- and Cl-amidine imparts a greater than 200-fold enhancement in kinact/KI ([26, 51] and unpublished results), the amino-alkyl portion of ABFA and ABCA likely enhances inhibitor binding to a similar extent. Further elaboration of the alkyl chain should lead to the development of inhibitors with improved potency.
Fig. 9.

Inactivation kinetics of ABFA and ABCA. The amount of NCP produced with varying amounts of ABCA (A) and ABFA (B) was measured. The pseudo first order rate constants (kobs) for inactivation was plotted versus the concentration of ABCA (C) and ABFA (D) were plotted.
4. Conclusions
The near constant emergence of antibiotic resistance mechanisms to virtually all known antibiotics [52] highlights the priority that must be placed on the development of new and novel antibiotics. Advances in genomics have, and continue to, unveil cellular processes that are unique to bacteria and such pathways represent novel targets for antibiotic development because they limit potential side effects. Given that the deimination of agmatine to form N-carbamoyl putrescine is one such pathway, HpAgD represents a potentially novel target for antibiotic development. As such, we set out to characterize both the substrate specificity and the catalytic mechanism of HpAgD, to determine its structure, and to develop HpAgD inhibitors. The mechanistic and structural studies described herein demonstrate that HpAgD is a bona fide agmatine deiminase that catalyzes the hydrolytic conversion of agmatine to NCP and ammonia, in a fashion that is analogous to mechanism employed by PAD4.
Although this enzyme is currently annotated in structural and genomic databases as a peptidyl arginine deiminase, our results definitively establish that this enzyme is, in fact, an AgD. This study highlights how important it is to obtain hard experimental data to validate or revise (as in this case) automatic annotations based on amino acid sequence alone. Given that dysregulated PAD activity is strongly associated with the onset and progression of Rheumatoid Arthritis [53, 54], the demonstration that HpAgD is not a PAD is significant because it rules out this enzyme as a possible trigger for this disease – a long held, but unproven, hypothesis in the rheumatoid arthritis community is that this disease is caused by an infectious agent of unknown etiology [55].
Given the mechanistic similarities between these two enzymes, and given the difference in substrate scope, we set out to design HpAgD inactivators using the same methodology that we have successfully employed to generate PAD4 inactivators, i.e. the use of a haloacetamidine reactive moiety in place of the guanidinium moiety of a peptidyl-arginine residue [35, 36, 50]. Based on this methodology, the replacement of the guanidinium moiety of agmatine with either a fluoro- or chloroacetamidine moiety led to the development of HpAgD inhibitors that are significantly more potent than current AgD inhibitors, e.g. arcaine (1,4-diguanidinobutane) [44]. The fact that the chloro-containing compound (i.e., ABCA) displays greater potency than ABFA is most easily attributed to the higher propensity of chloride to be displaced. These inhibitors represent chemical probes that can be used to discern the physiological function(s) of HpAgD, and possibly represent lead compounds for the treatment of the disease pathologies associated with H. pylori and other pathogenic bacteria that contain an AgD. Future work will include efforts to increase potency, and explore the in vivo efficacy of these compounds.
Supplementary Material
Acknowledgments
The original HpAgD clone was generously provided by SGX Pharmaceuticals, Inc.. We thank members of the Thompson laboratory for critical reading of the manuscript.
Footnotes
Coordinates and structure factors have been deposited with the PDB (accession code 3HVM).
The abbreviations used are: PPI, proton pump inhibitor; AgD, agmatine deiminase; NCP, N-carbamoyl putrescine; PAD, protein arginine deiminase; Cit, citrulline; ABFA, N-(4-aminobutyl)-2-fluoro-ethanimidamide; ABCA, N-(4-aminobutyl)-2-chloro-ethanimidamide; BAEE, benzoyl L-arginine ethyl ester; BAA, benzoyl L-arginine amide; BAME, N-α-Benzoyl-L-arginine methyl ester; DTT, dithiothreitol; GST, glutathione S-transferase; TCEP, Tris(2-carboxyethyl)phosphine hydrochloride; HEPES, N-(2-hydroxyethyl)piperazine-N’-(2-ethanesulfonic acid); Orn, Ornithine.
This work was supported in part by the University Of South Carolina Research Foundation (P.R.T) and NIH grant GM079357 to PRT.
Supplementary Table S1 and Figures S1, S2, and S3 are available. A complete description of the HpAgD purification protocol is provided in the supplementary material.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lembo A, Caradonna L, Magrone T, Mastronardi ML, Caccavo D, Jirillo E, Amati L. Curr Drug Targets Immune Endocr Metabol Disord. 2001;1(3):199–208. doi: 10.2174/1568008013341190. [DOI] [PubMed] [Google Scholar]
- 2.Atherton JC. Annu Rev Pathol. 2006;1:63–96. doi: 10.1146/annurev.pathol.1.110304.100125. [DOI] [PubMed] [Google Scholar]
- 3.Merrell DS, Falkow S. Nature. 2004;430(6996):250–256. doi: 10.1038/nature02760. [DOI] [PubMed] [Google Scholar]
- 4.Vakil N, Megraud F. Gastroenterology. 2007;133(3):985–1001. doi: 10.1053/j.gastro.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 5.Meyer JM, Silliman NP, Wang W, Siepman NY, Sugg JE, Morris D, Zhang J, Bhattacharyya H, King EC, Hopkins RJ. Ann Intern Med. 2002;136(1):13–24. doi: 10.7326/0003-4819-136-1-200201010-00008. [DOI] [PubMed] [Google Scholar]
- 6.Fischbach LA, van Zanten S, Dickason J. Aliment Pharmacol Ther. 2004;20(10):1071–1082. doi: 10.1111/j.1365-2036.2004.02248.x. [DOI] [PubMed] [Google Scholar]
- 7.Telford JL, Covacci A, Rappuoli R, Chiara P. Curr Opin Immunol. 1997;9(4):498–503. doi: 10.1016/s0952-7915(97)80101-x. [DOI] [PubMed] [Google Scholar]
- 8.Coleman CS, Hu G, Pegg AE. Biochem J. 2004;379(Pt 3):849–855. doi: 10.1042/BJ20040035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li G, Regunathan S, Barrow CJ, Eshraghi J, Cooper R, Reis DJ. Science. 1994;263(5149):966–969. doi: 10.1126/science.7906055. [DOI] [PubMed] [Google Scholar]
- 10.Regunathan S, Reis DJ. Annu Rev Pharmacol Toxicol. 1996;36:511–544. doi: 10.1146/annurev.pa.36.040196.002455. [DOI] [PubMed] [Google Scholar]
- 11.Raasch W, Regunathan S, Li G, Reis DJ. Life Sci. 1995;56(26):2319–2330. doi: 10.1016/0024-3205(95)00226-v. [DOI] [PubMed] [Google Scholar]
- 12.Llacer JL, Polo LM, Tavarez S, Alarcon B, Hilario R, Rubio V. J Bacteriol. 2007;189(4):1254–1265. doi: 10.1128/JB.01216-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shirai H, Mokrab Y, Mizuguchi K. Proteins. 2006;64(4):1010–1023. doi: 10.1002/prot.20863. [DOI] [PubMed] [Google Scholar]
- 14.Griswold AR, Jameson-Lee M, Burne RA. J Bacteriol. 2006;188(3):834–841. doi: 10.1128/JB.188.3.834-841.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Griswold AR, Nascimento MM, Burne RA. Oral Microbiol Immunol. 2009;24(1):79–82. doi: 10.1111/j.1399-302X.2008.00459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Satriano J. Amino Acids. 2004;26(4):321–329. doi: 10.1007/s00726-004-0078-4. [DOI] [PubMed] [Google Scholar]
- 17.Joshi MS, Ferguson TB, Jr, Johnson FK, Johnson RA, Parthasarathy S, Lancaster JR., Jr Proc Natl Acad Sci U S A. 2007;104(24):9982–9987. doi: 10.1073/pnas.0506824104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joshi MS, Ferguson TB, Jr, Johnson FK, Johnson RA, Parthasarathy S, Lancaster JR., Jr Proc Natl Acad Sci U S A. 2007;104(24):9982–9987. doi: 10.1073/pnas.0506824104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su CH, Liu IM, Chung HH, Cheng JT. Neurosci Lett. 2009;457(3):125–128. doi: 10.1016/j.neulet.2009.03.093. [DOI] [PubMed] [Google Scholar]
- 20.Wolf C, Bruss M, Hanisch B, Gothert M, von Kugelgen I, Molderings GJ. Mol Pharmacol. 2007;71(1):276–283. doi: 10.1124/mol.106.028449. [DOI] [PubMed] [Google Scholar]
- 21.Bussiere FI, Chaturvedi R, Cheng Y, Gobert AP, Asim M, Blumberg DR, Xu H, Kim PY, Hacker A, Casero RA, Jr, Wilson KT. J Biol Chem. 2005;280(4):2409–2412. doi: 10.1074/jbc.C400498200. [DOI] [PubMed] [Google Scholar]
- 22.Molderings GJ, Burian M, Homann J, Nilius M, Gothert M. Dig Dis Sci. 1999;44(12):2397–2404. doi: 10.1023/a:1026662316750. [DOI] [PubMed] [Google Scholar]
- 23.Molderings GJ, Kribben B, Heinen A, Schroder D, Bruss M, Gothert M. Cancer. 2004;101(4):858–868. doi: 10.1002/cncr.20407. [DOI] [PubMed] [Google Scholar]
- 24.Gobert AP, McGee DJ, Akhtar M, Mendz GL, Newton JC, Cheng Y, Mobley HL, Wilson KT. Proc Natl Acad Sci U S A. 2001;98(24):13844–13849. doi: 10.1073/pnas.241443798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shirai H, Blundell TL, Mizuguchi K. Trends Biochem Sci. 2001;26(8):465–468. doi: 10.1016/s0968-0004(01)01906-5. [DOI] [PubMed] [Google Scholar]
- 26.Knuckley B, Bhatia M, Thompson PR. Biochemistry. 2007;46(22):6578–6587. doi: 10.1021/bi700095s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knuckley B, Bhatia M, Thompson PR. Biochemistry. 2007;46(22):6578–6587. doi: 10.1021/bi700095s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakada Y, Itoh Y. Microbiology. 2003;149(Pt 3):707–714. doi: 10.1099/mic.0.26009-0. [DOI] [PubMed] [Google Scholar]
- 29.Thompson PR, Kurooka H, Nakatani Y, Cole PA. J Biol Chem. 2001;276(36):33721–33729. doi: 10.1074/jbc.M104736200. [DOI] [PubMed] [Google Scholar]
- 30.Kearney PL, Bhatia M, Jones NG, Yuan L, Glascock MC, Catchings KL, Yamada M, Thompson PR. Biochemistry. 2005;44(31):10570–10582. doi: 10.1021/bi050292m. [DOI] [PubMed] [Google Scholar]
- 31.Knipp M, Vasak M. Anal Biochem. 2000;286(2):257–264. doi: 10.1006/abio.2000.4805. [DOI] [PubMed] [Google Scholar]
- 32.Leatherbarrow RJ. Grafit Ver 5.0. Erathicus Software; Staines, UK: 2004. [Google Scholar]
- 33.Sugawara K, Oyama F. J Biochem (Tokyo) 1981;89(3):771–774. doi: 10.1093/oxfordjournals.jbchem.a133257. [DOI] [PubMed] [Google Scholar]
- 34.Kearney PL, Bhatia M, Jones NG, Luo Y, Glascock MC, Catchings KL, Yamada M, Thompson PR. Biochemistry. 2005;44(31):10570–10582. doi: 10.1021/bi050292m. [DOI] [PubMed] [Google Scholar]
- 35.Luo Y, Arita K, Bhatia M, Knuckley B, Lee YH, Stallcup MR, Sato M, Thompson PR. Biochemistry. 2006;45(39):11727–11736. doi: 10.1021/bi061180d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo Y, Knuckley B, Lee YH, Stallcup MR, Thompson PR. J Am Chem Soc. 2006;128(4):1092–1093. doi: 10.1021/ja0576233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 38.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Acta Cryst. 2005;D61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 39.Collaborative computational project. Acta Cryst. 1994;D50:760–763. [Google Scholar]
- 40.Roussel A, Cambillau C. 1991 [Google Scholar]
- 41.Murshudov GN, Vagin AA, Dodson EJ. Acta Cryst Sect D: Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 42.Janowitz T, Kneifel H, Piotrowski M. FEBS Lett. 2003;544(1–3):258–261. doi: 10.1016/s0014-5793(03)00515-5. [DOI] [PubMed] [Google Scholar]
- 43.Baumann S, Sander A, Gurnon JR, Yanai-Balser GM, Van Etten JL, Piotrowski M. Virology. 2007;360(1):209–217. doi: 10.1016/j.virol.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Llacer JL, Polo LM, Tavarez S, Alarcon B, Hilario R, Rubio V. J Bacteriol. 2007;189(4):1254–1265. doi: 10.1128/JB.01216-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stone EM, Costello AL, Tierney DL, Fast W. Biochemistry. 2006;45(17):5618–5630. doi: 10.1021/bi052595m. [DOI] [PubMed] [Google Scholar]
- 46.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 47.Arita K, Hashimoto H, Shimizu T, Nakashima K, Yamada M, Sato M. Nat Struct Mol Biol. 2004;11(8):777–783. doi: 10.1038/nsmb799. [DOI] [PubMed] [Google Scholar]
- 48.Kabsch W. Acta Cryst. 1976;A32:922–923. [Google Scholar]
- 49.Lu X, Li L, Wu R, Feng X, Li Z, Yang H, Wang C, Guo H, Galkin A, Herzberg O, Mariano PS, Martin BM, Dunaway-Mariano D. Biochemistry. 2006;45(4):1162–1172. doi: 10.1021/bi051591e. [DOI] [PubMed] [Google Scholar]
- 50.Luo Y, Knuckley B, Bhatia M, Pellechia PJ, Thompson PR. J Am Chem Soc. 2006;128(45):14468–14469. doi: 10.1021/ja0656907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stone EM, Schaller TH, Bianchi H, Person MD, Fast W. Biochemistry. 2005;44(42):13744–13752. doi: 10.1021/bi051341y. [DOI] [PubMed] [Google Scholar]
- 52.D’Costa VM, McGrann KM, Hughes DW, Wright GD. Science. 2006;311(5759):374–377. doi: 10.1126/science.1120800. [DOI] [PubMed] [Google Scholar]
- 53.Vossenaar ER, Van Venrooij WJ. Arthritis Res Ther. 2004;6(3):107–111. doi: 10.1186/ar1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vossenaar ER, Zendman AJ, van Venrooij WJ, Pruijn GJ. Bioessays. 2003;25(11):1106–1118. doi: 10.1002/bies.10357. [DOI] [PubMed] [Google Scholar]
- 55.Liao F, Li Z, Wang Y, Shi B, Gong Z, Cheng X. Med Hypotheses. 2009;72(6):732–735. doi: 10.1016/j.mehy.2008.12.040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.